

Abstract
Mass-spectrometry based assays in structural biology studies measure either intact or digested proteins. Typically, different mass spectrometers are dedicated for such measurements: those optimized for rapid analysis of peptides or those designed for high molecular weight analysis. A commercial trapped ion mobility-quadrupole-time of flight (TIMS-Q-TOF) platform is widely utilized for proteomics and metabolomics, with ion mobility providing a separation dimension in addition to liquid chromatography. The ability to perform high-quality native mass spectrometry of protein complexes, however, remains largely uninvestigated. Here, we evaluate a commercial TIMS-Q-TOF platform for analyzing non-covalent protein complexes by utilizing the instrument’s full range of ion mobility, MS, and MS/MS (both in-source activation and collision cell CID) capabilities. The TIMS analyzer is able to be tuned gently to yield collision cross sections on native-like complexes comparable to those previously reported on various instrument platforms. In-source activation and collision cell CID were robust for both small and large complexes. TIMS-CID was performed on protein complexes streptavidin (53 kDa), avidin (68 kDa), and cholera toxin B (CTB, 58 kDa). Complexes pyruvate kinase (237 kDa) and GroEL (801 kDa) were beyond the trapping capabilities of the commercial TIMS analyzer, but mass spectra could be acquired. The presented results indicate that the commercial TIMS-Q-TOF platform can be used for both omics and native mass spectrometry applications; however, modifications to the commercial RF drivers for both the TIMS analyzer and quadrupole (currently limited to m/z 3,000) are necessary to mobility analyze protein complexes greater than about 60 kDa.
Keywords: timsTOF, native MS, protein complexes, ion mobility
Graphical Abstract
Introduction
Determining and understanding protein structure is critical to unraveling key insights into protein function and malfunction. The field of structural biology utilizes several complementary to investigate the structures of biological assemblies. Those structures can then be integrated with physical theories and computational models.1 Traditional biophysical techniques employed for protein structure determination include X-ray crystallography, small angle X-ray scattering (SAXS), cryo-electron microscopy (Cryo-EM), and nuclear magnetic resonance (NMR) spectroscopy.1 Native mass spectrometry (nMS) is an additional analytical technique increasingly being utilized to answer structural biology questions, or provide complementary information to the preceding techniques.2–4 Electrospray ionization (ESI) allows peptides, proteins, and protein complexes to be gently transferred into the gas phase, by maintaining kinetically trapped solution-like structures and thereby expanding MS platform capabilities beyond only mass measurements to a tool capable of structural characterization.5–8
With early-generation mass spectrometers developed mostly for the analysis of small molecules, several instrument modifications are required to expand to high-mass capabilities.9–11 These modifications, originally performed primarily by individual research groups, have become more prevalent with instrument manufacturers, expanding nMS capabilities to a larger number of research institutions and industry partners.12–13 Modifications to mass spectrometers for nMS applications include the implementation of nanoelectrospray ionization (nESI) sources capable of desolvating and desalting biological samples, low-frequency (high m/z) RF drivers to extend the mass ranges of devices such as selection quadrupoles and ion mobility cells, and mass analyzers capable of measuring and resolving high mass species.5–7, 9–11
While tandem MS experiments provide mass measurements and structural information, ion mobility spectrometry (IMS) is capable of probing gas-phase three-dimensional structures of biological molecules.14–17 IMS is a gas-phase separation technique in which ions are separated based on their mobility (a function of an ion’s mass, charge, and rotationally averaged cross section or shape) in a weak electric field in the presence of a background gas. For Trapped Ion Mobility Spectrometry (TIMS), an electric field that opposes ion forward motion holds ions stationary against a background gas that pushes ions along the TIMS analyzer (an ion funnel). Ions of different mobilities are trapped at different points (potentials) along the ion optical axis field using an electric field gradient and are eluted from the analyzer as the TIMS potential gradient is reduced over time.18–23 TIMS (or an alternate ion mobility approach of high-field asymmetric waveform ion mobility, FAIMS) is often coupled after liquid chromatography in “omics” based mass spectrometry experiments, such as proteomics or metabolomics, in which complex biological mixtures require orthogonal modes of separation to identify the numerous, structurally similar components.18–20, 24–25 In addition to TIMS serving as a separation technique, the collision cross section (CCS) can be determined from the measured mobility value, providing another dimension of molecular identification as well as insight into protein shape and conformation.26–27 As a result, mass spectrometers equipped with IMS capabilities are highly desirable for various biological applications.
Recent work exploring the capabilities of TIMS technology for analyzing biomolecules, notably proteins and their non-covalent assemblies, has shown promise for adapting the technology beyond its initial application of small molecule analysis. However, most of this work has been performed on modified instrument platforms. Specifically, Fernandez-Lima and co-workers have mobility-analyzed macromolecular complexes up to 19,000 m/z by modifying the geometries of electrodes (and thus the trapping pseudopotentials) in the TIMS analyzer in a custom Q-TOF platform.28 Bleiholder and co-workers have demonstrated the value of implementing dual TIMS funnels (tandem-TIMS-MS) on a Q-TOF to characterize the heavily glycosylated protein complex avidin and performed structurally informative top-down experiments with both collision-induced dissociation (CID) and ultraviolet photon dissociation (UVPD).29–31 On a modified Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer, Wysocki and co-workers combined TIMS with surface-induced dissociation (SID) to provide connectivity information of protein complexes via structurally informative dissociation.32 While these works highlight the benefits and potential capabilities of TIMS for native MS analyses, in-house modified instrument platforms are not available to the broader nMS and structural biology communities. Here, we evaluate a commercial TIMS-Q-TOF MS platform initially marketed for small molecule and peptide analysis as a potential platform for nMS studies. The availability of commercial mass spectrometers able to span multiple applications, including both -omics and native experiments, allows investigators to perform more expansive and numerous experiments (such as bottom-up proteomics, top-down proteomics, complex-down mass spectrometry, and CCS measurements of an intact protein or complex) to provide insights to the structures of biologically relevant systems.33–34
Experimental Section
Materials
Ammonium acetate, cytochrome C, triethylammonium acetate (TEAA), and cholera toxin B (CTB) were purchased from Sigma Aldrich (St. Louis, MO). GroEL lyophilized powder was also purchased from Sigma and prepared via a refolding procedure described elsewhere.35 Avidin from hen egg-white and streptavidin from Streptomyces avidinii were purchased from Thermo Scientific Pierce Biotechnology (Rockford, IL). Pyruvate kinase from rabbit was purchased from Lee Biosolutions (Maryland Heights, MO).
Avidin, streptavidin, CTB, GroEL, and pyruvate kinase samples were buffer-exchanged into 200 mM ammonium acetate (pH ~6.8) with size exclusion chromatography spin columns with a 6 kDa cutoff (Micro Bio-Spin 6, Bio-Rad, Hercules, CA) and were further diluted with 200 mM ammonium acetate to 5–10 μM protein complex concentration. For experiments performed under charge-reducing conditions, TEAA was added to the protein solutions at a final concentration of 40 mM TEAA and 160 mM ammonium acetate. Cytochrome C was prepared at a protein concentration of 30 μM in 10 mM aqueous ammonium acetate (the difference in preparation for Cytochrome C is described in the instrumentation section below). A summary of the proteins, their masses, expected multimers, and multimeric masses can be found in the Supporting Information, Table S1.
