

Abstract
Integral membrane proteins isolated from cellular environment often lose activity and native conformation required for functional analyses and structural studies. Even in their native state, they lack sufficient surfaces to form crystal contacts. Furthermore, most of them are too small for cryo-EM detection and too big for solution NMR. To overcome these difficulties, we recently developed a strategy to stabilize the folded state of membrane proteins by restraining their two termini with a self-assembling protein coupler. The termini-restrained membrane proteins from distinct functional families retain their activities and show increased stability and yield. This strategy enables their structure determination at near-atomic resolution by facilitating the entire pipeline from crystallization, crystal identification, diffraction enhancement, phase determination, to electron density improvement. Furthermore, stabilization of membrane proteins allows their biochemical and biophysical characterizations. Here, we present the protocol of membrane protein engineering (1 week), quality assessment (1-2 weeks), protein production (1 to 4 weeks), crystallization (1-2 weeks), diffraction improvement (1-3 months), and crystallographic data analysis (1 week). This protocol is intended not only for structural biologists, but also for biochemists, biophysicists and pharmaceutical scientists whose research focuses on membrane proteins.
EDITORIAL SUMMARY
This protocol describes a strategy to stabilize integral membrane proteins by fusing their two termini to a self-assembling coupler protein, which also enables crystallization and facilitates every step of obtaining high-resolution structures.
Introduction
Membrane proteins constitute ~20-30% of the proteomes of living organisms and more than half of drug targets1–4. However, obtaining high-resolution structures of membrane proteins and maintaining in vitro their function for biochemical and biophysical studies remain prohibitively challenging. The activities and native conformations of these proteins are frequently compromised during overexpression in cells and after isolation from membranes5,6. Consequently, they are either deemed unsuitable for the intended studies or require extensive trial-and-error attempts to improve their behavior in vitro7–10. For structure determination, most membrane proteins are too small for cryo-electron microscopy (cryo-EM) and too big for solution NMR11. Even in successful cases, many cryo-EM and NMR structures do not reach the near-atomic resolution that crystal structures may afford12. Crystallographic studies are also difficult to perform with membrane proteins because their detergent-exposed regions hardly present enough crystal contacting interactions. Additionally, solving the crystallographic phase problem often requires tremendous efforts.
To overcome these challenges, we recently developed a termini-restraining approach that enables structural and functional studies of unstable membrane proteins13–17 (Fig. 1a). We stabilize these proteins by tethering their two termini to a coupler formed of two associable protein entities, taking advantage of the general folding topology of membrane proteins, most of which contain an even number of transmembrane helices (TMs)18,19. The N- and C- termini of these proteins are therefore located at the same side of the membrane, allowing the two parts of the fused coupler to assemble. Because the two termini are often flexible loops, the coupler is loosely tethered to the membrane protein, permitting conformational changes that are required for protein functions. On the other hand, the tethering provides a mild restraint that largely precludes drastic TM motions occurring during protein unfolding, thus favoring the folded state of membrane proteins. During protein overexpression and/or heterologous expression in cells, we found that the restraining significantly lowers the fraction of misfolded or aggregated protein13. Furthermore, the restrained proteins are less prone to denature and aggregate during isolation from native membranes and during subsequent purification in detergent solutions. In the end, membrane proteins with higher thermostability and larger yield can be obtained, facilitating subsequent structural and functional analyses.
Fig. 1. |. Overview of the concept and protocol of termini restraining.
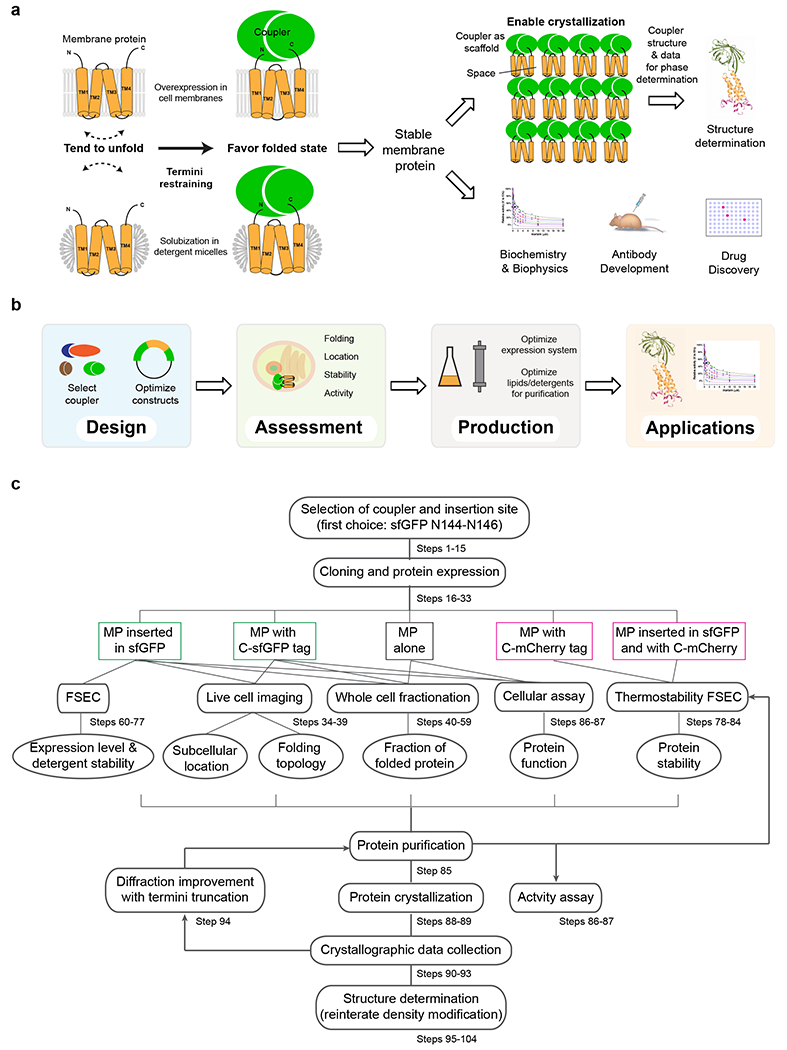
a, Membrane proteins (orange) are prone to unfold during overexpression in cells and after solubilization in detergents. The unfolding involves large motions of TMs (dashed arrow). To restrict such motions and stabilize the folded state, a self-assembling coupler protein (green) is fused to the flexible N and C termini of the membrane protein (orange) to provide a mild restraint. For structure determination, the coupler serves as a crystallization scaffold that promotes regular crystal packing, and increases the diffraction mass by lowering the relative space between non-contacting membrane regions, which is occupied by solvent and lipid/detergent. Existing structure and diffraction data of the coupler are used for solving the phase problem. Furthermore, stabilized membrane proteins are used in many biochemical and biophysical studies and pharmaceutical applications. b, The protocol consists of following stages: design and generation of restrained constructs, assessment of protein quality, large-scale expression and purification, and applications. c, Detailed workflow of the termini restraining protocol. MP: membrane protein.
Introducing the coupler enables the crystallization of membrane proteins and facilitates the entire pipeline of their structure determination. The coupler provides a large and stable hydrophilic surface to form crystal contacts, overcoming a major difficulty in membrane protein crystallization; without this additional surface, membrane proteins are mostly buried in the detergent micelles and often present small exposed regions or large flexible loops that are unsuitable for making crystal contacts. Furthermore, the sizable coupler provides a large portion of the diffraction mass and lowers the relative size of the space created by the non-contacting membrane portion, resulting in tightly packed, well diffracting crystals. The coupler can be a fluorescent or colored protein (e.g., flavoprotein, chromoproteins of GFP family) that facilitates detection of initial crystallization hits, which helps to avoid months of (sometimes futile) effort to confirm crystallization of the target membrane protein. The coupler is linked to the membrane protein via the two flexible termini or additional peptide linkers, increasing the chance of interactions that allow regular crystal packing to form. On the other hand, shortening the termini or linkers restricts the number of possible orientations between the coupler and membrane protein, effectively changing the crystal packing pattern to improve the diffraction limit. During structure determination, the known structure of the coupler is used for the molecular replacement (MR) phasing, and the MR solution can be readily obtained owing to the relatively large size of the coupler. After density modification of the partial model phases, the density maps at the unknown membrane protein region are usually sufficient for building a nearly complete model. In difficult cases, the diffraction data and structure of the coupler (both usually available in Protein Data Bank) can be used for cross-crystal averaging20 with those of the coupler-fused membrane protein to significantly improve the overall phases and density map.
Development of the protocol
Maintaining the native structure and function of membrane proteins is paramount for successful in vitro studies. Traditionally, to accelerate the characterization of a membrane protein family, large screens of homologous proteins are performed to identify very few that are sufficiently stable and monodispersed in detergent solutions. If a particular protein is of interest (e.g., a human protein needs to be studied), its instability and flexibility can be modified in several ways. Systematic mutagenesis can be performed on the entire membrane protein to identify multiple mutations that synergistically enhance the protein’s thermostability8–10. Another approach is to search for conformation-specific antibodies that stabilize a flexible region of the membrane protein when bound. The antibody also serves as a scaffold for co-crystallization with the membrane protein21 or for cryo–EM analyses by increasing the overall size to facilitate particle recognition and alignment22. Scaffold proteins have been particularly useful to obtain structures of G-protein coupled receptors (GPCRs), for which an intracellular loop is replaced with a soluble protein to stabilize their conformation and aid their crystallization23,24. There are, however, limited reports of applying this method to obtain structures of membrane proteins from other families.
The termini restraining protocol was designed to be broadly applicable to membrane proteins from multiple families, requiring only that they contain an even number of TMs. With current molecular biology tools, fusing a coupler to the two termini of membrane protein can be readily accomplished, minimizing trial-and-error attempts and avoiding large screens. We have selected superfolder green fluorescence protein25,26 (sfGFP) as an effective coupler to stabilize and crystallize small membrane proteins (2 to 6 TM). The choice of coupler is however not limiting, as many soluble proteins are known to permit splitting27–29. Even for poorly characterized couplers, the majority that we have tested (Supplementary Table 1) not only tolerated the splitting but also allowed the proper folding of the inserted membrane protein.
This new approach has yielded 14 structures of membrane proteins from humans and other vertebrates, all of which had resisted traditional approaches13–15. Among these proteins, the structure of CD53 is the first one ever captured of the partner-interacting state for the entire tetraspanin family15. The structure of SPCS1, a major therapeutic target of Zika virus30, suggests the regulatory role of this signal peptidase subunit during the viral maturation process. The structures of oxidoreductases VKOR and VKORL elucidate almost their entire catalytic cycle and their mechanism of inhibition by commonly used anticoagulants14. Overall, various functional and conformational states have been captured for membrane proteins from distinct functional families, demonstrating the versatility of this new method. Furthermore, stabilized VKOR and VKORL enabled their drug inhibition kinetics to be properly quantified for the first time16,17.
Comparison with traditional approaches
Termini restraining can enable structure determination of membrane proteins that have been out of reach by cryo-EM, NMR and traditional crystallographic methods. Fusing a coupler protein is a novel way of introducing a scaffold for the crystal packing of various membrane proteins, applicable to those with different molecular shapes and surface charges13. Use of split sfGFP as the coupler allows initial crystal hits to be identified under a fluorescence microscope; traditionally, it takes few months to optimize the initial hits and grow large enough crystals to confirm the presence of the target protein by silver stain or x-ray diffraction. The crystallographic phase problem is solved by using an existing model and diffraction data of the coupler protein; there is no need for experimental phasing, which is a tedious and sometimes unsuccessful process.
Solution NMR approaches have been developed for studying only small transmembrane domains31, because the combined molecular mass of a membrane protein and detergent micelles or lipid discs often exceeds the fundamental size limitations (~ 100 kDa) of solution NMR; a previous paper in Nature Protocols32 estimated that membrane proteins larger than 7 TMs cannot be de novo determined solely from solution NMR data. On the other hand, cryo-EM studies of membrane proteins, although rapidly evolving, are currently striving towards a minimum size of ~ 100 kDa. Most membrane proteins, however, are below this size and therefore lack enough contrast for cryo-EM detection. If they are not constitutive subunits of a large and stable assembly, determining their structures requires tremendous efforts, such as identifying a suitable antibody to form a complex, to overcome the size limitation.
Use of a coupler as the crystallization scaffold circumvents the search for suitable monoclonal antibodies to provide crystal contacts or increase the size for cryo-EM detection. Structural studies also require the antibodies to be specific to a membrane protein conformation, instead of recognizing a sequence segment. Identifying such antibodies, however, is beyond the means of most research labs because it is highly fortuitous, time consuming and expensive.
