

Abstract
The ability to undergo self-renewal is a defining characteristic of stem cells. Self-replenishing activity sustains tissue homeostasis and regeneration. In addition, stem cell therapy strategies require a heightened understanding of the basis of the self-renewal process to enable researchers and clinicians to obtain sufficient numbers of undifferentiated stem cells for cell and gene therapy. Here, we used postnatal muscle-derived stem cells to test the basic biological assumption of unlimited stem cell replication. Muscle-derived stem cells (MDSCs) expanded for 300 population doublings (PDs) showed no indication of replicative senescence. MDSCs preserved their phenotype (ScaI+/CD34+/desminlow) for 200 PDs and were capable of serial transplantation into the skeletal muscle of mdx mice, which model Duchenne muscular dystrophy. MDSCs expanded to this level exhibited high skeletal muscle regeneration comparable with that exhibited by minimally expanded cells. Expansion beyond 200 PDs resulted in lower muscle regeneration, loss of CD34 expression, loss of myogenic activity, and increased growth on soft agar, suggestive of inevitable cell aging attributable to expansion and possible transformation of the MDSCs. Although these results raise questions as to whether cellular transformations derive from cell culturing or provide evidence of cancer stem cells, they establish the remarkable long-term self-renewal and regeneration capacity of postnatal MDSCs.
INTRODUCTION
The majority of stem cell research efforts are focused on the promising applications of multilineage plasticity of putative stem cells. The equally important characteristic of self-renewal capacity is often overlooked but is increasingly becoming a focus of stem cell biology (Molofsky et al., 2004; Zipori, 2004) from both basic science and clinical perspectives. The potential use of stem cells for gene therapy and functional tissue engineering will require the expansion of undifferentiated stem cell populations to achieve sufficient numbers for use in clinical applications. It will be necessary to develop cellular expansion techniques that regulate the extrinsic and intrinsic biostimuli that lead to self-renewal and differentiation, or symmetric and asymmetric divisions. In addition, practical scalable culture systems with corresponding models for prediction of output numbers will be required. Finally, the need to pay careful attention to any changes in cell quality attributable to extensive expansion or, more generally, to cellular aging adds another layer of complexity to the development of efficient expansion techniques.
Hayflick first reported in 1961 that normal human fibroblasts undergo a limited number of divisions in vitro (Hayflick and Moorhead, 1961); he estimated the maximum number of population doublings (PDs) to be ∼50. After that initial report, many other investigators have verified that euploid cells in culture undergo a finite number of PDs. In contrast, the current consensus is that stem cells possess a unique proliferation capacity that is either several times greater than that displayed by euploid cells, or possibly unlimited (Potten and Morris, 1988; Rubin, 1997; Reyes et al., 2001; Rubin, 2002).
The capacity for long-term proliferation is regarded as an identifying characteristic of stem cells. Reports of stem cell replicative potentials show variability related to the developmental stage of the cell source, the species, and the culturing conditions. Human embryonic stem (ES) cells expanded for 250 PDs (Amit et al., 2000) or 130 PDs (Xu et al., 2001) have maintained their pluripotency and normal karyo-type. However, due both to ethical issues surrounding the use of embryonic cells and to reports that the cells form teratomas in vivo, research investigating stem cells derived postnatally has grown. Nonembryonic stem cells, such as human hematopoietic stem cells (HSCs), seem to exhibit a slower doubling rate than ES cells, although researchers have expanded some of these cell populations 2 × 106-fold (20 PDs) over a 6-mo period (Piacibello et al., 1997) and ∼1400-fold (10 PDs) over a 3-mo period (Gilmore et al., 2000). Varying degrees of expansion are reported for mesenchymal stem cells (MSCs), bone marrow stromal cells, and multipotent adult progenitor cells—all originating from the bone marrow—109-fold (30 PDs) (Colter et al., 2000), 50–70 PDs (Bianchi et al., 2003), and 120 PDs (Schwartz et al., 2002; Jiang et al., 2003), respectively.
In light of these findings, the generation of large numbers of cells via the expansion of stem cells seems feasible. However, numerous studies performed in the area of cellular aging have shown that cells with a high replicative age often exhibit chromosomal abnormalities. Incomplete DNA replication at the telomeres (i.e., the G-rich repeat units at the ends of chromosomes) leads to telomere shortening with each cell cycle; cellular senescence occurs after the telomeres reach a critical length. The serial propagation or aging of cells in culture also leads to other degenerative cellular changes, including abnormal structures in the cytoplasm, changes in metabolism, loss of methyl groups, and reiterated sequences from DNA, and reduction in replicative efficiency and growth rate (Rubin, 1997). In addition to the concern that such expanded and aged populations might have a reduced regeneration capacity, the accumulation of damage over the cells' lifetimes increases the probability of genetic instability that can culminate in apoptosis or tumorigenic behavior.