Instrumentation
Experiments were performed on commercial Bruker TIMS-Q-TOF (timsTOF Pro) mass spectrometers without any hardware modifications at both The Ohio State University (OSU) and Florida State University (FSU). Cytochrome C was prepared at a higher concentration and directly infused at a flow rate of 180 μL/hr using an Apollo II ESI ion source at FSU. All other proteins and protein complexes were directly infused using nanoelectrospray ionization (nESI) with a custom Bruker nESI source at OSU. We have included both ESI and nESI to guide users who do not have access to a compatible nESI source. We have previously used the nanospray source on a SolariX 15T T-ICR mass spectrometer and easily transferred it to the timsTOF Pro platform due to similarities of the source region.32,36–38 Briefly, the source consists of a linear positioning station in which a glass capillary containing the protein solution is brought into direct contact with a grounded platinum wire while a DC voltage is applied to the counter entrance electrode of the mass spectrometer. Borosilicate glass capillaries (with filament, Sutter Instruments) were prepared in-house using a Sutter Instruments P-97 pipette puller (Novato, CA) and protein samples were ionized using capillary voltages of −0.5–1.0 kV.
To transmit protein complexes, the RF amplitudes for ion transfer elements were increased relative to typical settings for small molecules while also taking care to prevent unintended ion activation. A basic schematic of the instrument is shown in Figure 1.
Figure 1.
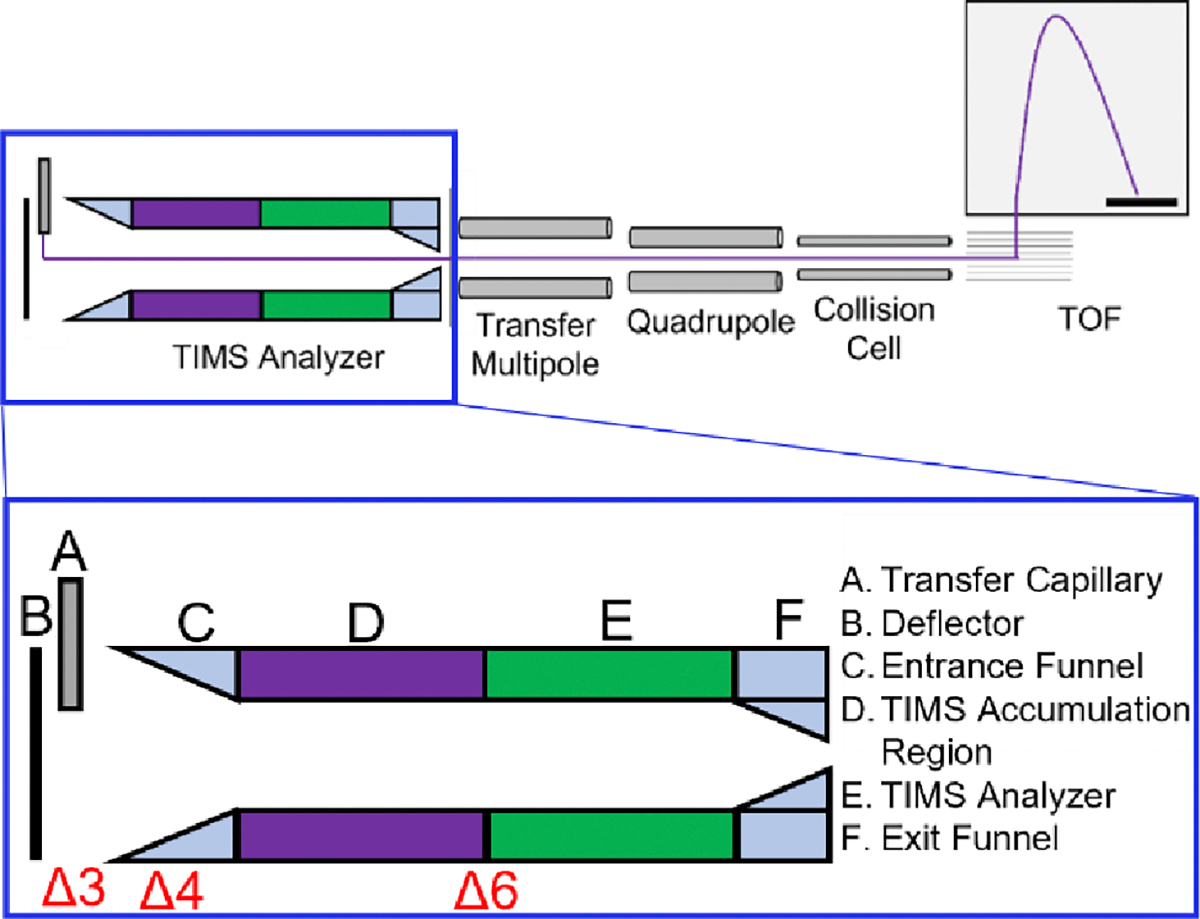
Overview of the timsTOF Pro instrument. Mobility analysis is performed in the TIMS analyzer, followed by a transfer multipole, quadrupole (selection up to 3000 m/z), collision cell, and Time-of-Flight (TOF) mass analyzer. The purple trace indicates the path of ion motion. Delta values represent potentials set in the source/TIMS region, with Δ3 being the voltage difference between the Deflector and Entrance Funnel, Δ4 the voltage across the entrance funnel, and Δ6 being the voltage applied between the exit of the TIMS accumulation region (purple) and the entrance of the TIMS analyzer region (green). Optimization of these potentials for high molecular weight operation is discussed in the main text.
A comparison of instrument settings typically used for bottom-up proteomics experiments on a commercial TIMS-Q-TOF instrument and how these values changed for the analysis of native-like protein complexes can be found in Table S2 of the Supporting Information. When performing TIMS of monomeric proteins, the TIMS funnel RF was set to 300 Vpp, and for protein complexes, the maximum value of 350 Vpp was used. The transfer multipole RF was set to 500 Vpp (out of 600 Vpp maximum), and the collision cell RF was set between 3000–4000 Vpp (out of 4000 Vpp maximum), depending on the mass of the analyte ions. When tuning for the highest m/z species, the tetradecameric GroEL, the optimum RF amplitudes were not achieved by the commercial hardware, so all RF amplitudes were set to the maximum values to evaluate instrument performance. The collision cell pre-pulse storage time and transfer time were increased to 15–25 μs and 120–140 μs, , respectively. Settings that can potentially disrupt native-like structures are further discussed in Results and Discussion: Preservation of Native-like Structures. Collision Cell gas flow rate was set to 55–95%. Mass calibration and TIMS mobility calibration were performed using Agilent ESI-L Low Concentration Tuning Mix (Santa Clara, CA). Collision cross sections (CCS) were calculated using the Mason-Schamp equation with the measured reduced mobilities (Ko) following calibration with Tune Mix.39,40 Single CCS measurements are presented in this work.
