

Abstract
The C–H arylation of 2-quinolinecarboxyamide bearing a C–Br bond at the N-aryl moiety is carried out with a palladium catalyst. The reaction proceeds at the C–H bond on the pyridine ring adjacent to the amide group in the presence of 10 mol % Pd(OAc)2 at 110 °C to afford the cyclized product in 42% yield. The yield is improved to 94% when the reaction is performed with PPh3 as a ligand of palladium. The reaction is examined with amides derived from unsubstituted picoline, 6-methylpicoline, and 2,6-pyridinedicarboxylic acid in a similar manner to afford the cyclized products in 70%, 77%, and 87% yield, respectively. The related reaction is also carried out with amides of non-pyridine derivatives terephthal- and benzamides to afford multiply fused heterocyclic compounds in 81% and 89% yields, respectively.
Keywords: intramolecular C–H arylation, multiply fused heterocycles, palladium acetate, phosphine ligand, pyridine amides
Introduction
Transition-metal-catalyzed synthetic reactions have recently attracted much attention in synthetic organic chemistry [1–2]. C–H Arylation reactions catalyzed by a transition metal are of particular interest because these reactions involve rather superior efficiencies in atom economy [3–4]. The extension of the reaction to an intramolecular version represents a viable approach for the construction of several fused-ring skeletons [5]. Such ring structures containing heterocyclic rings would be of crucial importance because heterocycle-fused ring structures [6–7] are found in a variety of advanced materials [8–9] and biologically important molecules [10–12]. A wide range of pyridine derivatives have been employed as extractants of metal ions through chelation [13]. Phenanthrolines, a class of pyridine derivatives, have attracted attention for the efficient and selective extraction of lanthanides and actinides and, furthermore, a number of heterocycles involving pyridine rings have been reported to exhibit biological activities [14–22]. We have recently reported, as shown in Figure 1, that the introduction of a multiply fused structure toward a phenanthroline diamide (Phen-2,9-diamide) [23] can be achieved by employing a palladium-catalyzed intramolecular C–H arylation [24–28]. One of the thus obtained products exhibited a remarkable extraction performance for a lanthanide ion, in which a metal-specific extraction was found despite the similarities in the lanthanide series [23]. Chakravorty and co-workers reported that a similar arylation reaction gave access to the fused skeleton of the diamide of 2,6-pyridinedicarboxlic acid (Py-2,6-diamide) [29]. Our interest has thus turned to extend the substrate scope of the palladium-catalyzed C–H arylation of phenanthroline to other nitrogen-containing heteroaromatic compounds. It is therefore intriguing to demonstrate the advantage of the palladium-catalyzed intramolecular C–H arylation compared to other protocols for the construction of related ring structures [30–32]. We herein report the palladium-catalyzed intramolecular C–H arylation of several pyridine and non-pyridine amides to afford multiply fused heterocyclic compounds.
Figure 1.

Structures of multiply fused heterocyclic compounds composed of pyridine rings.
Results and Discussion
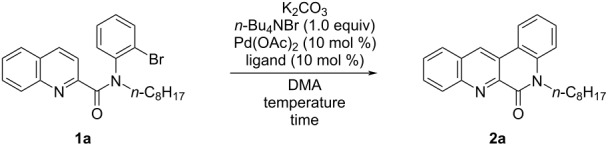
First, we started with the synthesis of the cyclization precursors 1a–c that was carried out by the reaction of the corresponding heteroaromatic carboxylic acids with thionyl chloride followed by treatment with N-octyl-2-bromoaniline [15]. The reactions proceeded smoothly affording products 1a–c in good yields as shown in Scheme 1.
Scheme 1.

Synthesis of C–H arylation precursors 1a–c.
We then studied the reaction of quinoline amide 1a under several conditions. We carried out the palladium-catalyzed intramolecular coupling reaction of precursor 1a under similar conditions [23], which afforded smooth reaction with phenanthroline bisamide, with 10 mol % of palladium acetate as a catalyst in the presence of potassium carbonate and tetra-n-butylammonium bromide in N,N-dimethylacetamide (DMA). Table 1 summarizes the results. The yield of the reaction improved as the temperature was increased from 90 °C to 130 °C (Table 1, entries 1–3). When the reaction was carried out at 150 °C, the yield decreased to 27%. A longer reaction period of 72 h at 130 °C also resulted in a decreased yield (27%) (Table 1, entries 4 and 5). It was found that increasing the amount of potassium carbonate to a three-fold excess improved the yield of 2a to 59% in the reaction at 110 °C shown in entry 6 of Table 1. Next, the effect of the ligand of the palladium catalyst was examined. The addition of ligand improved the yield of 2a as shown in Table 1, entries 7–10. Among several ligands, including Buchwald-type phosphines L1–L4 [33] examined, it was found that the use CyJohnPhos (L3) afforded the cyclized product in 90% yield and the reaction with PPh3 (L4) as a ligand was also effective to afford 2a in 94% yield.