Although extensively expanded stem cells may become targets of transformation due to accumulated damage, there may be a preexisting relationship to cancer. The self-renewal pathway may share some signals with pathways implicated in oncogenesis. Recent research has shown that the PTEN, notch, sonic hedgehog, and wnt signaling pathways may play a role in the self-renewal of HSCs (Karanu et al., 2000; Varnum-Finney et al., 2000; Bhardwaj et al., 2001; Reya et al., 2001; van Noort and Clevers, 2002; Stiles et al., 2004); these same pathways have been linked to oncogenicity. Furthermore, HSCs are thought to be more likely than committed progenitors to undergo transformation (Reya et al., 2001). Together, these findings clearly indicate the need to carefully examine the quality of expanded stem cell populations before using them as the basis for cell therapy and tissue engineering applications.
To test the long-term self-renewal potential of muscle-derived stem cells (MDSCs) and to identify any limits to the expansion of an MDSC population, we extensively expanded muscle stem cells for >300 PDs under normal culture conditions (225 d of expansion) and examined the cells to identify any in vitro phenotypic alterations, signs indicative of cellular aging and transformation, changes in the cells' capacity to differentiate, and changes in their capacity to regenerate skeletal muscle in vivo.
MATERIALS AND METHODS
Proliferation Kinetics and Expansion of MDSCs in Culture
MDSCs were obtained, as described previously, from normal (C57BL/6J) 3-wk-old mice (Qu-Petersen et al., 2002). Cells were cultured in DME medium supplemented with 10% fetal bovine serum, 10% horse serum (HS), 1% penicillin/streptomycin, and 0.5% chick embryo extract (Invitrogen, Carlsbad, CA) at an initial seeding density of 225 cells/cm2. After a sufficient number of cells was obtained (∼3–4 wk after isolation), 5600 cells (225 cells/cm2) were plated in 25-cm2 collagen-coated flasks, and routine cell passaging was performed every 2 or 3 d. After 2 or 3 d of growth, the cells were trypsinized, counted, and replated at a density of 225 cells/cm2. This process was repeated for a 6-mo period.
The cell counts and cellular dilution factor were recorded at each passage and used to calculate the expansion potential or theoretical yield, number of population doublings, and doubling time. The number of population doublings was calculated by solving the exponential equation Ni = N02(ti/PDT) for ti /PDT, which is the number of population doublings at time ti or no. of PDs = log2(Ni /No). Daily population doubling times were calculated using the following equation:
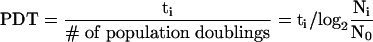 |
Local moving average PDT was calculated by fitting an exponential trend line to several measurements of N over a 1-wk period. The regression method provides a fitted curve of the form Ni = N0ekti. Because PDT is defined as the time, ti, such that Ni = 2No, k = ln2/PDT and PDT = ln2/k.
Cellular division time (DT) was measured directly by visual observation of time-lapsed images acquired at 10-min intervals over a 4-d period (Deasy et al., 2003). The mitotic fraction, or the fraction of daughter cells that are dividing (α or FD), was calculated by using the measured PDT and DT at the given doubling level and solving the rearranged Sherley model for α (using Mathematica 4.2; Wolfram Research, Champaign, IL) as described previously (Sherley et al., 1995; Deasy et al., 2003):
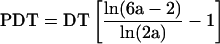 |
Morphological Analysis
Phase contrast light microscopy images were acquired every 2 wk in three random fields. Northern Eclipse software package was used to collect morphometric data by outlining or tracing 20–30 cells per image or time point, with five to eight time points per doubling grouping. Quantitative measurements for cell area (number of pixels calibrated to microns squared), diameter (maximal cell length in micrometers), cell elongation (ratio of major axis to minor axis), and roundness (perimeter2/4 * π * area) were compared using nonparametric one-way analysis of variance (ANOVA) and Dunn's method for multiple comparisons.
Sca-1 and CD34 Expression by Flow Cytometry
MDSCs were labeled with rat anti-mouse Sca-1 (phycoerythrin; BD Biosciences PharMingen, San Diego, CA) and CD34 (biotin; BD Biosciences PharMingen) monoclonal antibodies at 2-wk intervals. A separate portion of cells was treated with equivalent amounts of isotype control antibodies. Both fractions then were washed and labeled with streptavidin-allophycocyanin. 7-Amino-actinomycin D was added to exclude nonviable cells from the analysis. Appropriate gating was performed to determine Sca-1 and CD34 expression via flow cytometry with a FACStar Plus (BD Biosciences, San Jose, CA). One-way ANOVA was used to test for differences at various doubling levels; pairwise multiple comparisons to identify differences in expression were performed using the Tukey test with α = 0.05 and power = 0.997.
