

Abstract
Ferroptosis, an intricately regulated form of cell death characterized by uncontrolled lipid peroxidation, has garnered substantial interest since 2012 the term coined. Recent years have witnessed remarkable progress in elucidating the detailed molecular mechanisms governing ferroptosis induction and defense, with particular emphasis on the roles of heterogeneity and plasticity. Within the molecular ecosystem of ferroptosis, present and future advancements promise to unlock safe and effective therapeutic strategies across a broad spectrum of diseases.
Keywords: antioxidant, cell death, disease, ferroptosis, lipid peroxidation
Introduction
Ferroptosis, coined in 2012, is a distinct form of regulated cell death observed in cancer cells, relying on iron but differing from apoptosis and necroptosis1. Unlike lytic cell death mechanisms dependent on pore-forming proteins, ferroptosis is driven by toxic, oxidized lipids and their byproducts, notably 4-hydroxynonenal (4HNE)2, along with lipidated proteins formed through covalent binding to electrophilic lipid peroxidation breakdown products3.
Ferroptosis has significant implications in preclinical studies across diseases, including cancer, neurodegenerative disorders, and conditions associated with ischemia-reperfusion (I/R) injury. It offers promise as a therapeutic approach against drug-resistant cancer cells deficient in apoptosis4, 5, while its inhibition holds potential for managing infection-related diseases, sterile inflammation linked to iron overload or lipid toxicity6–8. Additionally, ferroptosis plays a vital role in tissue homeostasis and development8–10.
In this review, our aim is to offer an updated overview of ferroptosis, covering its fundamental mechanisms, heterogeneity, and plasticity. We will also delve into the integrated antioxidant and membrane system’s role in regulating ferroptotic sensitivity, along with discussing disease implications, therapeutic prospects, and associated challenges.
The core mechanism of ferroptosis
Erastin and RSL3 are common small molecules used to induce ferroptosis. Originally discovered in screens targeting RAS mutant cancer cells, these compounds trigger a non-apoptotic, iron-dependent form of cell death, leading to the term ‘ferroptosis’1, 11, 12. At the same time, genetic inactivation of GPX4 was found to induce oxidative, non-apoptotic cell death13, and overexpression of system xc− to protect cells from a similar non-apoptotic cell death14, highlighting the generality of this process as a potential cancer therapy targeting RAS mutations while sparing normal cells.
Further research has revealed that ferroptosis is highly context-dependent. Metal ions like zinc and copper, in addition to iron, can induce ferroptosis in specific conditions15, 16. Both RAS wild-type and mutant cells, including cancer and non-cancer cells, can undergo ferroptotic death. Conditional knockout of Gpx4 in various (e.g., kidney9) or cells (e.g., T cells8 or B cells10) can cause ferroptotic damage, highlighting its role in developmental biology.
Ferroptosis is closely linked to autophagy, and heightened autophagy levels often correlate with increased ferroptosis sensitivity17. Specific types of selective autophagy, such as ferritinophagy18, 19, lipophagy20, and clockophagy21, can lead to iron accumulation and lipid peroxidation, inducing ferroptosis. Genome-wide CRISPRi/a screens in human neurons revealed that so-called ATG (autophagy related) family members (e.g., BECN1 [beclin 1]) and lysosomal proteins (e.g., PSAP [prosaposin]) are involved in ferroptosis by triggering the formation of lipofuscin or increasing iron accumulation22. In certain conditions, the depletion of ATG genes has no effect on cell death, including ferroptosis.
These findings underscore the adaptable and context-dependent nature of ferroptosis, but its initiation involves three essential elements, which will be discussed below.
Reactive oxygen species
The first crucial element in ferroptosis induction is the presence of initiation signals that stimulate the production of ROS from various sources (Fig. 1):
Figure 1. The production of ROS in ferroptosis.
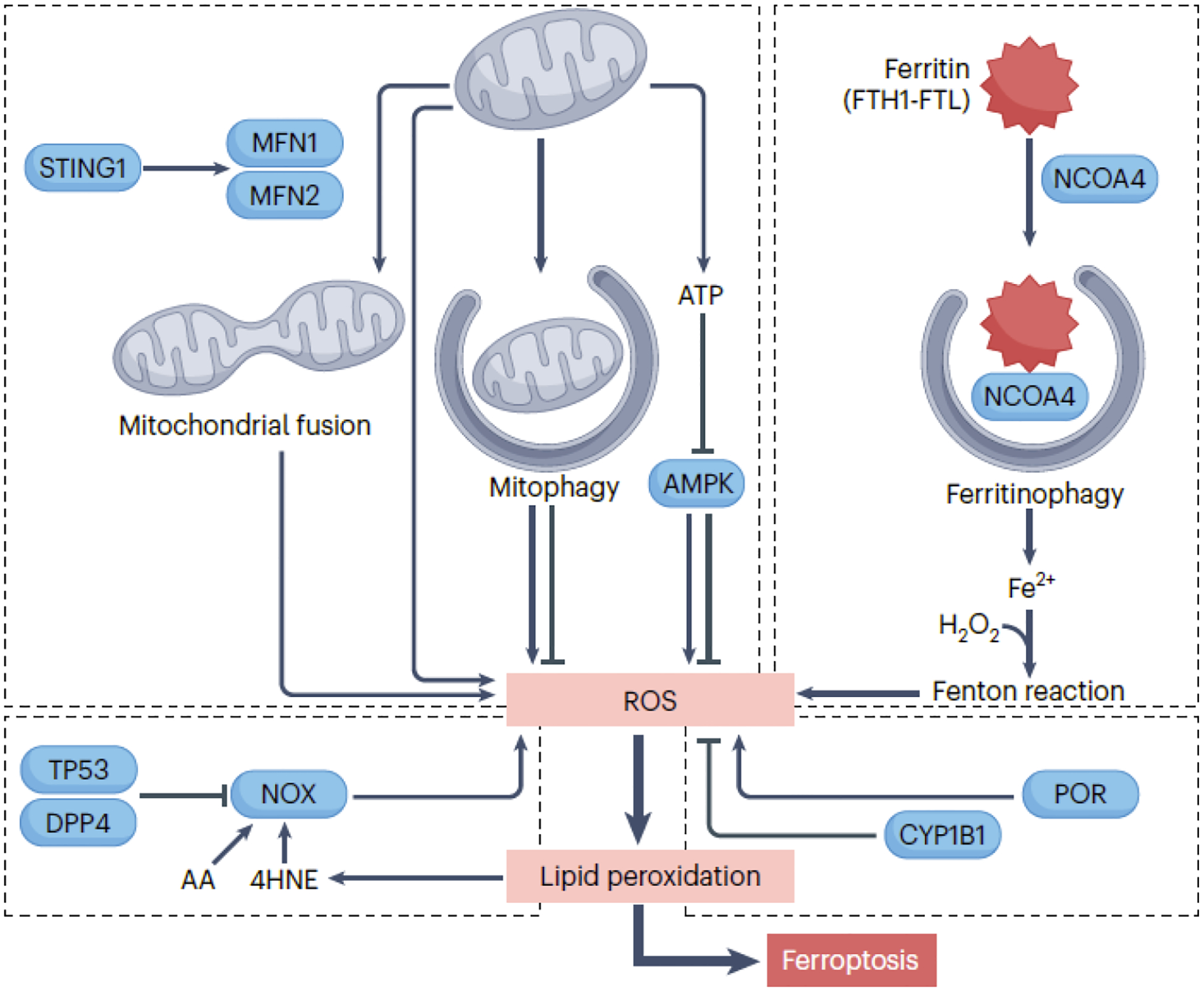
The initiation of ferroptosis requires an oxidative environment, facilitated by diverse sources of ROS. Mitochondrial ROS, primarily generated through the electron transport chain, can trigger ferroptosis in specific conditions. Mitophagy, involved in removing damaged mitochondria, has a dual role in promoting or inhibiting ferroptosis, while mitochondrial fusion increases cellular sensitivity to ferroptosis. Activation of the mitochondrial STING1 (stimulator of interferon response cGAMP interactor 1) may promote mitochondrial fusion, leading to ROS production implicated in ferroptosis. Mitochondrial energy stress activates AMPK, which can promote or inhibit ferroptosis by phosphorylating different substrates. NOX (NADPH oxidase) enzymes in cell membranes play a crucial role in generating ROS in ferroptosis. TP53 inhibits NOX-mediated ferroptosis by binding to DPP4 (dipeptidyl peptidase 4), while arachidonic acid (AA) and 4HNE enhance NOX1 activity to promote ROS production. POR (cytochrome p450 oxidoreductase) promotes ROS production and ferroptosis, whereas CYP1B1 (cytochrome P450 family 1 subfamily B member 1) inhibits ferroptosis. Ferritinophagy involves the degradation of the iron storage protein ferritin, releasing Fe2+ that triggers ROS production through the Fenton reaction.