Termini restraining is a universal protein engineering method. As a comparison, crystal structures of GPCRs are routinely obtained with the loop replacement method. This method, however, has proved difficult to apply to other membrane proteins, because without any structural information, which flexible loops to replace is unknown and the replacement may affect the membrane protein folding. Further, the replacement may be restricted by the size of the soluble protein, whose two ends also needs to roughly match the unknown distance between the two connecting TMs. Furthermore, inserting a stable protein to replace the intracellular loop in GPCRs provides only local stabilization between the two TMs joined by this loop. In contrast, termini restraining affords global stabilization of all TMs because the entire membrane protein is “inserted” into a highly stable coupler protein, enhancing the overall stability of the restrained constructs.
Termini restraining requires minimal construct screening, unlike conventional trial-and-error methods used in membrane protein studies, such as large screens of homologous proteins from different species and systematic mutagenesis of the entire protein. Termini restraining is a rational way of stabilizing the overall folding of membrane proteins and it increases the stability of membrane proteins much more than a single mutation does8–10,13. In addition, the membrane protein is more faithfully maintained in its native state without the multiple mutations.
Applications of the method
Termini restraining is a tool to improve the stability of membrane proteins for a broad range of studies. A major application is to obtain high-resolution structures of membrane proteins, which can be essential to mechanistic understandings of protein functions. Moreover, numerous biochemical and biophysical studies require the use of pure and stable membrane proteins. As an example, we recently showed that using stabilized membrane proteins in such assays ensure their proper enzymatic characterization, whereas using unstable proteins in non-native state has generated misleading data for decades16,17,33–36. The applications in biophysical studies include single molecule, electron paramagnetic resonance, surface plasmon resonance, isothermal titration calorimetry, fluorescence, mass spectrometry based footprinting, and small angle X-ray scattering. The stabilized membrane proteins also allow biophysical analyses under a wide range of experimental conditions, such as raised temperature and prolonged time. The stabilized membrane proteins can also be used for compound screening and drug development, or as antigens to generate antibodies for scientific uses.
The idea of termini restraining may inspire future developments in structural methods and protein engineering. For instance, this strategy may find applications in cryo-EM through restraining membrane proteins by a large scaffold and with a rigid linker to rapidly determine numerous novel structures. Moreover, the strategy can be expanded to stabilize soluble proteins or engineer novel high-order assemblies.
Limitations and adaptations
Caution should be exercised if the coupler is located proximally to the functional region of membrane protein. For instance, the coupler may sterically block protein-protein interactions. Likewise, membrane protein oligomerization may be affected by the presence of the coupler, and in such cases, use of a smaller coupler protein may be considered. Restraining by the coupler may also affect protein functions that require extremely large conformational changes. Nevertheless, the restraining may still allow individual conformational states to be captured.
Our previous studies have focused on relatively small membrane proteins with even number of TMs (2–6). Further research may demonstrate the application to membrane proteins with 8 or more TMs. The restraining is also potentially applicable to proteins with odd numbers of TMs by fusing one component of the coupler to an internal loop. Moreover, the coupler concept can be modified to use other self-assembling entities, such as protein oligomers, complexes, or intein-mediated polypeptide joining37, extending its application to a broad range of membrane proteins.
Experimental Design
Overview of the procedure
The procedure described in this protocol consists of four stages (Fig. 1b, c). First, the coupler protein and insertion site are selected, and we recommend inserting the target membrane protein into Asn144-Asn146 of sfGFP as the first choice (Steps 1-14). The fusion constructs are generated with a simplified seamless cloning method38 (Steps 15-31). Next, the membrane folding topology and cellular location of fusion protein are assessed by fluorescence imaging (Steps 33-38). The fraction of properly folded membrane protein is determined with differential detergent extraction (Steps 39-58), and protein thermostability is measured by fluorescence-detected size exclusion chromatography (FSEC)39 (Steps 59-83). Subsequently, the restrained membrane protein is expressed on a large scale and purified under optimized conditions (Supplementary Methods Steps 1-52), and its cellular and in vitro activities are confirmed (Steps 85-86). Finally, the purified protein is crystallized in lipid cubic phase and diffraction data are collected at a synchrotron beamline (Steps 87-92 and Supplementary Methods Steps 53-84). The crystal diffraction is optimized using constructs with serial termini truncations (Steps 93), and the crystal structure is determined with the aid of reiterative density improvement approaches (Steps 94-103 and Supplementary Methods Steps 85-105). Overall, these series of experiments require novel protein engineering design, assessment of protein stability, growth and optimization of crystals in lipid cubic phase, and challenging computational methods in crystallography. Technical details of these experiments are provided here for effective stabilization of membrane proteins and rapid determination of their structures.
Membrane protein engineering by termini restraining
To increase the overall stability of the TM protein after restraining, it is necessary to choose a highly stable soluble protein as the coupler. The coupler can be an engineered protein with increased thermostability, such as the sfGFP25,26, or a protein from a hyperthermophile species. To facilitate the formation of crystal contacts, couplers known to be highly crystallizable, such as thioredoxin and T4 lysozyme, can be used. Another factor to consider is that the insertion site on the coupler needs to be free of steric hindrance; in other words, one should avoid choosing an insertion site located in a concave area surrounded by stable structural elements. Nevertheless, multiple insertion sites can be typically identified in a single coupler protein. For instance, a common split site in sfGFP is on a loop between its first β-strand and rest of the structure25,26. For structure determination of membrane proteins13–15, however, we have selected a split site located at the end of a central β-strand of sfGFP25. In this protocol, we will focus on the sfGFP restraining because its fluorescence facilitates the assessments of cellular location, folding topology and thermostability, and allows rapid identification of protein crystals. Furthermore, using sfGFP as the coupler has proved successful for crystal structure determination of several small membrane proteins16,17.
To demonstrate the general applicability of the termini restraining strategy, we have selected many coupler proteins and fused them to various membrane proteins (Supplementary Table 1). We generally insert the entire membrane protein sequence into a coupler, without truncations at the two termini or addition of linkers. This fusion strategy requires the split N- and C-terminal parts of the coupler to fold independently and these two parts to assemble. Moreover, the membrane protein region, with coupler sequences added to both ends, needs to insert into membranes in the correct folding topology. Remarkably, such a complicated folding scenario can be well handled by both prokaryotic and eukaryotic cells. For instance, we have used sfGFP, thioredoxin, and other couplers to restrain membrane proteins from different species, such as human JAGN1, pufferfish VKORL, and E. coli DsbB13–15 (Supplementary Table 1). In most cases, the fusion proteins can be readily expressed in different systems using E. coli, Pichia and HEK293 cells, and properly folded, functional proteins can be purified in detergent. Overall, the choices of couplers and insertion sites are very broad, but we recommend focusing on loop regions of a rigidly structured protein that has been thoroughly characterized.
Quality assessment
Quality control (verification of accurate folding and activity) of stabilized membrane proteins is essential to subsequent structural, biochemical and biophysical studies. The correct membrane folding topology can be confirmed by fluorescence imaging of the cells overexpressing membrane proteins restrained by split sfGFP (Fig. 2a), because its reconstituted fluorescence indicates that membrane proteins with even numbers of TMs fold into their predicted topologies40. The GFP signal can be used also to assess the proper subcellular location of the protein constructs by fluorescence imaging and to detect whether protein aggregation occurs during the overexpression. Furthermore, the GFP signal allows the measurement of protein thermostability by FSEC using detergent-solubilized membranes or purified membrane proteins. For purified proteins, alternative methods of accessing the thermostability are also available8,41. Next, the fraction of correctly folded membrane protein is determined by performing Western blots of whole-cell lysates extracted with sodium dodecyl sulfate (SDS) and/or urea, which dissolve most proteins including aggregates and insoluble proteins, and extracted with n-dodecyl-β-D-maltoside (DDM), which preferably solubilizes well-folded membrane proteins42 (Fig. 2b). Finally, case-specific cellular and in vitro assays are often available to measure the transport activity, enzymatic activity or molecular interaction of termini-restrained membrane proteins. To illustrate this, we use sfGFP-restrained VKORL to show that the activity of this integral membrane enzyme is retained in a cellular assay (Fig. 2a). After being purified in detergent, the specific activity of restrained VKORL is much higher than that of the unrestrained protein (Fig. 2c). Furthermore, the native membrane folding topology of restrained VKORL is maintained (Fig. 2a), the fraction of well-folded protein is largely increased during overexpression (Fig. 2b), and the thermostability of purified protein is markedly improved (Fig. 2c).
Fig. 2. |. Quality assessment of restrained membrane proteins.
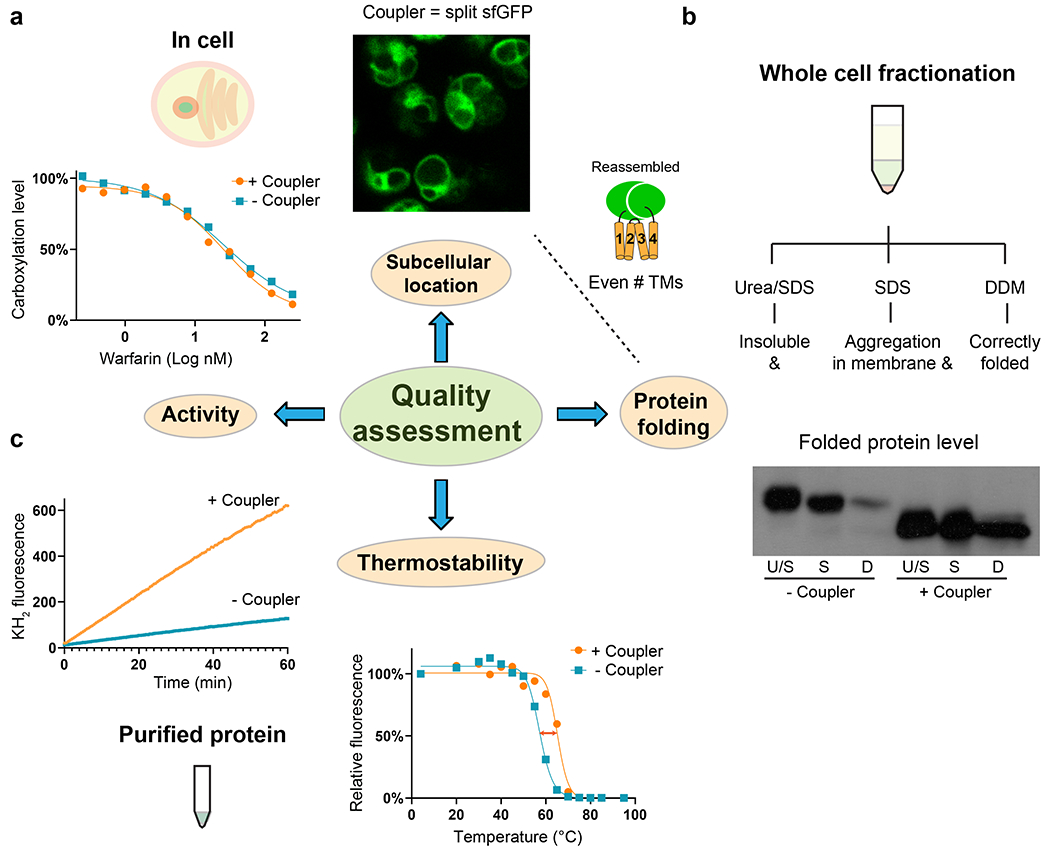
a, Cellular assays using sfGFP-restrained VKORL protein as an example. Left, VKORL is an integral membrane enzyme whose activity and inhibition by warfarin can be readily analyzed with a cellular γ-carboxylation assay. Right, Fluorescence imaging shows that restrained VKORL is correctly located in the endoplasmic reticulum membrane after overexpression in Pichia cells. Furthermore, the reconstituted GFP fluorescence indicates that restrained VKORL folds in the expected 4-TM (even numbered) folding topology. b, Whole-cell fractionation measurements of the folded protein level. The unrestrained and restrained VKORL are compared on Western blot. U/S: urea/SDS, S: SDS, D:DDM. Restrained VKORL shows a much larger DDM-extractable (well-folded) fraction. c, Verification of the activity of purified protein and measurement of improved thermostability. Left, The epoxide reductase activity of VKORL is monitored by increased fluorescence of the reaction product, vitamin K hydroquinone (KH2). Right, FSEC measurements show increased Tm with termini restraining.
