

Abstract
Bacterial infections cause high morbidity and mortality worldwide, and the emergence of drug-resistant bacteria further complicates the treatment of infections. Therefore, it is necessary to continuously develop new treatment methods. Polymyxin B (PMB), as the last line of defense, can combat most aerobic Gram-negative bacilli including common drug-resistant bacteria in clinical practice. However, the suboptimal lung tissue concentration of PMB and dose-dependent nephrotoxicity and neurotoxicity limit its clinical application. The nanodrug delivery system offers several key advantages, including high drug loading capacity, excellent biocompatibility, controlled release mechanisms, and targeted delivery. These features enhance the bioavailability of drugs while simultaneously reducing their toxicity and minimizing side effects. In this study, we designed a targeted nanodrug delivery system (PMB@HMnO2@NM) consisting of hollow mesoporous manganese dioxide (HMnO2) coated with neutrophil membrane (NM). In a mouse model of acute pneumonia induced by multidrug-resistant Pseudomonas aeruginosa, treatment with PMB@HMnO2@NM demonstrated the ability to target bacterial aggregation and specifically deliver the drug to the infected lung tissue. This targeted approach resulted in improved survival rates and reduced inflammatory damage without causing adverse effects. The findings of this study suggest the potential for developing a new class of multifunctional nanodrugs, providing new therapeutic strategies for multidrug-resistant (MDR) bacterial infections. Furthermore, these results provide a solid foundation for the design of biomimetic nanosized antibacterial drugs.
Neutrophil membrane-encapsulated manganese dioxide nanoparticles for the treatment of pneumonia caused by multidrug-resistant Pseudomonas aeruginosa.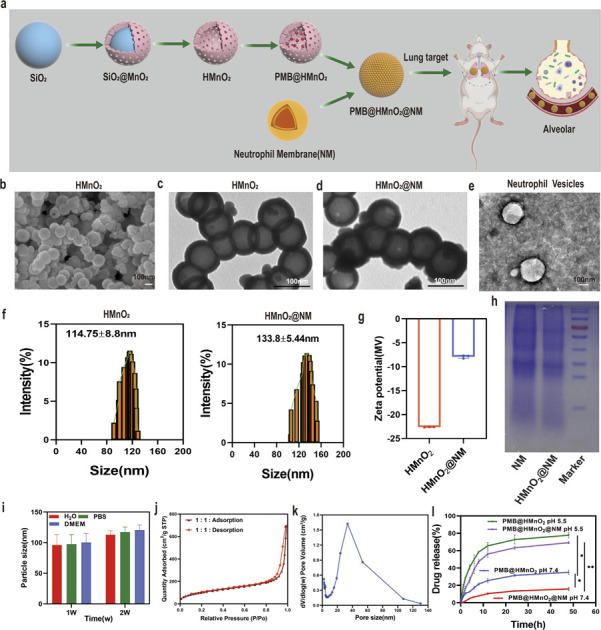
1. Introduction
Bacterial infections represent a major global health threat, leading to high incidence rates and mortality.1 Since the discovery of antibiotics, they have become the main means of bacterial infection treatment. However, the prolonged and widespread use of antibiotics has led to the development of bacterial resistance, especially the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) bacteria, posing significant challenges to effective clinical treatment.2 The development of new antibiotics is a lengthy and costly process, and the rate at which new drug-resistant strains emerge often surpasses the development of new antibiotics. Therefore, it is imperative to explore novel strategies and optimize existing drugs to delay the onset of drug resistance, thereby broadening the available arsenal of treatments for clinicians.
Polymyxins, a class of non-ribosomal polypeptide antimicrobials, exist in five structural forms, with polymyxin B (PMB) and colistin being the only ones currently used in clinical practice.3 The antimicrobial mechanism of polymyxins involves the binding of their polycationic rings to the phosphate groups on the cell membranes of Gram-negative bacteria, increasing membrane permeability and ultimately leading to bacterial swelling, lysis, and cell death.4–6 Despite their high sensitivity against common Gram-negative bacilli, polymyxins suffer from low lung tissue concentrations and dose-dependent nephrotoxicity and neurotoxicity,7 necessitating the exploration of effective strategies to increase the concentration of the drug in lung tissue or to mitigate the associated adverse effects.