Table 1.
Studies on the reaction conditions for 2a from 1a.
| |||||
|
| |||||
| Entry | Temp. (°C) | K2CO3 (equiv) | Time (h) | Ligand | Yield (%)a |
|
| |||||
| 1 | 90 | 1.0 | 24 | none | 7b |
| 2 | 110 | 1.0 | 24 | none | 42 |
| 3 | 130 | 1.0 | 24 | none | 49 |
| 4 | 150 | 1.0 | 24 | none | 27 |
| 5 | 130 | 1.0 | 72 | none | 27b |
| 6 | 110 | 3.0 | 24 | none | 59 |
| 7 | 110 | 3.0 | 24 | L1 c | 58 |
| 8 | 110 | 3.0 | 24 | L2 d | 69 |
| 9 | 110 | 3.0 | 24 | L3 e | 90 |
| 10 | 110 | 3.0 | 24 | L4 f | 94 (87b) |
aYield determined by 1H NMR with 1,1,2,2-tetrachloroethane as an internal standard; bisolated yield; cL1: SPhos = 2-dicyclohexylphosphino-2’,6’-dimethoxybiphenyl; dL2: PCy3 = tricyclohexylphosphine; eL3: CyJohnPhos = 2-(dicyclohexylphosphino)biphenyl; fL4: PPh3 = triphenylphosphine.
The reaction was then carried out with several pyridine derivatives including amide derivatives composed of 6-methylpicoline (1b) and unsubstituted picoline (1c) as summarized in Table 2. When the reaction was examined in the absence of a phosphine ligand, the yields of the cyclized products 2b and 2c were much worse compared to the same reaction of 1a. In the latter case product 2a was obtained in 59% yield, whereas the yields for 2b and 2c were only 18% and 5%, respectively. The use of PPh3 (L4) as a ligand slightly improved the yields of 2b and 2c to 58% and 24%, respectively. The highest yield of 2b was obtained in the presence of CyJohnPhos (L3) as ligand, while tricyclohexylphosphine (L2) gave the best yield in the reaction of 1c. Concerning the reaction of 1c, the use of tetra-n-butylammonium bromide and pivalic acid as additives and PCy3 (L2) as a ligand further improved the yield to 77%. Chakravorty and co-workers showed that a smooth reaction proceeded with pyridine 2,6-dicarboxylic acid bisamide 3 [29] and we thus compared the reaction of 3 under similar conditions to that of 1a. The reaction afforded product 4 in 87% yield, which was found to be comparable with the case of 1a. The reactivity toward the palladium-catalyzed cyclization was thus shown as 3 ≈ 1a >> 1b > 1c. The related trend was also observed in the reaction of phenanthroline monoamide 5a and diamide 5b. The reaction of 5a afforded the cyclized product in 51% yield, which contrasted with our previous result for the cyclization of 5b to afford the doubly cyclized product 6b (reported yield: 85% [23]), suggesting that the superior reactivity was found for bifunctional bisamides compared to monoamides.
Table 2.
Pd-catalyzed C–H arylation of heteroarenes.