Myogenic Marker Expression by Immunocytochemistry
Expression of the myogenic marker desmin was examined at 2-wk intervals via immunocytochemistry. In brief, after cold methanol fixation the cells were blocked in 5% HS, and then they were incubated with antibodies for desmin (mouse monoclonal desmin, 1:250; Sigma-Aldrich, St. Louis, MO), secondary biotinylated IgG (1:250; Vector Laboratories, Burlingame, CA), and streptavidin-Cy3 (1:500; Sigma-Aldrich) to fluorescently label the antigenic binding and to determine the percentage of myogenic cells within the population. After ANOVA, pairwise multiple comparisons to identify differences in desmin expression were performed using the Tukey test with α = 0.05 and power = 0.87.
In Vitro Myogenic Differentiation
MDSCs were plated at 1000 cells/cm2 in normal 20% serum DME medium for 3 d, and then they were placed in 2% serum DME medium for an additional 4 d to induce myogenic differentiation. On day 7, methanol-fixed cultures were blocked with 5% HS and were incubated with monoclonal mouse fast myosin heavy chain (anti-MyHC, 1:250; Sigma-Aldrich), biotinylated IgG (1:250; Vector Laboratories), and streptavidin-Cy3 (1:500). Differentiation efficiency was calculated as the ratio of myogenic nuclei to total nuclei. ANOVA and pairwise multiple comparisons to identify differences in differentiation were performed using the Tukey test with α = 0.05 and power = 0.848.
In Vivo Self-Renewal
MDSCs were retrovirally transduced to express green fluorescent protein (GFP) and neomycin resistance genes. The labeled cells (30–40 PDs) were transplanted into the gastrocnemius muscles of dystrophic primary recipient mice (n = 2). Each transplantation involved 0.2–1.0 × 106 donor cells. After 2 wk in vivo in the primary recipient, the cells were reharvested, selected in medium containing G418 (3 mg of active compound per milliliter of Geneticin, an analogue of neomycin; Invitrogen), and expanded for <3 wk. The MDSCs were subsequently transplanted into skeletal muscles of secondary recipients (n = 2). Reisolation of the serially transplanted MDSCs from secondary recipients was performed as described above. Although these experiments were semiquantitative in nature, we estimated that the percentage of GFP-positive cells in the primary muscle biopsy was 0.5–1% of the skeletal muscle biopsy (gastrocnemius) and isolated as obtained through the preplate technique. The GFP cells were recovered from preplates 1, 2, and 3. These cells were tested for GFP expression and the ability to differentiate into myotubes in vitro. The contralateral muscles of both the primary and secondary recipients were harvested, sectioned, and examined for GFP expression.
In Vivo Muscle Regeneration
Regeneration efficiency was examined as a function of cell age by transplantation of 1–3 × 105 cells into the gastrocnemius muscles of 5- to 8-wk-old mdx/SCID mice as described previously (Jankowski et al., 2002). Two weeks after transplantation, the mice were killed and the muscles were sectioned by cryostat (10 μm). Immunohistochemistry was performed to identify dystrophin-positive myofibers. Tissues sections were fixed with cold methanol, and immunostaining was performed using the mouse-on-mouse (M.O.M.) kit (Vector Laboratories) with DYS2 antibody (1:50; Novocastra, New Castle, United Kingdom). Populations of various doubling levels were grouped into six categories (0–50 PDs, 51–100 PDs, 101–150 PDs, 151–200 PDs, 201–300 PDs, and >300 PDs) and were compared with ANOVA. Pairwise multiple comparisons were performed using the Tukey test with α = 0.05 and power =1.0.
Transformation Analysis
Soft agar growth in populations from six doubling levels was examined by plating 2000 cells per 9.6-cm2 well in 0.35% low melting point agar as a top layer, with a bottom layer of 0.7% agar in 20% serum medium. Colonies were allowed to grow for 21 d at 37°C, at which time Northern Eclipse imaging software was used to score the colonies for both number and size. After ANOVA, pairwise multiple comparisons of the number of colonies were performed using the Tukey test with α = 0.05 and power = 1.0.