1) Mitochondria: Mitochondria serve as a major source of ROS, primarily superoxide anion/O2•− during oxidative phosphorylation. Mitochondrial SOD converts superoxide into other ROS, including hydrogen peroxide (H2O2). Mitochondrial ROS can trigger ferroptosis, with glutaminolysis promoting ferroptosis induced by cyst(e)ine deprivation cyst(e)ine deprivation23, 24. Mitochondrial quality is regulated by mitophagy, which has a dual role in ferroptosis. Whereas mitochondrial fission promotes apoptosis, mitochondrial fusion can increase cellular sensitivity to ferroptosis25. Mitochondrial energy stress inhibits ferroptosis through AMPK-mediated phosphorylation of ACACA/ACC (acetyl-CoA carboxylase alpha)26, but AMPK can also promote ferroptosis by targeting BECN127 or by disrupting pyrimidinosome assembly, hindering pyrimidine intermediate synthesis28.
2) NOX (NADPH oxidase): Overexpression of NOX increases ROS levels, heightening ferroptosis sensitivity. The activity of NOX in ferroptosis is regulated by multiple factors, such as TP53 (tumor protein p53)29 and ALDH1B1 (aldehyde dehydrogenase 1 family member B1)2. Trp53/TP53 deficiency promotes the accumulation of DPP4 (dipeptidyl peptidase 4) on the cell membrane, forming a complex with NOX1 and causing ferroptotic death29. ALDH1B1 inhibits the ferroptosis-inducing effect of NOX1 activity by catalyzing the oxidation of aldehydes, converting them into carboxylic acids2.
3) Enzymatic reactions: ROS can be byproducts of enzymatic reactions, such as cytochrome P450 and its reductase involved in drug metabolism. POR (cytochrome P450 oxidoreductase), a flavoprotein, induces lipid peroxidation and ferroptosis by generating superoxide radicals30, 31.
4) The Fenton reaction. This reaction involves the interaction between H2O2 and a transition metal, typically iron (Fe2+), leading to the generation of highly reactive hydroxyl radicals/•OH. An extensively studied iron metabolism mechanism during ferroptosis is ferritinophagy, where autophagy degrades the iron storage protein ferritin. This liberates free iron, converting one ROS type into another, thereby inducing ferroptosis in both cancer and non-cancer cells18, 19.
Oxidizable lipids
The second key element in ferroptosis is the presence of easily oxidizable polyunsaturated lipids (Fig. 2). Cell membranes, the primary target of oxidative damage in ferroptosis, can be influenced by metabolic pathways that promote lipid synthesis, particularly the generation of polyunsaturated fatty acids (PUFAs), increasing cell sensitivity to ferroptotic inducers. While the exact threshold for PUFA breakdown required to initiate ferroptosis remains obscure, one well-established positive regulator is ACSL4. ACSL4 activates long-chain fatty acids by converting them into acyl-CoA esters, facilitating their entry into various metabolic pathways32–35.
Figure 2. Lipid resources for ferroptosis.
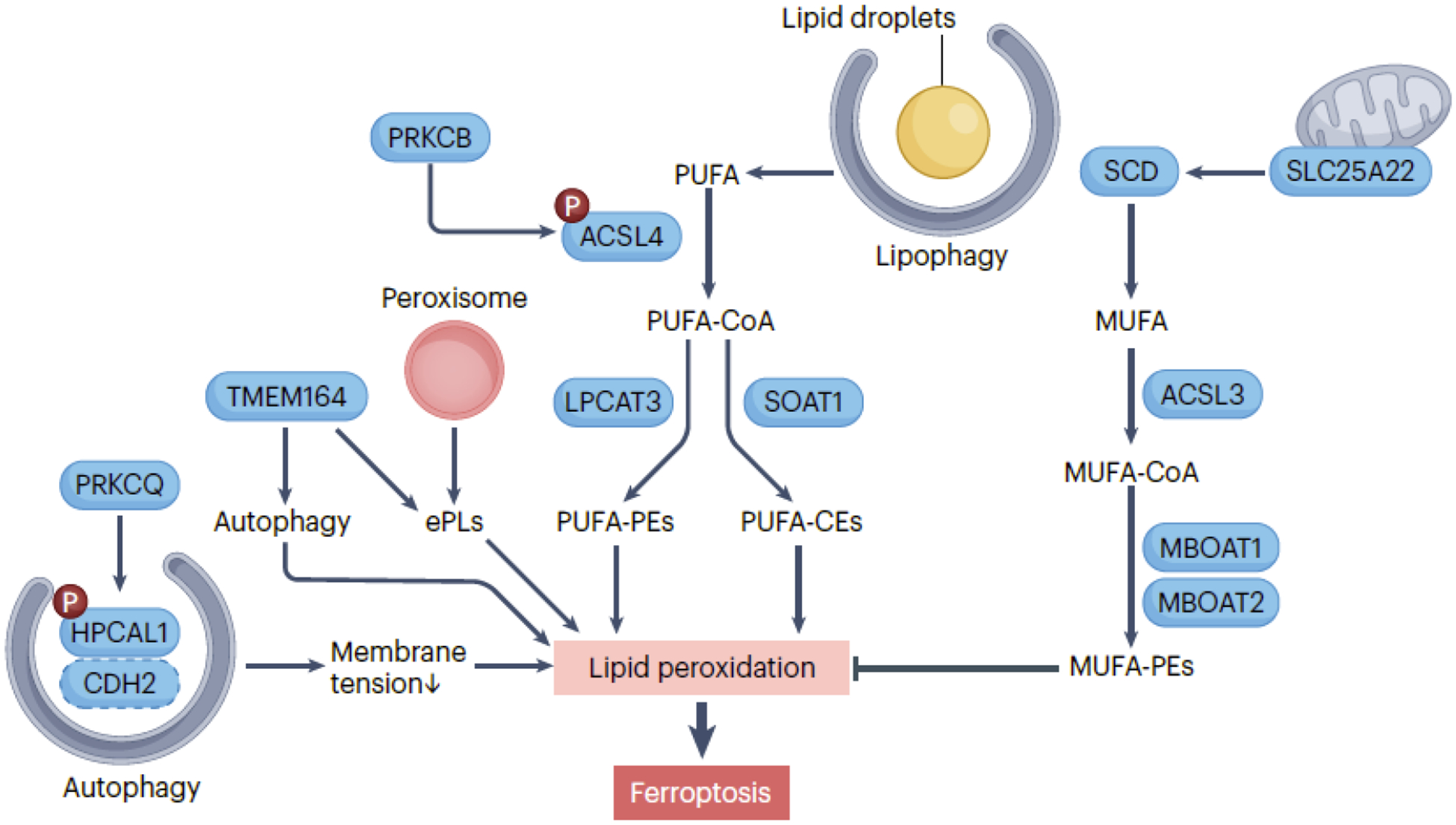
Cell membranes are the primary target of oxidative damage in ferroptosis, influenced by processes and metabolic pathways that promote lipid synthesis. ACSL4 (acyl-CoA synthetase long chain family member 4) plays a critical role in activating polyunsaturated fatty acid (PUFA) by converting them into acyl-CoA esters (PUFA-CoA), which serve as substrates for lipid peroxidation, contributing to the initiation of ferroptosis. Two downstream pathways involve LPCAT3 (lysophosphatidylcholine acyltransferase 3)-mediated PUFA-PEs and SOAT1 (sterol O-acyltransferase 1)-mediated PUFA-CEs. The activity of ACSL4 in ferroptosis is further enhanced by PRKCB (protein kinase C beta)-mediated ACSL4 phosphorylation. HPCAL1 (hippocalcin like 1) phosphorylation by PRKCQ (protein kinase C theta) promotes ferroptosis by inducing autophagic degradation of CDH2 (cadherin 2), leading to alterations in membrane tension in cancer cells. Monounsaturated fatty acid (MUFA) synthesis mediated by SCD (stearoyl-CoA desaturase) and ACSL3 (acyl-CoA synthetase long chain family member 3) counteracts the initiation of ferroptosis by protecting against PUFA peroxidation. The mitochondrial transporter SLC25A22 (solute carrier family 25 member 22) plays a role in inhibiting ferroptosis by facilitating the production of SCD-mediated MUFA. MBOAT1 (membrane bound O-acyltransferase domain containing 1) and MBOAT2 inhibit ferroptosis by remodeling the cellular phospholipid profile to produce MUFA-PEs. Peroxisomes contribute to the biosynthesis of ether phospholipids (ePLs), which are vulnerable to lipid peroxidation. TMEM164 (transmembrane protein 164) functions as an acyltransferase involved in ePLs synthesis or promotes the formation of autophagosomes. Lipophagy, the degradation of lipid droplets, releases lipids that can undergo peroxidation, increasing the susceptibility of cells to ferroptosis.
