

Abstract
NI006 (aka ALXN2220) is a therapeutic antibody candidate in phase III clinical development for the depletion of amyloid transthyretin (ATTR) in patients with ATTR cardiomyopathy, an infiltrative cardiomyopathy leading to increased left ventricular wall thickness (LVWT). The mode‐of‐action consists in removal of disease‐causing amyloid accumulations by activating phagocytic immune cells, a mechanism without precedent in cardiology. To select a safe and potentially efficacious dose range and treatment duration for a combined first‐in‐human and proof‐of‐concept clinical phase Ib study, we developed a mechanistic pharmacokinetic and pharmacodynamic (PK/PD) model that can predict NI006 exposure, its effects on cardiac amyloid load and on LWVT, which is a predictor of heart failure in this disease. The PK/PD model predictions supported 0.3 mg/kg monthly dosing as a safe starting dose and identified 10–60 mg/kg monthly as the potentially efficacious dose range with substantial and dose dependent cardiac amyloid burden reduction within 4 months for 60 mg/kg and 10 months for 10 mg/kg. These predictions were in good agreement with the observed primary results of the clinical phase Ib study where amyloid burden was measured by imaging. This novel translational PK/PD model provided important predictions to guide the design of the phase Ib study of NI006, indicating the value of this approach to integrate preclinical results into clinical trial design and increase translational success.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Antibody‐mediated amyloid depletion is a pharmacological mode‐of‐action without precedent in cardiology. There was no prior example to guide the design of the first‐in‐human study of NI006 in amyloid cardiomyopathy.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study addressed the selection of a safe starting dose for a first‐in‐human study, and the selection of doses and treatment durations expected to show pharmacological activity in patients based on preclinical data.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Mechanistic PK/PD modeling predicted a safe starting dose, and doses and treatment durations associated with reductions in cardiac amyloid surrogates. These predictions were validated by the clinical study results.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
We provide an example of successful translation of preclinical results and literature data into phase I study design. This supports further use of quantitative system pharmacology to capture the mechanistic understanding of the disease and treatment mode‐of‐action.
Amyloid transthyretin (ATTR) amyloidosis is a rare, progressive, degenerative, and fatal disease caused by the systemic deposition in tissues and organs of amyloid fibrils composed of misfolded transthyretin (TTR) protein, an abundant plasma protein secreted primarily by the liver. 1 The misfolding, aggregation, deposition, and accumulation of ATTR is responsible for the two major manifestations of the disease, where amyloid accumulation in cardiac tissues causes cardiomyopathy, whereas amyloid deposition in nerve fibers causes polyneuropathy. 2 , 3
In ATTR amyloidosis with cardiomyopathy (ATTR‐CM), the extracellular accumulation of insoluble ATTR fibrils in the cardiac extracellular space (interstitium) progressively displaces normal tissue leading to changes in heart morphology, including increased left ventricular wall thickness (LVWT), and stiffening of the heart muscle. These molecular and morphological changes cause a progressive impairment of cardiac function leading to heart failure and death. Current disease‐specific treatments rely on TTR‐stabilizing or TTR‐silencing therapies, which slow down the amyloid formation process and the corresponding disease progression, but do not directly target pre‐existing amyloid that accumulates over time in the heart and causes morphological enlargement and cardiac dysfunction. 4 Spontaneous amyloid removal is thought to exist but is very slow or extremely rare. 5 Reduction in cardiac amyloid load could be expected to reduce the mechanical and physiological constraints on the myocardium and result in improved heart function.
NI006 is an investigational human monoclonal antibody specifically targeting insoluble ATTR fibrils. It binds a cryptic linear epitope, which is not accessible in the native TTR protein fold but becomes surface‐exposed in the structurally reorganized amyloid fold. Upon binding, NI006 activates the elimination of ATTR‐antibody complexes by phagocytic immune cells, a fundamental mechanism of the adaptative immune system, which is highly conserved across mammalian species. NI006 depleter activity was established preclinically in a mouse fibril‐graft model and on patient myocardium tissues in the presence of human macrophages. 6 This amyloid depletion mode‐of‐action is a new concept in cardiology, and no prior information was available to assess safety, tolerability and dose–response in patients and guide the design of a combined first‐in‐human (FIH) and proof‐of‐concept clinical study with NI006. We therefore integrated preclinical results and published data to develop a novel mechanistic pharmacokinetic and pharmacodynamic (PK/PD) model of NI006‐dependent cardiac amyloid depletion in ATTR‐CM patients.
The model described hereafter is a three‐compartmental model, which predicts the antibody concentration‐dependent degree of ATTR elimination over time and its impact on the LVWT. The LVWT increases in ATTR‐CM patients together with ATTR fibril deposition and is prognostic for the risk of heart failure and death. 7 , 8 Thus, in this context, the LVWT was used as a clinical proxy for cardiac amyloid load and disease severity. It allowed linking the degree of ATTR reduction to clinically meaningful outcome and guided the design of the clinical phase I study.
METHODS
Rat PK study, modeling, and allometric scaling to human
NI006 PK was established in a study conducted with 36 rats receiving either a single dose of NI006 at 3 or 30 mg/kg IV, or 3 weekly doses at 300 mg/kg, with 6 male and 6 female animals per dose group. NI006 serum concentration was measured at different timepoints from 5 minutes up to 28 days post dose using a GLP‐qualified assay. A two‐compartmental PK model with linear first‐order elimination was used to describe the data. Population parameters were estimated using the stochastic approximation of expectation and maximization (SAEM) algorithm implemented in Monolix.
NI006 human PK parameters were derived from the rat using allometric scaling. The affinity of IgG1 molecules to rat FcRn, the key factor for IgG1 PK, is sufficiently close to the human FcRn 9 to make the rat a valid PK species as it has been demonstrated previously. 10 , 11 Scaling coefficients of 1 were used for the volumes of distribution V1 and V2, 0.75 for the clearance (Cl) and 2/3 for the intercompartmental clearance (Q), using body weights of 0.3 kg for rat and 70 kg for human.