In recent years, nanoparticle-based drugs have garnered extensive attention, which can overcome the limitations of traditional antimicrobials, including antibiotic resistance.8,9 Nanoparticles have been designed both as self-therapeutic agents and as delivery platforms for antimicrobial cargo to eliminate bacterial infections.9 Numerous studies have shown that nano-drug delivery systems offer several advantages, such as high drug loading capacity, controlled release, and targeted delivery, significantly improving the bioavailability of antibacterial agents.10–12 Among them, nanostructured manganese dioxide (MnO2) has garnered widespread interest due to its distinctive physical and chemical properties, which encompass pH-responsive degradability in acidic environments, elevated levels of glutathione and H2O2, superior catalytic properties, as well as favourable biocompatibility.13–16 However, the lack of active targeting capability in MnO2 nanoparticles and their vulnerability to immune system clearance significantly hinders efficient drug delivery. To address this issue, researchers have tried to decorate nanoparticles in a biomimetic manner, enabling them to camouflage and evade detection by the immune system.17,18 Among various biomimetic modification methods, cell membrane-coated nanoparticles have surfaced as a versatile therapeutic platform suitable for a wide range of applications. They are designed to minimize immune system clearance while imparting unique functionalities.19,20 Neutrophils, which are the most numerous leucocytes in the body, engage in diverse inflammatory responses and serve as the initial line of immune defense against invading pathogens or damaged tissues.21,22 The neutrophil-derived membrane coating facilitates targeted delivery of encapsulated drugs to the inflamed, infected tissues while neutralizing lipopolysaccharides (LPS) released by bacteria, thereby effectively mitigating the exacerbation of inflammatory responses caused by these toxins.23,24
Herein, we present neutrophil membrane-coated hollow MnO2 (HMnO2@NM) nanoparticles for loading PMB and further explore their role in treating pneumonia caused by MDR Pseudomonas aeruginosa (PA). This study offered a new idea for the treatment of MDR bacterial infections and lays the foundation for the design of biomimetic antimicrobial nanomedicines.
2. Materials and methods
2.1. Materials and reagents
Tetraethyl orthosilicate (TEOS) was sourced from Aladdin Reagent Co., Ltd in Shanghai, China. Potassium permanganate (KMnO4), methanol, and sodium carbonate (Na2CO3) were acquired from Sinopharm Chemical Reagent Co., Ltd. The SYTO-9/PI Live and Dead Bacteria Stain Kit was obtained from Zeye Co., Ltd, also located in Shanghai. DiO fluorescent dye, a Peripheral Blood Neutrophil Separation Kit, an MTT cell viability kit, and a BCA protein assay kit were all purchased from Beyotime Biotechnology in Shanghai. LB medium and agar were supplied by BD Difco in the USA. ELISA kits for IL-6 and TNF-α were provided by AiFang Biological Co., Ltd in Wuhan, China.
2.2. Preparation of solid silica nanospheres (sSiO2)
Solid silica NPs (sSiO2) were prepared using the previous method with some modifications.25 A mixture of 25 mL of absolute ethanol, 0.5 mL of distilled water, and 1.5 mL of aqueous ammonia was agitated at 500 rpm for 10 min. Subsequently, 0.75 mL of TEOS was gradually introduced and the mixture was stirred at a reduced speed of 150 rpm at 40 °C for 12 h. Following this, sSiO2 was isolated through centrifugation at 10 000 rpm for 20 min and then rinsed three times with both ethanol and distilled water.
2.3. Synthesis of HMnO2 and PMB@HMnO2
Potassium permanganate solution (KMnO4) (150 mg) was added to sSiO2 (20 mg) and the resulting sSiO2 nanoparticles were utilized as hard templates to synthesize sSiO2@MnO2 nanoparticles with core/shell structures. Finally, the sSiO2 core of sSiO2@MnO2 was etched away using Na2CO3 solution (2 mol L−1) at 60 °C for 12 h, resulting in the production of hollow MnO2 (HMnO2) nanoparticles. For loading PMB, the HMnO2 solution (0.2 mg mL−1) was incubated with various concentrations (0.1, 0.2, 0.4, 0.6 mg mL−1) of PMB and stirred for 24 h at 4 °C to form PMB@HMnO2 nanoparticles.
2.4. Synthesis of PMB@HMnO2@NM
150 mL of blood samples were collected from 20 healthy adults using anticoagulant tubes. This experiment was approved by the Second Affiliated Hospital of Nanjing Medical University (Approval No: 2023-KY-165-01), and informed consent was obtained from all participants. Neutrophil membranes (NMs) were prepared using the reported method with some modifications.24,26 At first, neutrophil cells were isolated using specific kits and then lysed in a buffer containing Tris–HCl, mannitol, sucrose, ethylenediaminetetraacetic acid and protease and phosphatase inhibitors. Homogenization was performed in an ice bath using a Dounce homogenizer. The homogenized suspension was centrifuged at 10 000 g for 10 min at 4 °C to remove organelles. The supernatant was then centrifuged at 20 000 g for 60 minutes at 4 °C to collect cell membranes. Cell membranes were resuspended in PBS and neutrophil cell membrane vesicles (NMV) were obtained by extruding 40 times through a 400 nm porous polycarbonate membrane using an Avanti mini-extruder. Membrane content was determined by quantifying membrane surface proteins using a BCA kit (membrane volume = 2 × membrane protein volume). To prepare PMB@HMnO2@NM, 0.5 mg per mL PMB@HMnO2 was mixed with neutrophil cell membrane (1 mg mL−1) and coextruded for 40 passes using the Avanti-mini extruder to acquire neutrophil cell membrane-coated nanoparticles.
