

Abstract
The goal of the study was to describe the population pharmacokinetics of trimethoprim, sulfamethoxazole, and N‐acetyl sulfamethoxazole in hospitalized patients. Furthermore, this study used the model to optimize dosing regimens of cotrimoxazole for Pneumocystis jirovecii pneumonia and in patients with renal insufficiency or with continuous renal replacement therapy (CRRT). This was a retrospective multicenter observational cohort study based on therapeutic drug monitoring (TDM) data from hospitalized patients treated with cotrimoxazole. We developed two population pharmacokinetic (POPPK) models: a model of trimethoprim and an integrated model with both sulfamethoxazole and N‐acetyl sulfamethoxazole concentrations. Monte Carlo simulations were performed to determine the optimal dosing regimen. A total of 348 measurements from 168 patients were available. The estimated glomerular filtration rate (eGFR) and CRRT were included as covariates on the clearance of all three compounds. Cotrimoxazole TID 1,920 mg and b.i.d. 2,400 mg led to sufficient exposure for infections with P. jirovecii in patients without renal insufficiency. To reach equivalent exposure, a dose reduction of 33.3% is needed in patients with an eGFR of 10 mL/minute/1.73 m2 and of 16.7% for an eGFR of 30 mL/minute/1.73 m2. N‐acetyl sulfamethoxazole accumulates in patients with a reduced eGFR. CRRT increased the clearance of sulfamethoxazole, but not trimethoprim or N‐acetyl sulfamethoxazole, compared with the median clearance in the population. Doubling the sulfamethoxazole dose is needed for patients on CRRT to reach equivalent exposure.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Cotrimoxazole is an antibiotic combination of trimethoprim and sulfamethoxazole, effective against both Gram‐positive and Gram‐negative pathogens. Sulfamethoxazole peak concentrations above 100 mg/L and trimethoprim concentrations >5 mg/L have been associated with clinical effectiveness for Pneumocystis jirovecii pneumonia. Concentrations of sulfamethoxazole >200 mg/L, its metabolite N‐acetyl sulfamethoxazole >75 mg/L, and trimethoprim >15 mg/L are associated with toxicity. All three components are cleared to some extent by renal clearance.
WHAT QUESTION DID THIS STUDY ADDRESS?
Which dosing regimens of trimethoprim/sulfamethoxazole should be used to reach the target concentrations for Pneumocystis jirovecii pneumonia and which dose adjustments are needed for patients with renal insufficiency or with continuous renal replacement therapy (CRRT).
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
TID 1,920 mg and b.i.d. 2,400 mg lead to sufficient exposure for infections with P. jirovecii in patients without renal insufficiency. To reach equivalent exposure a dose reduction of 33.3% is needed in patients with an eGFR of 10 mL/minute/1.73 m2 and of 16.7% for an eGFR of 30 mL/minute/1.73 m2. N‐acetyl sulfamethoxazole accumulates in patients with a reduced eGFR. CRRT increased the clearance of sulfamethoxazole, but not trimethoprim or N‐acetyl sulfamethoxazole, compared with the median clearance in the population.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The dosing regimens suggested in this study can enhance the utilization of cotrimoxazole in hospitalized patients. The population pharmacokinetic models developed in this study can also be utilized to optimize treatment with cotrimoxazole for other infectious diseases.
Cotrimoxazole is an antibiotic combination of trimethoprim and sulfamethoxazole, effective against both Gram‐positive and Gram‐negative pathogens. 1 The drug disrupts the production of essential nucleic acids needed for bacterial proliferation and replication by synergistically inhibiting two consecutive enzymatic steps involved in bacterial folinic acid synthesis. 1 It has excellent tissue penetration and is therefore used for the treatment of various infections, ranging from infections caused by more resistant pathogens (ESBL+ E. coli, MRSA) to Pneumocystis jirovecii pneumonia (PCP). 1 , 2
Trimethoprim is primarily eliminated through nonionic renal diffusion, with 40–75% of the drug recovered unchanged in the urine within 24 h. 3 , 4 Sulfamethoxazole is extensively metabolized into inactive metabolites which are renally excreted. Approximately, 16% of sulfamethoxazole is eliminated unchanged and 46% as N‐acetyl sulfamethoxazole, the predominant metabolite. 5 , 6 , 7 Although N‐acetyl sulfamethoxazole lacks relevant antibacterial activity, this metabolite has been associated with concentration‐dependent toxicity due to accumulation in patients with renal impairment. 4 , 5 Therapeutic drug monitoring (TDM) is therefore recommended in patients with impaired renal function receiving high‐dose cotrimoxazole.