Disease progression model
An increased LVWT is a hallmark of ATTR‐CM and is correlated with the accumulation of ATTR in the cardiac tissue. Data from untreated ATTR‐CM patients aged from 30 to 76 years showed that the LVWT was continuously increasing over the entire duration of the disease. 12 Because mass and volume are directly related, the ATTR load‐LVWT relationship was selected to link the drug target with a disease relevant morphological feature of the heart.
LVWT values from healthy individuals, from untreated ATTR‐CM patients aged from 30 to 76 years, and from patients treated with patisiran, a liposomal siRNA silencing TTR expression and thus ATTR formation, in the APOLLO Phase III study (NCT03997383) were used to establish a disease progression model describing the LVWT as net result of cardiac amyloid formation and spontaneous elimination. 12 , 13 , 14 The APOLLO study reported an 80% reduction of TTR plasma levels with patisiran and a reduction of the LVWT supporting the hypothesis that inhibition of ATTR accumulation by TTR silencing and spontaneous elimination of ATTR resulted in a net reduction in LVWT. Based on this evidence, the LVWT in ATTR‐CM patients was modeled with a turnover model with constant rate kgrowth, reflecting the close to constant plasma TTR levels over age, and a first‐order elimination process with rate kel with the following equation:
A LVWT baseline of 8.5 mm was used for healthy subjects. Patisiran‐treated patients, who presented on average 80% reduction in plasma TTR levels, were assumed to present a corresponding 80% reduction in the amyloid formation rate and associated LVWT growth rate (kgrowth). LVWT data from the natural history study in untreated ATTR‐CM patients and from the phase III clinical study with patisiran were simultaneously fitted using least squares minimization with the Nelder–Mead optimization algorithm in R.
Tissue amyloid depletion assay
Frozen human heart tissue sections presenting ATTR amyloidosis were incubated with human‐derived macrophages in presence of vehicle or NI006 at 0.01, 0.1, 1, and 10 nM. After 14 days of incubation, the remaining tissue amyloid was stained with thioflavin‐s and quantified using automated microscopy and image analysis. Method details were previously reported. 6
Fibril xenograft model
ATTR amyloid fibrils were extracted from patient cardiac tissues and implanted in mice by subcutaneous injection. Mice then received a single dose of mouse chimeric NI006 (ch.NI006) at 0.05, 0.5, 5, or 50 mg/kg, or the isotype control antibody at 50 mg/kg. One mouse from each group was sacrificed 6 hours and the remaining mice 96 hours after drug administration and the remaining amount of ATTR in the graft was quantified by immunohistochemistry. Method details were previously reported. 6
Estimation of NI006 concentration‐dependent amyloid elimination rate
Results from NI006 dose response in the tissue amyloid depletion assay and the fibril xenograft model were fitted with an ATTR‐binding and elimination model to determine the NI006 concentration‐dependent amyloid elimination rates in these assays. A first‐order elimination kinetic model was used, with an elimination rate proportional to the ATTR fraction bound by NI006. The maximum elimination rate, noted kelmax, was the elimination rate occurring with 100% ATTR occupancy by NI006. Initial ATTR concentration was estimated by the model, and it was further assumed that target‐bound NI006 was eliminated together with ATTR at the same rate. NI006‐to‐ATTR binding was described with the quasi‐equilibrium binding approximation and a dissociation constant (KD) obtained from surface plasmon resonance (SPR) experiments. 6 Data were fitted using least squares minimization with the Nelder–Mead optimization algorithm in R.
PK/PD model for predictions in ATTR‐CM
The PK/PD model included a central compartment with linear elimination and distribution into a peripheral compartment to describe the plasma PK of NI006. A third compartment, representing the heart interstitium, was linked to the central compartment. NI006 distribution into the heart interstitium was described by a bio‐distribution coefficient (10.2%) typical for an IgG1 molecule, and the volume of distribution (0.0488 L) was set to the heart interstitial space. 15 , 16
ATTR was modeled only in the heart. The cardiac ATTR mass was estimated by fitting a binding model to the experimental data with patient cardiac tissue incubated with NI006. The reported excess left ventricle wall mass in ATTR‐CM patients compared with healthy individuals was 136 g. 14 , 17 It was assumed that this increase in mass was not only due to ATTR accumulation but also due to secondary effects that include fibrotic tissue formation and ventricular hypertrophy because of increased tissue stiffness.
The fitting of the NI006 dose responses in the tissue amyloid depletion assay and fibril xenograft model with a binding and elimination model, as described above, led to an estimated ATTR mass accessible to antibody binding of ~ 1% of the heart tissue mass used in these experiments. Combined, this led to an estimate of 1.36 g (1% of 136 g) ATTR protein mass accessible to NI006 binding in the heart of ATTR‐CM patients.
The ATTR accumulation over time was described as the net result of ATTR deposition and spontaneous elimination according to the disease progression model based on LVWT data, described above. Heart ATTR mass was linked with the disease progression model by assuming a linear relationship between heart ATTR mass and LVWT. LVWT above the typical value in healthy individuals of 8.5 mm was assumed to be proportional to ATTR mass. 17 A LVWT value of 16.6 mm reported for ATTR‐CM patients was used as initial value for the simulations in ATTR‐CM patients and corresponded to the estimated ATTR mass of 1.36 g. 14 , 17 We thus had:
ATTR was represented as homogenously distributed in the heart interstitial space with all ATTR subunits (TTR monomers) equally accessible to NI006 binding. NI006 binding equilibrium with ATTR was described with a quasi‐equilibrium (QE) approximation using an equilibrium dissociation constant (KD) of 2.5 nM measured in vitro. 6 The QE approximation was justified for such a high affinity antibody as binding occurs within minutes to hours while distribution and elimination processes occur over days to weeks. NI006 bound to ATTR was eliminated as part of the NI006‐ATTR complex. The parameter values used for simulations and their sources are listed in Table S1 .