Results and Discussion
Several model proteins and protein complexes were chosen to evaluate the performance of the commercial, un-modified TIMS-Q-TOF for transmission of high molecular weight species, efficient trapping within the ion mobility analyzer, and efficient dissociation of protein complexes. These characterized systems are ideal for comparing with data obtained across multiple mass spectrometry vendors and platforms. TIMS was performed for Cytochrome C, avidin, streptavidin, and CTB. Protein complexes pyruvate kinase (237 kDa) and GroEL (801 kDa) exceeded the mobility range and trapping of the TIMS analyzer, however, the full mass spectra are shown below to showcase the transmission and detection of high mass complexes and efficiency of in-source activation for improving spectral appearance and enhancing the ability to more confidently identify and determine accurate masses for peaks convolved with heavy salt adduction, often present in the analysis of biological analytes by MS.
Preservation of Native-like Protein Structures
To preserve a protein close to its native structure in a TIMS measurement, it is imperative to minimize the heating of the protein in the ion source, throughout the TIMS analyzer, and in post-TIMS ion optics. Parameters affecting ion heating and native-like preservation for TIMS measurements have been published in greater detail previously, and we direct the readers to these fruitful discussions for critical insights beyond the general tuning parameters presented here.23, 41, 42 Reducing the thermal heating and unintended ion activation during ESI can be achieved by employing a low drying gas temperature and a reduced electrospray voltage. Retention of even weakly-bound peptide clusters in post-TIMS ion optics can be accomplished by reducing DC electric fields as described.21 Minimizing vibrational ion heating in the TIMS ion optics can be accomplished by minimizing translational-vibrational energy uptake due to (1) the axial DC electric field; (2) the radial RF electric field; and (3) space-charge effects and ion-ion interactions.22,23 In our experience, the most critical aspect is to minimize all DC electric fields in the regions prior to the mobility separation in TIMS. Hence, we apply a low DC bias between the deflector plate and entrance funnel (Δ3), across the entrance funnel (Δ4), and between the accumulation and mobility separation regions in the analyzer (Δ6). Radial confinement of elevated ion densities in TIMS can increase the ion translational energy due to long-range ion-ion interactions and power absorption from the RF electric field. At the same time, sufficient radial confinement of ions via the RF electric field is often required to ensure ion transmission through the TIMS analyzer. Hence, as a compromise between minimizing ion heating and optimizing ion transmission, we typically reduce the ion density by using low accumulation times and applying moderate RF amplitudes when possible.
The specific settings that will maintain the structure of a given protein close to its native structure must generally be optimized for the system of interest. To provide a set of “soft” TIMS settings that can be used as a starting point for researchers optimizing their own tuning, we discuss the retention of the 12.4 kDa protein cytochrome c close to its native structure (Figure 2). To minimize thermal activation during ESI, we employed a drying gas temperature less than 50° C. We minimized collisional activation prior to TIMS analysis by setting the DC voltage bias between the deflector and entrance funnel (Δ3), across the entrance funnel (Δ4), and between the accumulation and mobility separation regions in the analyzer (Δ6) to Δ3= 20 V, Δ4= 10 V, and Δ6=5 V, respectively. We used an RF peak-to-peak amplitude of 300 V in the TIMS analyzer with an accumulation time of 70 msec. We stress, however, that the values for these experimental settings, especially the RF amplitude and accumulation times, are not universally valid and should not be applied to other protein samples without optimizing tuning. Instead, to produce a “soft” spectrum for a specific protein system, the operator is generally required to optimize their setting by following the principles of reducing ion heating described above and elsewhere.22, 23
Figure 2.
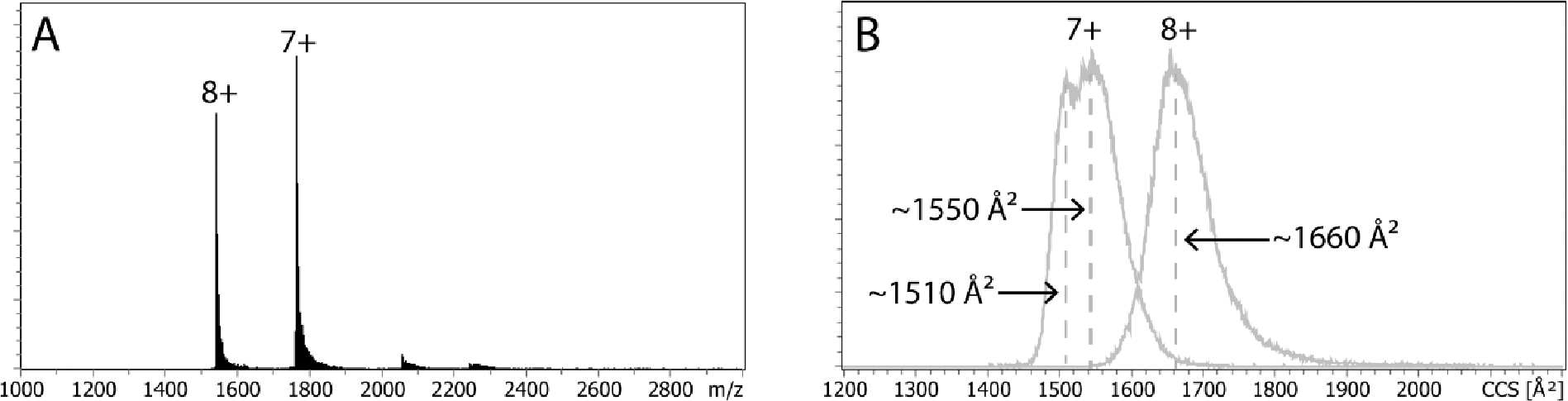
Full ESI mass spectra (A) and extracted mobility distributions (B) for 30 μM cytochrome c in 10 mM aqueous ammonium acetate solution.
Figure 2A shows the mass spectrum obtained for cytochrome c. In agreement with a previous report from McLean et al,16 charge states 7+ and 8+ dominate the spectrum. The ion mobility spectrum of charge state 7+ shows a peak centered at ~1550 Å2 with a shoulder at approximately 1510 Å2 (Figure 2B). These values are similar to cross sections of ~1550 Å2 and ~1590 Å2 observed by drift tube IMS16, 17 and calculated for the x-ray crystal structure (~1565 Å2).16 The ion mobility spectrum recorded for charge state 8+ displays a single feature centered at ~1660 Å2, within 6% of the cross section expected for the x-ray structure. Additionally, the experimental cross sections agree well with those obtained on the TIMS-Q-TOF platform when sprayed from native-like conditions with instrument parameters, specifically the Δ6 potential, tuned gently to preserve the solution-like structures.42 We stress that, in contrast to prior reports using a drift tube16 and TIMS43, peaks with cross sections in the range of 1800 Å2 to 2300 Å2 corresponding to unfolded cytochrome c structures are not present in Figure 2B. This finding indicates that TIMS operated with these suggested conditions yielded “as soft” spectra as drift tubes.44 The peaks corresponding to cytochrome c charge states 7+ and 8+ in the ion mobility spectra are much broader (FWHM ~100 Å2) than expected for a single conformation from the instrumental resolving power. As shown in previous reports for small monomeric proteins ubiquitin and cytochrome c using tandem-ion mobility spectrometry,45–47 the broad mobility distributions stem from the presence of multiple kinetically stable conformations that are not resolved at the given instrumental resolving power. These distinct, stable structures potentially originate from proteins’ conformational heterogeneity in the solution phase. Agreement between our data and reference 42 stresses reproducibility for native-like results between multiple researchers on different instruments, and presents a compelling case for future technological development, whereas previously there existed ambiguity for native capabilities in the literature.