2.5. Characterization of HMnO2@NM nanoparticles
Dynamic Light Scattering (Malvern Instruments, UK) was used to assess the size distribution and zeta potentials of the nanoparticles. The elemental composition and morphology of HMnO2 were determined utilizing transmission electron microscopy (TEM, FEI Tecnai F20) and scanning electron microscopy (SEM, Zeiss Sigma 300). Brunauer–Emmett–Teller (BET) analysis determined the surface area and pore size of HMnO2 in the HMnO2@NM composite. UV-vis spectra were obtained using a PerkinElmer Lambda 750 spectrophotometer. SDS-PAGE was used to identify the protein content in the membrane component of the nanoparticles.
2.6. In vitro encapsulation and loading of PMB
1 mmol of PMB and 5 mmol of Fluorescein Isothiocyanate (FITC) were mixed and stirred overnight at 4 °C and purified using dialysis to obtain FITC-labeled PMB. The PMB content in PMB@HMnO2@NM nanoparticles was measured using a UV-vis spectrometer at 490 nm. The drug loading and encapsulation efficiencies were calculated as follows:loading efficiency (%) = (the total weight of drug − the weight of free drug)/total weight of nanoparticles × 100
2.7. Minimum inhibitory concentration (MIC) evaluation
MDR Pseudomonas aeruginosa (PA) was kindly provided by the Department of Laboratory Medicine, the First Affiliated Hospital of Bengbu Medical University, for our experiments. The drug sensitivity of bacterial strains is shown in Table S1.† Samples (HMnO2@NM, free PMB, PMB@HMnO2 and PMB@HMnO2@NM) were 2-fold diluted from their initial concentration (2048 μg mL−1). 100 μL of the bacterial culture (1 × 107 CFU mL−1) was added to a 96-well plate and incubated (37 °C) with 100 μL of each sample. The MIC value was determined by evaluating the visible growth of microorganisms.
2.8. In vitro macrophage phagocytosis
RAW 264.7 (1 × 106) was inoculated into 12-well plates and incubated at 37 °C for 12 h. Then, 100 μL HMnO2@NM (10 μg per mL HMnO2), free PMB, PMB@HMnO2 (10 μg mL−1) and PMB@HMnO2@NM were added to each well. After 4 h of incubation, the cells were washed, fixed with 4% paraformaldehyde at room temperature, stained with DAPI, and then observed under a confocal microscope to assess macrophage phagocytosis.
2.9. Determination of intracellular bacterial content
SYTO-9-labeled PA and Hoechst 33 342-stained RAW 264.7 cells were cultured in antibiotic-free DMEM medium at an MOI of 10 : 1 and incubated at 37 °C for 2 h. The cells were washed three times with saline to remove non-phagocytosed bacteria. Fresh DMEM medium containing HMnO2@NM, free PMB, PMB@HMnO2 and PMB@HMnO2@NM (10 μg per mL HMnO2) were added to the RAW 264.7 cells and co-incubated for 2 h. Then, the cells were observed and photographed using a laser confocal microscope. In order to further quantify the effect of bacterial removal, 1% Trillatone X-100 was used to destroy RAW 264.7 cells, and 100 μL of appropriately diluted bacterial suspension was applied to LB plates, and the bacteria in different treatment groups were counted.
2.10. Crystalline violet assay
100 μL PMB@HMnO2, PMB@HMnO2@NM nanoparticles (100 μg mL−1) and free PMB (47.08 μg mL−1) were added to the bacterial suspension (100 μL, 1 × 107 CFU mL−1). The control group received 100 μL of PBS. After incubation for 24 h, the wells were collected and washed three times and then 0.1% (v/v) crystal violet solution was added. One hour later, 200 μL of 95% (v/v) ethanol was utilized to dissolve the crystal violet. The experiment was conducted in triplicate, and the absorbance was determined at 595 nm using a microplate reader. Bacterial biofilm inhibition was calculated according to the following formula:Inhibition of bacterial biofilm formation (%) = [1 − (absorbance of drug-treated cells/absorbance of control cells in the control group)] × 100.
2.11. Cell cytotoxicity assay
BEAS-2B cells were plated in 96-well (1 × 105 cells per well) and incubated for 12 h. After throwing away the medium, the cells were washed with PBS. Different concentrations (3.125, 6.25, 12.5, 25, 50, 100, and 200 μg mL−1) of PMB@HMnO2@NM in a complete medium were then added into the wells, and further incubated for 24 h. MTT assay was used to evaluate cell viability.