ACSL4 mediates two downstream pathways, yielding different PUFA-related acyl-CoA esters. One involves LPCAT3 (lysophosphatidylcholine acyltransferase 3), incorporating PUFA into phosphatidylethanolamines (PEs)32, 35, while the other activates SOAT1 (sterol O-acyltransferase 1), producing PUFA-cholesteryl esters (CEs) instead of PUFA-PEs36. Both pathways contribute to lipid peroxidation, acting as substrates depending on the context. In the lipid flippase SLC47A1-deficient human pancreatic cancer cells, ACSL4-driven PUFA-CE production is particularly relevant36. ACSL4 activation is a strategy to enhance chemotherapy or immunotherapy efficacy by inducing ferroptosis in solid cancers37. PRKCB/PKCβII enhances ACSL4 activity via Thr328 phosphorylation38, while HPCAL1 phosphorylation at Thr149 by PRKCQ induces ferroptosis by autophagic degradation of CDH2, altering membrane tension in cancer cells39.
ACSL3 synthesizes monounsaturated fatty acids (MUFAs), which may competitively inhibit PUFA peroxidation, protecting against ferroptosis initiation40, 41. The mitochondrial glutamate transporter SLC25A22 inhibits ferroptosis in pancreatic cancer cells by enhancing GSH and MUFA synthesis42. MBOAT1 (membrane bound O-acyltransferase domain containing 1) and MBOAT2, upregulated by sex hormone receptors, inhibit ferroptosis in cancer cells by remodeling the cellular phospholipid profile to produce MUFA-containing phospholipids43. ACSL4-independent pathways add complexity to the understanding of lipid metabolism in cell death regulation44.
Peroxisomes, involved in fatty acid breakdown, hydrogen peroxide production, and PUFA plasmalogen biosynthesis, can increase ferroptosis sensitivity45. They also contain antioxidant enzymes like CAT, which inhibit ferroptosis, as well as MUFA plasmalogens, which prevent ferroptosis46. Thus, peroxisomes or plasmalogens influence ferroptosis positively or negatively depending on the context.
Lipophagy selectively degrades lipid droplets, releasing lipids for peroxidation, making cells, especially hepatocellular carcinoma cells, more susceptible to ferroptosis20. Increased lipid storage in lipid droplets by ACSL3 can limit ferroptosis in clear cell renal cell carcinoma cells47.
Furthermore, TMEM164 acts as a positive regulator of ferroptosis by functioning as an acyltransferase, synthesizing C20:4 ether phospholipids48, and promoting the formation of membrane-driven phagophores49. These phagophores are essential for the subsequent creation of autophagosomes in pancreatic cancer cells in response to ferroptotic stimuli, rather than nutrient starvation49.
Lipid peroxidation
Several enzymes, including ALOXs, PTGS/cyclooxygenase, and cytochrome P450 enzymes, play a key role in catalyzing lipid peroxidation during ferroptosis (Fig. 3).
Figure 3. Lipid peroxidation in ferroptosis.
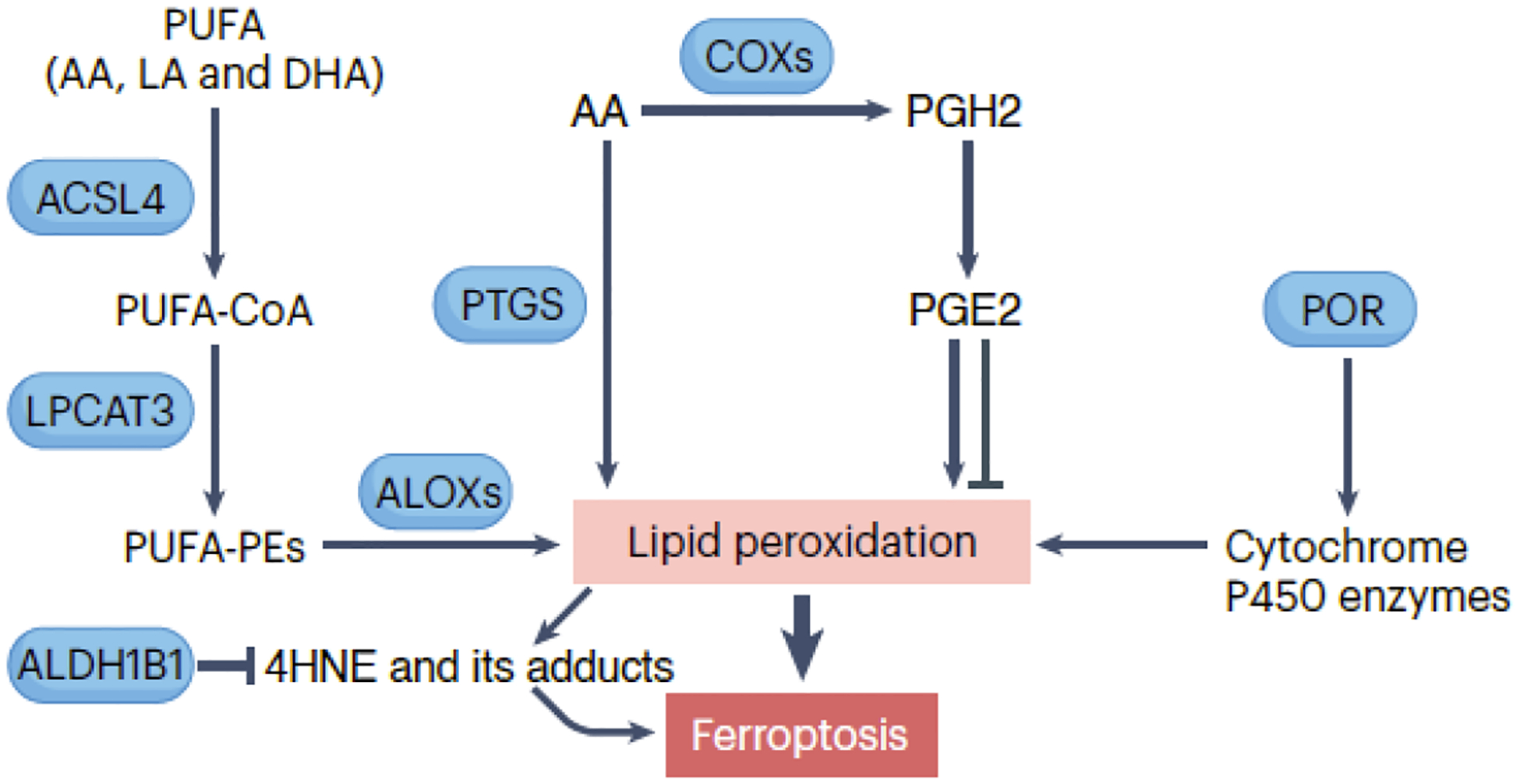
Several key enzymes participate in lipid peroxidation, including ALOX/lipoxygenase, PTGS/cyclooxygenase, and cytochrome P450 enzymes. ALOXs are a family of enzymes that catalyze the oxygenation of polyunsaturated fatty acids (PUFAs), such as arachidonic acid (AA), linoleic acid (LA), and docosahexaenoic acid (DHA), leading to the formation of lipid hydroperoxides. PTGS/cyclooxygenase enzymes are involved in prostaglandin synthesis but can also catalyze lipid peroxidation. The production of prostaglandin H2 (PGH2) and subsequently prostaglandin E2 (PGE2) promotes or inhibits ferroptosis in a context-dependent manner. Additionally, POR plays a role by supplying electrons to cytochrome P450 enzymes involved in the production of lipid hydroperoxides. These hydroperoxides can undergo further reactions, such as decomposition and rearrangement, generating highly reactive lipid radicals. Ultimately, this cascade of reactions can disrupt membrane integrity and contribute to ferroptotic cell death.