DNA content was examined in the same populations via flow cytometry. Cells were fixed in ethanol for 2 h and then resuspended in 0.1% (vol/vol) Triton X-100 (Sigma-Aldrich) in phosphate-buffered saline with 0.2 mg/ml DNase-free RNase A (Sigma-Aldrich) and 20 ng/ml propidium iodide (Sigma-Aldrich) solution. Experiments were performed three times, and results were averaged. One-way ANOVA was used to identify differences among the doubling levels for the 2N, 4N, and >4N peaks. Numerical chromosomal analysis was performed through the use of standard Geimsa staining and banding techniques as described previously (Lee et al., 2000). To test for the transformation of the highly expanded cells, subcutaneous injections into both SCID and syngeneic mice were performed. MDSCs that had been expanded to 15 PDs and MDSCs that had been expanded to 300 PDs were transplanted subcutaneously into the lower abdomen of both C57BL/6J-Prkdc SCID mice (0.3–0.4 × 106 cells per site; n = 4) and C57/BL6J (0.75–1.0 × 106 cells per site; n = 4). Tumor growth was followed by palpation and radiography, and mice were killed 130 d (SCID) and 150 d (syngeneic) after injection. Killed animals were dissected and were evaluated for growths at the site of injection and gross enlargement of spleen and lymph tissues. Hematoxylin and eosin staining was performed on 8-μm–thick sections of snap-frozen tissue.
RESULTS
To examine the effects of long-term expansion, we isolated highly purified populations of MDSCs from 3-wk-old normal mice (C57BL/6J), as described previously (Qu-Petersen et al., 2002) and maintained the cells in continuous culture for 6 mo. We cultured MDSCs at <30% visual confluence or a density of 2 × 103–104 (Figure 1A), and periodically analyzed the stem cell characteristics of MDSCs at different doubling levels.
Figure 1.

MDSC expansion. (A) Schematic representation of cell expansion method. Low growth density was maintained by routine passaging every 2 to 3 d. (B) MDSCs in culture were expanded for >300 PDs over a 6-mo period. (C) Closer examination of the cells during the first 6 wk (42 d) showed very slow growth. After ∼4 wk, the number of PDs (blue ▪) was ∼12 and the PDT (red ♦) had dropped to 52 h. Before the 4-wk time point, the average PDT was much longer than 52 h and is not shown on the scale of this graph. After this period, cell death seemed to subside; the PDT at 6 wk was 42 h and the steadily increasing number of PDs had reached ∼22 PDs. (D) Shown are the moving local average PDT (blue ▪, smoothed line) and the individual PDT measurements taken every 2 or 3 d (green ♦). (E) Cellular DT did not change significantly during the expansion. Median division times (blue ▪) are shown for MDSCs expanded for 15 PDs (13 h), 25 PDs (12 h), 75 PDs (14 h), 125 PDs (14 h), 170 PDs (14 h), and 300 PDs (14 h). Mitotic fractions (i.e., the fractions of daughter cells that were actively dividing) (red ♦) for MDSCs expanded for 15 PDs (0.37), 25 PDs (0.46), 75 PDs (0.75), 125 PDs (0.98), 170 PDs (0.88), and 300 PDs (0.83).
Expansion Potential of MDSCs
We were able to expand the MDSCs for >300 PDs (n = 3; Figure 1B) over a 6-mo period and obtained a theoretical yield of >10100 cells (>1 googol cells). We expanded a total of four different populations to a high doubling number: mdsc1 (300 PDs; n = 3), mdsc2 (300 PDs; n = 2), mdsc3 (200 PDs; n = 1), and mdsc4 (200 PDs; n = 2). We focused our study on the population that displayed the best engraftment efficiency in skeletal muscle: the mdsc1 population (hereafter referred to simply as “MDSCs”).
It is typical to observe very long doubling times early in the isolation process. Because cell death is common in cell populations obtained immediately after isolation, the population size actually tends to decrease during this initial period. For this reason, obtaining workable quantities of MDSCs can require several weeks, often leaving freshly isolated MDSCs in the same isolation flask for up to 2 wk. Measurements of doubling rate during the early isolation period are based on visual counts rather than hematocytometer counts.
We observed this expected lag in population growth during the first 4–6 wk after cell isolation (Figure 1C). Initially, the PDT was infinitely long, but by the 4-wk time point (28 d, as shown in the figure) the PDT had dropped sharply to ∼52 h, whereas the number of PDs increased concurrently (Figure 1C). After the lag phase, we observed no significant differences (p = 0.11) between the growth rates of cells in culture for 2 d and those in culture for 3 d (schematic shown in Figure 1A). Two-day growth periods resulted in an average of 3 PDs (1.5 ± 0.35 PDs/d), whereas 3-d growth periods resulted in an average of ∼5 PDs (1.6 ± 0.39 PDs/d). The population kinetics of the MDSCs cultured continuously for 6 mo indicate that the cells had an extended replicative lifetime (Figure 1, B and D). The PDT after the lag phase dropped sharply to ∼14–16 h, and then it remained relatively constant for the remainder of the expansion in culture.