2.12. Animal studies
Female BALB/c mice (18–20 g) were purchased from Yangzhou University Laboratory Animal Center and raised under specific pathogen-free (SPF) conditions and provided with enough food and water. All animal experiments were performed in accordance with the Guidelines of the National Institutes of Health for the Care and Use of Laboratory Animals and approved by the Ethics Committee of Animal Care and Welfare, Nanjing Medical University (Approval Number: 2404092). Mice were allocated and randomly divided into five groups: control, HMnO2@NM, free PMB, PMB@HMnO2 and PMB@HMnO2@NM groups (n = 6 per group). After the mice were anesthetized, each mouse was inoculated via endotracheal inoculation with 100 μL of PBS containing 4 × 107 CFU of clinical MDR PA to induce acute pneumonitis. 2 h later, mice were injected i.v. with 100 μL of saline, HMnO2@NM, free PMB, PMB@HMnO2, and PMB@HMnO2@NM (2 mg per kg PMB). The procedure was repeated on the third day, followed by euthanasia of the mice on the fifth day. Blood and lung tissue samples were collected to quantify IL-6 and TNF-α levels via ELISA and assess bacterial burden.
2.13. Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 software. Results were reported as mean ± SD. Significance was evaluated through Student's t-test or one-way ANOVA, where ns denoted non-significance, *P < 0.05, **P < 0.01, and ***P < 0.001.
3. Results and discussion
3.1. Preparation and characterization of HMnO2@NM nanoparticles (NPs)
HMnO2 nanoparticles were synthesized by a three-step reaction process (Fig. 1a). In brief, solid SiO2 nanoparticles (Fig. S1a†) were synthesized using the Stöber method.25 A uniform coating of mesoporous MnO2 was subsequently deposited on the SiO2 nanoparticle surface through the addition of KMnO4 to form SiO2@MnO2 nanoparticles (Fig. S1b†). In the end, SiO2 was etched away to obtain HMnO2 particles. The scanning electron microscope (SEM) and transmission electron microscopy (TEM) images of HMnO2 showed a uniform spherical hollow structure with an average particle diameter of 100 nm (Fig. 1b and c). After coating with neutrophil membrane (NM), a lipid layer structure could be seen on the surface of HMnO2 (Fig. 1d and e). As measured by dynamic light scattering (DLS), the hydrodynamic size of the membrane-cloaked nanoparticles increased from 114.75 ± 8.8 nm to 133.8 ± 5.44 nm after coating with the NM layer (Fig. 1f). The zeta potential changed from −22.64 ± 0.05 mV to −7.93 ± 0.32 mV (Fig. 1g). The SDS-PAGE analysis confirmed that the membrane proteins originating from NM were well retained on the surface of HMnO2@NM nanoparticles after coating (Fig. 1h). Furthermore, HMnO2@NM exhibited excellent colloidal stability and dispersion after 2 weeks of incubation in water, pH 7.4 phosphate buffer solution (PBS) and DMEM medium (Fig. 1i).
Fig. 1. Synthesis and characterization of HMnO2@NM nanoparticles. (a) Scheme of synthetic procedure of HMnO2@NM. (b) SEM images of HMnO2 scale bar: 100 nm. (c) TEM images of HMnO2, (d) HMnO2@NM and (e) neutrophil membrane vesicles. (f) Hydrodynamic sizes of HMnO2 and HMnO2@NM measured by DLS (n = 3). (g) Zeta potentials of HMnO2 and HMnO2@NM were detected by DLS. (h) Protein profiles of the neutrophil membrane vesicles and HMnO2@NM nanoparticles as analyzed using SDS-PAGE. (i) In vitro stability of HMnO2@NM nanoparticles maintained in H2O, PBS, and DMEM cell culture medium at 4 °C for 2 weeks (n = 3). (j) N2 adsorption/desorption isotherms. (k) Pore-size distribution curve of HMnO2 nanoparticles. (l) In vitro profiles of cumulative release for PMB released from PMB@HMnO2 and PMB@HMnO2@NM nanoparticles in PBS at pH 7.4 and 5.5. Data are presented as the mean ± SD (n = 3). *p < 0.05, **p < 0.01.