ALOXs are enzymes catalyzing PUFA oxygenation, initiating lipid peroxidation by introducing hydroperoxy-groups (-OOH) into fatty acid chains. Humans have six ALOX isoforms (ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3) with distinct substrate preferences and catalytic activities, contributing to ferroptosis in various cells or tissues41, 44, 50, 51. PEBP1 (phosphatidylethanolamine binding protein 1) forms catalytic complexes with ALOX15, efficiently peroxidizing PUFA-PE52. Inhibitors targeting ALOX15-PEBP1 complexes effectively prevent phospholipid peroxidation and mitigate injuries from total body irradiation in vivo53. However, the deletion of Alox15 does not prevent Gpx4 deletion-driven ferroptosis in kidney or T cells8, 9. Therefore, profiling ALOX expression in experimental models is crucial to assess the requirement of different ALOX members in ferroptosis.
PTGS/cyclooxygenase enzymes catalyze lipid peroxidation by oxygenating free PUFAs, generating lipid hydroperoxides. However, their primary function is prostaglandin synthesis, playing a secondary role in lipid peroxidation. PGE2 production can inhibit ferroptosis through PTGER1 and PTGER2 in cerebral I/R54, but promote it in acute kidney injury55.
Cytochrome P450 enzymes, involved in drug metabolism, can catalyze lipid peroxidation by introducing oxygen into fatty acid chains, generating lipid hydroperoxides and 4HNE, known ferroptosis mediators. As discussed earlier, POR plays a role by supplying electrons to molecular oxygen, facilitating H2O2 production for ferroptosis induction30, 31.
Regardless of the enzyme catalyzing lipid peroxidation, lipid hydroperoxides initiate a chain reaction. They undergo cleavage reactions, often catalyzed by transition metals like iron, generating highly reactive lipid radicals. These radicals react with nearby lipids, amplifying lipid peroxidation in a self-propagating process56. Electrophilic, oxidatively-truncated phospholipid variants then form, reacting with amino acid residues in proteins to induce protein lipoxidation3. This series of reactions damages cell membranes, altering membrane tension, compromising membrane repair, and ultimately leading to ferroptotic plasma membrane permeabilization57–59. The ER is proposed as the initial site that could potentially result in subsequent oxidative membrane damage in other organelles60.
Antioxidant systems in ferroptosis
Enzymatic antioxidants
The key enzyme involved in the antioxidant defense against ferroptosis is GPX4, which reduces lipid hydroperoxides to alcohols in biological membranes61 (Fig. 4). GPX4’s active center contains selenocysteine62, 63. Low selenium levels lead to ribosome stalling at GPX4’s inefficiently decoded selenocysteine UGA codon, causing ribosome collisions, premature translation termination, and proteasomal clearance of the N-terminal GPX4 fragment64. The molecular chaperone HSPA5 directly stabilizes GPX4 protein65, while autophagy66, 67 or the ubiquitin-proteasome system68 mediate GPX4 protein degradation, increasing ferroptosis sensitivity. CKB-mediated phosphorylation of GPX4 at serine residue 104 inhibits autophagy-mediated GPX4 degradation and subsequent ferroptosis67.
Figure 4. Enzymatic antioxidants in ferroptosis.
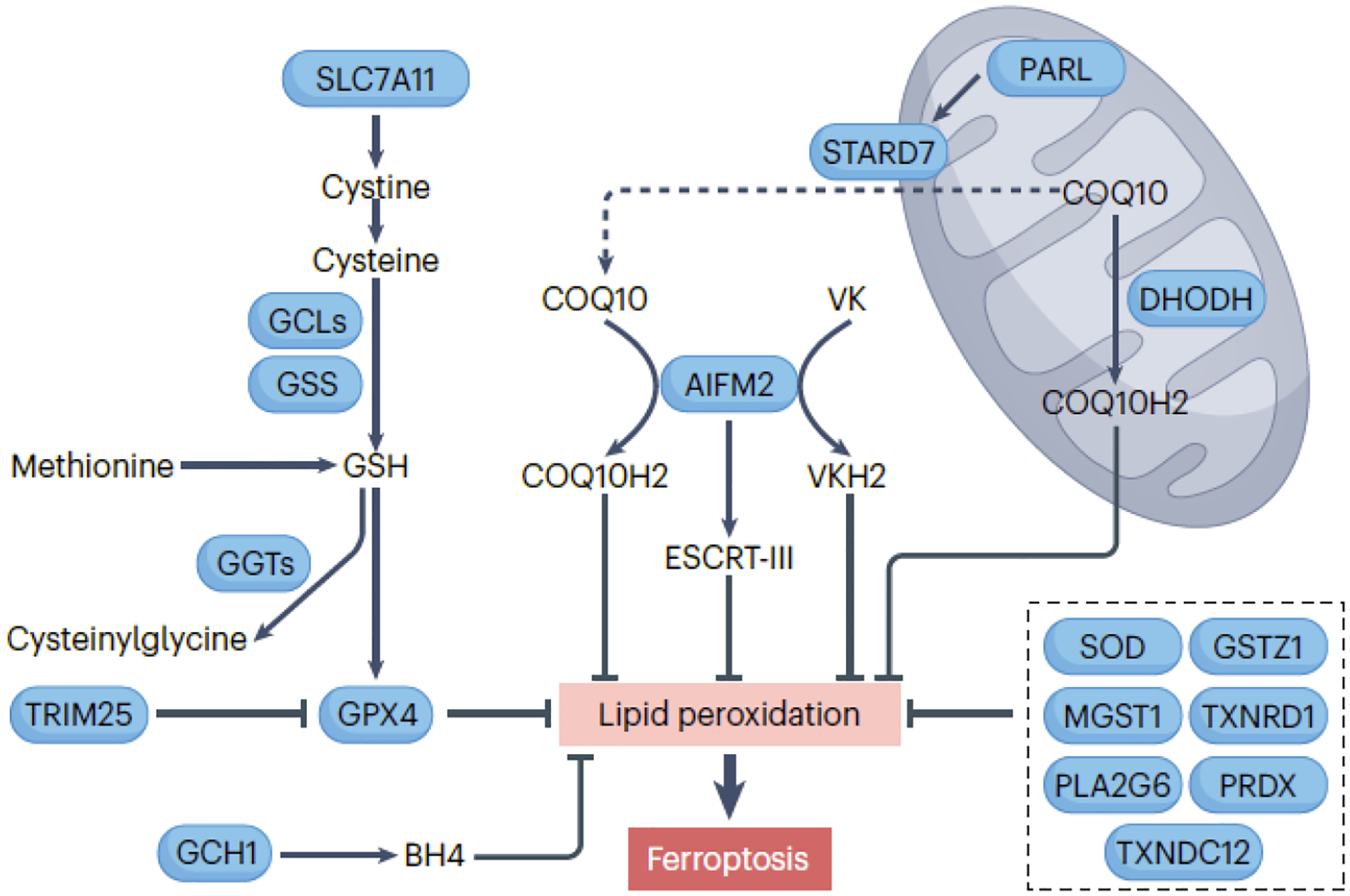
The main enzyme central to the antioxidant defense against ferroptosis is GPX4 (glutathione peroxidase 4), which requires the tripeptide cofactor glutathione (GSH), composed of glutamate, cysteine, and glycine. SLC7A11 (solute carrier family 7 member 11) is a key component of the cystine/glutamate antiporter system xc−, responsible for allowing the uptake of cystine, which is then reduced to cysteine within the cells. The synthesis of the majority of cellular GSH involves the rate-limiting substrate cysteine, catalyzed by GCLC (glutamate-cysteine ligase catalytic subunit) and GSS (glutathione synthetase). Cysteine can also be derived from the metabolism of methionine. A family of enzymes called GGT (gamma-glutamyltransferase) catalyze the breakdown of GSH into cysteinylglycine and free amino acids. AIFM2 (apoptosis inducing factor mitochondria associated 2) and DHODH dihydroorotate dehydrogenase (quinone)) play pivotal roles in the reduction of COQ10 (coenzyme Q10) to its antioxidant form, COQ10H2, in the plasma membrane/cytoplasm and mitochondria, respectively. The cleavage of STARD7 (StAR related lipid transfer domain containing 7) by the rhomboid protease PARL (presenilin associated rhomboid like) is essential for the synthesis and transport of COQ10 to the plasma membrane/cytoplasm, thereby inhibiting ferroptosis. Furthermore, AIFM2-mediated membrane repair and vitamin K (VK) reduction also contribute to its antiferroptotic activity. GCH1 (GTP cyclohydrolase 1) participates in the biosynthesis of tetrahydrobiopterin (BH4), a cofactor that helps maintain cellular redox balance and antioxidant defenses, thereby inhibiting susceptibility to ferroptotic cell death. Several other enzymes, such as SOD2 (superoxide dismutase) family, MGST1 (microsomal glutathione S-transferase 1), GSTZ1 (glutathione S-transferase zeta 1), TXNRD1 (thioredoxin reductase 1), PLA2G6 (phospholipase A2 group VI), and PRDX (peroxiredoxin) family inhibit ferroptosis in some cases.