Human PK/PD simulations were run in R using the mlxR library. PK/PD simulations were population‐typical predictions without between‐individual variability.
Comparison of PK/PD predictions with clinical study results
NI006 was evaluated in a phase I (Ia–Ib), double‐blind, placebo‐controlled, international, multicenter, combined single‐ascending‐dose and multiple‐ascending‐dose, randomized clinical trial with an open‐label extension phase, which primary results are available (NCT04360434). 18 This study (NI006‐101) was conducted in ATTR‐CM patients with early‐ to mid‐disease progression, which was comparable to patients enrolled in ATTR‐CM phase III studies of TTR stabilizers and expression silencers. NI006‐101 investigated the safety and exploratory efficacy of NI006 dosing at 0.3, 1, 3, 10, 30, and 60 mg/kg q4w IV, using fixed doses compared with placebo during the first 4 months, and placebo switch to treatment and dose up‐titrations during the following 8 months. Extracellular volume (ECV) determined by MRI and heart‐to‐whole‐body ratio using diphosphonate scintigraphy were used as surrogates for cardiac amyloid load monitoring, with measurements at baseline and after 4 and 12 months.
To compare clinically observed and model predicted dose–responses, the relative changes from baseline in the proxies used for cardiac amyloid load were plotted as a function of NI006 exposures, overlaying observed and predicted responses on the same graphs. Predicted effects were taken from the original preclinical model without any model recalibration to the clinical data. Predicted NI006 exposure from the preclinical model was calculated for a standard 70‐kg individual without between‐individual variability. Because the sampling in the NI006‐101 study did not allow to compute the total AUC over 4 and 12 months, a phase I population PK model was used to determine the post hoc individual parameters and calculate the total AUC from the simulated individual PK profiles. The actual individual dosing history was used for the simulations of each patient.
Ethics statement
The NI006‐101 study protocol was approved by local ethics committees and health authorities (EU Clinical Trials Register number, 2019‐001932‐80).
RESULTS
NI006 pharmacokinetics
NI006 PK was determined in a rat study, where NI006 serum concentration was monitored for 28 days after single doses at 3 and 30 mg/kg i.v. and after the first and last of three weekly doses at 300 mg/kg. The individual NI006 serum concentration profiles in rat showed two distinct elimination phases suggesting 2‐compartmental kinetics (Figure 1 ). NI006 serum concentration was dose‐proportional, and the population PK parameters estimated from these data were in the typical range for a human IgG1 antibody in rat (Table 1 ).
Figure 1.
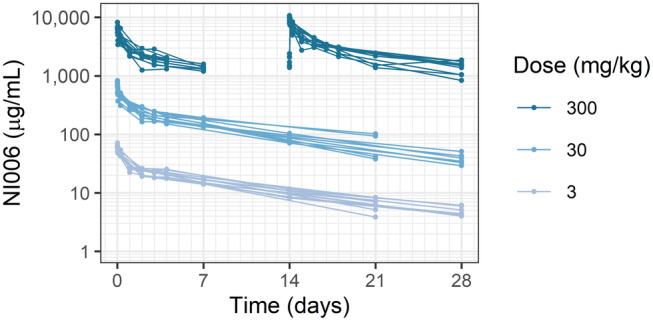
NI006 serum concentration in rats after single administrations at 3 and 30 mg/kg and three weekly dosing at 300 mg/kg i.v. At this high dose, serum samples were collected after the first and the third administrations but not after the second one. Each line represents one animal, with 6 male and 6 female animals per dose level.
Table 1.
NI006 rat and human PK parameters. Between‐animal variability as %CV in brackets
| Parameter (unit) | Rat parameter values | Human value scaled from rat |
|---|---|---|
| Cl (L/day) | 0.00245 (21%) | 0.146 |
| V1 (L) | 0.0144 (10%) | 3.37 |
| V2 (L) | 0.0189 (15%) | 4.42 |
| Q (L/day) | 0.0178 (18%) | 0.676 |
The rat PK parameters were allometric scaled based on the body weight to obtain the human PK parameters (Table 1 ).
Disease progression model
ATTR‐CM patients present an increase in LVWT over time that is associated with increasing amyloid deposition in the heart and disease progression; it is routinely measured by echocardiography at diagnosis and during patient follow‐up. The LVWT values from ATTR‐CM patients aged from 30 to 76 years and from the APOLLO Phase III study testing TTR gene silencing with patisiran were used for disease modeling. 12 , 14 Using these data in combination with a healthy LVWT of 8.5 mm as baseline, the LVWT growth rate in ATTR‐CM patients was estimated to 0.43 mm/year and the LVWT contraction rate was estimated to 0.053 per year (Figure S1 and Table S2 ). This contraction rate would correspond to a spontaneous reduction in excess LVWT by 50% within 13 years if amyloid production were entirely stopped. This LVWT growth rate estimate of 0.43 mm/year proved well in line with recent longitudinal measurements in ATTRwt and ATTRv patients, which determined an interventricular septum thickness growth of 0.39 mm per year, without significant difference between WT and variant carriers. 19
NI006 concentration‐dependent amyloid depletion rate
NI006 binding to ATTR results in antibody‐target complex that is eliminated by phagocytic immune cells. NI006 binding to ATTR in the heart was modeled using the same affinity as measured in vitro, corresponding to an equilibrium dissociation constant (KD) of 2.5 nM. 6 The elimination rate of the antibody‐target complex was estimated from a tissue amyloid depletion assay, where heart tissue sections from patients were incubated with human‐derived macrophages in presence of NI006, leading to the concentration‐dependent reduction in amyloid deposits. 6 The NI006‐ATTR complex elimination rate was estimated to 0.066 per day in this assay, corresponding to an elimination half‐life of 10.5 days (Figure 2 a ). An in vivo model was also developed, consisting of a subcutaneous graft of patient derived ATTR fibrils in mice. 6 Fibril elimination in this xenograft model was faster, with an elimination rate estimated to 0.37 per day, corresponding to an elimination half‐life of 1.9 days (Figure 2 b ). The slower elimination rate measured in the tissue amyloid depletion assay was used for simulations as it presented the amyloid in its native context, embedded within cardiac tissue, and tested the activity of human‐derived macrophages. The faster elimination measured in vivo possibly resulted from higher activity of the mouse immune system and amyloid presentation outside of the native context through fibril extraction and purification. It was used to test model sensitivity to different elimination rates, presented below.