Compared with other antibiotics, the pharmacodynamics of cotrimoxazole are poorly understood. It is unclear which pharmacokinetic/pharmacodynamic (PK/PD) index (fT > MIC, C max > MIC, or AUC/MIC) best correlates with the efficacy of cotrimoxazole. 8 , 9 Target concentrations for trimethoprim and sulfamethoxazole are predominantly based on theoretical assumptions and in vitro measurements rather than an association with clinical outcomes. For the treatment of PCP, target concentrations for sulfamethoxazole and trimethoprim have been suggested and used in both research and TDM in clinical practice. 8 , 10 , 11 Specifically, sulfamethoxazole peak concentrations above 100 mg/L and trimethoprim concentrations above 5 mg/L are recommended. These targets are originally based on two small studies. The first study included 26 children with PCP and found that 3–5 mg/L for trimethoprim and 100–150 mg/L for sulfamethoxazole resulted in clinical effectiveness. 10 The other study included eight adults with PCP and reported that trimethoprim concentrations below 5.5 mg/L were linked to treatment failure. 11 The concentrations associated with toxicity have been studied more extensively: concentrations of sulfamethoxazole >200 mg/L, N‐acetyl sulfamethoxazole >75 mg/L, and trimethoprim >15 mg/L are associated with toxicity both in healthy individuals as in patients with PCP. concentration‐dependent adverse events that have been described include hematological toxicity, hyperkalemia, renal failure and central nervous system adverse effects. 12 , 13 , 14 , 15
Studies assessing the optimal initial dosing regimen to reach the target concentrations for PCP are lacking. Moreover, limited information is available regarding the need for dose reductions in patients with reduced renal function or on continuous renal replacement therapy (CRRT). Several dosing regimens have been proposed for patients with altered renal function, but these recommendations lack support from population pharmacokinetic (POPPK) simulations that consider both effectiveness and toxicity. 5 , 7 , 16 , 17
To address these gaps, we conducted a retrospective cohort study in hospitalized patients treated with cotrimoxazole. Based on the measured serum concentrations, a model for trimethoprim and a combined population pharmacokinetic model for sulfamethoxazole and its metabolite N‐acetyl sulfamethoxazole were developed. These models enabled us to simulate dose regimens and dose adjustments for patients with altered renal function or those receiving CRRT for PCP.
METHODS
Study design
This multicenter, retrospective cohort study was conducted in three university medical centers (Leiden University Medical Center, Erasmus MC, and University Medical Center Groningen) in The Netherlands. It was approved by the Medical Research Ethics Committee Leiden, Den Haag & Delft (reference number: G21.153) and by the boards of the participating hospitals. The need for informed consent was waived by the Medical Ethics Committee.
Participants and data collection
Patients aged ≥18 years were included when they received therapeutic dosages of intravenous or oral cotrimoxazole between January 2016 and December 2021, had plasma concentrations of sulfamethoxazole, and/or trimethoprim measured during treatment and at least one serum creatinine measurement was available. Patients with registered objection for the use of their data and patients treated with extracorporeal membrane oxygenation (ECMO) treatment or intermittent hemodialysis during cotrimoxazole treatment were excluded.
Cotrimoxazole measurement
All cotrimoxazole concentrations were measured as part of routine TDM in patients with high‐dose cotrimoxazole. The plasma concentrations were quantified using validated high‐performance liquid chromatography with diode‐array detection (HPLC‐DAD) or liquid chromatography with tandem mass spectrometry (LC–MS/MS) in ISO15189 accredited laboratories. The method used in the UMCG was an LC–MS/MS method and was previously validated. 18 LUMC used an LC–MS/MS method and Erasmus MC a HPLC‐DAD method, these methods are described in the supplement file. All laboratories participated in a proficiency testing program for trimethoprim and sulfamethoxazole assuring interchangeability of the methods.
Pharmacokinetic modeling
Nonlinear mixed‐effects modeling (NONMEM) with first‐order conditional estimation method with interaction (FOCE‐I) was used for POPPK model building. Two different POPPK models were developed. The first model described the population pharmacokinetics of trimethoprim, and the second model was an integrated model describing both the pharmacokinetics of sulfamethoxazole and N‐acetyl sulfamethoxazole. For all three components, one‐ and two‐compartment models with first‐order elimination were explored during the structural model development. Oral absorption was modeled using first‐order absorption and the biological availability was estimated. A lag time on oral absorption was evaluated. No urine concentrations were available for sulfamethoxazole or N‐acetyl sulfamethoxazole; therefore, 40% of the total sulfamethoxazole clearance was estimated to be converted to N‐acetyl sulfamethoxazole. 19 Additive, proportional, and combined error models were tested for residual variability in drug concentrations. Inter‐individual variation was tested for all PK parameters. The structural model selection was based on reduction of the objective function value (OFV) (approximation of a χ2 distribution for nested models, with a ΔOFV of 3.84 corresponding to a P‐value of 0.05), goodness‐of‐fit (GOF) plots, shrinkage, and precision of pharmacokinetic parameter estimates.
To investigate potential factors that may impact the pharmacokinetics of the three components, biologically plausible patient characteristics were tested for inclusion as covariates. This included age, body weight, body mass index, estimated glomerular filtration rate (eGFR, calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) formula) and the use of CRRT. Continuous covariates were modeled using linear, exponential, and power functions. For body weight, the allometric rule standardized to an average adult of 70 kg was also considered. The inclusion of a covariate in the final model was determined by assessing if its effect was biologically plausible, if it produced a clinically relevant reduction in the inter‐individual variability (IIV) of the parameter, and if the OFV was decreased by at least 3.84 (P < 0.05) in forward inclusion and 6.63 (P < 0.01) in backward deletion.
The final model was evaluated using GOF plots and 500 prediction‐corrected visual predictive checks (pcVPCs). Parameter estimates and confidence intervals were assessed using nonparametric bootstrapping using 500 resampled data sets. Population pharmacokinetic modeling was carried out using NONMEM (v7.4; Icon Development Solutions, Ellicott City, MD, USA) and Perl Speaks NONMEM (v.5.0.0). Pirana (v.2.9.8) and R statistics (v.4.2.3) were used for interpretation and visualization of the pharmacokinetic models.
Dosing simulation
Monte Carlo simulations (n = 1,000) were conducted using the final model to assess the impact of cotrimoxazole dosing regimen, renal function, and use of CRRT on the probability of target attainment during the first 5 days of cotrimoxazole treatment. For both the toxicity threshold and target concentrations the maximal concentration during the dose interval (C max) was used. The minimal target concentrations for the treatment of PCP were 100 mg/L for sulfamethoxazole and 5 mg/L for trimethoprim. The following concentrations were used as upper limit of the therapeutic range: 200 mg/L for sulfamethoxazole, 75 mg/L for N‐acetyl sulfamethoxazole, and 15 mg/L for trimethoprim.