Transmission and TIMS of Protein Complexes
Protein complexes avidin, streptavidin, and CTB were used to evaluate the spectral and IMS quality/capabilities of the TIMS-Q-TOF MS (Figure 3). Mobility distributions were extracted for the entire m/z envelope of individual charge states. CCS values were calculated using the 1/Ko value from the apex of the mobility distribution.
Figure 3.
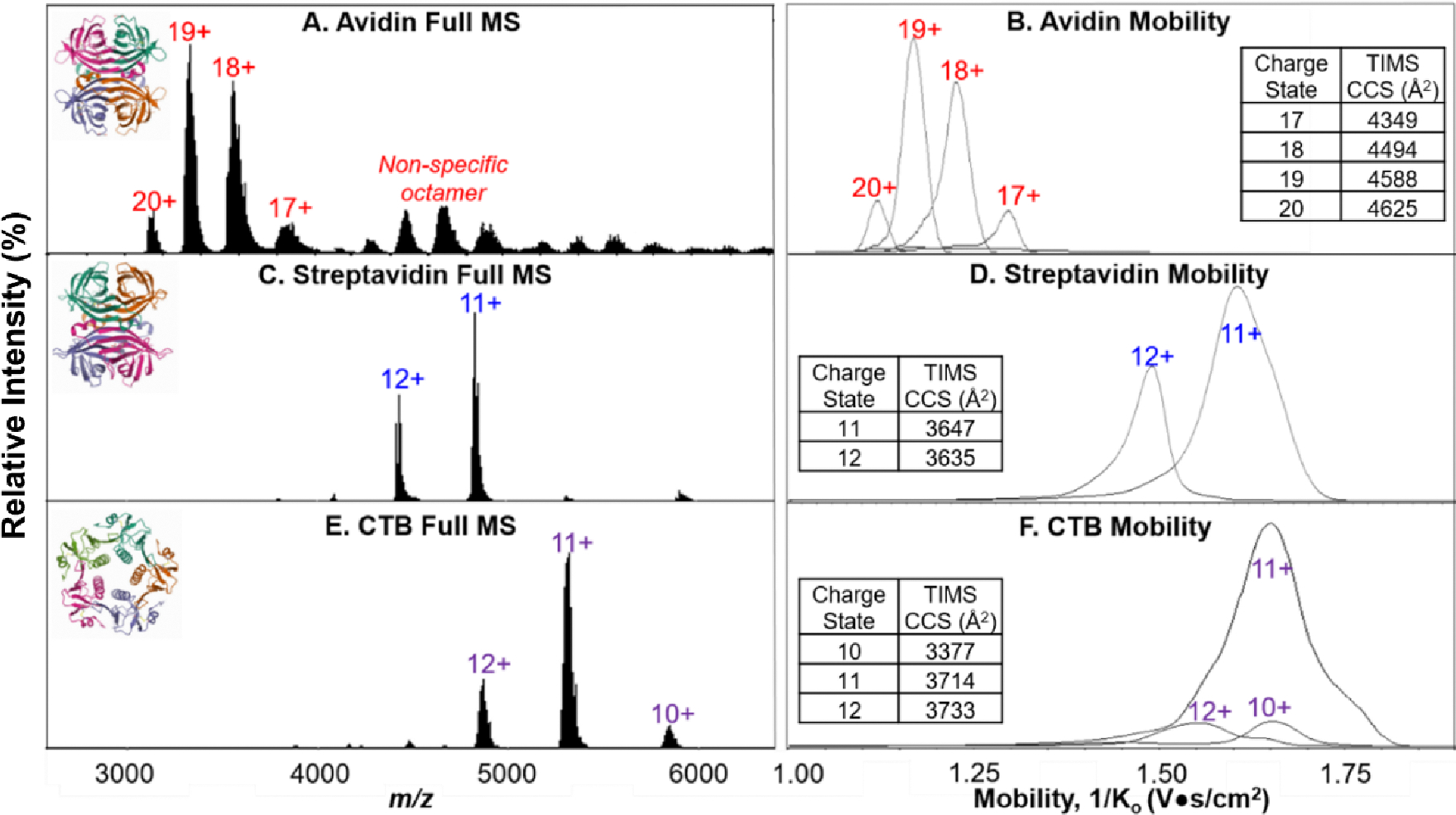
Full mass spectra and extracted mobility distributions with experimental CCS values (single measurements) for 20 μM avidin (A, B, PDB 1AVE) in 200 mM ammonium acetate, 5 μM streptavidin (C, D, PDB 1SWB) in 160 mM ammonium acetate and 40 mM TEAA, and 5 μM CTB (E, F, PDB 1FGB) in 160 mM ammonium acetate and 40 mM TEAA.
Avidin, a 64 kDa homotetramer, was prepared in 200 mM ammonium acetate at a final protein concentration of 20 μM. While this concentration is higher than those for other protein complexes, we have included it here to showcase the transmission of non-specific oligomers at a higher mass-to-charge range (both octamer and 12-mer were observed, Figure 3A). TIMS analysis of avidin yielded well-defined and resolved mobility distributions for the tetrameric charge states (Figure 3B). It should be noted that avidin is a heavily glycosylated protein complex, and hence the mass spectrum contains “wider” peaks (broader m/z range because of multiple glycoforms present) when compared to streptavidin and CTB.29, 48 We do caution users to optimize the TIMS duty cycle (TIMS accumulation time as a function of TIMS ramp time) for proteins and complexes from the standard proteomics settings (100% duty cycle with 100–166 ms accumulation times) to approximately 330–50% duty cycle to avoid overfilling the TIMS device with too many charges. For the data shown here, we do not attribute the wide mobility distributions to overfilling the TIMS analyzer (i.e., charge-charge repulsion) but predominately due to the insufficient pseudopotentials required to confine large molecular weight ions in the TIMS analyzer.28,49 Comparing the mobility distributions for 5 μM and 2 μM CTB did not show significant differences for the 11+ pentamer (Figure S1). Additionally, for 2 μM streptavidin, we did not see significant mobility peak broadening for the 11+ tetramer when increasing the duty cycle from 50 to 70 to 100%, encouraging users to also use lower sample amounts when possible (Figure S2).