Figure 2.
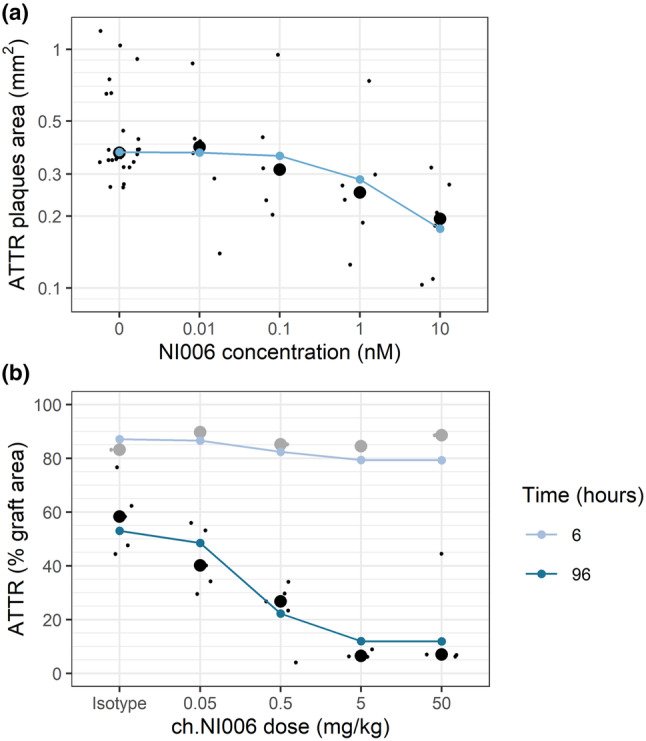
Determination of NI006‐ATTR complex elimination rates observed in (a) the tissue amyloid depletion assay using NI006, and (b) the patient‐derived amyloid xenograft model in mice using a mouse chimeric variant of NI006 (ch.NI006). Black dots: experimental measurements, individual values (small dots) and group median (large dots). Blue dots connected by line: modeling results.
ATTR‐CM patient PK/PD model structure
These results were integrated in a mechanistic PK/PD model for NI006‐dependent cardiac amyloid depletion, designed as a three‐compartmental model with three volumes representing respectively the blood serum (V1), the interstitial fluid of all peripheric organs and tissues except the heart (V2), and the cardiac interstitial fluid (V3) (Figure 3 ). The model described ATTR amyloid formation and accumulation in the heart, the key organ affected by the disease. The NI006 human PK parameters, disease progression rate, target binding affinity, and antibody‐target complex elimination rate described above were used. In addition, the biodistribution coefficient (BC) and the intercompartmental clearance (Q2) between serum and heart interstitium, specific for human immunoglobulin G (IgG), and the volume V3 were obtained from the literature. 15 , 16
Figure 3.
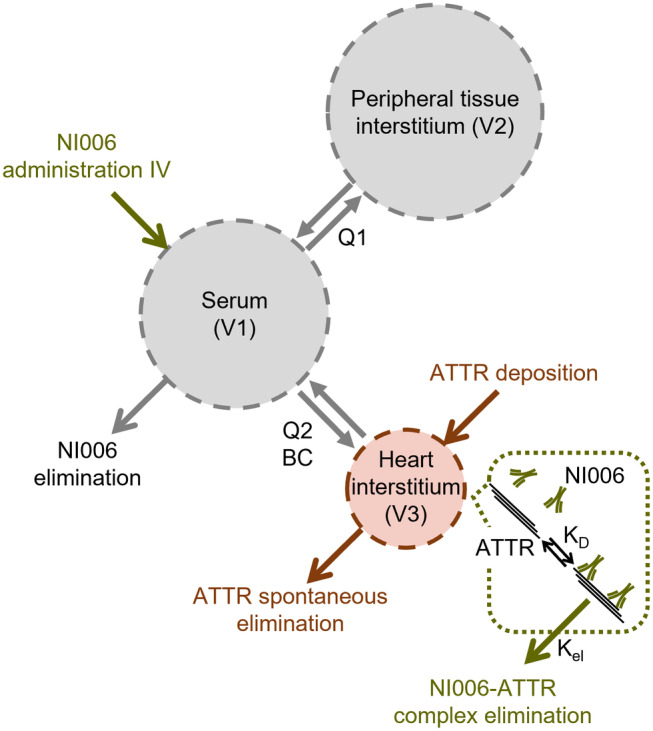
Schematic representation of key model elements: two‐compartmental PK model (in gray) with a third volume capturing ATTR deposition in the heart (in red) and ATTR elimination by NI006 and immune cells (in green).
Determination of a safe starting dose
The first objective for PK/PD modeling was to predict a safe starting dose for the first‐in‐human study with NI006. Because of the novel mechanism of action and the immune system activating properties of NI006, a conservative starting dose was selected based on the NI006 serum concentrations and percent ATTR target occupancy predicted by the PK/PD model. Plasma exposure and ATTR target occupancy were predicted for doses of 0.3, 1, 3, 10, 30 and 60 mg/kg administered every 4 weeks (q4w) for 12 months as a 2‐hour intravenous (IV) infusion in patients with a baseline LVWT of 16.6 mm.