Multiple dosing regimens were simulated to evaluate the optimal dose for PCP infections for different renal functions. Considering the approximate 100% biological availability of both antibacterial components, we used intravenous administration for the simulations. The following dosing regimens were simulated: twice daily 800 mg sulfamethoxazole + 160 mg trimethoprim, twice daily 1,200 mg + 240 mg, twice daily 1,600 mg + 320 mg, twice daily 2,000 mg + 400 mg and three times daily 1,600 mg + 320 mg.
Furthermore, we simulated the dose reductions required for patients with a reduced eGFR or receiving CRRT that resulted in similar exposure compared with patients without renal insufficiency. We performed simulations based on different eGFRs (10, 30, 50, and 70 mL/minute/1.73 m2), as well as a simulation considering the concomitant use of CRRT. The percentage dose reduction was calculated as well as the minimal dose leading to 25% of the simulated patients reaching concentrations of either sulfamethoxazole, N‐acetyl sulfamethoxazole, or trimethoprim, above the upper limit of the target range.
RESULTS
Population
A total of 168 (16 LUMC, 116 Erasmus MC and 36 UMCG) patients were included in this study. The population was predominantly male (64.3%) and the median age was 58 years. The median eGFR, based on the CKD‐EPI formula, was 70 mL/minute/1.73 m2 and 18 patients (10.7%) were concomitantly treated with CRRT. The most frequently used initial dosing regimen was cotrimoxazole TID 1,920 mg (48.2%) and intravenous infusion was the most common route of administration (55.4%). Baseline patient characteristics are presented in Table 1 .
Table 1.
Baseline characteristics
| All patients | Patients with measured trimethoprim concentrations | |
|---|---|---|
| n = 168 | n = 52 | |
| Patient characteristics | ||
| Males (% of total) | 108 (64.3) | 33 (63.4) |
| Age (years) (mean, SD) | 58.2 ± 15.2 | 60.3 ± 11.9 |
| Weight (kg) (mean, SD) | 76.7 ± 16.2 | 80.6 ± 18.5 |
| Height (cm) (mean, SD) | 174.0 ± 10.3 | 173.8 ± 11.1 |
| BMI (kg/m2) (mean, SD) | 25.3 ± 5.0 | 26.5 ± 5.0 |
| Laboratory values | ||
| Serum creatinine (μmol/L) (mean, SD) | 119.7 ± 91.8 | 170.1 ± 101.5 |
| Serum albumin (g/L) (mean, SD) | 26.6 ± 6.8 | 29.1 ± 7.2 |
| eGFR (mL/minute/1.73 m2) (mean, SD) | 70.8 ± 33.2 | 49.2 ± 34.7 |
| Initial route of administration (% of total) | ||
| Oral | 75 (44.6) | 22 (42.3) |
| Intravenous | 93 (55.4) | 30 (57.7) |
| Cotrimoxazole daily starting dose (% of total) (mg) | ||
| ≤960 | 10 (6.0) | 6 (11.5) |
| 1,920–2,400 | 27 (16.1) | 19 (36.6) |
| 2,880 | 15 (8.9) | 6 (11.5) |
| 3,840–4,800 | 35 (20.8) | 7 (13.5) |
| 5,760 | 81 (48.2) | 14 (27.0) |
| Co‐medication | ||
| Corticosteroids (% of total) | 62 (36.9) | 18 (34.6) |
| Comorbidities | ||
| Continuous renal replacement therapy (count, %) | 18 (10.7) | 14 (27%) |
| Stem‐cell transplantation (% of total) | 13 (7.7) | 3 (5.8) |
| Solid organ transplantation (% of total) | 35 (20.8) | 24 (46.2) |
| Malignancy (% of total) | 23 (13.7) | 6 (11.5) |
| HIV (% of total) | 19 (11.3) | 0 (0.0) |
In total, 348 sulfamethoxazole and N‐acetyl sulfamethoxazole concentrations were available for analysis. Trimethoprim concentrations were only measured in two of the three participating hospitals, with 137 concentrations from 52 patients (16 LUMC and 36 UMCG) available for the trimethoprim model. The concentrations included both peak and through concentrations. Sulfamethoxazole concentrations ranged between 2 and 380 mg/L, N‐acetyl sulfamethoxazole between 2 and 173.4 mg/L, and trimethoprim concentrations were between 0.2 and 15.6 mg/L.
Pharmacokinetic modeling
The pharmacokinetics of trimethoprim were adequately described by a one‐compartment model (Table 2 ). Elimination followed first‐order kinetics and a proportional error model described the residual error. Biological availability was estimated at first, but as this was ~100%, fixing it to 100% led to similar model performance. The estimation of the absorption constant resulted in a relatively large residual error. IIV was included in the clearance and volume of distribution of trimethoprim. eGFR and CRRT were included as covariates. As the variation in clearance was small for patients on CRRT, we estimated the IIV for the clearance for patients treated with CRRT and patients without CRRT.
Table 2.