The R152H mutation in GPX4 can cause Sedaghatian-type spinal metaphyseal dysplasia/SSMD, a rare and fatal disease in newborns69. In vitro studies suggest that this R152H mutation does not affect the catalytic activity of the enzyme in a direct fashion but rather interferes with its allosteric activation by cardiolipin70. Further examination is necessary to determine if excessive cardiolipin peroxidation by dysfunctional mitochondrial GPX4 contributes to the disease’s development.
Constitutive knockout of the Gpx4 gene in mice leads to embryonic death around 7.5–8.5 days71. In vivo evidence linking Gpx4 deficiency to ferroptosis was first observed in mice with a conditional knockout of Gpx4 in the kidney, combined with a vitamin E-deficient diet, leading to kidney damage9. This phenotype was reversed by vitamin E supplementation or the ferroptosis inhibitor liproxstatin-19. Similarly, ferroptosis of activated T cells in the absence of Gpx4 in mice is prevented by a vitamin E enriched diet8. Under normal breeding conditions and chow feeding, conditional knockout of Gpx4 in several cell types (e.g., myeloid, pancreatic epithelial cells or hepatocytes) is not lethal72–74. However, the inducible conditional knockout of Gpx4 in neurons or homozygous conditional deletion of Gpx4 in gut epithelium under the standard chow diet is lethal75, 76. Thus, GPX4 and its defense against lipid peroxidation play a context-dependent role in regulating tissue development.
GSH, a tripeptide composed of glutamate, cysteine, and glycine, acts as a GPX4 cofactor. Cysteine, a critical precursor for GSH synthesis, can limit GSH production and is derived from methionine metabolism. In addition, and more importantly, cells import extracellular cystine via the cystine/glutamate antiporter system xc−, composed of SLC7A11 and SLC3A2 subunits. Imported cystine is subsequently reduced to cysteine. Pharmacological agents like erastin or sulfasalazine can inhibit system xc− 1, 77, 78. At high concentrations, sorafenib reportedly inhibits the activity of system xc− in an indirect fashion77, but a recent study indicated that sorafenib fails only to induce ferroptosis in certain cancer cells79. GSH is primarily synthesized in the cytosol through enzymatic reactions80 and system xc− is crucial for maintaining GSH levels to prevent ferroptosis before it begins, as GSH synthesis during ferroptosis onset is too slow.
Whereas GSH depletion contributes to ferroptosis, GPX4 is not the exclusive target of GSH, suggesting the existence of GPX4-independent protective pathways against ferroptosis (Fig. 4). Among them, AIFM2/FSP1 relocates from mitochondria to the cell membrane in Gpx4-deficient cells, reducing COQ10 and inhibiting ferroptosis81, 82. STARD7 (StAR related lipid transfer domain containing 7), found in both mitochondrial intermembrane space and cytosol after cleavage by PARL (presenilin associated rhomboid like), participates in COQ10 synthesis and transport to the plasma membrane, also hindering ferroptosis83. Additionally, AIFM2 contributes to membrane repair84 and the canonical vitamin K cycle85, enhancing its antiferroptotic effects. AIFM2’s activity in ferroptosis relies on phase separation and can be initiated by N-terminal myristoylation, facilitated by compound icFSP186.
DHODH (dihydroorotate dehydrogenase (quinone)) is a mitochondrial enzyme involved in pyrimidine biosynthesis, crucial for DNA and RNA formation. The activity of DHODH has an influence on the ferroptotic susceptibility of cancer cells expressing low levels of GPX4, likely due the DHODH-catalyzed utilization of COQ10 as an electron acceptor87. Inhibiting DHODH reduces COQ10, increasing susceptibility to lipid peroxidation and ferroptosis. However, DHODH inhibitors’ potential off-target effects on AIFM2 remain debated88, 89.
Several antioxidant enzymes beyond GPX4, AIFM2, and DHODH play roles in suppressing ferroptosis. GCH1 (GTP cyclohydrolase 1) is involved in tetrahydrobiopterin/BH4 biosynthesis, contributing to cellular redox balance and ferroptosis inhibition90. Mitochondrial SOD2 defends against heat-stress-induced ferroptosis91. NOS2/iNOS (nitric oxide synthase 2) represses ferroptosis in macrophages by suppressing ALOX15-mediated lipid peroxidation92. NFE2L2/NRF2-mediated upregulation of MGST1 aids cellular detoxification in pancreatic cancer cells in response to ferroptotic activators93. GSTZ1/maleylacetoacetate isomerase (glutathione S-transferase zeta 1) inhibits ferroptosis in bladder cancer cells94, while TXNRD1 (thioredoxin reductase 1), TXNDC12 (thioredoxin domain containing 12), and peroxiredoxins (PRDX) also have context-dependent roles in ferroptosis inhibition95, 96. Additionally, Ca2+-independent PLA2G6/iPLA2β/PNPLA9 (phospholipase A2 group VI) plays a role in eliminating ferroptotic death signals by hydrolyzing peroxidized membrane phospholipids, potentially mediated by TP53 regulation97, 98. Understanding the synergistic effects of different antioxidant systems in ferroptosis remains a central theme or challenge in translational medicine.