Model predictions indicated that a dose of 0.3 mg/kg would result in a maximum serum concentration (C max) of 8.4 μg/mL and a predicted ATTR occupancy by NI006 in the heart below 0.1% after the fourth administration. In contrast, the highest tentative dose for the FIH study of 60 mg/kg would lead to an ATTR occupancy reaching 30% after the fourth dose and 100% after 8 doses (Figure 4 a,b ). In the absence of prior clinical experience with amyloid depleter antibodies in heart failure patients with amyloid cardiomyopathy, a conservative starting dose of 0.3 mg/kg was selected corresponding to less than 0.1% target occupancy. The starting dose of 0.3 mg/kg was also supported by data from in vitro cytokine release assays and antibody‐dependent cell cytotoxicity and complement‐dependent cytotoxicity assays and allowed an efficient dose escalation with a limited number of cohorts into a clinically efficacious dose range.
Figure 4.
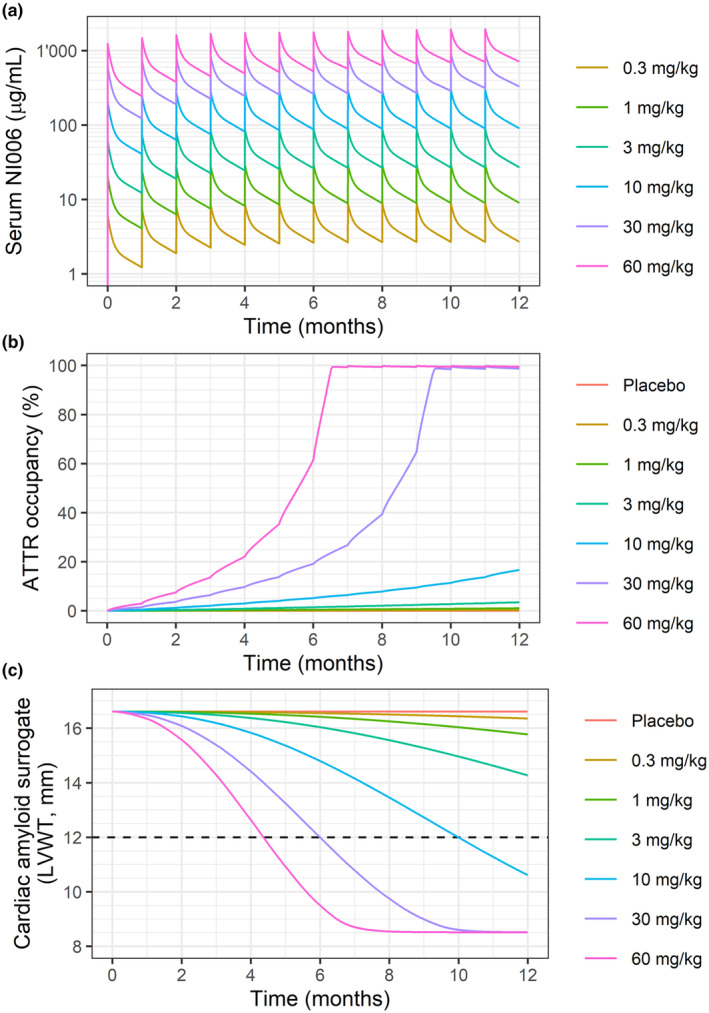
Model predictions for (a) NI006 serum concentration, (b) ATTR occupancy in the heart and (c) Cardiac amyloid surrogate (LVWT) over a 12‐month treatment period with doses every 4 weeks; the horizontal dashed line represents target LVWT for clinical effect. Predictions are for a typical individual.
Determination of time to clinical effect
The second objective was to predict the time to clinical effect for chronic monthly doses of up to 60 mg/kg. Clinical effect was defined as reduction in LVWT to below 12 mm, a threshold previously associated with ATTR‐CM patient survival. 8 Time to clinical effect was predicted to range from 4.4 months for monthly dosing at 60 mg/kg to 10 months for monthly dosing at 10 mg/kg. In contrast, time to clinical effect increased to more than 18 months for monthly dosing at 3 mg/kg and below. In other words, the model predictions identified doses at and above 10 mg/kg to trigger a substantial clinical benefit within a year or less, whereas doses below 10 mg/kg would not provide a clinically meaningful benefit in this time frame (Figure 4 c ).
Sensitivity to ATTR load and elimination rate
ATTR load in the heart and the rate of amyloid depletion by immune cells were both expected to vary between patients; their possible effects on treatment were investigated using the PK/PD model.
Baseline cardiac ATTR load was increased 5 times and 10 times (respectively 1.36, 6.8, and 13.6 g). ATTR occupancy after the fourth monthly NI006 administration at 60 mg/kg was predicted to decrease from 22% to 1.8% with a 10‐fold increase in ATTR mass. Because of the lower ATTR occupancy at higher cardiac ATTR load, a 5× increase in ATTR mass approximately doubled the time to clinical effect irrespective of the dose, and a 10× increase approximately tripled that time (Figure 5 a ).
Figure 5.