Model parameters trimethoprim
| Parameter | Estimate | RSE (%) | Bootstrap median | 95% CI |
|---|---|---|---|---|
| Trimethoprim | ||||
| Biological availability | 1 Fixed | 1 Fixed | ||
| Absorption rate constant | 0.337 | 54 | 0.364 | 0.095–0.877 |
| Apparent clearance (L/hour)a | 4.21 | 13 | 4.23 | 3.21–5.42 |
| eGFR on CL | 0.317 | 36 | 0.319 | 0.049–0.558 |
| CRRT on CL | 1.12 | 16 | 1.13 | 0.77–1.57 |
| Volume of distribution (L) | 134 | 10 | 132 | 105–161 |
| IIV CL (%) | 40.1 | 12 | 39.2 | 26.9–48.5 |
| IIV CL patients on CRRT (%) | 32.9 | 32 | 31.5 | 7.54–48.7 |
| IIV Vd (%) | 31.9 | 32 | 29.8 | 9.77–49.2 |
| Proportional error | ||||
| Trimethoprim | 0.169 | 10 | 0.167 | 0.131–0.198 |
aFormula for clearance: in case of no CRRT = 4.21 × (EGFR/68)0.317 in case of CRRT: 4.21 × 1.12.
A one‐compartment integrated model for sulfamethoxazole and N‐acetyl sulfamethoxazole described the data best (Table 3 ). Elimination followed first‐order kinetics, and a proportional error model was used to describe the residual error. Similarly to trimethoprim, the oral bioavailability was first estimated and then fixed at 100%. Inter‐individual variability (IIV) was included in the clearance of both components and in the volume of distribution of sulfamethoxazole. Both eGFR and the use of CRRT were included as covariates on the clearance of sulfamethoxazole and N‐acetyl sulfamethoxazole. The clearance of sulfamethoxazole in patients treated with CRRT was 2.2 (95% CI 2.0–2.4) times higher compared with the clearance of patients with a median eGFR of 70 mL/minute/1.73 m2 (Figure S6 ); however, the clearance of the N‐acetyl sulfamethoxazole was 0.68 (95% CI 0.53–0.83) times lower in patients with CRRT.
Table 3.
Model parameters sulfamethoxazole
| Parameter | Estimate | RSE (%) | Bootstrap median | 95% CI |
|---|---|---|---|---|
| Sulfamethoxazole | ||||
| Biological availability | 1 Fixed | 1 Fixed | ||
| Absorption rate constant | 0.978 | 45 | 0.95 | 0.25–8.61 |
| Apparent clearance (L/hour)a | 0.97 | 4 | 0.97 | 1.06 |
| eGFR on CL | 0.27 | 20 | 0.28 | 0.16–0.38 |
| CRRT on CL | 2.2 | 10 | 2.2 | 1.8–2.6 |
| Volume of distribution (L) | 37.0 | 6 | 36.8 | 21.5–43.3 |
| IIV CL (%) | 36.3 | 8 | 36.5 | 29.6–42.4 |
| IIV Vd (%) | 62.9 | 11 | 60.2 | 37.4–79.4 |
| N‐acetyl sulfamethoxazole | ||||
| Conversion parent metabolite | 0.4 × CL | |||
| Apparent clearance (L/hour)a | 1.34 | 4 | 1.33 | 1.21–1.43 |
| eGFR on CL | 0.797 | 7 | 0.79 | 0.65–0.92 |
| CRRT on CL | 0.683 | 11 | 0.68 | 0.54–0.86 |
| Volume of distribution (L) | 3.98 | 24 | 3.87 | 1.55–6.33 |
| IIV CL (%) | 40.7 | 8 | 40.9 | 34.5–48.3 |
| Proportional error | ||||
| Sulfamethoxazole | 0.181 | 8 | 0.178 | 0.144–0.208 |
| N‐acetyl sulfamethoxazole | 0.201 | 8 | 0.194 | 0.155–0.230 |
aFormula for clearance: in case of no CRRT = 0.97 × (EGFR/68)0.27 in case of CRRT: 1.34 × 2.2.
Formula for clearance of the metabolite in case of no CRRT = 1.34 × (EGFR/68)0.797 in case of CRRT: 1.34 × 0.683.
Internal validation
The GOF plots and VPCs are visualized in Figures S1 – S5 and indicate that the observed concentrations were adequately described by both models. The bootstrap results are presented in Tables 2 and 3 and indicate good stability of the POPPK parameters of the final model.
Dosing simulations for PCP
The results show that in a median patient with an eGFR of 70 mL/minute/1.73 m2, a dosing regimen of b.i.d. 2,400 mg resulted in exposure in 89.0% of the patients within, 3.0% below and 8.0% above the target range for trimethoprim and 68.7% within, 10.9% below and 20.4% above for sulfamethoxazole and N‐acetyl sulfamethoxazole. TID 1,920 mg resulted in 90.9% within, 1.8% below, and 7.3% above for trimethoprim and 63.0% within, 6.2% below, and 30.8% above the target range for sulfamethoxazole.
In patients with a reduced eGFR, the likelihood of reaching concentrations above the target range increases. Figures 1 and 2 illustrate simulated dose reductions for the target concentrations for the treatment of PCP. For patients with an eGFR of 10 mL/minute/1.73 m2, all dosing regimens except b.i.d. 960 mg resulted in >25% of the patients with sulfamethoxazole or N‐acetyl sulfamethoxazole concentrations above the upper limit of the target range, while b.i.d. 960 mg leads to 54.2% of patients with sulfamethoxazole underexposure.
Figure 1.