Many nMS experiments involve the addition of a charge reducing agent, in an attempt to limit any unintentional charge-mediated unfolding.50, 51 Therefore, streptavidin and CTB were both analyzed under charge-reducing conditions (Figure 3C, 3D). For streptavidin, a 53 kDa homotetramer, the expected native-like charge states were observed in the full mass spectrum, and well-resolved mobility distributions with fronting were obtained. Examples of a full mass spectrum and CID spectra of streptavidin with “raw” intensities (as opposed to relative) are shown in Supporting Information Figure S3. As shown in Figure 3, the mobility distributions for the streptavidin charge states are wider than for avidin despite being lower in mass and less subject to glycosylation. We attribute this to a combination of both the insufficient trapping potentials for large m/z ions of the commercial TIMS analyzer and protein complexes having numerous conformations with different mobilities. For streptavidin, we are approaching the upper m/z limit of the TIMS device, and thus, the trapping efficiency of the analyzer is decreasing at this RF frequency (~800 kHz). Ions of higher m/z are not as well confined toward the center of the TIMS funnel and, therefore, elute from the analyzer as more dispersed ion packets. As shown in Figure 3, the mobility peak broadening becomes more severe for the lower mobility ions, further confirming the need of higher electric fields for trapping high molecular weight ions in the TIMS analyzer. This is also observed for CTB (Figure 3F), a 58 kDa homopentamer, which also resulted in broad mobility distributions for individual charge states that were not resolved from one another. With the mass limit of the commercial TIMS analyzer reached with relatively small protein complexes, an RF driver of lower frequency would be recommended to analyze analytes of this size or larger. Previously acquired TIMS data for streptavidin and CTB on an FT-ICR platform yielded narrower mobility distributions than observed here when a custom-built low frequency RF driver in the TIMS analyzer (450 kHz) and a different TIMS electrode geometry (as discussed below) was used.32 The FT-ICR experiments were also performed with TEAA added to protein solutions. Therefore we do not attribute the mobility broadening to charge-reduction agents being utilized with TIMS. The experimental CCS values align with previously reported literature for avidin and streptavidin, suggesting native-like structures are maintained for these tetrameric protein complexes.52 The CCS values for CTB (3370 Å2 here), specifically the 10+ pentamer, is lower than that previously reported in literature (compared to 3910 Å2)51 which we mainly attribute to the inefficient trapping in the TIMS device resulting in a broad ion packet in the analyzer that may not reside at the accurate position along the electric field gradient but toward the exit of the funnel (that is, yielding a lower mobility or Ko value). . We must also consider that reduced experimental CCS values may suggest structural collapse of the pentameric complex as the TIMS RF Vpp is set to the maximum value of 350 V in attempt to improve trapping efficiency. Recent work altering the trapping pseudopotentials of TIMS by changing the commercial electrode geometry from concave to convex electrodes allowed for trapping of molecular weight assemblies up to 14,000 m/z.28 While the convex TIMS funnel is not available commercially yet, it shows promise for future incorporation.
Collision-induced Dissociation of Streptavidin
Tandem mass spectrometry is commonly utilized to gain structural information in both small and large molecule analysis. Because protein complexes are large in mass, and therefore high in degrees of freedom, high CID energies are required to achieve dissociation, particularly for charge-reduced species. Here, we performed CID in the commercial TIMS-Q-TOF collision cell to evaluate the efficiency of dissociation for charge-reduced streptavidin (Figure 4).
Figure 4.
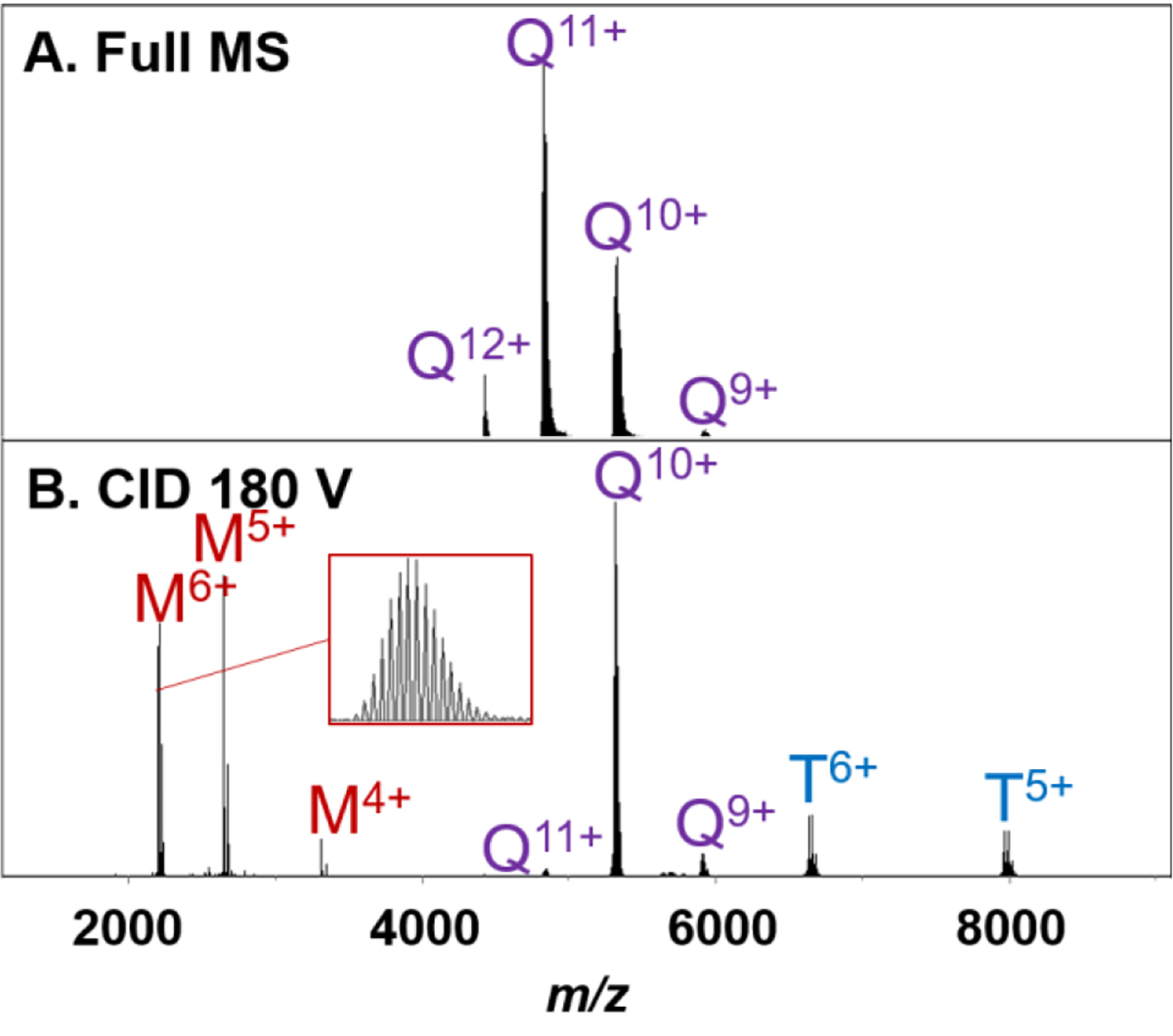
Streptavidin tetramer (5 μM) in 160 mM ammonium acetate with 40 mM TEAA full MS (A). Collision cell CID 180 V of the entire tetramer charge state distribution (B). Initial precursor ions in panel A are labeled Q for Tetramer with their corresponding charge state. In panel B, remaining tetramers are also labeled Q, with fragment ions labeled M for monomeric ions or T for trimeric ions with corresponding charge states.
Because we are beyond the upper limit of the analytical quadrupole’s rf driver, limited by the manufacturer to m/z 3,000, no mass selection was performed prior to activation. Collision cell CID of the entire streptavidin charge state distribution yielded the expected CID product ions. The tetramer fragmented to produce highly charged monomer and the correspondingly charged trimer. Dissociation was observed for CID energies as low as 150 V (out of a 200 V maximum). CID at greater than 200 V may be required to dissociate protein complexes higher in mass. Also, isotopic resolution was observed for the monomeric subunits, as shown in Figure 4B. It was also observed that the collision cell transfer time/pre pulse storage time must be increased from default settings (60 μs/10 μs default to120 μs/15 μs) to allow for the transfer of larger m/z ions from the collision cell to the TOF.