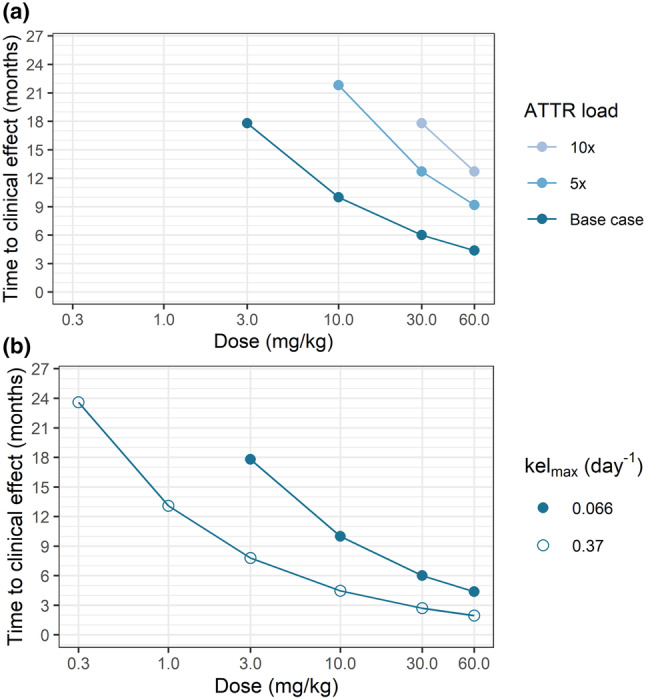
Predicted time to clinical effect as a function of (a) cardiac ATTR load and (b) NI006‐ATTR complex elimination rate (kelmax). The absence of data point at a given dose means that time to clinical effect was predicted to be longer than 26 months.
Additional simulations were run using the NI006‐ATTR complex elimination rate of 0.37 day−1 observed in fibril xenograft model. The 5.6× faster elimination rate observed in mice resulted in an approximately two‐fold decrease in the predicted time to clinical effect (Figure 5 b ).
Model prediction and phase I study results
These model predictions guided the selection of starting dose, dose escalation, dosing frequency and treatment duration for the Phase Ib study, which primary results were published last year. 18 The starting dose of 0.3 mg/kg Q4W proved safe, and, overall, no dose‐limiting toxicities were observed up to the highest dose tested (60 mg/kg Q4W).
Instead of LVWT, the clinical study used extracellular volume (ECV) determined by MRI and heart‐to‐whole‐body ratio using diphosphonate scintigraphy as more direct markers for cardiac amyloid load monitoring. To accommodate for these different readouts, the model‐predicted and clinically observed dose responses were compared using relative change from baseline, without recalibration of the model. The clinically observed reductions in these imaging proxies of cardiac amyloid load occurred in a dose‐ and time‐dependent manner that were qualitatively and quantitatively in agreement with the PK/PD model predictions, both at 4 and 12 months of treatment duration (Figure 6 ). Doses at and above 10 mg/kg resulted in reductions in cardiac amyloid surrogate markers after 12 months of treatment, whereas doses at 3 mg/kg and below had no quantifiable effects.
Figure 6.
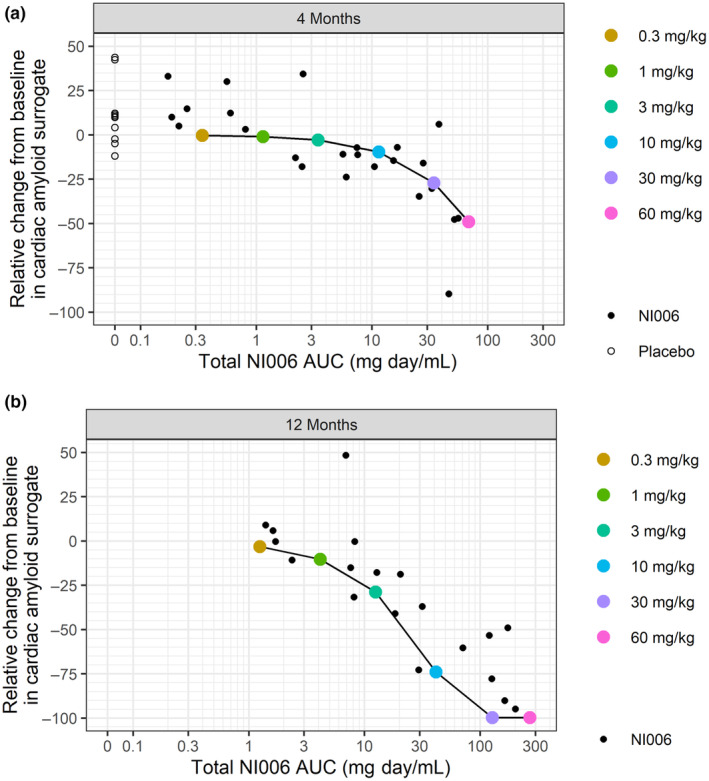
Relative change in cardiac amyloid surrogate plotted vs. cumulative NI006 exposure after (a) 4 and (b) 12 months of treatment. Observations from the phase Ib study are depicted with small black dots, model predictions with large colored‐coded dots connected by a line.
DISCUSSION
This modeling work supported the design of a combined first‐in‐human and proof‐of‐concept phase I study of a novel antibody for cardiac ATTR amyloidosis. In vitro and in vivo results were integrated in a mechanistic and quantitative manner with published data to generate a translational PK/PD model of NI006 concentration‐dependent cardiac amyloid depletion by immune cells.
With the model, 0.3 mg/kg was identified as a safe starting dose based on low maximum serum concentration (8.4 μg/mL) and low target occupancy (< 0.1%) in the heart. Supported by in vitro safety data in various assays, this low starting dose/target occupancy was selected given the absence of prior clinical experience with amyloid depleters in heart failure patients with amyloid cardiomyopathy. Patients treated with this starting dose had no drug‐related adverse events, enabling dose escalation.