3.2. pH-Dependent drug behaviours
Brunauer–Emmett–Teller (BET) results showed that HMnO2 had large specific surface area of 370.82 m2 g−1 (Fig. 1j) and average pore size of 10.91 nm (Fig. 1k), which was beneficial for drug loading and delivery. To prepare PMB-loaded nanoshells, FITC labeled PMB was used to establish a standard curve, plotting various concentrations of lysostaphin protein against their corresponding optical absorption intensities. UV-vis spectra of FITC-labeled PMB exhibited an absorption peak for FITC at 490 nm, indicating successful labeling of PMB (Figs. S2a–c†). We mixed HMnO2 nanoparticles with different concentrations of PMB under overnight stirring and sequentially coated with the cell membrane. At the PMB : HMnO2 ratio of 3 : 1, the PMB loading efficiency of HMnO2@NM reached 47.08% (Fig. S3†). The responsiveness of HMnO2@NM to the microenvironment within biofilms is a pivotal characteristic, enabling targeted drug release directly at the site of infection. PMB@HMnO2 and PMB@HMnO2@NM exhibited quite different drug release behaviours under varied pH (Fig. 1l). In a neutral solution (pH 7.4), approximately 35.2% of PMB was released from PMB@HMnO2 after 48 h. However, only 15.9% of PMB was released from PMB@HMnO2@NM, suggesting that the presence of cell membranes can partially shield the encapsulated drugs from leakage. Whilst in an acidic solution (pH 5.5), the decomposition of HMnO2 could induce about 77.8% and 69.2% PMB release of PMB@HMnO2 and PMB@HMnO2@NM within 48 h, respectively.
3.3. In vitro antibacterial effect of PMB@HMnO2@NM
In order to clarify the specific recognition function of HMnO2@NM for bacteria with the assistance of neutrophil membrane, HMnO2 and HMnO2@NM nanoparticles were co-incubated with PA for 4 h and observed by SEM. Fig. 2a shows that a large number of nanoparticles were attached to the surface after the exposure to HMnO2@NM, while few nanoparticles were found on the surface of PA when exposed to HMnO2 nanoparticles. This indicated that NM promoted the binding of HMnO2@NM to PA and had a targeting effect for bacteria.
Fig. 2. Bactericidal activity in vitro. (a) SEM images of PA after treatment with PBS, HMnO2 and HMnO2@NM nanoparticles. (b) Bacteria-killing kinetics of PBS (control), HMnO2@NM, PMB and PMB@HMnO2@NM. (c) Illustrative fluorescence images for live/dead bacterial staining assay of MDR PA after treatment with PBS (control), HMnO2@NM, PMB and PMB@HMnO2@NM. Scale bar: 20 μm. (d) Representative SEM images of MDR PA at 4 h after treatment with PBS (control), HMnO2@NM, PMB and PMB@HMnO2@NM (47.08 μg mL−1 of PMB).

To evaluate the influence of encapsulating PMB within HMnO2 on its enzymatic activity, we determined the minimal inhibitory concentration (MIC) for HMnO2@NM, PMB@HMnO2@NM and free PMB. Notably, PMB@HMnO2@NM demonstrated an MIC value of 4 μg mL−1, which was equivalent to that of free PMB. Conversely, HMnO2@NM alone exhibited no bactericidal activity (Table 1).
Assessment of the minimum inhibitory concentration (MIC) of HMnO2@NM, PMB@HMnO2, PMB@HMnO2@NM nanoparticles and free PMB.
| Bacterial strains | MIC (μg mL−1) | |||
|---|---|---|---|---|
| HMnO2@NM | PMB@HMnO2 | PMB@HMnO2@NM | PMB | |
| MDR-PA | No inhibition | 4 | 4 | 4 |
These findings indicate that encapsulation within nanoparticles does not compromise the antibacterial efficacy of the loaded PMB. As shown in Fig. 2b, PMB@HMnO2@NM eliminated the bacteria at a faster rate than free PMB or HMnO2@NM alone. After 4 h of incubation, PMB@HMnO2@NM completely eliminated all bacteria, whereas samples treated with PMB alone still contained live bacteria in the same time range. These results of the live/dead staining further revealed that bacterial cells treated with PBS and HMnO2@NM displayed mostly green fluorescence (indicating live bacteria). In contrast, cells exposed to PMB and PMB@HMnO2@NM exhibited intense red fluorescence, signifying the disruption of their cell membranes and subsequent permeability to PI (Fig. 2c). Bacteria treated with PBS or HMnO2@NM exhibited intact cell walls, smooth cell membranes, and a dense, uniform cytoplasm. Conversely, bacteria exposed to PMB or PMB@HMnO2@NM displayed disrupted or damaged cell walls, along with wrinkled and deformed cell membranes. These findings indicated that PMB encapsulated within PMB@HMnO2@NM was equally efficacious as free PMB in eliminating PA by disrupting its cell wall structure and ultimately causing bacterial lysis (Fig. 2d).
To investigate the bactericidal effect of PMB@HMnO2@NM on MDR PA within macrophages, we labeled PA and RAW 264.7 cells with SYTO-9 and Hoechst 33342, respectively, and subsequently observed them using Confocal Laser Scanning Microscopy (CLSM). Macrophages treated with PMB@HMnO2@NM exhibited superior PA clearance capability compared to other groups, evidenced by a decrease in green fluorescence spots within the macrophages (Fig. 3a). To quantify this effect further, we lysed the cells using detergent and enumerated the bacterial colony-forming units (CFU). The reduction in CFU numbers in the PA-infected macrophage cells following PMB@HMnO2@NM treatment confirmed the enhanced bacterial clearance ability (Fig. 3b and c).