Production and purification of restrained membrane proteins
Restrained membrane proteins need to be expressed in a suitable cellular system. For eukaryotic proteins, we perform initial expression test in Pichia pastoris cells because this system is cost and time effective, and often gives high protein yield (milligrams of protein per liter of culture). Membrane proteins from mammals often need to be expressed in a Bacmam system, a procedure previously described in Nature Protocols43. For prokaryotic membrane proteins, we usually use C41(DE3) and C43(DE3) E. coli cells, which are known as “Walker strains” that have been selected for improved membrane protein overexpression44,45. Before large-scale expression and protein purification, we conduct FSEC to assess the expression level, elution profile, detergent stability and thermostability of restrained membrane proteins. For the protein purification, selection of proper detergents, addition of lipids, and use of conformational specific ligands afford further stabilization to membrane proteins.
Crystallization and crystal handling
The coupler protein enables the crystallization of membrane proteins and allows optimization of crystals to a near-atomic diffraction. The restrained membrane proteins can be crystallized in detergent or in lipid cubic phase (LCP). The LCP method improves crystal diffraction by promoting additional crystal packing between transmembrane regions; we use a procedure modified from that previously described in Nature Protocols46. With sfGFP as the coupler, the initial hits of tiny LCP crystals can be readily identified under a fluorescence microscope (Fig. 3a). In addition, LCP crystals in mesophase bolus can be frozen directly without additional cryoprotectants (Fig. 3b–c). Moreover, crystal diffraction is significantly improved by systematically shortening the two termini of the membrane protein linking to the coupler (Fig. 3d–f). Due to the small size (often < 50 μm) of LCP crystals, data collection needs to be conducted at a synchrotron beamline and requires special handling that involves centering crystals by diffraction-guided scans and minimization of radiation damage.
Fig. 3. |. Crystal identification and diffraction improvement of restrained membrane protein.
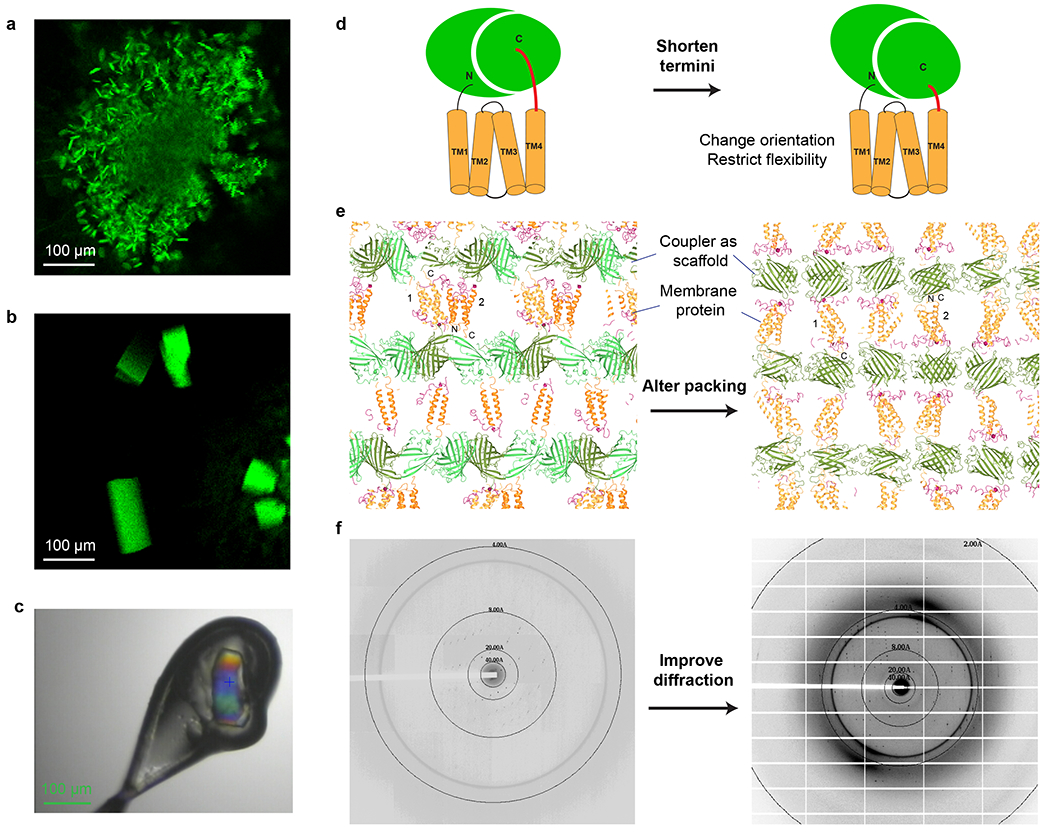
a, Identification of initial crystallization hits by fluorescence imaging. The sfGFP-restrained VKORL is used as an example. b, Large LCP crystals after optimization of buffer conditions and constructs. c, Crystal identification (green color) for data collection at synchrotron beamline. d, Shortening the termini of restrained VKORL changes its orientation relative to the sfGFP coupler and/or restricts the flexibility of the fused molecule. e, Crystal packing interactions and pattern are altered after the termini shortening. The termini of two representative VKORL molecules (1 and 2) in the different crystal forms are indicated. f, Crystal diffraction is dramatically improved.
Structure determination
The addition of the coupler protein enables rapid structure determination of restrained membrane proteins (Fig. 4). Structural coordinates of the coupler protein are used to obtain molecular replacement solution. After rigid body refinement of this model, the partial model phases are used for automatic solvent flattening and non-crystallographic symmetry (NCS) averaging. The density-modified maps are usually sufficient for automatic model building. The updated model is used for model phasing and fine-tuning density modification, and the improved map is used for further automatic or manual model building. Reiteration of this process is usually sufficient to generate a nearly complete model of the membrane protein region. In difficult cases, cross-crystal averaging is applied between the fused protein and the coupler protein alone to dramatically improve the electron density map for model building.
Fig. 4. |. Structure determination using the coupler structure and diffraction data.
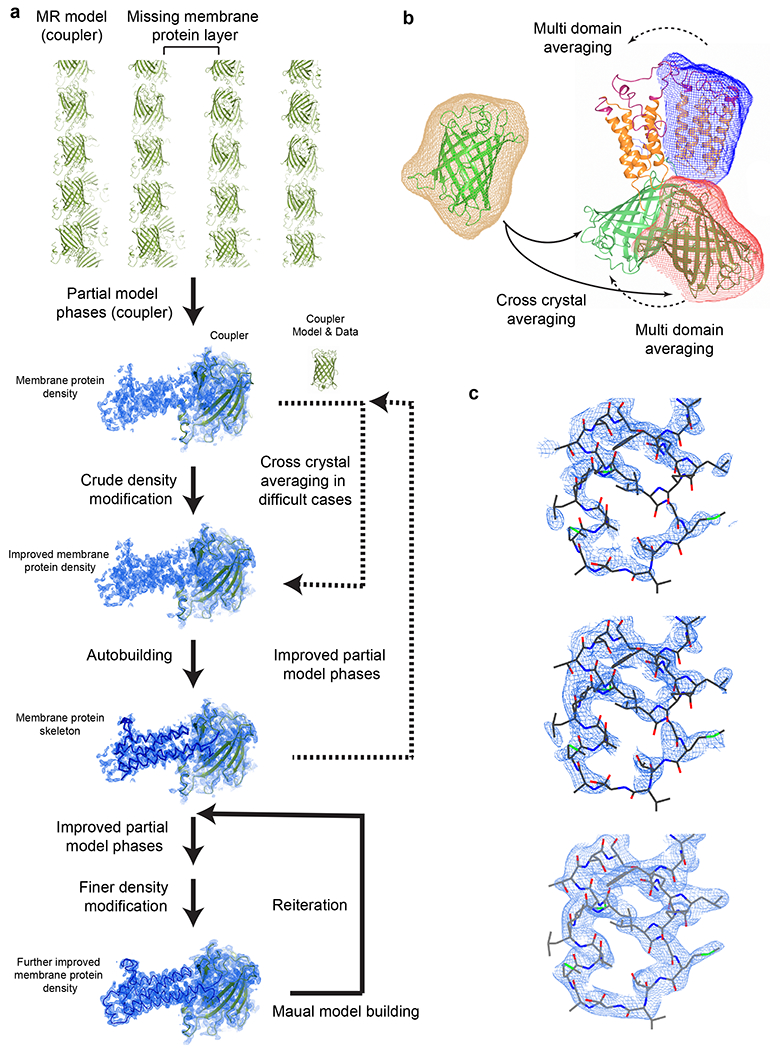
a, Structure determination with reiterative density modification procedures. Top to Bottom, molecular replacement locates the coupler molecule in the crystal and the partial model phases are used for initial density modification. This electron density map generally allows automatic model building that generates a large part of the membrane protein skeleton. Advanced density modification strategies (Steps 96-102) and reiterations of phasing and model building generate the final structure. Dashed arrows indicate optional steps. Electron density maps and models of sfGFP-restrained VKORL are shown as an example. b, Averaging strategies. In difficult cases, the structure and diffraction data of the coupler can be used for cross crystal averaging with those of the coupler-fused membrane protein. Only partial phases from the molecular replacement model are required initially (i.e., without knowledge of the membrane protein portion). For NCS related molecules, multi domain averaging is often required to account for the relative movement between the coupler and the membrane protein. The masks on each domain are shown in different colors. c, Zoom view of density maps of the membrane protein region during the structure determination process. Top, 2FoFc map after rigid body refinement of the molecular replacement model (coupler only). Middle, PARROT density modification map using the partial model phase (coupler only). Bottom, 2FoFc map after reiterating model building and density modification. All maps are contoured at 1 σ.
Materials
Biological materials
DH5α competent cells (New England Biolabs, cat. no. C2987H)
SMD1163 yeast strain (Invitrogen, Genbank accession no. XM_002489831)
HEK293S GnTI− cells (ATCC, cat. no. CRL-3022, RRID: CVCL_A785) https://scicrunch.org/resolver/CVCL_A785
-
HEK293T cells (Sigma-Aldrich, cat. no. 12022001, RRID: CVCL_0063) https://scicrunch.org/resolver/CVCL_0063
! CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
pBud4.1 vector (Thermo Fisher, cat. no. V53220)
pEG_Bacmam vector (Addgene, cat. no. 160451)
pPICZ vectors (Thermo Fisher, cat. no. V19020)
sfGFP (1-230 AA) with His tag in a pPICZ-derivatized vector (Addgene, cat. no. 174095)
sfGFP-restrained human VKOR in a pPICZ-derivatized vector (Addgene, cat. no. 174097)
sfGFP-restrained DsbB in pET28b vector (Addgene, cat. no. 174099)
Reagents
Anti-Mouse IgG peroxidase antibody (Sigma-Aldrich, cat. no. A4416, RRID: AB_258167) https://scicrunch.org/resolver/AB_258167
Can Get Signal immunoreaction enhancer solution (Cosmo Bio, cat. no. TYB-NKB-101)
Dulbecco’s modified eagle medium (DMEM; high glucose; Thermo Fisher, cat. no. 11965084)
n-dodecyl-β-D-maltopyranoside (DDM; Anatrace, cat. no. D310S)
DpnI restriction enzyme (New England Biolabs, cat. no. R0176L)
ECL western blotting substrate (Thermo Fisher, cat. no. 32109)
ER-Tracker blue-white DPX (Invitrogen, cat. no E12353)
Ethylenediaminetetraacetic acid (EDTA; disodium salt; Sigma-Aldrich, cat. no. E5134)
Fetal bovine serum (FBS; Thermo Fisher, cat. no. 26140079)
-
FM 4-64 (Invitrogen, cat. no. T13320)
His-tag antibody (mouse monoclonal; GenScript, cat. no. A00186, https://scicrunch.org/resolver/AB_914704)
MitoTracker red FM (Invitrogen, cat. no. M22425)
Penicillin-Streptomycin (Thermo Fisher, cat. no. 15140122)
Phusion® high-fidelity DNA polymerase (New England Biolabs, cat. no. M0530L)
Phosphate-buffered saline (PBS; Sigma-Aldrich, cat. no. D8537)
Protease inhibitor cocktail (cOmplete™ EDTA-free tablets; Roche, cat. no. 11873580001)
Sodium chloride (NaCl; Sigma-Aldrich, cat. no. S3014)
Trypsin-EDTA (0.05%; Thermo Fisher, cat. no. 25300054)
Tween 20 (Sigma-Aldrich, cat. no. S6501)
Triton X-100 (Sigma-Aldrich, cat. no. X100)
Urea (Sigma-Aldrich, cat. no. U5378)
Zeocin (100 mg/mL; Thermo Fisher, cat. no. R25005)
Equipment
Thermal cycler
Water bath
Heat block
SDS-PAGE electrophoresis system
Western blotting transfer system
Shakers
Cell density meter (VWR, Ultrospec 10 model)
CO2 incubators
Tube rotator (Thermo Scientific, cat. no. 56264-306)
Refrigerated incubator shaker with 12 X 2 L platform (New Brunswick, Excella E-25R model)
Mixer mill (Retsch, MM 400 model)
Glass beads (0.5mm; Fisher, cat. no. NC0386496)
Tabletop centrifuge
Tabletop ultracentrifuge (Beckman Coulter, Optima Max-TL model)
Microfuge tube (Beckman Coulter, cat. no. 357448)
Glass bottom culture dishes (Thermo Scientific, cat. no 150680)
FSEC system (Shimadzu, LC-2030C model with RF-20AXS fluorescence detector and autosampler)
Superose 6 increase column (Cytiva, cat. no. 29091596)
Confocal microscope (Zeiss, LSM 880 II Airyscan model)
Synchrotron radiation beamline (e.g., 24-ID-C beamline, Advanced Photon Source, Argonne National Laboratory)
Software
-
CCP4 Program Suite:
BUCCANEER (http://legacy.ccp4.ac.uk/html/cbuccaneer.html)
DM (http://www.ysbl.york.ac.uk/~cowtan/ccp4wiki/wiki66.html)
DMMULTI (http://legacy.ccp4.ac.uk/html/dmmulti.html)
PARROT (http://www.ysbl.york.ac.uk/~cowtan/parrot/cparrot.html)
Graphpad Prism (https://www.graphpad.com/scientific-software/prism/)
HKL2000 (https://www.hkl-xray.com/)
PolyPhobius (https://phobius.sbc.su.se/poly.html)
PSIPRED and MEMSAT (http://bioinf.cs.ucl.ac.uk/psipred/)
SnapGene viewer (https://www.snapgene.com/)
XDS (https://xds.mr.mpg.de/)
Reagent setup
DDM (16% wt/vol)
Dissolve 163 mg DDM in 900 μL FSEC lysis buffer to make 1 mL 16% (wt/vol) stock. Prepare fresh solution before use.