| ||||
|
| ||||
| Substrate | Conc.a | Ligand | Product | Yieldb |
|
| ||||
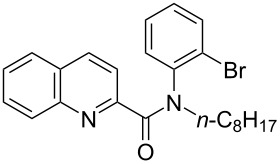
1a |
0.033 | none |
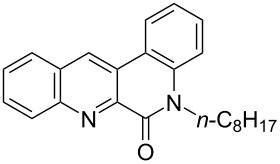
2a |
59% |
| 1a | 0.033 | L4 | 2a | 94% (87%) |
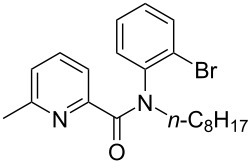
1b |
0.033 | none |
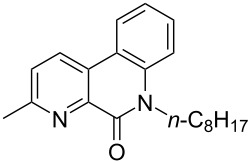
2b |
18% |
| 1b | 0.033 | L2 | 2b | 47% |
| 1b | 0.033 | L4 | 2b | 58% |
| 1b | 0.033 | L3 | 2b | 70% (52%) |
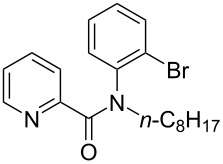
1c |
0.033 | none |
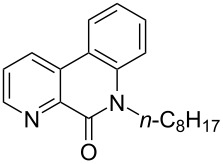
2c |
(5%) |
| 1c | 0.067 | none | 2c | 11% |
| 1c | 0.067 | L4 | 2c | 24% |
| 1c | 0.067 | L3 | 2c | 33% |
| 1c | 0.067 | L2 | 2c | 42% |
| 1c | 0.067 | L2 | 2c | 77%c (62%) |
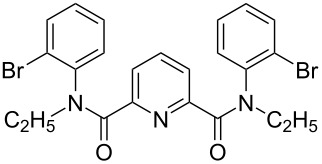
3 |
0.032 | none |
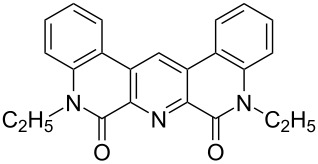
4 |
87%d |
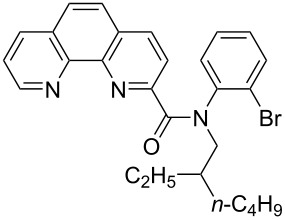
5a |
0.032 | none |
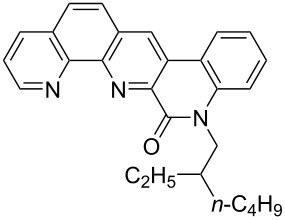
6a |
(51%) |
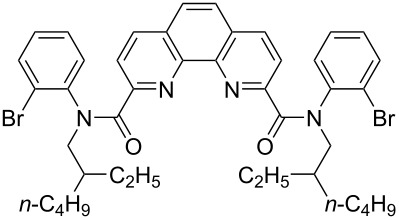
5b |
0.032 | none |
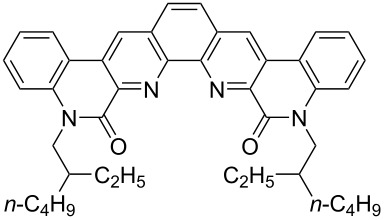
6b |
85%d,e |
aSubstrate/DMA (mol/L); byield determined by 1H NMR with 1,1,2,2-tetrachloroethane as an internal standard and isolated yield is given in parenthesis; cn-Bu4NBr/t-BuCOOH 1:1 was used as an additive; d2.0 equiv of n-Bu4NBr was used; eresult taken from [23].
It was also found that the reaction also is applicable to a carbocyclic amide derivative. When the reaction was carried out with 7a under similar conditions, the cyclization occurred to afford 8a in 81% yield as shown in Scheme 2. The formation of 8a was confirmed by X-ray crystallographic analysis (CCDC 2227450). The related monofunctionalized analog 7b also smoothly underwent cyclization to afford 8b in 89% yield under similar conditions, in which the result of carbocyclic amide (7a vs 7b) contrasted with the case of heterocyclic ones, 1c vs 3 and 5a vs 5b.
Scheme 2.

Palladium-catalyzed intramolecular direct arylation for synthesizing 8a and 8b and the X-ray crystallographic structure of 8a.
Conclusion
We have shown the facile synthesis of fused nitrogen-containing heterocycles and extended the scope of the intramolecular palladium catalyzed C–H arylation to pyridine derivatives. The cyclization reaction proceeded in a moderate to excellent yield when an appropriate phosphine ligand was employed. The reaction is expected to be useful for the synthesis of functional materials, and bioactive molecules in a facile manner.