Fig. 3. (a) Confocal fluorescence images of PA-infected RAW264.7 cells treated with PBS (control), HMnO2@NM, PMB and PMB@HMnO2@NM. Scale bar, 20 μm. (b) Photographs of CFU and CFU counting (c) of PA released from macrophages under different treatments. (d) Crystal violet staining photographs of biofilms after different treatments. (e) Inhibition rates of biofilms after treatment with PBS (control), HMnO2@NM, PMB and PMB@HMnO2@NM (47.08 μg mL−1 of PMB) (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, n.s. means no significance. Data are expressed in mean ± SD).

Infections caused by PA often manifest as biofilms, which feature an extracellular polymer matrix that significantly impedes antibiotic penetration, leading to a notable increase in bacterial resistance, ranging from 100 to 1000 fold.27 Thus, we further evaluated the in vitro biofilm eradication activities of PMB@HMnO2@NM. Through crystal violet staining, we observed that biofilms treated with PBS and HMnO2@NM remained largely viable and structurally intact, indicative of negligible antibiofilm activity. Conversely, the PMB@HMnO2@NM group showed substantial biomass loss (Fig. 3d and e), highlighting its potent anti-biofilm potential.
3.4. Biodistribution of HMnO2@NM in vivo
To verify the lung targeting ability of HMnO2@NM, we constructed ICG@HMnO2 and ICG@HMnO2@NM nanoparticles loaded with the fluorescent dye ICG and injected them via the tail vein into mice with pneumonia caused by MDR PA. Representative time-dependent in vivo imaging was then performed using the Xenogen IVIS system. As shown in Fig. 4a, the ICG@HMnO2@NM group displayed a higher level of fluorescence intensity in the lungs compared to the ICG@HMnO2 group, indicating that ICG@HMnO2@NM could accumulate in the lungs. This accumulating ability was essential for targeting the delivery of drugs to the lungs.
Fig. 4. Bactericidal activity in vivo. (a) In vivo fluorescence images of MDR PA lung-infected mice at various time points post-intravenous injection of ICG@HMnO2 and ICG@HMnO2@NM. (b) Lung bacterial loads in mice post-treatment with saline (control), HMnO2@NM, PMB and PMB@ HMnO2@NM. (c) Giemsa staining results of lung tissue after treatment with saline (control), HMnO2@NM, PMB and PMB@HMnO2@NM. Scale bars: 20 μm. (d) H & E staining of lung tissue after treatment with saline (control), HMnO2@NM, PMB and PMB@HMnO2@NM. Scale bars: 20 μm. (e) Serum levels of IL-6 and TNF-α collected after treated with saline (control), HMnnO2@NM, PMB and PMB@HMnO2@NM. (f) Percentage survival of pneumonia mice treated with saline (control), HMnO2@NM, PMB and PMB@HMnO2@NM (n = 6, *p < 0.05, **p < 0.01, ***p < 0.001, n.s. means no significance. Data are expressed in mean ± SD).

3.5. PMB@HMnO2@NM reduces bacterial load in lung tissue and alleviates the inflammatory response
Given the potent bactericidal activity demonstrated in vitro, along with the lung targeting capability of PMB@HMnO2@NM, we assessed its therapeutic efficacy in mice with acute pneumonia caused by MDR PA. The mice were inoculated intratracheally (i.t.) with MDR PA (4 × 107 CFU) to induce acute pneumonia. After 2 h of post-inoculation, the mice were intravenously administered either saline (control), HMnO2@NM, PMB, or PMB@HMnO2@NM. Notably, the CFU counts in mice treated with PMB@HMnO2@NM were significantly lower than those in other groups at 48 h of post-intravenous injection, indicating superior in vivo bactericidal activity (Fig. 4b). This enhanced therapeutic effect may be attributed to the higher concentration of PMB achieved in the alveoli of mice treated with PMB@HMnO2@NM compared to those receiving free PMB. Meanwhile, the results of Giemsa staining showed the lowest bacterial residues in the lung tissue after treatment of PMB@HMnO2@NM (Fig. 4c). We further assessed lung inflammation in mice subjected to various treatments to demonstrate the therapeutic benefits of PMB@HMnO2@NM in acute pneumonia caused by MDR PA. Histological analysis of lung tissues in the control group showed severe alveolar damage and widespread infiltration of inflammatory cells, indicative of extensive lung pathology (Fig. 4d). Mice treated with free PMB exhibited a decrease in alveolar count, incomplete alveolar expansion, and alveolar fusion. Compared to the control, the PMB@HMnO2@NM treatment markedly alleviated lung injury, exhibiting a notable decrease in inflammatory cell infiltration, hemorrhage, and alveolar damage. Next, we assessed cytokine production in mouse serum using ELISA assays. As illustrated in Fig. 4e, mice treated with PMB@HMnO2@NM exhibited the lowest production of proinflammatory cytokines, including tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6). To evaluate the impact of PMB@HMnO2@NM on survival rates in mice with pneumonia, we established a severe pneumonia model by inoculating high doses of PA (4 × 108 CFU per mouse). The outcomes demonstrated that all mice in the control and HMnO2@NM group died within 3 days, while the survival rate in the PMB@HMnO2@NM group was beyond 70% within 7 days (Fig. 4f). Collectively, these results demonstrate that PMB@HMnO2@NM treatment effectively reduces the bacterial burden in lung tissues, alleviating the inflammatory response, decreasing cytokine levels, and significantly improving the survival rates of mice with pneumonia.