! CAUTION wear a mask to avoid inhalation of DDM powder.
DMEM growth media with FBS
Add 50 mL FBS and 10 mL 100× Pen Strep to 1 L DMEM. This medium can be stored at 4°C for several weeks.
EDTA (500 mM)
Add 186.1 g of disodium EDTA to ddH2O, adjust the pH to 8.0 with NaOH and filter with membrane disc filter (0.2 μm). This solution can be stored at room temperature (20–22°C) for several months.
FSEC lysis buffer
Add 2.5 mL 1 M Tris-HCl stock pH 8.0, 1.5 mL 5 M NaCl, and 100 μL 0.5 M EDTA to ddH2O. Adjust the final volume to 50 mL. Add 1× protease inhibitor cocktail. Prepare fresh solution before use.
FSEC running buffer
Add 30 mL 5 M NaCl and 20 mL 1 M Tris-HCl pH 8.0 to 950 mL ddH2O. Add 500 mg DDM, dissolve and filter (0.2 μm). Prepare fresh solution before use.
NaCl stock (5 M)
Dissolve 292.5 g NaCl in ddH2O, adjust to 1 L final volume and filter (0.2 μm). This solution can be stored at room temperature for several months.
Pichia lysis buffer (1.5×)
Add 30 mL 1 M Tris-HCl pH 8.0 and 45 mL 5 M NaCl to ddH2O, and adjust final volume to 1 L. This solution can be stored at room temperature for several weeks.
Potassium phosphate (2 M, pH 6.0)
Dissolve 91.96 g K2HPO4, 472.52g KH2PO4 and 36 g KOH in ddH2O to a final volume of 1 L. This solution can be stored at room temperature for several months.
Tris buffered saline (TBS, 10×)
Dissolve 24 g Tris base and 88 g NaCl in 900 mL ddH2O. Adjust to pH 7.6 using 1 M HCl and to 1 L final volume. This solution can be stored at room temperature for several months.
Tris buffered saline with 0.1% (vol/vol) Tween 20 (TBST, 1×)
Mix 100 mL 10× TBS and 1 mL Tween 20 with 900 mL ddH2O. This solution can be stored at room temperature for several weeks.
Tris-HCl stock (1 M)
Dissolve 121.1 g Tris base in ddH2O, adjust to desired pH (7.5 or 8.0) with HCl and to 1 L final volume with ddH2O. After filtering (0.2 μm), this solution can be stored at room temperature for several months.
Tris-NaCl buffer
Add 30 mL 5 M NaCl and 20 mL 1 M Tris-HCl pH 7.5 or 8.0 to ddH2O, and adjust final volume to 1 L. This solution can be stored at room temperature for several weeks.
Procedure
Selection of suitable couplers and design of restrained constructs ● TIMING 1 d
-
Choose a target membrane protein for the following purposes.
Determine the structure of the target membrane protein. We suggest using sfGFP restraining as the primary choice to crystallize small membrane proteins (2 and 4 TMs). The restraining method may also be applied to membrane proteins with more than 4 TMs or heterodimeric membrane proteins whose structure determination has been unsuccessful despite extensive trials with traditional methods.
Stabilize the membrane protein for biochemical, biophysical and other studies. The restraining is intended for improving stability of proteins that show aggregation during overexpression or heterologous expression in various cell systems, or proteins that become denatured or aggregated during purification in a detergent solution.
-
Verify suitability of the termini restraining strategy by taking the following criteria into consideration.
The membrane protein contains an even number of TMs (Step 3).
Beware of membrane proteins whose functional site(s) is in proximity to the two termini; with such an architecture, introducing the coupler may interfere with the protein function.
Relatively small membrane proteins (TM2–6) are the best candidates and supported by our existing data. Applications of termini restraining to membrane proteins with more TMs are yet to be demonstrated.
Application to monomeric membrane proteins is supported by existing data. In principle, the approach is also applicable to oligomeric membrane proteins, however the fused coupler(s) may sterically interfere with the oligomerization. In such cases, smaller couplers will be more appropriate.
-
Analyze the folding topology of the target membrane protein to verify an even number of TMs.
Deduce the folding topology and number of TMs from the protein family that the membrane protein belongs to. For example, tetraspanins are known to have 4 TMs.
Alternatively, search the literature or databases (e.g., TOPDB47) for experimentally derived knowledge of folding topology.
Alternatively, use web servers to predict the folding topology. In our lab, we use MEMSAT-SVM48,49, PolyPhobius50 and TMHMM51 for the topology prediction. TMHMM is for the overall assessment of the hydrophobic profiles of TMs. PolyPhobius gives more accurate prediction by including homology information and distinguishing signal peptides from TMs. MEMSAT-SVM was reported to have further improved prediction accuracy owing to a machine learning algorithm49,52.
Experimentally verify the membrane folding topology. For simplicity, we recommend using fluorescence of the reconstituted sfGFP coupler as a direct readout53,54 for even number of TMs (Steps 33-38). If the membrane protein has odd number of TMs, however, the split sfGFP is not fluorescent because the two sfGFP halves are separated by the membrane and cannot reconstitute. Additionally, many experimental methods are available to determine the membrane topology, including protease protection, N-glycosylation in eukaryotic cells, or other fluorescence-based assays40.
Use sfGFP as the first choice of the coupler protein. Perform Steps 32-86 to assess its suitability; the sfGFP-restrained membrane protein should be well expressed, remain functional or active, and maintain proper folding and expected cellular location. As stated in Experimental Design, fluorescence of sfGFP facilitates protein quality assessment and crystal identification. For structure determination of the unknown membrane protein portion, both the crystal structure and diffraction data of sfGFP are available (PDB code 2B3P) and highly accurate (1.4 Å resolution). In addition, sfGFP has been engineered for increased stability and optimized for efficient self-association25,26.
-
If sfGFP is unsuccessful, identify another suitable coupler. We recommend starting with well characterized proteins, such as MBP, thioredoxin, and T4-lysozyme. Alternatively, perform advanced search in Protein Data Bank (PDB) (see below). Consider the following criteria to select a suitable coupler:
Structure of the coupler has been determined and deposited in PDB. Crystal structure is preferred.
Resolution of the reported coupler structure is higher than 3 Å. High-resolution structure usually suggests conformational rigidity and optimal quality of the coupler protein.
(Optional) Verify availability of the crystal diffraction data in PDB. The diffraction data of the coupler can be used for cross-crystal averaging (Step 102) to improve the electron density map.
The coupler protein should be highly stable, such as engineered proteins or proteins from hyperthermophile species. Alternatively, the coupler can be any highly crystallizable protein, such as thioredoxin and T4 lysozyme, or a protein that has been used as crystallization scaffold (e.g., MBP).
Compared to the membrane protein, the coupler should have a relatively large size to facilitate crystal packing (Fig. 3e) and structure determination (Fig. 4). As an example, sfGFP (27 kDa) has been successful for the structure determination of small membrane proteins that have 2-4 TMs and 11-22 kDa MW13. Stability improvement has been observed for membrane proteins of 2-6 TMs and 11-37 kDa using sfGFP as the coupler.
For a broad search of couplers, go to the advanced search page of Protein Data Bank (RCSB PDB). Specify the above criteria, i.e., high resolution cutoff, suitable molecular weight cutoff, and structural determination by crystallography. Inspect the structure of candidate couplers and avoid those with flexible structural domains.
-
Test insertion sites in sfGFP by following Steps 32-92.
Insert the membrane protein between Asn144 and Asn146 of sfGFP (Fig. 5a,b) as the first choice. Using this insertion site25, we have determined 15 structures of 6 different membrane proteins13.
Alternatively, test other reported split sites in sfGFP, such as Gln157-Lys158 or Glu172-Asp17355,56 (Fig. 5a). The sfGFP is a β-barrel protein that has no apparent steric hindrance at multiple insertion sites. For a complete list of the split sites, refer to Table 1 in Romei and Boxer, 201957.
-
Alternatively, identify a suitable insertion site in other couplers (Fig. 5c).
Use an insertion or split site that has already been reported in the literature27–29.
Alternatively, inspect the structure of the selected coupler protein. Identify a disordered loop region as a potential insertion site. Insertion of a membrane protein in such a flexible region often can be tolerated.
The insertion site should not be sterically blocked by another part of the coupler structure as this will hinder insertion of the membrane protein.
The insertion should not induce severe clash of the coupler structure with the membrane plane. For instance, membrane protein insertion at a shielded region of the coupler will likely cause the rest of the coupler structure to clash with the membrane plane. If the clash occurs, the fusion protein is unlikely to fold correctly. Use FSEC for rapid assessment of the level of well-folded protein (Steps 59-72).
Screen several insertion sites to identify a suitable one. Use FSEC for rapid assessment of the inserted constructs (Steps 59-72).
Alternatively, a heterodimeric coupler can be used with each subunit linked to one terminus of the membrane protein.
-
Select a linker region in the target membrane protein.
Identify the boundaries of the first and last TM from the knowledge of membrane folding topology (Step 3). Additionally, the TM boundaries are often defined by aromatic residues (aromatic belts).
Determine the lengths of the termini regions before the first TM and after the last TM. Short termini (e.g., <30 amino acids (AA) are unlikely to be structured. To confirm, analyze the termini for secondary structure (e.g., with PSIPRED58) or disordered region (e.g., with D2P259).
-
Use the full-length membrane protein as the first choice. This strategy has been successful for most proteins we have tested so far.
! CAUTION Some proteins may have a folded extramembrane domain at their N or C-terminal (typically at least 40 AA). Consider directly using this domain as the coupler by joining the two termini with intein37.
! CAUTION The flexible termini regions should be > 5 AA in length. This gives sufficient flexibility to allow proper folding of both the membrane protein and the coupler. If the termini regions are too short, add a flexible linker, e.g., with GSGGS sequence.
Follow Steps 15-31 to generate clones of the membrane protein inserted into the coupler (i.e., restrained) (Fig. 1c). As a positive control, generate a clone of the full-length membrane protein without the coupler (i.e., unrestrained) using the same vector. For thermostability FSEC, generate clones of restrained and unrestrained membrane protein with a C-terminal mCherry tag.
Evaluate the DDM extracted protein level and protein elution profile with FSEC (Steps 59-72). Because DDM preferably extracts well folded proteins, high extraction level and good elution profile are indicators that the fusion proteins are properly folded. Compare the FSEC profiles of coupler-restrained and unrestrained membrane protein.
Compare the activity of the coupler-restrained and unrestrained membrane protein (Steps 85-86).