Experimental
Typical experimental procedure for the C–H arylation of pyridine derivative 5-octyldibenzo[b,f][1,7]naphthyridin-6(5H)-one (2a): To a screw-capped test tube equipped with a magnetic stirring bar were added amide 1a (44.1 mg, 0.100 mmol), potassium carbonate (42.0 mg, 0.304 mmol), tetrabutylammonium bromide (31.7 mg, 0.098 mmol), Pd(OAc)2 (2.2 mg, 10 mol %), and triphenylphosphine (2.8 mg, 10 mol %). The mixture was dissolved in 3.1 mL of DMA and stirring was continued at 110 °C for 24 h. Then, water (3 mL) was added after cooling to room temperature. The product was extracted with dichloromethane (2 mL) three times. The combined organic extracts were repeatedly washed with water (20 mL) and brine (20 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give a crude material, which was purified by silica gel column chromatography (hexane/MeOAc 1:1) to give 31.0 mg (87% yield) of 2a as a colorless solid. (NMR yield: 94%); mp 85.1–86.6 °C; 1H NMR (CDCl3) δ 8.98 (s, 1H), 8.43 (d, J = 8.4 Hz, 1H), 8.31 (dd, J = 8.0, 1.2 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.76 (ddd, J = 8.0, 7.6, 1.2 Hz, 1H), 7.62 (ddd, J = 7.6, 7.6, 1.2 Hz, 1H), 7.54 (ddd, J = 8.4, 8.0, 1.2 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.30 (dd, J = 8.0, 7.6 Hz, 1H), 4.40 (dd, J = 8.0, 7.6 Hz, 2H), 1.76–1.88 (m, 2H), 1.44–1.56 (m, 2H), 1.18–1.42 (m, 8H), 0.86 (t, J = 6.8 Hz, 3H); 13C{1H} NMR (CDCl3) δ 160.1, 148.4, 142.0, 136.7, 131.1, 130.5, 130.2, 130.1, 129.1, 128.7, 127.7, 126.3, 123.8, 122.7, 118.3, 115.4, 43.3, 31.9, 29.5, 29.3, 27.3, 27.1, 22.7, 14.2; IR (ATR): 2959, 2929, 2856, 1661, 751 cm−1; HRMS–DART+ (m/z): [M + H]+ calcd for C24H27N2O, 359.2123; found, 359.2134.
Supporting Information
Accession code CCDC 2227450 contains the supplementary crystallographic data for 8a. This data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures, or by emailing data_request@ccdc.cam.ac.uk, or by contacting Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Additional experimental details and copies of 1H and 13C{1H} NMR spectra.
Acknowledgments
We thank Professor Shigeki Mori and Ms. Rimi Konishi (Ehime University) for the X-ray crystallographic analysis.
Data Availability
Additional research data generated and analyzed during this study is not shared.
References
- 1.Diederich F, Stang P J, editors. Metal-Catalyzed Cross-Coupling Reactions. Weinheim, Germany: Wiley-VCH; 1998. [DOI] [Google Scholar]
- 2.de Meijere A, Bräse S, Oestreich M, editors. Metal-Catalyzed Cross-Coupling Reactions and More. Weinheim, Germany: Wiley-VCH; 2014. [DOI] [Google Scholar]
- 3.Bohra H, Wang M. J Mater Chem A. 2017;5:11550–11571. doi: 10.1039/c7ta00617a. [DOI] [Google Scholar]
- 4.Albano G, Punzi A, Capozzi M A M, Farinola G M. Green Chem. 2022;24:1809–1894. doi: 10.1039/d1gc03168f. [DOI] [Google Scholar]
- 5.Hartwig J F. Pure Appl Chem. 1999;71:1417–1423. doi: 10.1351/pac199971081417. [DOI] [Google Scholar]
- 6.Ganguly A K, Wang C H, David M, Bartner P, Chan T M. Tetrahedron Lett. 2002;43:6865–6868. doi: 10.1016/s0040-4039(02)01537-x. [DOI] [Google Scholar]
- 7.Das S, Kundu S, Metya A, Maji M S. Chem Sci. 2024;15:13466–13474. doi: 10.1039/d4sc03438d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Z, Yan C, Zhu W-H. Angew Chem, Int Ed. 2020;59(25):9812–9825. doi: 10.1002/anie.201913249. [DOI] [PubMed] [Google Scholar]
- 9.Yu C P, Yamamoto A, Kumagai S, Takeya J, Okamoto T. Angew Chem, Int Ed. 2023;62:e202206417. doi: 10.1002/anie.202206417. [DOI] [PubMed] [Google Scholar]
- 10.Michael J P. Nat Prod Rep. 2002;19:742–760. doi: 10.1039/b104971m. [DOI] [PubMed] [Google Scholar]