Non-enzymatic antioxidants
Non-enzymatic antioxidants counteract harmful ROS and protect cells from oxidative damage, maintaining cellular redox balance. Examples in ferroptosis include vitamin E9, vitamin K99, GSH1, COQ1081, 82, 87, and NADPH100. They collaborate with enzymatic antioxidants to prevent or alleviate oxidative stress. Antioxidants scavenge radicals when reduced, but their oxidized form may increase oxidative stress, emphasizing the importance of monitoring redox reactions dynamically.
Metal chelators
Metal ions like iron and copper participate in Fenton or Haber-Weiss reactions, producing highly reactive hydroxyl radicals. Metal-binding proteins, such as TF (transferrin) and ferritin, sequester free iron to prevent these damaging reactions18, 19. Intracellular metal homeostasis is tightly regulated by specialized proteins, including metal chaperones that deliver metals to their target proteins101. Metallothioneins also help control metal ion availability, reducing their contribution to oxidative damage and ferroptosis78. Additionally, metal chelator drugs like deferoxamine, deferiprone, deferasirox, and ciclopirox, used in clinical settings, have shown promise in regulating ferroptosis by countering lipid peroxidation processes.
Transcriptional regulators
NFE2L2: In response to oxidative stress or exposure to electrophilic compounds, NFE2L2 is released from KEAP1 and translocates into the nucleus. SQSTM1 (sequestosome 1)-mediated protein degradation regulates the levels of KEAP1, and impaired autophagy leads to SQSTM1 accumulation, resulting in KEAP1 degradation and increased NFE2L2 protein stability102. In the nucleus, NFE2L2 binds to specific DNA sequences known as antioxidant response elements/AREs or electrophile response elements/EpREs in the promoter regions of target genes. This binding activates the transcription of a set of genes involved in both GPX4-dependent and GPX4-independent pathways to inhibit ferroptosis103, 104. A key unanswered question is how NFE2L2 selectively activates target genes to inhibit ferroptosis rather than other types of cell death.
TP53: TP53 has a dual role in regulating ferroptosis susceptibility. For instance, the acetylation-deficient TP53 variant, TP53[3KR], lacks the ability to induce apoptosis and cell cycle arrest. However, it retains its capacity for tumor suppression similar to wild-type TP53 by suppressing SLC7A11 expression, thereby increasing ferroptosis sensitivity in certain cancer cells105. TP53-mediated downregulation of VKORC1L1 also increases ferroptosis sensitivity in cancer cells through vitamin K metabolism106. Additionally, TP53 positively regulates ferroptosis by inducing the expression of SAT1, a rate-limiting enzyme in polyamine catabolism that can produce ROS107. Conversely, under certain conditions, TP53 inhibits ferroptosis. For instance, in human colorectal cancer cells, TP53 deletion increases sensitivity to erastin-triggered ferroptosis through the activation of the DPP4-NOX1 pathway on the cell membrane29. The classical TP53-inducible gene, CDKN1A/p21, also inhibits ferroptosis in cancer cells108. Furthermore, TP53 mutation (R175H) yields a modified TP53 protein that functions as a suppressor of ferroptosis by preventing BACH1-mediated downregulation of SLC7A11, thus promoting tumor growth109. These findings underscore the wide implications of TP53 in the modulation of ferroptosis.
ATF4: ATF4 (activating transcription factor 4) plays a crucial role in ER stress and amino acid metabolism. ATF4 activation by ER stress upregulates anti-ferroptotic genes, such as HSPA565, SLC7A11110, or TXNDC1296. This pathway protects against ferroptosis in cancer cells and mitochondrial cardiomyopathy111, 112. Sublethal cytochrome c release induced by pro-apoptotic BH3 mimetics (ABT-737 and S63845) can lead to ATF4-dependent chemotherapy resistance in cancer cells113. Considering the importance of the ER as a critical organelle for ferroptosis60, ATF4 likely plays a specific role in transcriptional regulation, preserving cellular viability and conferring ferroptosis resistance.
Other important transcription factors, including HIF1A114, NFKB/NF-κB115, YAP1116, 117, WWTR1116, 117, and SREBF1118, also play a context-dependent role in shaping the ferroptotic response through multiple targeted genes.
Membrane repair system
Ca2+ is the key initiator of the membrane repair response. When the plasma membrane is damaged, Ca2+ enters the cytoplasm from outside sources, signaling downstream repair processes, such as endosomal sorting complexes required for transport (ESCRT)-III58, 59 and exocytosis119, thereby enhancing ferroptosis resistance. Efficient membrane repair is vital for cell function, and its disruption may be irreversible. However, Ca2+ signaling from different organelles has a dual role in the control of ferroptosis sensitivity, underscoring the importance of timely monitoring.
Therapeutic opportunities and challenges
Therapeutic opportunities
Preclinical studies suggest that targeting ferroptosis has broad implications for various diseases, notably in oncology, neurodegenerative disorders, and I/R injury, as elaborated below.
Cancer cells often undergo metabolic changes that disrupt redox balance and increase their reliance on antioxidants, making them vulnerable to ferroptosis induction. Targeting ferroptosis offers a novel approach to overcome treatment limitations105, 120–124, despite occasional resistance mechanisms (e.g., due to enhanced biosynthesis of pyrimidines28 or hydropersulfides125). Furthermore, specific mutations in genes like KRAS and TP53 in certain solid cancers are associated with ferroptosis sensitivity, offering potential for precision medicine strategies1, 105, 109.
Neurodegenerative disorders, such as Alzheimer, Parkinson, and Huntington diseases, involve neuronal destruction and protein aggregation in the brain. Oxidative stress plays a key role in this degeneration, leading to lipid peroxidation and ferroptotic cell death. Therapies targeting ferroptosis inhibition aim to reduce oxidative damage and enhance neuron survival62, 126. Modulating ferroptosis pathways may help mitigate the accumulation of harmful byproducts like lipid peroxides and reactive aldehydes, potentially slowing neurodegeneration, including in conditions like multiple sclerosis127.
I/R events trigger oxidative stress and cell death, making ferroptosis-targeting therapies promising for mitigating oxidative damage and preserving tissue function in conditions like stroke, myocardial infarction, and kidney and liver injuries. Combining ferroptosis and necroptosis inhibition has shown particular effectiveness128, 129. For kidney tubules, ferroptotic cell death propagation follows a unique pattern that has been referred to as a “wave-of-death” and has since also been described in other systems56. These studies highlight the therapeutic potential of ferroptosis inhibitors in I/R-related diseases.
Therapeutic challenges
Specificity and selectivity: High specificity and selectivity are needed to minimize off-target effects and potential toxicity. For instance, there are concerns about off-target effects of RSL3 and ML162 on the TXNRD1 protein130. Imidazole ketone erastin (IKE) is a widely used in vivo ferroptosis inducer131, but its activity relative to other in vitro activators needs further study. Additionally, inhibiting ferroptosis through antioxidant mechanisms may impact non-ferroptotic pathways, including apoptosis132 and necroptosis128, 133.
Drug delivery: Developing targeted drug delivery systems is essential to enhance therapeutic effectiveness and reduce systemic side effects. Recent research has shown promise in using nanoparticles, including liposomes, micelles, and polymer-based carriers, to address these challenges. Nanoparticles provide advantages like enhanced drug stability, solubility, and targeted delivery.
Biomarker identification: Several biomarkers, such as TFRC134, ACSL434, and PTGS261, hyperoxidized PRDX3135, have been measured at the mRNA or protein levels to monitor ferroptosis responses. Theoretically, blood-based biomarkers have strong translational potential for clinical use, particularly danger signals like HMGB1136, ATP137, SQSTM1138, and DCN (decorin)139, which can indicate plasma membrane rupture during ferroptosis. DCN is notable for its ability to distinguish ferroptosis from other cell death types, especially in early stages139. LC-MS-based redox lipidomics is a valuable tool for characterizing ferroptotic biomarkers in vivo, especially in various disease conditions3.