Desolvation and De-salting of Pyruvate Kinase
One challenge with analyzing large protein complexes using nMS is the presence of non-specific salt adducts, which broaden peaks in the resulting mass spectrum, resulting in poor apparent mass accuracy and poor apparent resolution (unresolved peaks). Therefore, in-source activation (isCID) is commonly used to knock off these adducts. The commercial TIMS-Q-TOF is equipped with “in-source activation” capabilities, which are demonstrated below by using pyruvate kinase in 200 mM ammonium acetate (Figure 5). It is important to distinguish that with the current instrument configuration, when applying the in-source activation setting, the activation occurs after the TIMS analyzer, and therefore post-mobility analysis. Note that isCID can cause restructuring of protein complexes if the voltage is set too high. After identification/accurate mass measurements are performed, we recommend using CID or other activation methods without in-source dissociation when performing MS/MS experiments to determine subunit connectivity or localization of ligands in a complex.
Figure 5.

Full mass spectra of Pyruvate kinase (5 μM) in 200 mM ammonium acetate with increasing amounts of in-source activation (isCID) to remove non-specific adducts. As adducts were lost, the apex of each charge state decreased as demonstrated by the red line placed at the apex of 38+ for no activation (A-C). At high isCID, GBP adductions began to be resolved.
Pyruvate kinase is a 237 kDa tetramer that adducts with the allosteric regulator 2,5-anhydro-D-glucitol, 1, 6-bisphosphate (GBP, 324 Da), which is retained even with collisional activation.53 Without any isCID, the peaks corresponding to each individual charge state of pyruvate kinase are relatively wide (~ 200 m/z), with ligand/salt-adduction apparent (Figure 5A). However, as isCID is increased, the center of each peak corresponding to a given charge state shifts to lower m/z, indicating the loss in mass from salt adductions (Figure 5B and C). At 150 V isCID, GBP additions began to be resolved. These results suggest that the isCID provided on the commercial TIMS-Q-TOF MS is sufficient for the de-salting of large protein complexes- though the authors acknowledge that the spectral resolution is not of the same quality achieved on FT-ICR or Orbitrap platforms.54–56
Transmission of a High Molecular Weight Protein Complex
Higher m/z ion transmission was tested using GroEL, a 801 kDa 14-mer. The full MS for GroEL is shown in Figure 6. The full mass spectrum of GroEL consists of well-resolved charge states, even at the highest m/z region we analyzed.
Figure 6.
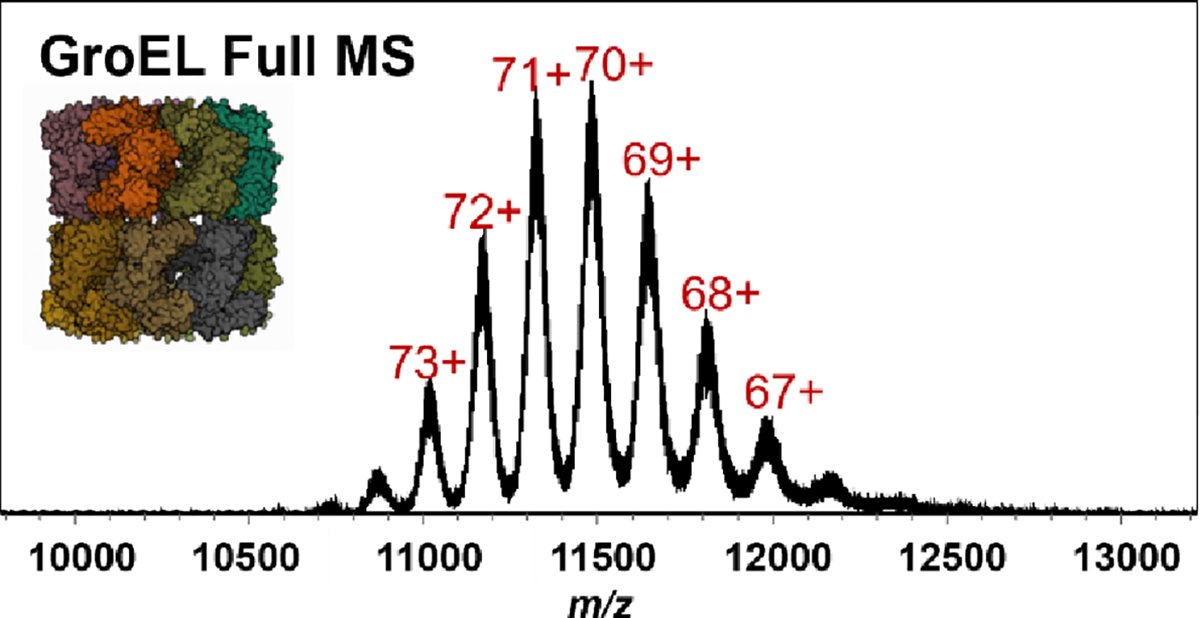
Full MS of GroEL (5 μM) in 200 mM ammonium acetate.
GroEL was able to be transferred throughout all ion optics in the instrument using all maximum RF Vpp and lowest RF frequency settings available. However, GroEL, like pyruvate kinase, was outside the trapping range of the commercial TIMS analyzer and would require instrument modifications for ion mobility analysis of large complexes.57
Conclusions
In this work we evaluated the use of a commercially available timsTOF Pro for native mass spectrometry applications. Collision cross section values for cytochrome c obtained with carefully tuned TIMS measurements agreed with literature and crystal structure values. Multiple protein complexes, including avidin, streptavidin, and cholera toxin B were analyzed for both efficient ion trapping and transmission, yielding full MS spectra with high signal-to-noise and TIMS mobility distributions with adequate separation of multiple charge states for avidin and streptavidin. Charge-reduced streptavidin and CTB began to reach the upper trapping limit of the TIMS device. CID performed in the collision cell dissociates these 53–58 kDa protein complexes, allowing for MS/MS experiments on native samples. Larger protein complexes pyruvate kinase and GroEL were outside the TIMS trapping capabilities of the commercial instrument. However, the complexes were still transferred throughout the instrument optics and native mass spectra acquired, even demonstrating the applicability of in-source activation for sample clean-up to obtain accurate mass measurements. The Bruker timsTOF Pro, without complicated modifications, can be used to analyze native-like proteins and relatively small protein complexes, essentially serving as a dual “omics” and native mass spectrometer to provide complementary structural biology information. The commercial quadrupole, however, is limited in m/z selection up to 3000, therefore limiting the range of proteins and the complexes that can be selectively characterized by CID. Here, we show the use of both an in-house nESI source and the commercially available ESI source for the analysis of native-like proteins. Separate work is in progress to lower the frequency of the RF drivers for both the TIMS and quadrupole to extend the range of the platform to larger protein complexes (greater than a few tens of kDa).