- 11.Shang X-F, Morris‐Natschke S L, Liu Y-Q, Guo X, Xu X-S, Goto M, Li J-C, Yang G-Z, Lee K-H. Med Res Rev. 2018;38(3):775–828. doi: 10.1002/med.21466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yadav T T, Murahari M, Peters G J, YC M. Eur J Med Chem. 2022;239:114527. doi: 10.1016/j.ejmech.2022.114527. [DOI] [PubMed] [Google Scholar]
- 13.Shimada A, Yaita T, Narita H, Tachimori S, Kimura T, Okuno K, Nakano Y. Solvent Extr Res Dev, Jpn. 2004;11:1–10. [Google Scholar]
- 14.Xiao C-L, Wang C-Z, Yuan L-Y, Li B, He H, Wang S, Zhao Y-L, Chai Z-F, Shi W-Q. Inorg Chem. 2014;53(3):1712–1720. doi: 10.1021/ic402784c. [DOI] [PubMed] [Google Scholar]
- 15.Alyapyshev M, Ashina J, Dar’in D, Kenf E, Kirsanov D, Tkachenko L, Legin A, Starova G, Babain V. RSC Adv. 2016;6(73):68642–68652. doi: 10.1039/c6ra08946a. [DOI] [Google Scholar]
- 16.Nakase M, Kobayashi T, Shiwaku H, Suzuki S, Grimes T S, Mincher B J, Yaita T. Solvent Extr Ion Exch. 2018;36:633–646. doi: 10.1080/07366299.2018.1532137. [DOI] [Google Scholar]
- 17.Healy M R, Ivanov A S, Karslyan Y, Bryantsev V S, Moyer B A, Jansone‐Popova S. Chem – Eur J. 2019;25(25):6326–6331. doi: 10.1002/chem.201806443. [DOI] [PubMed] [Google Scholar]
- 18.Simonnet M, Suzuki S, Miyazaki Y, Kobayashi T, Yokoyama K, Yaita T. Solvent Extr Ion Exch. 2020;38:430–440. doi: 10.1080/07366299.2020.1744806. [DOI] [Google Scholar]
- 19.Simonnet M, Kobayashi T, Shimojo K, Yokoyama K, Yaita T. Inorg Chem. 2021;60:13409–13418. doi: 10.1021/acs.inorgchem.1c01729. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Yang X, Xu L, Miao Y, Yang X, Xiao C. Ind Eng Chem Res. 2023;62:15613–15624. doi: 10.1021/acs.iecr.3c02101. [DOI] [Google Scholar]
- 21.Wang S, Yang X, Xu L, Xiao C. Ind Eng Chem Res. 2024;63:10773–10781. doi: 10.1021/acs.iecr.4c01205. [DOI] [Google Scholar]
- 22.Pramanik S, Li B, Driscoll D M, Johnson K R, Evans B R, Damron J T, Ivanov A S, Jiang D-e, Einkauf J, Popovs I, et al. J Am Chem Soc. 2024;146(37):25669–25679. doi: 10.1021/jacs.4c07332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asano T, Nakanishi Y, Sugita S, Okano K, Narita H, Kobayashi T, Yaita T, Mori A. ChemRxiv. 2024 doi: 10.26434/chemrxiv-2024-8lpmz. [DOI] [Google Scholar]
- 24.García-Cuadrado D, Braga A A C, Maseras F, Echavarren A M. J Am Chem Soc. 2006;128:1066–1067. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]
- 25.Takagi K, Maeda A, Tsunekawa R. Results Chem. 2022;4:100432. doi: 10.1016/j.rechem.2022.100432. [DOI] [Google Scholar]
- 26.Campeau L-C, Parisien M, Leblanc M, Fagnou K. J Am Chem Soc. 2004;126:9186–9187. doi: 10.1021/ja049017y. [DOI] [PubMed] [Google Scholar]
- 27.Campeau L-C, Parisien M, Jean A, Fagnou K. J Am Chem Soc. 2006;128:581–590. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- 28.Liégault B, Petrov I, Gorelsky S I, Fagnou K. J Org Chem. 2010;75(4):1047–1060. doi: 10.1021/jo902515z. [DOI] [PubMed] [Google Scholar]
- 29.Majumdar K C, De N, Chakravorty S. Synth Commun. 2010;41:121–130. doi: 10.1080/00397910903531870. [DOI] [Google Scholar]
- 30.Benson S C, Gross J L, Snyder J K. J Org Chem. 1990;55:3257–3269. doi: 10.1021/jo00297a050. [DOI] [Google Scholar]
- 31.Chen S, Priebbenow D L, Somkhit J, Scullino C V, Agama K, Pommier Y, Flynn B L. Chem – Eur J. 2022;28:e202201925. doi: 10.1002/chem.202201925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hassanien A E. Synth Commun. 2024;54:1652–1664. doi: 10.1080/00397911.2024.2397814. [DOI] [Google Scholar]
- 33.Surry D S, Buchwald S L. Angew Chem, Int Ed. 2008;47:6338–6361. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional experimental details and copies of 1H and 13C{1H} NMR spectra.
Data Availability Statement
Additional research data generated and analyzed during this study is not shared.