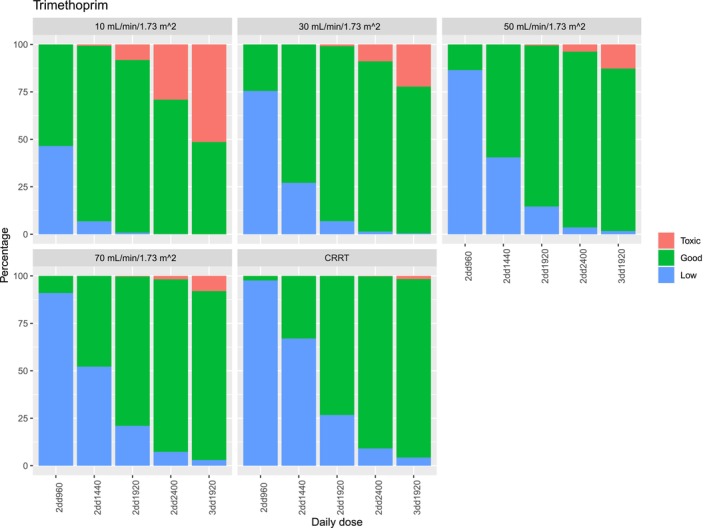
Target attainment of trimethoprim for infections with Pneumocystis jirovecii pneumonia for different dosing regimens and different eGFRs. Low is determined as a C max below 5 mg/L, toxic is determined as a C max above 15 mg/L.
Figure 2.
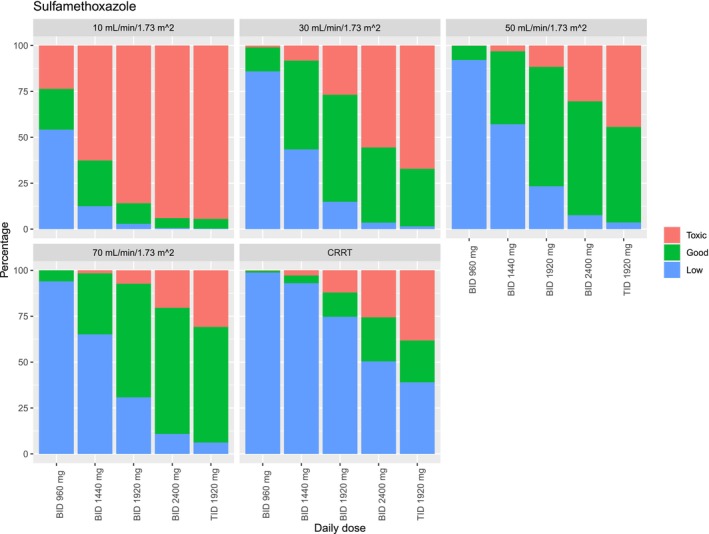
Target attainment of sulfamethoxazole for infections with Pneumocystis jirovecii pneumonia for different dosing regimens and different eGFRs. Low is determined as a C max of sulfamethoxazole below 100 mg/L and toxic is determined as a sulfamethoxazole C max above 200 mg/L or an N‐acetyl sulfamethoxazole concentration above 75 mg/L.
For patients with an eGFR of 30 mL/minute/1.73 m2, b.i.d. 1,920 mg results in 92.2% of the simulated patients on‐target, 6.9% low, and 0.9% high concentrations for trimethoprim and 58.3% on‐target, 14.9% low, and 26.8% high sulfamethoxazole concentrations. b.i.d. 1,440 mg leads to 72.9% within, 27.1% low exposure and no concentrations above the therapeutic range for trimethoprim and 48.4% within 43.4% below and 8.2% above the target range for sulfamethoxazole and N‐acetyl sulfamethoxazole.
Similarly, for patients with an eGFR of 50 mL/minute/1.73 m2, b.i.d. 1,920 mg leads to 84.9% on‐target, 14.6% low, and 0.5% high trimethoprim concentrations, and 65.1% of the patients on‐target, 23.3% low, and 11.6% high concentrations for sulfamethoxazole and N‐acetyl sulfamethoxazole.
In patients on CRRT trimethoprim, TID 1,920 mg resulted in 94.0% within, 4.3% below, and 1.7% above the target range for trimethoprim. Sulfamethoxazole concentrations were below the target range for more than 50% of the simulated patients using all dosing regimens lower than TID 1,920 mg; furthermore, accumulation of N‐acetyl sulfamethoxazole occurred in patients receiving TID 1,920 mg leading to toxic concentrations in 38.2% of the patients (Figure 1 ). Hence, none of the current dosing regimens achieved adequate target attainment for most simulated patients for sulfamethoxazole.
Dose reductions to reach equivalent exposure
In Table 4 , the doses required to reach equivalent exposure compared with a patient with an eGFR of 70 mL/minute/1.73 m2 (the median eGFR in the study population) are shown and the lowest dose that leads to >25% of the simulated patients reaching concentrations above the upper limit of the target range. In Figures S6 and S7 the exposure reached with different dosing regimens is visualized. The dose reductions were similar for all simulated eGFRs for both trimethoprim and sulfamethoxazole for patients with a low eGFR. To reach equivalent exposure a dose reduction of 16.7% is needed in patients with an eGFR of 30 mL/minute/1.73 m2 and of 33.3% for an eGFR of 30 mL/minute/1.73 m2. For CRRT the dose adjustments were not similar for trimethoprim and sulfamethoxazole. A 100% increased dose is needed for sulfamethoxazole, while for trimethoprim no dose adjustments are required. The risk of concentrations above the target range increases with increasing dose for patients with reduced renal function.
Table 4.