For biochemistry or biophysics studies, avoid further manipulating the full-length protein if the native activity is retained or improved after the restraining. If the activity is compromised, however, consider addition of linkers (Step 14) to avoid over restraining the membrane protein.
-
For structural studies, perform serial truncations to improve the protein yield and/or crystal diffraction.
Limit the truncations to the flexible termini regions and do not extend into the first and last TMs.
Generate combinations of 5 or 3 residue truncations. As an example, human VKOR is a 163 AA protein and PolyPhobius predicts that the first TM starts at residue 12 and the last TM ends at residue 149. Therefore, the following constructs can be generated: 6-163, 11-163, 1-158, 1-153, 6-158, 11-158, 6-153, and 11-153.
Evaluate these truncations by FSEC profile (Steps 59-72). Low protein level after DDM extraction indicates that the membrane protein is overly truncated.
Generate constructs with finer truncations in 1-2 AA steps. Evaluate the constructs by FSEC and an activity assay suitable for the target TM protein. Identify the approximate limit of the N- and C-terminal truncations.
-
Select a few constructs with shortened termini for crystallization trials (Steps 87-88). Shortened constructs often result in structures of improved resolution13.
▲ CRITCAL STEP In several cases we have tested, inserting the full-length membrane protein between Asn144/Asn146 of sfGFP readily generates a good yield of proteins and high-resolution structures. For example, the structures of CD53 and JAGN1 were determined to 2.9 Å or 2.25 Å, respectively, with the full-length protein insertion. For VKORL, only one truncation construct (5 AA at N-term) is required to obtain a 2.4 Å structure. Even in difficult cases, we estimate that less than 20 truncation variants need to be tested.
-
(Optional) Add linkers to the two termini if proper folding and function of certain membrane proteins are compromised.
We recommend starting with the linker length of five AA.
Add flexible linkers, such as GSGGS (or repeats of GSGGS), to lower the strain between the fused coupler and membrane protein.
Alternatively, add rigid linkers such as poly Ala or EAAAR (or repeats of EAAAR) to extend the first or last TM.
-
Test various linker lengths. Although we have not extensively investigated extended linkers, we estimate that fewer than 10 AA additions will be needed.
▲ CRITICAL STEP Refer to Chen et al. 2013 for a list of flexible and rigid linkers60.
Fig. 5. |. Expression vectors and construct generation.
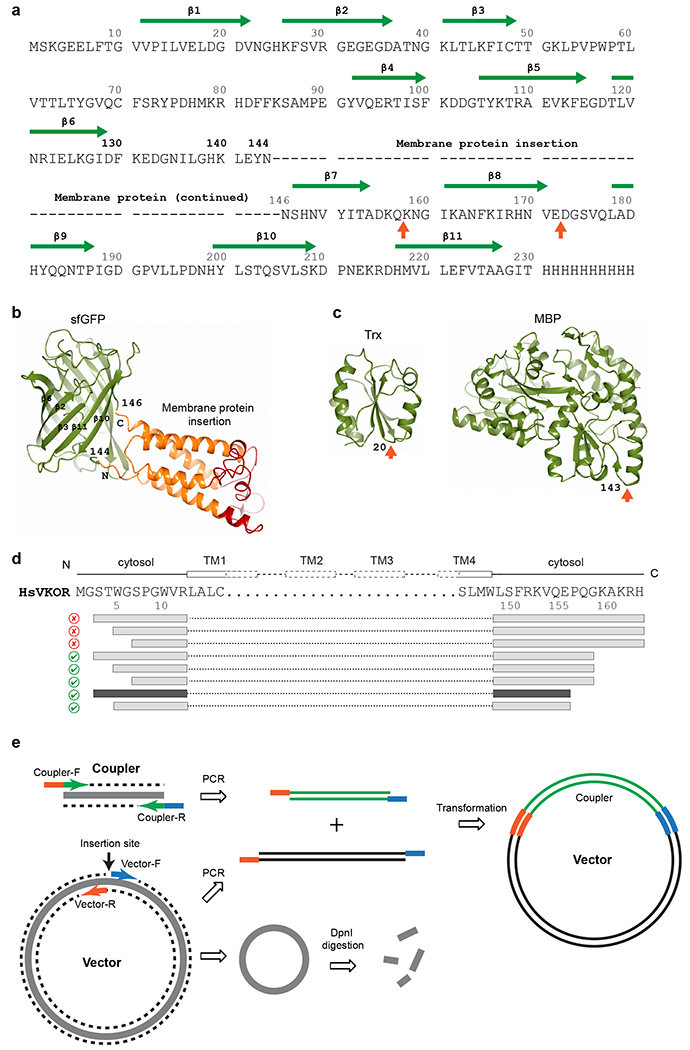
a, The sequence of sfGFP and insertion sites for a membrane protein. The first choice of insertion (dashes represent membrane protein sequence) is between Asn144 and Asn146 in sfGFP. Other potential insertion sites55,56 are indicated by orange arrows below the sequence. The β-strands and residue numbers of sfGFP are indicated above the sequence. The C-terminal 8 residues (231-238) of sfGFP have been removed to reduce its structural flexibility and to favor stable crystal packing. b, Structure of sfGFP (shown in green) with fused human VKOR (transmembrane region in orange and extramembrane region in red). The Asn144-Asn146 insertion site is indicated. c, Structures of other couplers, thioredoxin (Trx) and maltose binding protein (MBP), that we have tested. Insertion sites are indicated by orange arrows. d, Serial truncations exemplified by human VKOR constructs. The FSEC results are indicated by good (green checkmark) and bad (red cross) icons. Interestingly, truncations at the C-terminus (removing the ER retention signal, KAKRH) rescue the expression of restrained constructs. Further truncations result in the final construct (dark grey bars) that is used to obtain the high-resolution structure. e, Simplified seamless cloning38. The coupler coding sequence and the entire expression vector are PCR amplified. The primers for coupler contain sequences (orange and blue) overlapping with the primers for the vector. DpnI digestion removes the original methylated template. The two PCR fragments can assemble to form the coupler-inserted plasmid with considerable efficiency after transformation into DH5α E. coli cells.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solutions |
|---|---|---|---|
| 20 | No PCR product, low intensity band, or multiple bands are observed | Primers have low specificity PCR parameters are not optimal |
Increase the length of primers (can be > 25 bases) to increase specificity. Adjust primer or template concentration, change annealing or extension temperature. |
| 26 | No PCR band or wrong size of band is observed | Original template is incompletely digested Overlap sequence (between the primers for vector and coupler) has low Tm |
Lower template concentration, or PCR verify more colonies Redesign primers with increased length, introduce silent mutations (when possible) on the vector primer to make it GC rich. |
| 35 38 |
Restrained and unrestrained membrane proteins both show abnormal subcellular location | Membrane protein is heterologously expressed in Pichia cells Protein overexpression in mammalian cells interferes with protein sorting or results in protein degradation |
For mammalian proteins, analyze subcellular location in HEK393 cells, but purification and crystallization of the restrained membrane proteins expressed in Pichia are usually unaffected. |
| 74 | Restrained protein remains unstable in detergent | Membrane protein is highly unstable | Add high affinity ligand to stabilize protein conformation, add cholesterol derivative (e.g., cholesteryl hemisuccinate) |
| 97 | Density map at the membrane protein region is of poor quality | The dataset is of low resolution (worse than 3.5 Å) or poor quality (e.g., severe anisotropy) | Use multi-crystal averaging (Step 102), perform anisotropy correction |
Generation of fusion constructs of the membrane protein with the coupler by seamless cloning ● TIMING 2 weeks
▲ CRITICAL Generating the fusion constructs requires seamless cloning. A protocol is presented below to rapidly clone restrained constructs including those with serial termini truncations or linker additions (Steps 13-14). This simplified protocol does not require ligase, restriction enzymes or gel purification of the PCR products.
-
15
Select expression vectors for the fusion constructs. In our lab, we use pPICZ-derivatized vector (Supplementary Fig. 1a) for expression in Pichia system and pEG_Bacmam-derivatized vector for expression in mammalian cells (HEK293S). Both derivatized vectors contain coding sequences of a C-terminal PreScission protease cleavage site, followed by an eGFP or mCherry tag for florescence detection, and a His tag for protein purification. In addition, we use pBud4.1 vector for transient transfection in HEK293T cells. For prokaryotic membrane proteins, we prefer E. coli expression using pET28b vector (Supplementary Fig. 1b, kanamycin resistance).
-
16
Design primers for the expression vector (Fig. 5e). Start from the selected insertion site in the vector, copy 18-25 bases in the forward direction and use as the sequence of the forward primer on the vector (Vector-F). For the reverse primer (Vector-R), copy 18-25 bases in the reverse direction from the reverse complementary sequence starting from the insertion site. Use SnapGene viewer to facilitate the primer design.
-
17
Design PCR primers for the selected coupler and insertion site (Steps 4-7). Copy 18-25 bases from the beginning of coupler coding region and add to the 3’ end of the reverse complementary sequence of Vector-R. Use the combined sequence as the coupler forward primer (Coupler-F). For the coupler reverse primer (Coupler-R), copy 18-25 bases from the reverse complementary sequence from the end of coupler coding region and add to the 3’ end of the reverse complementary sequence of Vector-F. Synthesize all primers using a commercial supplier of your choice.
-
18
Use Vector-F and Vector-R as primers to amplify the entire expression vector (Step 15). Set up a 50 μL PCR reaction mixture containing 10-20 ng vector, 10 pmol of each primer, 1 unit (0.5 μL) Phusion polymerase, 10 μL 5× Phusion HF buffer, and ddH2O. Setup the following PCR program: 1 cycle of 98 °C 2 min; 18 cycles of 98 °C 10 s, 55 °C 30 s and 72 °C 2-4 min (extension time based on template size; use 30 s/kb); and 1 cycle of 72 °C 10 min.
▲ CRITICAL STEP Use less than 20 ng of template plasmid to reduce the amount of original methylated template not completely digested by DpnI (Step 21), which could generate false positive transformants (Steps 23-26). Use 16-18 cycles of PCR to reduce potential random mutations that can result from replication errors.
-
19
PCR amplify the coupler coding sequence. Use Coupler-F and Coupler-R as primers (Step 17) and an appropriate template containing the coupler coding sequence. Setup the PCR reaction as in Step 18. Adjust the extension time at 72 °C based on the length of the coupler coding sequence.
-
20
Verify the size and intensity of PCR products with agarose gel (1-1.2% wt/vol) electrophoresis.
▲ CRITICAL STEP The PCR amplified bands should be bright (i.e., abundant full-length product) to allow efficient cloning by this method (Steps 15-29). Optimize PCR reaction parameters if necessary.
? TROUBLESHOOTING
-
21
Add 20 units of DpnI restriction enzyme to each 50 μL PCR reaction mixture. To destroy original methylated DNA template, digest at 37 °C for at least 1 h (overnight digestion is preferred).
-
22
Mix PCR products of vector (Step 18) and coupler (Step 19) in equal volume (5 μL each). Incubate at room temperature for 2 min.
-
23
Transform the mixture into DH5α competent cells pre-thawed on ice. For each transformation, aliquot 50 μL cells in a 1.5 mL Eppendorf tube on ice. Add 2 μL mixed PCR products (Step 22). Gently mix by flicking and place the tube on ice for 30 min. Heat shock 45 s in 42 °C water bath.
! CAUTION Use standard sterile and cell culture techniques to manipulate all cells in this protocol.
-
24
Place the tube on ice for 2 min. Add 300 μL LB. Shake at 220 rpm and 37 °C for 45 min.
-
25
Spread the transformed cells onto LB agar plates containing appropriate antibiotic(s). Incubate at 37 °C for 14-16 h.
-
26
Verify the constructs by PCR and analyze by agarose gel electrophoresis. Pick 4 colonies and pipet mix each colony with 50 μL LB. Use 1 μL of mixture as template, and save the rest at 4 °C for Step 27. To ensure presence of the fusion construct, use one primer in the expression vector and the other primer in the coupler region. Setup PCR reactions as in Step 18.
? TROUBLESHOOTING
-
27
Inoculate 20 μL of verified colony mixture into 4 mL LB with appropriate antibiotic. Shake at 220 rpm and 37 °C for 12-16 h.
-
28
Isolate the plasmids by miniprep following manufacturer’s instructions. Measure the plasmid concentration by Nanodrop.
-
29
To verify the clone, sequence the plasmids using a sequencing facility.
-
30
Follow Steps 16-29 to insert membrane protein coding sequence into the selected site (Steps 6-7) of the coupler coding sequence that has been cloned in expression vector (Steps 16-29).