Supplementary Material
Acknowledgements
The authors would like to thank The Ohio State University Campus Chemical Instrument Center, Mass Spectrometry and Proteomics, for allowing us to use their Bruker timsTOF Pro mass spectrometer (1S10OD026945). We are also grateful to Yue (Linny) Ju and Guillaume Tremintin from Bruker Daltonics for providing insight on using the timsTOF Pro. We acknowledge support from the National Institutes of Health (Native Mass Spectrometry Guided Structural Biology Center, P41GM128577 and RM1GM149374 to VHW) and R01GM135682 to CB.
Footnotes
Author Information
The authors declare the following competing financial interest: EMP (currently), MER, and MAP are employees of Bruker, which manufactures and sells the Bruker timsTOF Pro evaluated in this work.
Supporting Information
Sample information, comparison of instrument tune settings for proteomics and protein complexes, comparison of sample concentration on mobility distribution, effect of TIMS duty cycle on mobility distribution, and raw mass spectra for full MS and CID of streptavidin.
References
- 1.Ward AB, Sali A, Wilson IA: Integrative Structural Biology. Science. 339, 913–915 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heck AJR: Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Met. 5, 11, 927–933 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Van den Heuval RHH and Heck AJR: Native protein mass spectrometry: from intact oligomers to functional machineries. Curr. Opin. Chem. Biol. 8, 5, 519–526 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Leney AC and Heck AJR: Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom. 28, 5–13 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM: Electrospray ionization for mass spectrometry of large biomolecules. Science. 246, 4926, 64–71 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Katta V and Chait BT: Observation of the heme-globin complex in native myoglobin by electrospray-ionization mass spectrometry. J. Am. Chem. Soc. 113, 22, 8534–8535 (1991). [Google Scholar]
- 7.Ganem B, Li YT, Henion JD: Detection of noncovalent receptor-ligand complexes by mass spectrometry. J. Am. Chem. Soc. 113, 16, 6294–6296 (1991). [Google Scholar]
- 8.Laganowsky A, Reading A, Hopper JTS, Robinson CV: Mass spectrometry of intact membrane protein complexes. Nat. Protoc. 8, 4, 639–651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benesch JLP, Ruotolo BT, Sobott F, Wildgoose J, Gilbert A, Bateman R, Robinson CV: Quadrupole-Time-of-Flight Mass Spectrometer Modified for Higher-Energy Dissociation Reduces Protein Assemblies to Peptide Fragments. Anal. Chem. 81, 1270–1274 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Chernushevich IV and Thomson BA: Collisional Cooling of Large Ions in Electrospray Mass Spectrometry. Anal. Chem. 76, 6, 1754–1760 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Sobott F, Hernádez H, McCammon MG, Tito MA, Robinson CV: A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal. Chem. 74, 6, 1402–1407 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Fort KL, van de Waterbeemd M, Boll D, Reinhardt-Szyba M; Belov ME, Sasaki E, Szchoche R, Hilvert D, Makarov AA, Heck AJR: Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst 143, 1, 100–105 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Van de Waterbeemd M, Fort KL, Boll D, Reinhardt-Szyba M, Routh A, Makarov AA, Heck AJR: High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat. Meth. 14, 3, 283–286 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH: Applications of a travelling wave-based radio-frequency-only stacked ring ion guide. Rap. Comm. Mass Spectrom. 18, 20, 2401–2414 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Campuzano IDG and Giles K: Historical, current, and future developments of travelling wave ion mobility mass spectrometry: a personal perspective. Trends in Anal. Chem. 120, 115620 (2019). [Google Scholar]
- 16.May JC, Jurneczko E, Stow SM, Kratochvil I, Kalkhof S, McLean JA; Conformational Landscapes of Ubiquitin, Cytochrome C, and Myoglobin: Uniform Field ion Mobility Measurements in Helium and Nitrogen Drift Gas. Int. J. Mass Spectrom. 427, 29–90 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT: Collision Cross Sections of Proteins and their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal. Chem. 82, 22, 9557–9565 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Meier F, Brunner AD, Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park MA, Bache N, Hoerning O, Cox J, Räther O, Mann M; Online Parallel Accumulation-Serial Fragmentation with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. & Cell Prot. 17, 2534–2545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spraggins JM, Djambazova KV, Rivera ES, Migas LG, Neumann EK, Fuetterer A, Goedecke N, Ly A, Van de Plas R, Caprioli RM: High-Performance Molecular Imaging with MALDI Trapped Ion Mobility Time-of-Flight (timsTOF) Mass Spectrometry. Anal. Chem. 91, 22, 14552–14560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steigenberger B, van den Toorn H, Bijl E, Greisch J, Räther O, Lubeck M, Pieters RJ, Heck AJR, Scheltema RA: Benefits of Collision Cross Section Assisted Precursor Selection (caps-PASEF) for Cross-linking Mass Spectrometry. Mol. & Cell Prot. Mcp.RA120.002094 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirk SR, Liu FC, Cropley TC, Carlock HR, Bleiholder C: On the Preservation of Non-covalent Peptide Assemblies in a Tandem-Trapped ion mobility Spectrometer-Mass Spectrometer (TIMS-TIMS-MS). J. Am. Soc. Mass Spectrom. 30, 7, 1204–1212 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Liu FC, Kirk SR, Bleiholder C: On the Structural Denaturation of Biological Analytes in Trapped Ion Mobility Spectrometry-Mass Spectrometry. Analyst 141, 3722–3730 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Bleiholder C, Liu FC, Cha M: Comment on Effective Temperature and Structural Rearrangement in Trapped Ion Mobility Spectrometry. Anal. Chem. 92, 24, 16329–16333 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hebert AS, Prasad S, Belford MW, Baile DJ, McAlister GC, Abbatiello SE, Hugert R, Woulters ER, Dunyach J, Brademan DR, Westphall MS, Coon JJ; Comprehensive Single-Shot Proteomics with FAIMS on Hybrid Orbitrap Mass Spectrometer. Anal. Chem. 90, 15, 9529–9537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bekker-Jensen DB, Martinez-Val A, Steigerwald S, Rüther P, Fort KL, Arrey TN, Harder A, Makarov A, Olsen JV: A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell Proteom. 19, 4, 716–729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bleiholder C, Liu FC, Chai M: Chapter: Calculation of Momentum Transfer Cross-Sections in Ion Mobility-Mass Spectrometry: Fundamentals and Applications, New Developments in Mass Spectrometry. Royal Society of Chemistry (2021). [Google Scholar]