3.6. Biosafety of PMB@HMnO2@NM in vitro and in vivo
Normal human bronchial epithelial cells (BEAS-2B) were utilized to assess the cytotoxicity of PMB@HMnO2@NM nanoparticles in vitro using MTT assay. PMB@HMnO2@NM displayed favourable biocompatibility, as evidenced by cell viabilities exceeding 80% following 24 h of incubation at various concentrations (Fig. 5a). To investigate the long-term safety profile, healthy mice were subjected to treatments with these compounds. Over a 28 days period, none of the mice died, and there were no significant changes in the body weight among the treated mice, indicating that PMB@HMnO2@NM does not exhibit systemic toxicity (Fig. 5b). Hematological parameters, including Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST), creatinine (Cre), and blood urea nitrogen (BUN) remained within normal ranges on 28th days (Fig. 5c–f). Furthermore, histological examinations of vital organs such as the heart, kidney, liver, spleen, and lungs revealed no discernible damage in mice treated with PMB@HMnO2@NM (Fig. 5g). These findings collectively underscore the exceptional biosafety of PMB@HMnO2@NM, indicating its absence of both acute and chronic toxicity, both locally and systemically.
Fig. 5. Biosafety of PMB@HMnO2@NM nanoparticles. (a) Cytotoxicity in BEAS-2B cells after being treated with various concentrations of PMB@HMnO2@NM for 24 h. (b) The body weight change of mice treated with saline (control), HMnO2@NM, PMB and PMB@HMnO2@NM during 28 days. (c–f) Analysis of blood biochemical parameters of mice in various treatment groups on day 28 post-administration, encompassing (c) ALT, (d) AST, (e) BUN and (f) creatinine (Cre). (g) H & E staining of vital organ sections of mice at day 28 after treated with saline (control), HMnO2@NM, PMB and PMB@HMnO2@NM. Scale bar,100 μm (n = 3).

4. Conclusions
In summary, we successfully developed a novel biomimetic nanoplatform, PMB@HMnO2@NM, which integrates an HMnO2 nanoparticle core with a neutrophil membrane coating for the targeted systemic treatment of pneumonia caused by multidrug-resistant Pseudomonas aeruginosa. This nanoplatform demonstrated efficacy in delivering antibacterial drugs directly to the infectious sites. Treatment with PMB@HMnO2@NM significantly enhanced survival rates, reduced bacterial loads, and mitigated inflammatory damage in infected lungs. These promising results underscore the clinical translation potential of PMB@HMnO2@NM as a novel therapeutic option for Pseudomonas aeruginosa-infected lung diseases, offering a significant advancement in the treatment of MDR bacterial infections.