For sfGFP restraining, we have deposited in Addgene the sfGFP-restrained human VKOR in a pPICZ-derivatized vector for Pichia pastoris expression and the sfGFP-restrained DsbB in pET28b vector for E. coli expression. Substitute VKOR or DsbB in these vectors (same cloning as the insertion) to restrain your target membrane proteins.
-
We use the following primers to PCR amplify the sfGFP-containing expression vector.
sfGFP-146-F, 5’-AACTCACACAATGTATAC-3’
sfGFP-144-R, 5’-GTTGTATTCGAGTTTGTGTC-3’
-
As an example, we use the following primers to amplify the VKOR coding sequence with N and C-terminal truncations (AA 3-155). The underlined sequences are reverse complementary to the sfGFP primers.
VKOR(3-155)-F: 5’-GACACAAACTCGAATACAACAGCACCTGGGGCAGCCCG-3’
VKOR(3-155)-R: 5’-GTATACATTGTGTGAGTTTTCCTGCACTTTGCGAAAGCTC-3’
-
To sequence the coding sequence of a membrane protein inserted into sfGFP, we use the following primers.
sfGFP-111-F: 5’-GAAGTCAAGTTTGAAGGTGATACC-3’
sfGFP-170-R: 5’-GTTGTGGCGAATTTTGAAGTTAGC-3’
-
31
For comparison with unrestrained membrane protein without the coupler, follow Steps 16-29 to insert the coding sequence of the membrane protein into the selected expression vector (Step 15).
Protein expression ● TIMING 1 d - 6 weeks
-
32
Use the most suitable method to express the coupled membrane proteins. Include unrestrained membrane proteins as a control. In our lab we express eukaryotic membrane proteins in Pichia pastoris cells and HEK293 cells, and prokaryotic membrane proteins in C41(DE3) and C43(DE3) E. coli cells. Detailed protocols for different expression systems are described in the Supplementary Method.
Live cell imaging of Pichia cells ● TIMING 4 h
-
33
Transfer 50 μL Pichia cells (Supplementary Methods Step 14) to a glass bottom culture dish. Add PBS to dilute cells and bring the final volume to 200 μL. Inspect cell density and morphology under a confocal microscope in differential interference contrast (DIC) mode. Repeat dilution and inspection until a single layer of cells is distributed at the glass bottom.
! CAUTION Cell shrinkage may occur due to overgrowth. Regrow cells if necessary.
-
34
Stain cells with different dyes according to the subcellular location of the target membrane protein. For proteins located in the ER, add Tracker blue-white DPX to 5 μM final concentration. For mitochondrial membrane proteins, add Mito-Tracker red FM to 10 μg/mL final concentration. For proteins located in the plasma membrane, add FM 4-64 to 10 μg/ml final concentration. Incubate 5 min at room temperature.
-
35
Inspect cells under confocal microscope in fluorescence mode and with 63× oil immersion objective.
Select and focus on cells that are not overlapping with the others.
Verify that target cellular compartment in selected cells can be clearly visualized with dye staining.
Adjust fluorescence magnitudes of sfGFP and the dye to similar levels for localization comparison.
For properly localized membrane proteins, the majority of cells expressing the protein should overlap with the dye staining. Conversely, missorted proteins may have different subcellular location and aggregated proteins may accumulate in vacuoles.
Compare sfGFP-restrained and C-sfGFP tagged membrane proteins for their localization and potential aggregation.
? TROUBLESHOOTING
Live cell imaging of HEK293 cells ● TIMING 2 d
-
36
Detach cells plated in 24-well plate after 16 h transfection (Supplementary Methods Steps 29-32). Replace media with 200 μL Trypsin-EDTA in well. Aspirate and immediately replace again with 200 μL Trypsin-EDTA. Remove most liquid and leave ~20 μL in well. Place plate in 37 °C CO2 incubator for 2-5 min. Then, add 200 μL DMEM FBS medium to wells and pipet to mix.
-
37
Transfer 1/10 detached cells to a glass bottom culture dish. Grow in 37 °C incubator for 12-20 h.
-
38
Before cell confluency reaches 90%, stain and inspect cells as in Steps 34-35. Use 40× oil objective for HEK293T cells.
Whole cell fractionation of Pichia cells ● TIMING 4 d
-
39
Prechill 125 mL steel jar and 6 steel balls in liquid nitrogen. Weigh 20 g frozen Pichia cells (Supplemental Method Step 18) and transfer to the jar. Wait until the frozen cells further cool down in liquid nitrogen; this low temperature keeps the cells in frozen form despite the heat generated from grinding (Step 40).
-
40
To break the Pichia cell wall, put steel balls in the jar. Grind cells in Ball mill at 400 rpm for 3 min, with switching between clockwise and counterclockwise every min. Cool down the entire jar in liquid nitrogen for 3 min. Repeat the grinding and cooling cycles for 3-4 times.
! CAUTION Wear face and hand protection when handling liquid nitrogen. Carefully operate the jar with forceps. To avoid explosion, ensure there is no liquid nitrogen inside the jar before fastening it in Ball mill.
-
41
Store cell powder at −80 °C for large-scale protein purification (Supplementary Methods Steps 19-28).
■ PAUSE POINT Cell powder can be stored at −80°C for many weeks.
-
42
Resuspend ~0.5 g cell powder in 50 mL TBS. Measure OD600 of the cell suspension with a cell density meter or spectrophotometer, and adjust OD600 to 0.1-1 with TBS (varying by expression level) to obtain comparable levels of Western blot signal (Steps 46-54) for restrained and unrestrained membrane proteins.
-
43
To break Pichia cell membranes, transfer the cell suspension to a 50 mL beaker in an ice-water bath. Sonicate at 25% power with cycles of 1 s on and 1.5 s off for 5 min total.
-
44
Aliquot 300 μL whole-cell lysates in three 1.5 mL Beckman Ultracentrifuge tubes. Add 180 mg urea and 100 μL 10% (wt/vol) SDS to tube 1. Add 100 μL 10% (wt/vol) SDS to tube 2, and 100 μL 10% (wt/vol) DDM to tube 3. To match to same volume (as 180 mg dissolved urea in tube 1), add 100 μL TBS in tubes 2 and 3. Add protease inhibitor cocktail (1× final) to each tube. Incubate 30 min at room temperature with gentle rotation.
-
45
Balance and transfer tubes to a precooled TLA-110 rotor. Centrifuge at 110,000g and 4 °C for 20 min in a tabletop ultracentrifuge.
-
46
To analyze extracted protein on a Western blot, take 80 μL supernatant from each tube and add 20 μL 5× SDS-PAGE loading buffer (containing 200 mM DTT). Heat at 90 °C for 10 min.
▲ CRITICAL STEP Many membrane proteins aggregate at high temperature and show abnormal mobility on SDS-PAGE after heating. If you observe this, test 65 °C heating and no heating after mixing the supernatant with SDS-PAGE loading buffer. In addition, test loading buffer without DTT. For a fast test, use purified protein (if available from Step 84).
-
47
Load 30 μL sample on 12% (wt/vol) SDS polyacrylamide gel. Run SDS-PAGE at 100 V for 2 h.
-
48
Transfer proteins to PVDF membrane by electroblotting.
-
49
Incubate membrane in 1× TBST at room temperature for 5 min with rotating. Repeat twice.
-
50
Incubate membrane with mouse His tag antibody (1:5,000) in Can Get Signal Enhancer Solution 1 at 4 °C overnight.
-
51
Repeat Step 49.
-
52
Incubate membrane with anti-mouse peroxidase antibody in Enhancer Solution 2 at room temperature for 1 h.
-
53
Repeat Step 49.
-
54
Expose and develop film with enhanced chemiluminescence substrate following manufacturer’s instructions.
Whole cell fractionation for HEK293 cells ● TIMING 2 d
-
55
Remove medium from 10 cm culture plate (Supplementary Methods Steps 29-32) and wash twice with 10 mL ice-cold PBS each time.
-
56
Scrape cells from plate and transfer to a 10 mL Falcon tube. Gently mix by pipetting and divide equal volume of cells to three 10 mL Falcon tubes. Centrifuge at 500g and 4 °C for 5 min.
-
57
Remove supernatant and resuspend the cell pellet with 100 μL TBS buffer containing different ingredients: tube 1 uses 6 M urea, 0.1% (wt/vol) SDS and 1% (vol/vol) triton X-100; tube 2 uses 0.1% (wt/vol) SDS and 1% (vol/vol) Triton X-100; and tube 3 uses 2% (wt/vol) DDM. Add protease inhibitor cocktail (1× final) and EDTA (1 mM final) to each tube. Incubate at 4 °C for 30 min with rotation.
▲ CRITICAL STEP For HEK293 cells, use the mixture of SDS and Triton X-100 to lower the interference from released nuclear content that significantly lowers the Western blot signal.
-
58
Follow Steps 46-54 to analyze extracted proteins with Western blot.
FSEC analysis of protein expression level in Pichia and HEK293 cells ● TIMING 5-7 d
-
59
For FSEC analysis of small-scale Pichia culture, select 2 colonies for each construct (Supplementary Methods Step 12). Inoculate each colony into 2.5 mL BMG medium with 200 μg/mL Zeocin in a 14 mL round-bottom capped Falcon tube. Shake at 220 rpm and 30 °C for 24-48 h.
! CAUTION Use less than 2.5 mL medium to avoid settling of Pichia cells during shaking.
-
60
Transfer 500 μL cells to a sterile cryovial and add 250 μL 50% (vol/vol) sterile glycerol. Mix well and store glycerol stocks at −80 °C.
-
61
For an FSEC expression test, centrifuge the remaining 2 mL cells at 1,200g and 4 °C for 5 min.
-
62
Resuspend the cell pellet in 2 mL BMM medium to induce protein expression. Shake at 220 rpm and 25 °C for 24 h.
-
63
Centrifuge the cells at 1,200g and 4 °C for 5 min.
■PAUSE POINT. Cell pellet can be stored at −80 °C for later analysis.
-
64
Resuspend the cell pellet in 750 μL FSEC lysis buffer and transfer to an Eppendorf tube. Add 200-250 μL of glass beads.
! CAUTION Wear a mask to avoid inhalation of small glass beads. Put a tray under the tube rack to collect spilled glass beads.
-
65
Break Pichia cell wall using a Mixer mill. Grind for 1.5 min with 30 Hz oscillation, and cool down on ice for 3 min. Repeat twice. (Optional) If mixer mill is not available, vortex at top speed for 1.5 min, cool down on ice for 3 min, and repeat twice.
-
66
Transfer 420 μL cell suspension to a new Eppendorf tube and add 60 μL 16% DDM (wt/vol) to lyse cell membrane. Incubate with rotation at 4 °C for at least 1 h.
-
67
(OPTIONAL) For FSEC analysis of protein expression level in HEK293T cells, skip Steps 59-66. Resuspend HEK293T cells (2×106) grown in 6-well plates in 400 μL per well FSEC lysis buffer with 2% DDM. Incubate with rotation at 4 °C for at least 1 h.
-
68
Centrifuge at 21,000g and 4°C for 1 h.
-
69
Transfer 320 μL supernatant to a sample vial supplied with the FSEC system. Load vial onto autosampler kept at 4°C.
-
70
Use FSEC system to equilibrate size-exclusion column (Superdex 200 or Superose 6) with at least 30 mL FSEC buffer.
-
71
Inject 100 μL sample from Step 69 with autosampler. Run isocratic flow at 0.5 mL/min for 60 min. Monitor eGFP fluorescence with excitation at 480 nm and emission at 520 nm.
-
72
Inspect elution profile for peak position, height, and distribution. Well-behaved restrained membrane protein should elute as a symmetrical and sharp peak at expected volume and with relatively high fluorescence.
-
73
Keep the glycerol stock of cells (Step 60) with best elution profile and dispose of the others.
-
74
(Optional) Screen additional 5-6 colonies (Steps 59-73) and select one with the highest protein expression level.
? TROUBLESHOOTING
-
75
To generate duplicate glycerol stocks, inoculate 200 μL glycerol stock from Step 73 to 10 mL YPD or BMG medium. Shake at 220 rpm and 30°C overnight.
-
76
Add 5 mL 50% (vol/vol) sterile glycerol to the culture. Aliquot and store at −80 °C.
Thermostability FSEC ● TIMING 2 d
-
77
With sfGFP used as the coupler, use the constructs with a C-terminal mCherry tag (Step 9) for both restrained and unrestrained constructs (as in Steps 16-29).