- 27.Larson EJ, Roberts DS, Melby JA, Buck KM, Zhu Y, Zhou S, Han L, Zhang Q, Ge Y: High-Throughput Multi-attribute Analysis of Antibody-Drug Conjugates Enabled by Trapped Ion Mobility Spectrometry and Top-Down Mass Spectrometry. Anal. Chem. 93, 29, 10013–10021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dit Fouque KJ, Garabedian A, Leng F, Tse-Dinh Y, Ridgeway ME, Park MA, Fernandez-Lima F: Trapped Ion Mobility Spectrometry of Native Macromolecular Assemblies. Anal. Chem. 93, 5, 2933–2941 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu FC, Cropley TC, Ridgeway ME, Park MA, Bleiholder C: Structural Analysis of the Glycoprotein Complex Avidin by Tandem-Trapped Ion Mobility Spectrometry-Mass Spectrometry (Tandem-TIMS/MS). Anal. Chem. 92, 6, 4459–4467 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu FC, Ridgeway ME, Windfred JSRV, Polfer NC, Lee J, Theisen A, Wootton CA, Park MA, Bleiholder C: Tandem-trapped ion mobility spectrometry/mass spectrometry coupled with ultraviolet photodissociation. Rapid Commun. Mass Spectrom. 35, 22, e9192 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu FC, Ridgeway ME, Park MA, Bleiholder C: Tandem-trapped ion mobility spectrometry/mass spectrometry (tTIMS/MS): a promising analytical method for investigating heterogenous samples. Analyst 147, 2317–2337 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panczyk EM, Snyder DT, Ridgeway ME, Somogyi A, Park MA, Wysocki VH: Surface-induced Dissociation of Protein Complexes Selected by Trapped Ion Mobility Spectrometry. Anal. Chem. 93, 13, 5513–5520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Nguyen HH, Ogorzalek Loo RR, Campuzano IDG, Loo JA: An Integrated native Mass Spectrometry and Top-Down Proteomics Method that Connects Sequence to Structure and Function of Macromolecular Complexes. Nat. Chem. 10, 2, 139–148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gault J, Liko I, Landreh M, Shutin D, Bolla JR, Jeffries D, Agasid M, Yen H, Ladds MJG, Lane DP, Khalid S, Mullen C, Remes P, Huguet R, McAlister G, Goodwin M, Vinter R, Syka J, Robinson CV: Combining Native and ‘omics’ Mass Spectrometry to Identify Endogenous Ligands Bound to Membrane Proteins. Nat. Methods 17, 5, 505–508 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou M, Jones CM, Wysocki VH: Dissecting the Large Noncovalent Protein Complex GroEL with Surface-Induced Dissociation and Ion Mobility-Mass Spectrometry. Anal. Chem. 85, 8262–8267 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Yan J, Zhou M, Gilbert JD, Wolff J, Somogyi A, Pedder RE, Quintyn RS, Morrison LJ, Easterling MJ, Pasa-Tolic L, Wysocki VH: Surface-Induced Dissociation of Protein Complexes in a Hybrid Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. Anal. Chem. 89, 895–901 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Snyder DT, Panczyk E, Stiving AQ, Gilbert JD, Somogyi A, Kaplan D, Wysocki V: Design and Performance of a Second-Generation Surface-Induced Dissociation Cell for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry of Native Protein Complexes. Anal. Chem. 91, 14049–14057 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snyder DT, Panczyk EM, Somogyi A, Kaplan DA, Wysocki V: Simple and Minimally Invasive SID Devices for Native Mass Spectrometry. Anal. Chem. 92, 11195–11203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Revercomb H and Mason EA: Theory of plasma chromatography/gaseous electrophoresis. Anal. Chem. 47, 970–983 (1975). [Google Scholar]
- 40.Mason EA and McDaniel EW: Transport Properties of Ions in Gases. Wiley: New York: (1988). [Google Scholar]
- 41.Morsa D, Hanozin E, Eppe G, Quinton L, Gabelica V, De Pauw E: Effective Temperature and Structural Rearrangement in Trapped Ion Mobility Spectrometry. Anal. Chem. 92, 6, 4573–4582 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Borotto NB; Osho KE; Kamakamakamae R; Graham KA: Collision-Induced Unfolding of Native-like Protein Ions Within a Tapped Ion Mobility Spectrometry Device. J. Am. Soc. Mass Spectrom. 33, 83–89 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Morsa D, Hanozin E, Gabelica V, De Pauw E: Response to Comment on Effective Temperature and Structural Rearrangement in Trapped Ion Mobility Spectrometry. Anal. Chem. 92, 24, 16334–16337 (2020). [DOI] [PubMed] [Google Scholar]
- 44.France AP, Migas LG, Sinclair E, Bellina B, Barran PE: Using Collision Cross Section Distributions to Assess the Distribution of Collision Cross Section Values. Anal. Chem. 92, 6, 4340–4348 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Koeniger SL, Merenbloom SI, and Clemmer DE: Evidence for Many Resolvable Structures within Conformation Types of Electrosprayed Ubiquitin Ions, J. Phys. Chem. B 2006, 110, 7017–7021. [DOI] [PubMed] [Google Scholar]
- 46.Allen SJ, Eaton RM, and Bush MF: Structural Dynamics of Native-Like Ions in the Gas Phase: Results from Tandem Ion Mobility of Cytochrome c, Anal. Chem. 2017, 89, 7527–7534. [DOI] [PubMed] [Google Scholar]
- 47.Cropley TC, Liu FC, Chai M, Bush MF, Bleiholder C: Metastability of Protein Solution Structures in the Absence of Solvent: Rugged Energy Landscape and Glass-Like Behavior, DOI: 10.26434/chemrxiv-2023-q09db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruch RC and White III HB: Computational and structural heterogeneity of avidin glycopeptides. Biochemistry 21, 21, 5334–5341 (1982). [DOI] [PubMed] [Google Scholar]
- 49.Benigni P, Marin R Molano-Arevalo JC., Garabedian A., Wolff JJ., Ridgeway ME., Park MA., Fernandez-Lima F.: Towards the Analysis of High Molecular Weight Proteins and Protein Complexes using TIMS-MS. Int. J. Ion Mobil. Spectrom. 19, 2, 95–104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehmood S, Marcoux J, Hopper JTS, Allison TM, Liko I, Borysik AJ, Robinson CV: Charge Reduction Stabilizes Intact Membrane Protein Complexes for Mass Spectrometry. J. Am. Chem. Soc. 136, 49, 17010–17012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma X, Loo JA, Wysocki VH: Surface induced dissociation yields substructure of Methanosarcina thermophila 20S proteosome complexes. Int. J. Mass Spectrom, 377, 201–204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stiving AQ, Jones BJ, Ujma J, Giles K, Wysocki VH: Collision Cross Sections of Charge-Reduced Proteins and Protein Complexes: A Database for Collision Cross Section Calibration. Anal. Chem. 92, 6, 4475–4483 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL: From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal. Chem. 85, 23, 11163–11173 (2013). [DOI] [PubMed] [Google Scholar]
- 54.McGee JP, Melani RD, Yip PF, Senko MW, Compton PD, Kafader JO, Kelleher NL: Isotopic Resolution of Protein Complexes up to 466 kDa Using Individual Ion Mass Spectrometry. Anal. Chem, 93, 5, 2723–2727 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schachner LF, Ives AN, McGee JP, Melani RD, Kafader JO, Compton PD, Patrie SM, Kelleher NL: Standard Proteoforms and Their Complexes for Native Mass Spectrometry. J. Am. Soc. Mass Spectrom, 30, 7, 1190–1198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Nguyen HH, Ogorzalek Loo RR, Campuzano IDG, Loo JA: An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem., 10, 2, 139–148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Y, Di Capua A, Panczyk E, Jones B, Ridgeway M, Somogyi A, Kaplan D, Park M, Wysocki V: Native Mass Spectrometry on a modified timsTOF Pro, in Proceedings of the 71st ASMS Conference on Mass Spectrometry and Allied Topics, (Houston, TX, 2023). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.