Side effects: Current widely used ferroptosis activators lack cell or tissue selectivity, potentially causing unintended cell death in various immune cell types, such as neutrophils140, CD8+ T cells141, 142, natural killer cells143 and dendritic cells144. Strategies are needed to selectively target tumor cells while preserving immune cell integrity and anticancer immune responses. A compound called N6F11 shows promise in selectively inducing ferroptosis in cancer cells, not immune cells, by triggering TRIM25-dependent GPX4 degradation68. Ferroptosis therapy can also lead to adverse effects like early-onset cachexia145, stem cell death146, bone marrow injury147, hematopoiesis disruption146, and inflammation-driven tumorigenesis73, 74, 112.
Clinical translation: While some FDA-approved drugs like sorafenib77, sulfasalazine77, artesunate148, and zalcitabine50 have shown potential in preclinical ferroptosis induction, their effects may be linked to adverse off-target effects. Identifying safe drugs for patients is crucial, as is considering co-administration of medications to mitigate systemic toxicity and exploring intermittent treatment regimens for better tolerability. Future research should address these aspects to understand ferroptosis in human diseases. Well-designed clinical trials are essential to evaluate the effectiveness, safety, and long-term outcomes of ferroptosis-targeting agents. These trials should enroll specific patient populations, identify sensitive ferroptosis biomarkers, and measure them alongside clinical outcomes.
Conclusion and outlook
In recent years, the field of ferroptosis research has witnessed a remarkable surge. This surge reflects the establishment of a genuine ferroptosis-focused research era149, 150. However, the initial definition of ferroptosis as Fe(II)-dependent regulated necrosis accompanied by lipid peroxidation is now recognized as incomplete. Although iron-induced oxidative stress remains a prominent trigger, other iron-independent stimuli or stresses are undoubtedly involved in ferroptosis. Considering that the core downstream feature of ferroptosis is structural damage to cellular membranes resulting from uncontrolled lipid peroxidation, the term “lipotoxicity” may also reflect its core mechanism.
Molecular mechanisms of ferroptosis have expanded beyond the original GPX4 regulatory pathway. This review explores the interplay between pro-ferroptotic and anti-ferroptotic mechanisms, categorized as GPX4-dependent and GPX4-independent, encompassing historical insights and recent findings. However, questions about when, where, and how these pathways activate persist.
Numerous regulatory molecules linked to ferroptosis also play roles in other types of cell death, emphasizing the complexity of intercellular crosstalk. Untangling these mechanisms requires well-designed experiments, stringent controls, and the validation of specific biomarkers. Understanding how physiological and pathological stressors influence ferroptosis in real-world situations remains a challenge. Additionally, the intricate connections between stress pathways leading to ferroptotic and non-ferroptotic cell death require further elucidation.
Despite occasional research limitations and conflicting hypotheses, we maintain optimism about the future prospects of ferroptosis. We believe that the principles of ferroptosis will eventually find clinical applications beyond their heuristic value.
Acknowledgements
The authors appreciate all of the pioneers in the field and our colleagues who contributed to the study of the process and function of ferroptosis. The authors apologize if they were unable to cite all of the important references in this field owing to space limitations.
Footnotes
Competing interests statement
B.R.S. is an inventor on patents and patent applications involving ferroptosis; co-founded and serves as a consultant to ProJenX, Inc. and Exarta Therapeutics; holds equity in Sonata Therapeutics; serves as a consultant to Weatherwax Biotechnologies Corporation and Akin Gump Strauss Hauer & Feld LLP; B.G. is an inventor on patent applications involving targeting ferroptosis in cancer therapy, and reports personal fees from Guidepoint Global, Cambridge Solutions, and NGM Bio; D.I.G. is an employee and shareholder of AstraZeneca; V.G.S. serves as an advisor to and/or has equity in Branch Biosciences, Ensoma, and Cellarity, all unrelated to the present work; L.G. is/has been holding research contracts with Lytix Biopharma, Promontory and Onxeo, has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Imvax, Sotio, Promontory, Noxopharm, EduCom, and the Luke Heller TECPR2 Foundation, and holds Promontory stock options; A.I.B. holds shares in Cogstate Ltd, Alterity Ltd and a profit share with Collaborative Medicinal Development LLC and acts as a paid consultant to Collaborative Medicinal Development LLC. The remaining authors declare no competing interests. X.J. holds inventorship of patents related to autophagy and cell death, and holds equity as well as consults for Exarta Therapeutics and Lime Therapeutics. G.K. has been holding research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, PharmaMar, Osasuna Therapeutics, Samsara Therapeutics, Sanofi, Tollys, and Vascage. G.K. is on the Board of Directors of the Bristol Myers Squibb Foundation France. G.K. is a scientific co-founder of everImmune, Osasuna Therapeutics, Samsara Therapeutics and Therafast Bio. G.K. is in the scientific advisory boards of Hevolution, Institut Servier and Longevity Vision Funds. G.K. is the inventor of patents covering therapeutic targeting of aging, cancer, cystic fibrosis and metabolic disorders. G.K.’s wife, Laurence Zitvogel, has held research contracts with Glaxo Smyth Kline, Incyte, Lytix, Kaleido, Innovate Pharma, Daiichi Sankyo, Pilege, Merus, Transgene, 9 m, Tusk and Roche, was on the on the Board of Directors of Transgene, is a cofounder of everImmune, and holds patents covering the treatment of cancer and the therapeutic manipulation of the microbiota. G.K.’s brother, Romano Kroemer, was an employee of Sanofi and now consults for Boehringer-Ingelheim. All other authors have disclosed no conflicts of interest, whether financial or non-financial. The funders were not involved in the preparation of the manuscript.
References
- 1.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X et al. A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun 13, 6318 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amoscato AA et al. Formation of protein adducts with Hydroperoxy-PE electrophilic cleavage products during ferroptosis. Redox Biol 63, 102758 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li P et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol 22, 1107–1117 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amaral EP et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med 216, 556–570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsushita M et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 212, 555–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedmann Angeli JP et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muri J, Thut H, Bornkamm GW & Kopf M B1 and Marginal Zone B Cells but Not Follicular B2 Cells Require Gpx4 to Prevent Lipid Peroxidation and Ferroptosis. Cell Rep 29, 2731–2744 e2734 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Dolma S, Lessnick SL, Hahn WC & Stockwell BR Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Yang WS & Stockwell BR Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15, 234–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seiler A et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Banjac A et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene 27, 1618–1628 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Chen PH et al. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis 12, 198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Q et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 19, 1982–1996 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 17, 3361–3374 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou W et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao M et al. Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai Y et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun 508, 997–1003 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Yang M et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv 5, eaaw2238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian R et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat Neurosci 24, 1020–1034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao M, Monian P, Quadri N, Ramasamy R & Jiang X Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59, 298–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao M et al. Role of Mitochondria in Ferroptosis. Mol Cell 73, 354–363 e353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Liu J, Hou W, Kang R & Tang D STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev Biol 9, 698679 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee H et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 22, 225–234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song X et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X(c)(−) Activity. Curr Biol 28, 2388–2399 e2385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang C et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat Cell Biol 25, 836–847 (2023). [DOI] [PubMed] [Google Scholar]
- 29.Xie Y et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep 20, 1692–1704 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Yan B et al. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell (2020). [DOI] [PubMed] [Google Scholar]
- 31.Zou Y et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16, 302–309 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kagan VE et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan H, Li X, Zhang X, Kang R & Tang D Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478, 1338–1343 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Dixon SJ et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 10, 1604–1609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Z et al. The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat Commun 13, 7965 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao P et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40, 365–378 e366 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang HL et al. PKCbetaII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol 24, 88–98 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Chen X et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy 19, 54–74 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magtanong L et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432 e429 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang WS et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y et al. SLC25A22 as a Key Mitochondrial Transporter Against Ferroptosis by Producing Glutathione and Monounsaturated Fatty Acids. Antioxid Redox Signal 39, 166–185 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang D et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 186, 2748–2764 e2722 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chu B et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21, 579–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou Y et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xin S et al. MS4A15 drives ferroptosis resistance through calcium-restricted lipid remodeling. Cell Death Differ 29, 670–686 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klasson TD et al. ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metab 10, 14 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reed A, Ware T, Li H, Fernando Bazan J & Cravatt BF TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids. Nat Chem Biol 19, 378–388 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J et al. TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy 19, 945–956 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li C et al. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 17, 948–960 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagasaki T et al. 15LO1 dictates glutathione redox changes in asthmatic airway epithelium to worsen type 2 inflammation. J Clin Invest 132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wenzel SE et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 171, 628–641 e626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dar HH et al. Discovering selective antiferroptotic inhibitors of the 15LOX/PEBP1 complex noninterfering with biosynthesis of lipid mediators. Proc Natl Acad Sci U S A 120, e2218896120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Y et al. COX-2/PGE2 Pathway Inhibits the Ferroptosis Induced by Cerebral Ischemia Reperfusion. Mol Neurobiol 59, 1619–1631 (2022). [DOI] [PubMed] [Google Scholar]