▲ CRITICAL STEP Use the mCherry tag to avoid Tm comparisons between split and intact sfGFPs; the Tm difference could not be properly determined if the C-terminal sfGFP tag were to be used for comparison, because C-sfGFP is an intact folded protein, whereas the split sfGFP coupler has weakened folding. With the C-mCherry tag, the sfGFP serves only as the coupler because the GFP fluorescence is not used and the Tm value is determined from the mCherry fluorescence.
-
78
Use either detergent-solubilized cells (Step 69) or purified protein (Step 84) for thermostability tests. For purified protein, adjust the protein concentrations of coupler-restrained and unrestrained membrane proteins to 1 mg/mL. Use a lower concentration (0.05-0.5 mg/mL) if the amount of purified protein is limited. For cell lysate, adjust restrained and unrestrained (without coupler) membrane protein to the same protein concentration based on fluorescence.
! CAUTION Use a relatively high protein concentration for mCherry-tagged proteins because quantum yield of mCherry is lower than GFP.
-
79
Aliquot 100 μL of each protein or cell lysate to 15× 0.2 mL PCR tubes.
-
80
Heat the PCR tubes in a thermal cycler for 10 min at 15 different temperatures: 4°C, 20°C, 30°C, 35°C, 40°C, 45°C, 50°C, 55°C, 60°C, 65°C, 70°C, 75°C, 80°C, 85°C, and 95°C. Simultaneously heat restrained and unrestrained membrane protein at each temperature.
-
81
Transfer the samples to Eppendorf tubes and dilute with 200 μL FSEC buffer. Centrifuge at 21,000g and 4°C for 1 h.
-
82
Analyze samples by FSEC as described in Steps 69-72. Use excitation wavelength at 587 nm and emission at 610 nm for mCherry.
▲ CRITICAL STEP For a precise Tm comparison, analyze consecutively the restrained and unrestrained membrane protein heated at the same temperature (Step 80).
-
83
To calculate Tm, use PeakFit for Gaussian peak fitting. Normalize peak heights at different temperatures to that of the 4°C sample. To obtain Tm values, plot normalized peak heights against temperature and fit with sigmoidal dose-response function installed in Graphpad Prism.
Protein purification ● TIMING 2 d
-
84
Use most suitable method to purify the coupled membrane protein. In our lab we have done this as described in Supplementary Methods Steps 19-28, 33-39 and 45-52.
Analyses of cellular function and activity of purified protein ● TIMING 3-7 d
-
85
If a cellular assay of the target membrane protein function is available, verify the activity of coupler-restrained membrane protein and compare with that of unstrained membrane protein. Transfect or transform the restrained and unrestrained constructs into the cells. Measure enzymatic activity, transporter activity, or molecular interaction of membrane proteins with different functions13. An example of measuring the cellular γ-carboxylation activity of VKOR is provided in Fig. 2a.
-
86
Determine the activity of purified coupler-restrained membrane protein with available assays, and compare it with the activity of the unrestrained membrane protein. An example of measuring the epoxide reductase activity of VKORL is provided in Fig. 2c.
Protein crystallization ● TIMING 1-2 weeks
-
87
Use most suitable method to crystallize the coupled membrane protein. The restrained membrane protein is compatible with LCP crystallization. In our lab we have done this as described in Supplementary Methods Steps 53-76.
-
88
Alternatively, perform vapor diffusion crystallization of the restrained membrane protein in a suitable detergent solution. We have also obtained crystals of sfGFP restrained membrane protein under such conditions.
Crystallographic data collection ● TIMING 1 d
-
89
Identify s suitable synchrotron beamline designed for macromolecular crystallography. Choose beamline equipped with insertion device and microfocus beam to handle LCP crystals.
-
90
Load and center the cryo loop following beamline instructions. Use the yellow or green color to find the crystal of sfGFP-restrained membrane proteins (Fig. 3c).
-
91
Perform a loop scan to identify a position with best diffraction. Rotate crystal by 90° and perform a vertical scan to center the diffraction position.
▲ CRITICAL STEP LCP crystals that are small or buried in lipid bolus are difficult to visualize and center. In some cases, there are multiple crystals in a loop. Locate a single crystal using the diffraction scans, using the green color of the crystal as an aid (Fig. 3c).
-
92
Take two orthogonal snapshots to confirm good diffraction in both directions.
! CAUTION Small LCP crystals require a relatively stronger beam for diffraction and therefore are particularly sensitive to radiation damage. Minimize test shots to lower radiation damage.
Diffraction improvement with termini truncation ● TIMING 1-3 months
-
93
To improve crystal diffraction and quality, generate constructs with a shortened linker sequence between the coupler and the membrane protein. Truncate the two termini of the membrane protein using the following strategy:
Stepwise, truncate 5 amino acids (AA) from the N-terminus and/or C-terminus of the membrane protein. Follow Step 30 to insert the coding sequence of the truncated membrane protein to the coupler in an expression vector. Alternatively, perform the deletion in the existing construct generated in Step 30 using nonoverlapping PCR61.
Express and purify the truncated fusion protein (Steps 32 and 84). Crystallize and assess the crystal diffraction and quality (Steps 87-92).
(Optional) For further improvement, narrow down the truncations to every 3 AA or 1AA.
(Optional) Truncate the coupler sequence flanking the membrane protein insertion site. If the insertion site is part of an unstructured loop, the loop can often be shortened.
Crystal structure determination ● TIMING 1-7 d
▲ CRITICAL Structure determination of coupler-fused membrane proteins often relies on extensive density modification procedures (Fig. 4a). This is because the membrane protein constitutes a significant fraction of the diffraction mass, and therefore the partial phases from molecular replacement solution of the coupler are sometimes insufficient for resolving the electron density of the membrane protein portion. This part of the density can be further improved by reiterating density modification and by a powerful density modification method, cross-crystal averaging, that takes advantage of the crystallographic data available for the coupler protein. In addition, noise reduction is often performed to improve the data quality of LCP crystals.
-
94
(Optional) Reduce noise in the dataset. The LCP crystals frozen in mesophase bolus often show a ring of lipid diffusion diffraction (~4.6 Å). For poorly diffracting crystal, this noise is particularly detrimental to data quality.
In HKL2000, under the top tab ‘Macros’, click [add Macro] in ‘During Scaling’ and enter ‘reject fraction #’ (default 0.75). Increase the reject fraction to reduce the noise from lipid diffusion diffraction.
Alternatively, in XDS input, add a line ‘WFAC1=#’ (default 1.5). Decrease WFAC1 to reduce the noise.
Test a few values of reject fraction or WFAC1. Inspect the change in the plot of I/sigma vs. resolution: the bump of increased diffraction intensity around 4.6 Å should be reduced.
-
95
Obtain molecular replacement solution of the coupler and perform rigid body refinement using the molecular replacement solution. We have done this as described in Supplementary Methods Steps 88-89.
-
96
Perform density modification using automatically generated solvent and averaging masks. We have done this with the PARROT program as described in Supplementary Methods Step 90.
-
97
Inspect the density modified map near the fused point of the coupler: electron densities representing helices from unknown transmembrane region should be clearly visible.
? TROUBLESHOOTING
-
98
Use your favorite program for automatic model building using the density modified map (from Step 96). We recommend BUCCANEER-autobuild/refine62 (described in Supplementary Methods Steps 92-94), because this program can handle maps at relatively low resolution. Confirm and rearrange protein chains. Repeat the autobuilding until there is no further improvement toward a more complete model (2-3 more rounds are generally sufficient).
-
99
Use the model from Step 98 for additional density modification to improve the overall phases and the density map (map from Step 96 may have missing or poor densities). Run rigid body refinement of the new model and perform density modification as in Steps 95-96.
▲ CRITICAL STEP The density maps (2FoFc and FoFc) from the rigid body refinement should be inspected and compared with the density modified maps. The rigid body maps are ‘original’. With the nearly complete model, the partial model phases from rigid body refinement are often sufficient for resolving certain ambiguities.
-
100
Manually generate averaging and solvent masks for fine tuning of density modification. Generate an NCS matrix for each additional molecule. Perform density modification using DM63. In our lab we perform these as described in Supplementary Methods Steps 96-99.
▲ CRITICAL STEP Multidomain averaging often needs to be applied with the coupler as one domain and the fused membrane protein as the other domain, addressing their relative movement in each NCS molecule. Use the preliminary model of the membrane protein molecules (from Step 98) to generate the NCS matrix for each molecule. Create a manually built solvent mask if the automatic mask generated by density modification programs results in artificial removal of the electron density at a certain region. In addition, changing solvent content to a lower value often can recover artificially pruned densities.
-
101
Inspect the DM map in Coot and compare with maps from Steps 96 and 99. If the map is improved, repeat the autobuild (Step 98) with the PHIDM and FOMDM phases. Alternatively, missing parts of the model can be manually built to fit the densities of DM map. Repeat Steps 98-101 until no further improvement in model building or density map.
-
102
(Optional) For low-resolution or low-quality data, improve density maps by multi crystal averaging.
Download the existing model and diffraction data of the coupler protein from PDB.
Create an averaging mask on one molecule of the coupler (as in Step 100).
(Optional) Create an averaging mask on one molecule of the preliminary model of the membrane protein if NCS is present in this coupler-fused crystal.
Generate an “NCS” matrix (as in Step 100) for each molecule in the two structural models: coupler alone and coupler fused with the membrane protein. Use separate NCS matrices for multi-domain cross-crystal averaging to account for the relative motions between coupler and membrane protein in different NCS molecules.
Generate the model phases for the coupler-only crystal using rigid body refinement (as in Step 95) with the downloaded model and data of the coupler as inputs.
Under the CCP4i program list, select the top tab ‘Density Improvement’ and ‘Run DmMulti’.
Select a phase extension scheme (as described in Supplementary Methods Step 99-ii).
In ‘Crystal number 1’, input the .mtz generated from rigid body refinement of the coupler crystal, using data labels FP, SIGFP, PHWT and FOM. In ‘Crystal number 2’, input the .mtz generated from rigid body refinement of the molecular replacement model of the coupler against the data of coupler fused with membrane protein (from Step 95), or the .mtz generated from the rigid body refinement of the model with membrane protein portion partially built (Step 98).
In ‘Describe domains’, enter the averaging masks.
In ‘Describe Crystals’ and ‘Describe Crystal 1’ enter the solvent content (calculated as in Supplementary Methods Step 87) and the NCS matrix of the coupler-only crystal (enter 0 numbers if there is only one molecule). In ‘Describe Crystal 2’, enter the solvent content for the crystal of the coupler fused with the membrane protein.
Inspect the NCS correlation (as in Supplementary Methods Step 99) for each domain in each crystal.
Compare DMMULTI maps generated with different phase extension schemes and parameters.
-
103
Manually build the model to completeness and refine the model. Use the 2FoFc and FoFc maps to correct the model or build missing part. Repeat until Rfree, Ramachandran, and other validation requirements are satisfactorily met.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Selection of suitable couplers and design of restrained constructs
Step 1-14: 1 d
Generation of fusion constructs of membrane protein with coupler by seamless cloning
Steps 15-29: 1 week
Steps 30-31: 1 week
Protein expression
Step 32: 1 d - 6 weeks
Live cell imaging of Pichia cells
Steps 33-35: 4 h
Live cell imaging of HEK293 cells
Steps 36-38: 2 d
Whole cell fractionation of Pichia cells
Steps 39-41: 2 h
Steps 42-54: 4 d
Whole cell fractionation for HEK293 cells
Steps 55-58: 2 d
FSEC analysis of protein expression level in Pichia and HEK293 cells
Steps 59-76: 5-7 d
Thermostability FSEC
Steps 77-83: 2 d
Protein purification
Step 84: 2 d
Analyses of cellular function and activity of purified protein
Steps 85-86: 3-7 d
Protein crystallization
Steps 87-88: 1-2 weeks
Crystallographic data collection
Steps 89-92: 1 d
Diffraction improvement with termini truncation
Step 93: 1-3 months
Crystal structure determination
Steps 94-103: 1-7 d
Anticipated results
Terminal-restrained membrane proteins should have increased stability and higher effective yield than their unrestrained counterparts, and their crystal structures at near-atomic resolution can often be obtained. As an example, we have restrained VKORL, an integral membrane oxidoreductase, with split sfGFP. We first conduct a cellular carboxylation assay to show that the restraining does not compromise VKORL activity and sensitivity to its inhibitor, warfarin (Fig. 2a). After protein purification, the activity and inhibition of VKORL is analyzed again by a fluorometric assay, which shows that the specific activity of restrained VKORL is five times higher than that of the unrestrained protein (Fig. 2c). To access potential protein misfolding or aggregation during overexpression in cells, we compared the sfGFP-restrained VKORL with C-terminal GFP-tagged VKORL by fluorescence imaging13. We found that the restraining does not change the localization of VKORL in the endoplasmic reticulum (ER) membrane (Fig. 2a). Moreover, the reconstituted fluorescence of sfGFP indicates that VKORL folds in its predicted 4-TM topology. The sfGFP-restrained VKORL distributes evenly at the ER membrane boundary, whereas C-GFP-tagged VKORL shows accumulation in the ER and/or vacuoles. Whole-cell extraction shows that a large portion of unrestrained VKORL is expressed in aggregated or insoluble form (Fig. 2b). In contrast, similar amount of the restrained VKORL are obtained with DDM, SDS and SDS/urea extractions, indicating that most restrained VKORL are well folded and the restraining has prevented protein aggregation during overexpression. Consistently, FSEC measurements show that the Tm of VKORL is increased by 8.8°C after the restraining; this increased thermostability indicates that restrained VKORL is more stably folded and less prone to aggregate.