Dose adjustments in patients with a reduced eGFR or treated with CRRT
| Estimated glomerular filtration rate | Dose required to reach equivalent exposure to a patient with an eGFR of 70 mL/minute/1.73 m2 | Lowest simulated dose with >25% of the simulated patients above the upper limit of the target concentrations |
|---|---|---|
| 50 mL/minute/1.73 m2 | 100% | TID 1,920 mg |
| 30 mL/minute/1.73 m2 | 83.3% | b.i.d. 1,920 mg |
| 10 mL/minute/1.73 m2 | 66.7% | b.i.d. 1,440 mg |
| CRRT |
200% for sulfamethoxazole 100% for trimethoprim |
b.i.d. 2,400 mg |
DISCUSSION
We conducted a population pharmacokinetic study of sulfamethoxazole and trimethoprim in hospitalized patients. To the best of our knowledge, this is the largest cohort study up until now and the only population pharmacokinetic analysis that included trimethoprim, sulfamethoxazole, and N‐acetyl sulfamethoxazole.
The pharmacokinetics of trimethoprim, sulfamethoxazole, and N‐acetyl sulfamethoxazole were accurately described by one‐compartment models. The clearance and volume of distribution observed in our population were similar compared with values published in the literature for trimethoprim and sulfamethoxazole in both healthy individuals and other cohorts of hospitalized patients. 7 , 12 , 15 , 16 , 20 , 21 To the set of our knowledge, there is no previously published information on the pharmacokinetics of N‐acetyl sulfamethoxazole in the currently available literature. The bioavailability of both trimethoprim and sulfamethoxazole was close to 100%, which aligns with previous studies and indicates that even in severely ill patients, oral administration of cotrimoxazole is a viable method. 22 Inter‐individual variation was large for both trimethoprim and sulfamethoxazole, which could be explained by the heterogeneous population in this study or other unmeasured variables affecting the pharmacokinetics. Generally, the model performance was good, except for some imprecision in the estimation of the absorption constant for trimethoprim. This discrepancy is likely due to the smaller sample size and the use of routinely collected data, which may introduce some uncertainty regarding the exact drug administration time.
eGFR and CRRT were included as covariates in both models. As expected, it was observed that the inclusion of eGFR had a more significant impact on the clearance of N‐acetyl sulfamethoxazole compared with sulfamethoxazole. The primary clearance route for sulfamethoxazole is liver metabolism, with less than 30% being recovered unchanged in urine. In contrast, N‐acetyl sulfamethoxazole is mainly cleared renally, leading to its accumulation in patients with reduced eGFR. 7 , 16 For trimethoprim, the association between eGFR and clearance was also as expected, since the primary elimination route is renal clearance, with only a small percentage of metabolism or biliary excretion. 19
The final model was used to generate dose recommendations for PCP and dose reductions in case of renal insufficiency. A starting dose of TID 1,920 mg or b.i.d. 2,400 mg for PCP will result in sufficient exposure in the majority of patients. TID 1,920 mg has a larger risk of concentrations above the target range while b.i.d. 2,400 mg has a larger risk of underexposure. Because of the large IIV, TDM should be considered to improve the percentage of patients within the target range. Patients with an eGFR of 30 mL/minute/1.73 m2 require a dose reduction of 16.7%, and patients with an eGFR of 10 mL/minute/1.73 m2 a dose reduction of 33.3% to reach similar exposure to patients without renal insufficiency. The risk of concentrations above the target range increases with doses in patients with a low eGFR. Therefore, for infections requiring high‐dose cotrimoxazole like PCP or infections caused by Nocardia spp. the risk of concentration‐dependent toxicity should be compared with the risk of low exposure when selecting an initial dosing regimen.
The clearance of sulfamethoxazole in patients on CRRT exceeds the median clearance of the patients in this study. For trimethoprim, the clearance in patients on CRRT was comparable to patients with an eGFR of 70 mL/minute/1.73 m2. N‐acetyl sulfamethoxazole is not rapidly eliminated by CRRT. The rapid clearance of sulfamethoxazole and the difference between sulfamethoxazole and N‐acetyl sulfamethoxazole clearance has been previously reported in small case series. 17 , 23 One possible explanation is that tubular reabsorption, which is significant for sulfamethoxazole, does not occur in patients treated with CRRT. 5 , 24 When filtrated via CRRT, sulfamethoxazole will be cleared directly from the body via the ultrafiltrate. This lack of tubular reabsorption of sulfamethoxazole could explain the increased clearance observed in patients treated with CRRT. In contrast, N‐acetyl sulfamethoxazole, being reabsorbed to a lesser extent, is less affected by CRRT. 21 , 24 This change in pharmacokinetics presents a risk of rapid clearance of active sulfamethoxazole in CRRT patients, while N‐acetyl sulfamethoxazole accumulates, potentially leading to underexposure or toxicity. The clinical impact is unclear as the exposure to trimethoprim is sufficient in these patients. To prevent toxicity, therapy for PCP could be started with TID 1,920 mg and doses can be subsequently individualized using early TDM. 17 For other infections requiring lower daily doses and thereby a lower risk of N‐acetyl sulfamethoxazole accumulation, doubling the cotrimoxazole dose should be considered to reach similar exposure of sulfamethoxazole compared with patients with a normal renal function.