- 55.Liu Y et al. PGE2 pathway mediates oxidative stress-induced ferroptosis in renal tubular epithelial cells. FEBS J 290, 533–549 (2023). [DOI] [PubMed] [Google Scholar]
- 56.Riegman M et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol 22, 1042–1048 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirata Y et al. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr Biol 33, 1282–1294 e1285 (2023). [DOI] [PubMed] [Google Scholar]
- 58.Pedrera L et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ 28, 1644–1657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai E, Meng L, Kang R, Wang X & Tang D ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun 522, 415–421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.von Krusenstiern AN et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol 19, 719–730 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ingold I et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Yao Y et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol 22, 1127–1139 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Li Z et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat Chem Biol 18, 751–761 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu S et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res 77, 2064–2077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Z et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A 116, 2996–3005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu K et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat Cell Biol 25, 714–725 (2023). [DOI] [PubMed] [Google Scholar]
- 68.Li J et al. Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med 15, eadg3049 (2023). [DOI] [PubMed] [Google Scholar]
- 69.Liu H et al. Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol 18, 91–100 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roveri A et al. Cardiolipin drives the catalytic activity of GPX4 on membranes: Insights from the R152H mutant. Redox Biol 64, 102806 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yant LJ et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34, 496–502 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Kang R et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24, 97–108 e104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dai E et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun 11, 6339 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conche C et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen L, Hambright WS, Na R & Ran Q Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem 290, 28097–28106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mayr L et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat Commun 11, 1775 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dixon SJ et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun X et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64, 488–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zheng J et al. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis 12, 698 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forman HJ, Zhang H & Rinna A Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 30, 1–12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doll S et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Bersuker K et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deshwal S et al. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat Cell Biol (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dai E et al. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun 523, 966–971 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Mishima E et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakamura T et al. Phase separation of FSP1 promotes ferroptosis. Nature 619, 371–377 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mao C et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mishima E et al. DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature 619, E9–E18 (2023). [DOI] [PubMed] [Google Scholar]
- 89.Mao C, Liu X, Yan Y, Olszewski K & Gan B Reply to: DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature 619, E19–E23 (2023). [DOI] [PubMed] [Google Scholar]
- 90.Kraft VAN et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu L, Wang M, Gong N, Tian P & Deng H Se improves GPX4 expression and SOD activity to alleviate heat-stress-induced ferroptosis-like death in goat mammary epithelial cells. Anim Cells Syst (Seoul) 25, 283–295 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kapralov AA et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol 16, 278–290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kuang F, Liu J, Xie Y, Tang D & Kang R MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol 28, 765–775 e765 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Wang Q et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis 12, 426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lovatt M et al. Peroxiredoxin-1 regulates lipid peroxidation in corneal endothelial cells. Redox Biol 30, 101417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tang L et al. TXNDC12 inhibits lipid peroxidation and ferroptosis. iSciense 132, 108449 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sun WY et al. Phospholipase iPLA(2)beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol 17, 465–476 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen D et al. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun 12, 3644 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kolbrink B et al. Vitamin K1 inhibits ferroptosis and counteracts a detrimental effect of phenprocoumon in experimental acute kidney injury. Cell Mol Life Sci 79, 387 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shimada K, Hayano M, Pagano NC & Stockwell BR Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol 23, 225–235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Protchenko O et al. Iron Chaperone Poly rC Binding Protein 1 Protects Mouse Liver From Lipid Peroxidation and Steatosis. Hepatology 73, 1176–1193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Komatsu M et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12, 213–223 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Sun X et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Anandhan A et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv 9, eade9585 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiang L et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang X et al. Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metab (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ou Y, Wang SJ, Li D, Chu B & Gu W Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A 113, E6806–E6812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tarangelo A et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22, 569–575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Su Z et al. Specific regulation of BACH1 by the hotspot mutant p53(R175H) reveals a distinct gain-of-function mechanism. Nat Cancer 4, 564–581 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen D et al. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 36, 5593–5608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ahola S et al. OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab 34, 1875–1891 e1877 (2022). [DOI] [PubMed] [Google Scholar]
- 112.He F et al. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J Hepatol 79, 362–377 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kalkavan H et al. Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185, 3356–3374 e3322 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang Z et al. HIF-1alpha drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep 42, 112945 (2023). [DOI] [PubMed] [Google Scholar]
- 115.Yao F et al. A targetable LIFR-NF-kappaB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat Commun 12, 7333 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu J et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang WH et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep 28, 2501–2508 e2504 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yi J, Zhu J, Wu J, Thompson CB & Jiang X Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A 117, 31189–31197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ralhan I et al. Autolysosomal exocytosis of lipids protect neurons from ferroptosis. J Cell Biol 222 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ubellacker JM et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang W et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lang X et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov 9, 1673–1685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Badgley MA et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20, 1181–1192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barayeu U et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat Chem Biol 19, 28–37 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun J et al. Midbrain dopamine oxidation links ubiquitination of glutathione peroxidase 4 to ferroptosis of dopaminergic neurons. J Clin Invest 133 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jia JN et al. Neuroprotective Effects of the Anti-cancer Drug Lapatinib Against Epileptic Seizures via Suppressing Glutathione Peroxidase 4-Dependent Ferroptosis. Front Pharmacol 11, 601572 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tonnus W et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun 12, 4402 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Linkermann A et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111, 16836–16841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cheff DM et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol 62, 102703 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang Y et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633 e629 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sun Y, Deng R & Zhang C Erastin induces apoptotic and ferroptotic cell death by inducing ROS accumulation by causing mitochondrial dysfunction in gastric cancer cell HGC-27. Mol Med Rep 22, 2826–2832 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Muller T et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci 74, 3631–3645 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Feng H et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep 30, 3411–3423 e3417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cui S et al. Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases. Mol Cell (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wen Q, Liu J, Kang R, Zhou B & Tang D The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun 510, 278–283 (2019). [DOI] [PubMed] [Google Scholar]
- 137.Efimova I et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. Journal for immunotherapy of cancer 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang L et al. Extracellular SQSTM1 exacerbates acute pancreatitis by activating autophagy-dependent ferroptosis. Autophagy, 1–12 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu J et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy 18, 2036–2049 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim R et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature 612, 338–346 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xu S et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577 e1567 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ma X et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab 33, 1001–1012 e1005 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Poznanski SM et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab 33, 1205–1220 e1205 (2021). [DOI] [PubMed] [Google Scholar]
- 144.Han L et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun 576, 33–39 (2021). [DOI] [PubMed] [Google Scholar]
- 145.Ferrer M et al. Ketogenic diet promotes tumor ferroptosis but induces relative corticosterone deficiency that accelerates cachexia. Cell Metab 35, 1147–1162 e1147 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhao J et al. Human hematopoietic stem cell vulnerability to ferroptosis. Cell 186, 732–747 e716 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Song X et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun 480, 443–449 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Eling N, Reuter L, Hazin J, Hamacher-Brady A & Brady NR Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2, 517–532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stockwell BR Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tang D, Chen X, Kang R & Kroemer G Ferroptosis: molecular mechanisms and health implications. Cell Res 31, 107–125 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]