VKORL fused to the sfGFP coupler has been readily crystallized with the LCP method. Fluorescence imaging identifies the initial crystallization hits (Fig. 3a), and optimization of crystallization buffer conditions leads to sizable crystals (Fig. 3b). Subsequently, the termini of VKORL are truncated to change the relative orientation and/or motion flexibility between VKORL and sfGFP (Fig. 3d), thereby altering crystal packing (Fig. 3e). The diffraction limit is improved from 4.3 Å to 2.4 Å after the truncation (Fig. 3f). The density map generated from the high-resolution data allows autobuilding of 98% of the structural model of restrained VKORL.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work is supported by the W. M. Keck Foundation (Forefront of Science Award), NHLBI (R01 HL121718), Children’s Discovery Institute (MCII 2020-854), NEI (R21 EY028705), NIGMS (R01 GM131008), and American Heart Association (20CSA35310354) to W. L. Crystallographic data collection used NE-CAT beamlines (GM124165), a Pilatus detector (RR029205) and an Eiger detector.
Footnotes
PROPOSED TWEET #NewNProt describes a strategy to stabilize integral membrane proteins for crystal structures and functional studies.
PROPOSED TEASER Termini restrain membrane protein for structures.
Competing interests
The authors declare no competing interests.
Data availability
The data presented in this protocol have been previously published13–17, and associated raw data are provided in the original articles.
References
- 1.Uhlen M et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419, doi: 10.1126/science.1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B & Hopkins AL How many drug targets are there? Nat Rev Drug Discov 5, 993–996, doi: 10.1038/nrd2199 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Fagerberg L, Jonasson K, von Heijne G, Uhlen M & Berglund L Prediction of the human membrane proteome. Proteomics 10, 1141–1149, doi: 10.1002/pmic.200900258 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Yildirim MA, Goh KI, Cusick ME, Barabasi AL & Vidal M Drug-target network. Nat Biotechnol 25, 1119–1126, doi: 10.1038/nbt1338 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Bill RM et al. Overcoming barriers to membrane protein structure determination. Nat Biotechnol 29, 335–340, doi: 10.1038/nbt.1833 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Carpenter EP, Beis K, Cameron AD & Iwata S Overcoming the challenges of membrane protein crystallography. Curr Opin Struct Biol 18, 581–586, doi: 10.1016/j.sbi.2008.07.001 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JL & Tate CG Engineering an ultra-thermostable beta(1)-adrenoceptor. J Mol Biol 413, 628–638, doi: 10.1016/j.jmb.2011.08.057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serrano-Vega MJ, Magnani F, Shibata Y & Tate CG Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. U. S. A 105, 877–882, doi: 10.1073/pnas.0711253105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tate CG A crystal clear solution for determining G-protein-coupled receptor structures. Trends Biochem. Sci 37, 343–352, doi: 10.1016/j.tibs.2012.06.003 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Magnani F et al. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat Protoc 11, 1554–1571, doi: 10.1038/nprot.2016.088 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Zorzi R, Mi W, Liao M & Walz T Single-particle electron microscopy in the study of membrane protein structure. Microscopy (Oxf) 65, 81–96, doi: 10.1093/jmicro/dfv058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gauto DF et al. Integrated NMR and cryo-EM atomic-resolution structure determination of a half-megadalton enzyme complex. Nat Commun 10, 2697, doi: 10.1038/s41467-019-10490-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu S, Li S, Yang Y & Li W Termini restraining of small membrane proteins enables structure determination at near-atomic resolution. Sci Adv 6, doi: 10.1126/sciadv.abe3717 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S et al. Structural basis of antagonizing the vitamin K catalytic cycle for anticoagulation. Science 371, doi: 10.1126/science.abc5667 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y et al. Open conformation of tetraspanins shapes interaction partner networks on cell membranes. EMBO J 39, e105246, doi: 10.15252/embj.2020105246 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S, Liu S, Liu XR, Zhang MM & Li W Competitive tight-binding inhibition of VKORC1 underlies warfarin dosage variation and antidotal efficacy. Blood Adv 4, 2202–2212, doi: 10.1182/bloodadvances.2020001750 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S, Liu S, Yang Y & Li W Characterization of Warfarin Inhibition Kinetics Requires Stabilization of Intramembrane Vitamin K Epoxide Reductases. J. Mol. Biol 432, 5197–5208, doi: 10.1016/j.jmb.2020.05.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daley DO et al. Global topology analysis of the Escherichia coli inner membrane proteome. Science (New York, N.Y.) 308, 1321–1323, doi: 10.1126/science.1109730 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Kim H, Melen K, Osterberg M & von Heijne G A global topology map of the Saccharomyces cerevisiae membrane proteome. Proceedings of the National Academy of Sciences 103, 11142–11147, doi: 10.1073/pnas.0604075103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W & Li F Cross-crystal averaging with search models to improve molecular replacement phases. Structure 19, 155–161, doi: 10.1016/j.str.2010.12.007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y, Morais-Cabral JH, Kaufman A & MacKinnon R Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 414, 43–48, doi: 10.1038/35102009 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Kim JM et al. Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature 517, 396–400, doi: 10.1038/nature13872 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenbaum DM et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273, doi: 10.1126/science.1150609 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Chun E et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976, doi: 10.1016/j.str.2012.04.010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baird GS, Zacharias DA & Tsien RY Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci U S A 96, 11241–11246, doi: 10.1073/pnas.96.20.11241 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cabantous S, Terwilliger TC & Waldo GS Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol 23, 102–107, doi: 10.1038/nbt1044 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Pierre B, Xiong T, Hayles L, Guntaka VR & Kim JR Stability of a guest protein depends on stability of a host protein in insertional fusion. Biotechnol. Bioeng 108, 1011–1020, doi: 10.1002/bit.23039 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Palanisamy N et al. Split intein-mediated selection of cells containing two plasmids using a single antibiotic. Nat Commun 10, 4967, doi: 10.1038/s41467-019-12911-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foit L et al. Optimizing protein stability in vivo. Mol Cell 36, 861–871, doi: 10.1016/j.molcel.2009.11.022 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang R et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535, 164–168, doi: 10.1038/nature18625 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu Q, Piai A, Chen W, Xia K & Chou JJ Structure determination protocol for transmembrane domain oligomers. Nat Protoc 14, 2483–2520, doi: 10.1038/s41596-019-0188-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagn F, Nasr ML & Wagner G Assembly of phospholipid nanodiscs of controlled size for structural studies of membrane proteins by NMR. Nat Protoc 13, 79–98, doi: 10.1038/nprot.2017.094 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watzka M et al. Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: insights into improved patient diagnosis and treatment. J. Thromb. Haemost 9, 109–118, doi: 10.1111/j.1538-7836.2010.04095.x (2011). [DOI] [PubMed] [Google Scholar]
- 34.Fasco MJ, Principe LM, Walsh W. a. & Friedman P. a. Warfarin inhibition of vitamin K 2,3-epoxide reductase in rat liver microsomes. Biochemistry 22, 5655–5660 (1983). [DOI] [PubMed] [Google Scholar]
- 35.Hodroge A, Longin-Sauvageon C, Fourel I, Benoit E & Lattard V Biochemical characterization of spontaneous mutants of rat VKORC1 involved in the resistance to antivitamin K anticoagulants. Arch. Biochem. Biophys 515, 14–20, doi: 10.1016/j.abb.2011.08.010 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Bevans CG et al. Determination of the warfarin inhibition constant Ki for vitamin K 2,3-epoxide reductase complex subunit-1 (VKORC1) using an in vitro DTT-driven assay. Biochim. Biophys. Acta 1830, 4202–4210, doi: 10.1016/j.bbagen.2013.04.018 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Schwarzer D & Cole PA Protein semisynthesis and expressed protein ligation: chasing a protein’s tail. Curr Opin Chem Biol 9, 561–569, doi: 10.1016/j.cbpa.2005.09.018 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Li C et al. FastCloning: A highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 11, 92–92, doi: 10.1186/1472-6750-11-92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawate T & Gouaux E Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure (London, England : 1993) 14, 673–681, doi: 10.1016/j.str.2006.01.013 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Lee H & Kim H Membrane topology of transmembrane proteins: determinants and experimental tools. Biochem. Biophys. Res. Commun 453, 268–276, doi: 10.1016/j.bbrc.2014.05.111 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Alexandrov AI, Mileni M, Chien EY, Hanson MA & Stevens RC Microscale fluorescent thermal stability assay for membrane proteins. Structure 16, 351–359, doi: 10.1016/j.str.2008.02.004 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Thomas JA & Tate CG Quality control in eukaryotic membrane protein overproduction. J Mol Biol 426, 4139–4154, doi: 10.1016/j.jmb.2014.10.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goehring A et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nature Protocols 9, 2574–2585, doi: 10.1038/nprot.2014.173 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miroux B & Walker JE Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260, 289–298, doi: 10.1006/jmbi.1996.0399 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Wagner S et al. Tuning Escherichia coli for membrane protein overexpression. Proc Natl Acad Sci U S A 105, 14371–14376, doi: 10.1073/pnas.0804090105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caffrey M & Cherezov V Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4, 706–731, doi: 10.1038/nprot.2009.31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tusnády GE, Kalmár L & Simon I TOPDB: Topology data bank of transmembrane proteins. Nucleic Acids Res. 36, doi: 10.1093/nar/gkm751 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones DT Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23, 538–544, doi: 10.1093/bioinformatics/btl677 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Nugent T & Jones DT Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics 10, 159, doi: 10.1186/1471-2105-10-159 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Käll L, Krogh A & Sonnhammer ELL Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 35, doi: 10.1093/nar/gkm256 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krogh a., Larsson B, von Heijne G & Sonnhammer EL Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol 305, 567–580, doi: 10.1006/jmbi.2000.4315 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Reddy A et al. Reliability of nine programs of topological predictions and their application to integral membrane channel and carrier proteins. J Mol Microbiol Biotechnol 24, 161–190, doi: 10.1159/000363506 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kulzer S, Petersen W, Baser A, Mandel K & Przyborski JM Use of self-assembling GFP to determine protein topology and compartmentalisation in the Plasmodium falciparum-infected erythrocyte. Mol Biochem Parasitol 187, 87–90, doi: 10.1016/j.molbiopara.2012.11.004 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Toddo S et al. Application of split-green fluorescent protein for topology mapping membrane proteins in Escherichia coli. Protein Sci 21, 1571–1576, doi: 10.1002/pro.2131 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doi N & Yanagawa H Design of generic biosensors based on green fluorescent proteins with allosteric sites by directed evolution. FEBS Lett 453, 305–307, doi: 10.1016/s0014-5793(99)00732-2 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Ghosh I, Hamilton AD & Regan L Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein [12]. J. Am. Chem. Soc 122, 5658–5659, doi: 10.1021/ja994421w (2000). [DOI] [Google Scholar]
- 57.Romei MG & Boxer SG Split Green Fluorescent Proteins: Scope, Limitations, and Outlook. Annu Rev Biophys 48, 19–44, doi: 10.1146/annurev-biophys-051013-022846 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones DT Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292, 195–202, doi: 10.1006/jmbi.1999.3091 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Oates ME et al. D(2)P(2): database of disordered protein predictions. Nucleic Acids Res 41, D508–516, doi: 10.1093/nar/gks1226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen X, Zaro JL & Shen WC Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 65, 1357–1369, doi: 10.1016/j.addr.2012.09.039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng L, Baumann U & Reymond JL An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32, e115, doi: 10.1093/nar/gnh110 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cowtan KD Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr 66, 470–478, doi: 10.1107/S090744490903947X (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cowtan KD ‘dm’: An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography 31, 34–38 (1994). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this protocol have been previously published13–17, and associated raw data are provided in the original articles.