We did not estimate the optimal dosing regimens for infections other than PCP. In the literature target concentrations have been suggested for infections caused by other pathogens; however, these target concentrations are based predominantly on in vitro studies and are not clinically validated. 8 For PCP, the evidence for the target range is also limited to small studies, therefore, larger studies are needed to establish the optimal concentrations for effectiveness. To further optimize the dosing regimens of cotrimoxazole, additional research is needed to investigate its PK/PD, PK/toxicity, post‐antibiotic effect, and tissue penetration. In a murine thigh model, AUC/MIC was found to best predict the effectiveness for MRSA infections. 9 However, also, T > MIC, C max > MIC and species‐dependent associations have been suggested. 9 , 25 , 26 , 27 Based on the specific association for a certain pathogen, alternative dosing regimens could be explored. For instance, beta‐lactam antibiotics are often administered via continuous infusion because T > MIC best predicts their effectiveness. If T > MIC is also a predictor of cotrimoxazole's effectiveness, shortening the dose interval or using continuous infusion might be beneficial. 28 If C max best predicts the effectiveness and there is sufficient post‐antibiotic effect, high doses with a prolonged dosing interval might be more effective. Furthermore, because of the high tissue penetration of both trimethoprim and sulfamethoxazole the target site concentrations might be higher compared with plasma concentrations and require alternative dosing regimens based on infection site. 19
This study has some limitations. First, we utilized retrospectively collected routine TDM data to build the model, which unavoidably introduced some uncertainties regarding exact dosing and sampling times. In addition, the number of samples per patient was limited. However, the chosen type of analysis, nonlinear mixed‐effects modeling, can still provide reliable pharmacokinetic parameter estimates even in sparse datasets. Secondly, renal function was estimated using the CKD‐EPI formula to calculate the eGFR. The CKD‐EPI formula is not ideal for assessing glomerular filtration in ICU patients. Furthermore, trimethoprim interferes with tubular creatinine secretion resulting in an approximate 10–30% elevation in serum creatinine concentrations and thereby falsely underestimated eGFR. 29 To address this issue, future studies should focus on alternative measures of GFR, such as cystatin C or iohexol clearance. 30 , 31 Third, because of the limited number of patients treated with CRRT, we were not able to investigate the influence of different CRRT regimens and settings on the pharmacokinetics. Lastly, we lacked information about N‐acetyltransferase or CYP2C19 genotypes, as well as urine pH, all of which are factors previously associated with the pharmacokinetics of cotrimoxazole.
CONCLUSION
Sulfamethoxazole and trimethoprim pharmacokinetics show large IIV in hospitalized patients, and are affected by eGFR and CRRT. The current recommended dose for PCP treatment leads to adequate exposure in general, but there is a risk of both under and over‐exposure. Dose reductions for patients with an eGFR <30 mL/minute/1.73 m2 are suggested to reach similar exposure compared with patients without renal insufficiency. The risk of concentrations above the target range increases in patients with lower eGFR and high cotrimoxazole doses. Notably, in patients treated with CRRT, sulfamethoxazole clearance is increased. Therefore, doubling the sulfamethoxazole dose is needed to reach similar exposure to patients without renal failure. Monitoring with TDM is necessary in patients treated with CRRT as high doses might lead to N‐acetyl sulfamethoxazole accumulation.
FUNDING
No funding was received for this work.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
E.L., L.B., E.W., L.V., D.T., B.d.W., M.d.B., J.v.P., C.v.d.B., J.v.P., T.v.G., and D.M. wrote the manuscript. E.L., L.B., T.v.G., D.T., D.M., and B.d.W. designed the research. L.B. and E.L. performed the research. E.L., L.B., and D.M. analyzed the data.
Supporting information
Data S1.
References
- 1. Kemnic, T.R. & Coleman, M. Trimethoprim Sulfamethoxazole. Treasure Island (FL) ineligible companies. Disclosure: Meghan Coleman declares no relevant financial relationships with ineligible companies. StatPearls Publishing Copyright © 2023, StatPearls Publishing LLC (2023).
- 2. Viaggi, B. et al. Tissue penetration of antimicrobials in intensive care unit patients: a systematic review‐part II. Antibiotics 11, 1164 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bushby, S.R. & Hitchings, G.H. Trimethoprim, a sulphonamide potentiator. Br. J. Pharmacol. Chemother. 33, 72–90 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sigel, C.W. , Kunin, C.M. , Grace, M.E. & Nichol, C.A. Metabolism of trimethoprim in man and measurement of a new metabolite: a new fluorescence assay. J. Infect. Dis. 128(Supplement_3), S580–S583 (1973). [DOI] [PubMed] [Google Scholar]
- 5. van der Ven, A.J. , Mantel, M.A. , Vree, T.B. , Koopmans, P.P. & van der Meer, J.W. Formation and elimination of sulphamethoxazole hydroxylamine after oral administration of sulphamethoxazole. Br. J. Clin. Pharmacol. 38, 147–150 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bergan, T. & Brodwall, E.K. The pharmacokinetic profile of co‐trimoxazole. Scand. J. Infect. Dis. Suppl. 8, 42–49 (1976). [PubMed] [Google Scholar]