Data availability
The authors confirm that the data supporting the study's findings are provided in the article, with some placed in the ESI.†
Author contributions
J. H. designed and performed experiments and analyzed data; X. H. and Y. L. performed the experiments; Y. W. and K. H. performed experiments and analyzed the data. C. Z. and L. X. wrote the paper and led the project. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This research was supported by the Clinical Medicine Translation Special Program of the Key Research and Development Plan in Anhui Province (202204295107020033).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07577c
References
- Averbuch D., Bacterial Infections, 2019, pp. 265–272 [Google Scholar]
- Rodrigo-Troyano A. Sibila O. Respirology. 2017;22:1288–1299. doi: 10.1111/resp.13115. [DOI] [PubMed] [Google Scholar]
- Molina J. Cordero E. Pachon J. Expet Opin. Pharmacother. 2009;10:2811–2828. doi: 10.1517/14656560903334185. [DOI] [PubMed] [Google Scholar]
- Rabanal F. Cajal Y. Nat. Prod. Rep. 2017;34:886–908. doi: 10.1039/c7np00023e. [DOI] [PubMed] [Google Scholar]
- Yin J. Meng Q. Cheng D. Fu J. Luo Q. Liu Y. Yu Z. Appl. Microbiol. Biotechnol. 2020;104:3771–3780. doi: 10.1007/s00253-020-10525-y. [DOI] [PubMed] [Google Scholar]
- Hussein M. Hu X. Paulin O. Crawford S. Tony Z. Q. Baker M. Schneider-Futschik E. K. Zhu Y. Li J. Velkov T. Comput. Struct. Biotechnol. J. 2020;18:2247–2258. doi: 10.1016/j.csbj.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q. J. Xiang B. X. Song M. H. Yang C. Y. Liang J. H. Xie Y. L. Zuo X. C. Pharmacotherapy. 2024;44:631–641. doi: 10.1002/phar.4601. [DOI] [PubMed] [Google Scholar]
- Solanki R. Makwana N. Kumar R. Joshi M. Patel A. Bhatia D. Sahoo D. K. RSC Adv. 2024;14:33568–33586. doi: 10.1039/d4ra06117a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A. Mumtaz S. Li C. H. Hussain I. Rotello V. M. Chem. Soc. Rev. 2019;48:415–427. doi: 10.1039/c7cs00748e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makabenta J. Nabawy A. Li C. H. Schmidt-Malan S. Patel R. Rotello V. M. Nat. Rev. Microbiol. 2021;19:23–36. doi: 10.1038/s41579-020-0420-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. H. Chen X. Landis R. F. Geng Y. Makabenta J. M. Lemnios W. Gupta A. Rotello V. M. ACS Infect. Dis. 2019;5:1590–1596. doi: 10.1021/acsinfecdis.9b00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virzi N. F. Greco V. Stracquadanio S. Jasim A. Greish K. Diaz-Rodriguez P. Rotondo N. P. Stefani S. Pittala V. Giuffrida A. RSC Adv. 2024;14:34066–34080. doi: 10.1039/d4ra04457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Zhang X. Khan S. A. Li W. Wan L. Front. Microbiol. 2021;12:761084. doi: 10.3389/fmicb.2021.761084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T. Chen S. Zhang J. Li T. Li P. Liu J. Du X. Wang S. Nanomaterials. 2020;10:1545. doi: 10.3390/nano10081545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Xiao Y. Zhu W. Chu J. Xu J. Zhao H. Shen F. Peng R. Liu Z. Small. 2020;16:e2000589. doi: 10.1002/smll.202000589. [DOI] [PubMed] [Google Scholar]
- Qian C. Zhao G. Huo M. Su M. Hu X. Liu Q. Wang L. RSC Adv. 2024;14:17612–17626. doi: 10.1039/d4ra00539b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Xia Z. Wang H. Wang D. Sun T. Hossain E. Pang X. Liu Y. Theranostics. 2023;13:3224–3244. doi: 10.7150/thno.81520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi W. Xiao P. Liu X. Zhao Z. Sun X. Wang J. Zhou L. Wang G. Cao H. Wang D. Li Y. Signal Transduct. Targeted Ther. 2022;7:386. doi: 10.1038/s41392-022-01250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X. Chen Y. Chen Z. Wei X. Dai Y. Yuan Z. Ultrason. Sonochem. 2022;84:105972. doi: 10.1016/j.ultsonch.2022.105972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y. Zhang L. Xu J. Xiang L. Biomed. Mater. 2024;19:4. doi: 10.1088/1748-605X/ad46d4. [DOI] [PubMed] [Google Scholar]
- Carnevale S. Di Ceglie I. Grieco G. Rigatelli A. Bonavita E. Jaillon S. Front. Immunol. 2023;14:1180810. doi: 10.3389/fimmu.2023.1180810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales C. J. Leukoc. Biol. 2020;108:377–396. doi: 10.1002/JLB.4MIR0220-574RR. [DOI] [PubMed] [Google Scholar]
- Li C. Gan Y. Li Z. Fu M. Li Y. Peng X. Yang Y. Tian G. B. Yang Y. Y. Yuan P. Ding X. Biomater. Res. 2023;27:30. doi: 10.1186/s40824-023-00372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Chen X. Xu L. Tu F. Rui X. Zhang L. Yan Z. Liu Y. Hu R. Nanomedicine. 2023;48:102640. doi: 10.1016/j.nano.2022.102640. [DOI] [PubMed] [Google Scholar]
- Stöber W. Fink A. Bohn E. J. Colloid Interface Sci. 1968;26(1):62–69. [Google Scholar]
- Wu X. Lin Z. Zhao C. Liu L. Zhang K. Lai J. Meng Q. F. Yao G. Huang Q. Zhao X. Z. Rao L. Biosens. Bioelectron. 2022;213:114425. doi: 10.1016/j.bios.2022.114425. [DOI] [PubMed] [Google Scholar]
- Feng W. Li G. Kang X. Wang R. Liu F. Zhao D. Li H. Bu F. Yu Y. Moriarty T. F. Ren Q. Wang X. Adv. Mater. 2022;34:e2109789. doi: 10.1002/adma.202109789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the study's findings are provided in the article, with some placed in the ESI.†