The model predicted quantitative, dose‐ and time‐dependent reductions in cardiac amyloid load achieved in less than 12 months at 10, 30, and 60 mg/kg Q4W, and minimal or virtually absent reductions for doses at and below 3 mg/kg (Figure 4 ). The predicted clinically efficacious doses were identified based on reductions in ATTR load and LVWT below the clinically relevant threshold of 12 mm within 6 months for dosing at 30 mg/kg Q4W, and within 4 months for dosing at 60 mg/kg. Based on these predictions, the dose range of 0.3 to 60 mg/kg was explored in 6 dose steps in a double‐blinded study of 4‐month duration followed by an 8‐month open‐label extension phase. The model was instrumental in devising an efficient dose escalation and providing guidance to explore dose levels and treatment durations sufficient to observe clinically relevant effects on cardiac ATTR load via imaging. Escalation decisions in the phase I study were based on safety and tolerability and overall, the use of NI006 up to 60 mg/kg, the highest dose tested, was associated with no apparent drug‐related serious adverse event. 18
The efficacy predictions were confirmed in the clinical trial: Without model recalibration, the predicted changes in cardiac amyloid load were in agreement with the observed changes in the imaging surrogates for cardiac amyloid load (ECV determined by MRI and diphosphonate scintigraphy) after 4 and 12 months of treatment (Figure 6 ). Thus, PK/PD modeling guided the design of a phase I study, which established the pharmacological activity of NI006. Moreover, once the correctness of the preclinical model predictions was confirmed, the model parameters were updated using the phase I study final results and the updated model was used for dose selection and the design of the ongoing phase III study (NCT06183931).
The PK/PD model captured ATTR accumulation in the heart and not in other organs and peripheral tissues. This choice was made because no clinical data was available that could be used to estimate the amyloid mass in the periphery. In contrast, LVWT is one of the key clinical features to confirm ATTR amyloidosis diagnosis. Therefore, it is routinely measured in patients with heart failure, reported in the literature, and known to increase together with amyloid deposition and disease progression. We used published, large LVWT data sets to model disease progression and estimate the amyloid formation and spontaneous elimination rates. 12 , 14
In the PK/PD model, the in vitro equilibrium dissociation constant was used to describe NI006 binding to ATTR. The agreement of the model predictions with the phase I study results supported this assumption. The NI006‐ATTR elimination rate by phagocytic immune cells was estimated using data from the tissue amyloid depletion assay (in vitro) and the patient‐derived fibril xenograft model (in mice). These two models gave different elimination rates, with > 5× faster elimination in vivo. We based our modeling efforts and instructed the clinical study design using the slow NI006‐ATTR elimination rate from the in vitro model, because this assay preserved better the amyloid conformation, its compact structure and complex proteomic composition as it is in patients. In contrast, the fibril graft model in mice made use of semi‐purified amyloid fibrils mechanically extracted from patient heart tissues, with a loosened presentation and potentially better epitope accessibility, and the fibrils were grafted in an animal species known to never present spontaneous amyloid formation. This model was deemed less translational than the in vitro assay.
Nonetheless, the in vivo NI006‐ATTR complex elimination rate guided the sensitivity analysis for this parameter. The 5.6× faster elimination rate resulted in model predictions indicating ~2× faster cardiac amyloid depletion at the same NI006 doses. With both elimination rates, model predictions presented a progressive saturation of the dose–response at high doses, suggesting the highest doses may not be necessary to achieve a rapid clinical effect (Figure 5 b ). Similarly, there was some uncertainty on our estimate of cardiac ATTR mass. The model predictions indicated that a clinical trial of 12‐month duration would remain able to detect treatment effect in patients with 5× more cardiac amyloid at 30 mg/kg and 10× more at 60 mg/kg (Figure 5 a ). Altogether, the results from this model sensitivity analyses suggested that a clinical study design testing doses up to 60 mg/kg for a duration of 12 months would have good chances of observing NI006 pharmacological activity despite uncertainties on the amyloid depletion rate and the cardiac amyloid load at baseline.
The spontaneous ATTR elimination rate was estimated from the observed LVWT decrease in patients treated with patisiran, a silencer of TTR expression, which was shown to reduce plasma TTR levels by 80% and prevent or reduce disease progression. As TTR tetramer dissociation into monomers is the rate limiting step for amyloid formation, it is expected that 80% reduction in plasma TTR levels translates into 80% reduction in ATTR deposition. NI006 was predicted to remove ATTR within months while the spontaneous removal was estimated to take 13 years to clear 50% of the ATTR if de novo ATTR formation would be 100% inhibited. Spontaneous ATTR removal was therefore deemed not relevant for the prediction of the NI006 effect. Conversely, TTR stabilizers, silencers and gene editing, which are targeting the amyloid production, would require very long treatment durations until ATTR accumulation would resolve spontaneously. Only limited data was available for the estimation of the spontaneous ATTR elimination rate, linking it with some degree of uncertainty. It will be possible to refine this estimate as additional clinical data for TTR silencers will become available.
The common approach to model antibody–target interaction is via mass action kinetics assuming a homogeneously distributed target. In the case of ATTR amyloidosis, this assumption is clearly inadequate. Cardiac ATTR forms packed, higher order fibrils and deposits in the interstitial space and in such structures, many monomers are likely to be inaccessible to binding. 20 Because the NI006‐ATTR elimination rate was obtained using data from an experimental model that presented ATTR in its native context, it is likely the estimated PK/PD model parameters accounted for the violation of the assumption. Thus, the correctly predicted effect on the surrogates of ATTR load suggested that even for targets in an insoluble, fibrous structure mass action kinetic is a valid approach.
The PK/PD model based the ATTR removal rate on the ATTR target occupancy. The downstream activation of phagocytic immune cells that remove ATTR was not modeled. This simplification neglected a potential rate limitation of phagocytic immune cells, where for example, phagocytic activity becomes exhausted over time. However, the simplified PK/PD model described well the ATTR removal in two time‐limited experimental models and in the clinical study after 4 and 12 months of treatment, suggesting that secondary regulation of phagocytic immune cells, including exhaustion, was not relevant and that ATTR removal was largely dependent on the degree of ATTR occupancy by NI006.