- 7. Siber, G.R. , Gorham, C.C. , Ericson, J.F. & Smith, A.L. Pharmacokinetics of intravenous trimethoprim‐sulfamethoxazole in children and adults with normal and impaired renal function. Rev. Infect. Dis. 4, 566–578 (1982). [DOI] [PubMed] [Google Scholar]
- 8. Brown, G.R. Cotrimoxazole—optimal dosing in the critically ill. Ann. Intensive Care 4, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hagihara, M. et al. The first report on pharmacokinetic/pharmacodynamic study of trimethoprim/sulfamethoxazole against Staphylococcus aureus with a neutropenic murine thigh infection model. Chemotherapy 64, 224–232 (2019). [DOI] [PubMed] [Google Scholar]
- 10. Hughes, W.T. , Feldman, S. , Chaudhary, S.C. , Ossi, M.J. , Cox, F. & Sanyal, S.K. Comparison of pentamidine isethionate and trimethoprim‐sulfamethoxazole in the treatment of pneumocystis carinii pneumonia. J. Pediatr. 92, 285–291 (1978). [DOI] [PubMed] [Google Scholar]
- 11. Lau, W.K. & Young, L.S. Trimethoprim‐sulfamethoxazole treatment of pneumocystis carinii pneumonia in adults. N. Engl. J. Med. 295, 716–718 (1976). [DOI] [PubMed] [Google Scholar]
- 12. Stevens, R.C. , Laizure, S.C. , Williams, C.L. & Stein, D.S. Pharmacokinetics and adverse effects of 20‐mg/kg/day trimethoprim and 100‐mg/kg/day sulfamethoxazole in healthy adult subjects. Antimicrob. Agents Chemother. 35, 1884–1890 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee, B.L. , Medina, I. , Benowitz, N.L. , Jacob, P. III , Wofsy, C.B. & Mills, J. Dapsone, trimethoprim, and sulfamethoxazole plasma levels during treatment of pneumocystis pneumonia in patients with the acquired immunodeficiency syndrome (AIDS). Evidence of drug interactions. Ann. Intern. Med. 110, 606–611 (1989). [DOI] [PubMed] [Google Scholar]
- 14. Bowden, F.J. , Harman, P.J. & Lucas, C.R. Serum trimethoprim and sulphamethoxazole levels in AIDS. Lancet 1, 853 (1986). [DOI] [PubMed] [Google Scholar]
- 15. Chin, T.W. , Vandenbroucke, A. & Fong, I.W. Pharmacokinetics of trimethoprim‐sulfamethoxazole in critically ill and non‐critically ill AIDS patients. Antimicrob. Agents Chemother. 39, 28–33 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baethke, R. , Golde, G. & Gahl, G. Sulphamethoxazole/trimethoprim: pharmacokinetic studies in patients with chronic renal failure. Eur. J. Clin. Pharmacol. 4, 233–240 (1972). [Google Scholar]
- 17. Curkovic, I. , Lüthi, B. , Franzen, D. , Ceschi, A. , Rudiger, A. & Corti, N. Trimethoprim/sulfamethoxazole pharmacokinetics in two patients undergoing continuous Venovenous Hemodiafiltration. Ann. Pharmacother. 44, 1669–1672 (2010). [DOI] [PubMed] [Google Scholar]
- 18. Dijkstra, J.A. , Alsaad, N.S. , Hateren, K. , Greijdanus, B. , Touw, D.J. & Alffenaar, J.W. Quantification of co‐trimoxazole in serum and plasma using MS/MS. Bioanalysis 7, 2741–2749 (2015). [DOI] [PubMed] [Google Scholar]
- 19. Grayson, M.L. Kucers' the Use of Antibiotics: A Clinical Review of Antibacterial, Antifungal, Antiparasitic and Antiviral Drugs 6th edn. (Hodder Arnold London, London, 2010). [Google Scholar]
- 20. Hess, M.M. et al. Trimethoprim‐sulfamethoxazole pharmacokinetics in trauma patients. Pharmacotherapy 13, 602–606 (1993). [PubMed] [Google Scholar]
- 21. Grose, W.E. , Bodey, G.P. & Loo, T.L. Clinical pharmacology of intravenously administered trimethoprim‐sulfamethoxazole. Antimicrob. Agents Chemother. 15, 447–451 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klepser, M.E. et al. Oral absorption of trimethoprim‐sulfamethoxazole in patients with AIDS. Pharmacotherapy 16, 656–662 (1996). [PubMed] [Google Scholar]
- 23. Clajus, C. et al. Cotrimoxazole plasma levels, dialyzer clearance and total removal by extended dialysis in a patient with acute kidney injury: risk of under‐dosing using current dosing recommendations. BMC Pharmacol. Toxicol. 14, 19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reeves, D.S. , Bint, A.J. & Bullock, D.W. Use of antibiotics. Sulphonamides, co‐trimoxazole, and tetracyclines. Br. Med. J. 2, 410–413 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burgess, D.S. , Frei, C.R. , Lewis Ii, J.S. , Fiebelkorn, K.R. & Jorgensen, J.H. The contribution of pharmacokinetic‐pharmacodynamic modelling with Monte Carlo simulation to the development of susceptibility breakpoints for Neisseria meningitidis . Clin. Microbiol. Infect. 13, 33–39 (2007). [DOI] [PubMed] [Google Scholar]
- 26. Close, S.J. , McBurney, C.R. , Garvin, C.G. , Chen, D.C. & Martin, S.J. Trimethoprim‐sulfamethoxazole activity and pharmacodynamics against glycopeptide‐intermediate Staphylococcus aureus . Pharmacotherapy 22, 983–989 (2002). [DOI] [PubMed] [Google Scholar]
- 27. Cheng, A.C. et al. Dosing regimens of cotrimoxazole (trimethoprim‐sulfamethoxazole) for melioidosis. Antimicrob. Agents Chemother. 53, 4193–4199 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morgan, D.J. & Raymond, K. Evaluation of slow infusions of co‐trimoxazole by using predictive pharmacokinetics. Antimicrob. Agents Chemother. 17, 132–137 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delanaye, P. , Mariat, C. , Cavalier, E. , Maillard, N. , Krzesinski, J.‐M. & White, C.A. Trimethoprim, creatinine and creatinine‐based equations. Nephron Clin. Pract. 119, c187–c194 (2011). [DOI] [PubMed] [Google Scholar]
- 30. Delanaye, P. et al. Iohexol plasma clearance for measuring glomerular filtration rate in clinical practice and research: a review. Part 2: why to measure glomerular filtration rate with iohexol? Clin. Kidney J. 9, 700–704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inker, L.A. et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N. Engl. J. Med. 367, 20–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.