One final simplification was that non‐cardiac ATTR was neglected. ATTR amyloidosis is a systemic disease potentially affecting many different tissues with varying degrees of deposits. Yet, the dominant clinical manifestation is often limited to a single organ such as the heart. The simplification was made because of the focus on ATTR‐CM together with the lack of data on ATTR load in other tissues. A consequence of this assumption was that the total ATTR body load was most likely underestimated. However, the agreement with the phase I study data suggested that this simplification was adequate for predicting the NI006 effect on cardiac ATTR load.
The excellent agreement of the predicted and observed NI006 dose‐ and time‐dependent effect on ATTR load supported the translatability of the preclinical models and the PK/PD modeling approach with the assumptions and simplifications. The model contributed to the identification of a safe starting dose and to the design of a phase I trial exploring an adequate dose range for an efficient dose escalation into the clinically efficacious range. Further, it justified a long study duration of 12 months required to observed meaningful effects at the different dose levels. This study design enabled the observation of substantial reductions in cardiac amyloid surrogates in a first‐in‐human study, generating important evidence on the pharmacological activity of NI006 in patients. 18 Despite the assumptions and simplifications, this work highlights the translational value of quantitative system pharmacology (QSP) to design first‐in‐patients clinical studies. With appropriate adaptations and the availability of appropriate data, this work supports the application of QSP to other diseases and treatments.
FUNDING
This work was funded by Neurimmune AG.
CONFLICT OF INTEREST
A.M., C.C., F.B., C.H., R.M.N., P.C.K., and J.G. are employees and shareholders of Neurimmune AG. A.M., C.H., P.C.K., and J.G. are inventors on patents relating to NI006. C.U. and M.F.M. are employees of Alexion, AstraZeneca Rare Disease. L.R. and M.M. declare no competing interest for this work.
AUTHOR CONTRIBUTIONS
A.M., L.R., and M.M. wrote the manuscript. A.M., L.R., and M.M. designed the research. A.M., L.R., and M.M. performed the research. A.M., L.R., and M.M. analyzed the data. A.M., C.C., C.U., M.F.M., F.B., C.H., R.M.N., P.C.K., and J.G. contributed analytical tools.
PRIOR PRESENTATIONS
This work was presented as a poster at the European Society of Cardiology, Heart Failure World Congress (ESC‐HF) in Lisbon, PT, May 11–14, 2024, and at the 19th International Symposium on Amyloidosis (ISA) in Rochester, Minnesota, USA, May 26–30, 2024.
Supporting information
Data S1
ACKNOWLEDGMENTS
We thank the patients and their family members for participating in the NI006‐101 clinical study.
References
- 1. Ando, Y. et al. Guideline of transthyretin‐related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 8, 31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruberg, F.L. & Maurer, M.S. Cardiac amyloidosis due to transthyretin protein: a review. JAMA 331, 778–791 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adams, D. et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 268, 2109–2122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vaishnav, J. , Brown, E. & Sharma, K. Advances in the diagnosis and treatment of transthyretin amyloid cardiomyopathy. Prog. Cardiovasc. Dis. 82, 113–124 (2024). [DOI] [PubMed] [Google Scholar]
- 5. Fontana, M. et al. Antibody‐associated reversal of ATTR amyloidosis–related cardiomyopathy. N. Engl. J. Med. 388, 2199–2201 (2023). [DOI] [PubMed] [Google Scholar]
- 6. Michalon, A. et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat. Commun. 12, 3142 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pucci, A. et al. Amyloid deposits and fibrosis on left ventricular endomyocardial biopsy correlate with extracellular volume in cardiac amyloidosis. J Am Heart Assoc 10, e020358 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rapezzi, C. et al. Role of 99mTc‐DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin‐related cardiac amyloidosis. JACC Cardiovasc. Imaging 4, 659–670 (2011). [DOI] [PubMed] [Google Scholar]
- 9. Neuber, T. et al. Characterization and screening of IgG binding to the neonatal fc receptor. mAbs 6, 928–942 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang, W. & Prueksarimont, T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm. Drug Dispos. 31, 253–263 (2010). [DOI] [PubMed] [Google Scholar]
- 11. Mahmood, I. A single animal species‐based prediction of human clearance and first‐in‐human dose of monoclonal antibodies: beyond monkey. Antibodies 10, 35 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arvidsson, S. , Pilebro, B. , Westermark, P. , Lindqvist, P. & Suhr, O.B. Amyloid cardiomyopathy in hereditary transthyretin V30M amyloidosis – impact of sex and amyloid fibril composition. PLoS One 10, e0143456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grogan, M. et al. Natural history of wild‐type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J. Am. Coll. Cardiol. 68, 1014–1020 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Solomon, S.D. et al. Effects of Patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin‐mediated amyloidosis: analysis of the APOLLO study. Circulation 139, 431–443 (2019). [DOI] [PubMed] [Google Scholar]
- 15. Shah, D.K. & Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 39, 67–86 (2012). [DOI] [PubMed] [Google Scholar]
- 16. Shah, D.K. & Betts, A.M. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 5, 297–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clay, S. , Alfakih, K. , Radjenovic, A. , Jones, T. & Ridgway, J.P. Normal range of human left ventricular volumes and mass using steady state free precession MRI in the radial long axis orientation. Magn. Reson. Mater. Phy. 19, 41–45 (2006). [DOI] [PubMed] [Google Scholar]
- 18. Garcia‐Pavia, P. et al. Phase 1 trial of antibody NI006 for depletion of cardiac transthyretin amyloid. N. Engl. J. Med. 389, 239–250 (2023). [DOI] [PubMed] [Google Scholar]
- 19. Henein, M.Y. , Pilebro, B. & Lindqvist, P. Disease progression in cardiac morphology and function in heart failure: ATTR cardiac amyloidosis versus hypertensive left ventricular hypertrophy. Heart Vessels 37, 1562–1569 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steinebrei, M. et al. Cryo‐EM structure of an ATTRwt amyloid fibril from systemic non‐hereditary transthyretin amyloidosis. Nat. Commun. 13, 6398 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1