We observed no indication of replicative senescence (Figure 1D) or significant changes in cellular DT during expansion of the MDSCs. We used a unique combinatorial cell culture system (Deasy et al., 2003) to obtain time-lapsed images to directly observe and measure median division time, which was similar for cells at 15 PDs (12 h), 25 PDs (12 h), 75 PDs (12 h), 125 PDs (14 h), 170 PDs (13 h), and 300 PDs (12.5 h; Figure 1E). We used the Sherley model (Sherley et al., 1995) to calculate the mitotic fraction (i.e., the fraction of actively dividing daughter cells), which increased from 0.37—shortly after cell isolation (15 PDs) and essentially during the isolation process—to 0.75–0.98 after 75 PDs (Figure 1E).
Morphological Analysis
Qualitative examination of cell morphology showed that the MDSC populations from early passages seemed to be similar to those from late passages. Figure 2 illustrates the cell density at which these cells were grown and the heterogeneous morphology of the cell populations. Most of the cells maintained a small round shape, although we did observe some well defined subpopulations, including a group of cells with a more spindle-shaped appearance (Figure 2A).
Figure 2.

MDSC morphology. (A) Images illustrate MDSC morphology and the range of cellular densities at particular expansion levels (PDs) throughout the 6-mo period of in vitro expansion (100×; bar, 250 μm) (B) Morphological cellular features of MDSC populations during expansion were measured from phase contrast images. Box plots show the median, 10th, 25th, 75th, and 90th percentiles as vertical boxes with error bars. Highly expanded populations had a significantly larger area and diameter and were less round and more elongated (p < 0.05). Median cross-sectional area is as follows: 0–50 PDs (610 μm2), 51–100 (690 μm2), 101–150 (680 μm2), 151–200 (644 μm2), 201–300 (1230 μm2), and >300 (1260 μm2). Median diameter is as follows: 0–50 PDs (39 μm), 51–100 (39 μm), 101–150 (36 μm2), 151–200 (40 μm), 201–300 (53 μm), and >300 (83 μm). Roundness values are as follows: 0–50 PDs (0.67), 51–100 (0.70), 101–150 (0.78), 151–200 (0.69), 201–300 (0.72), and >300 (0.45). Elongation values are as follows: 0–50 PDs (1.56), 51–100 (1.55), 101–150 (1.35), 151–200 (1.47), 201–300 (1.52), and >300 (3.06).
Morphological cellular features (cell area, diameter, and cell elongation and roundness) of MDSC populations were measured from phase contrast images. Figure 2B illustrates distribution of the measurements through the use of box plots with the median, 10th, 25th, 75th, and 90th percentiles as vertical boxes with error bars. The median cross-sectional area and diameter of cells expanded beyond 200 PDs were significantly larger than the area and diameter of cells in the other groups (p < 0.05; ANOVA). The roundness parameter is a measure of the shape of the cell, with values closest to 1 indicating the roundest cells. The elongation parameter is the ratio of the length of the major axis of the cell to that of its minor axis. The roundness and elongation measurements of MDSCs expanded beyond 300 PDs differed significantly from those obtained for all MDSCs expanded to a lesser degree; MDSCs expanded beyond 300 PDs were less round and more elongated. Cell populations expanded for 100–150 PDs were significantly rounder than the MDSCs in the other groups, although this difference was not qualitatively recognizable.
Stem Cell Marker Analysis
The use of flow cytometry to analyze expression of the putative muscle-derived stem cell markers CD34 and Sca-1 revealed that the MDSCs maintained the same general profile for the first 200 PDs. In MDSC populations between 0 and 50 PDs, 90 ± 13% of cells were positive for CD34 expression and 78 ± 15% were positive for Sca-1 expression. We observed no significant difference in the total expression of Sca-1 by MDSC populations expanded for various PDs up to 300 PDs (p = 0.10; one-way ANOVA). However, we observed a significant decrease in total CD34 expression after 200 PDs (65 ± 22% of MDSCs at 200 PDs vs. 94.7 ± 7% at 150–200 PDs; p <0.05) and after 300 PDs (36 ± 22%; p <0.05 vs. all other doubling levels; Figure 3A). Shifts in CD34 expression are apparent in the dot plots, but Sca-1 expression seems to be constant (Figure 3B). Stage-specific embryonic antigen 1 also was analyzed by flow cytometry, although MDSCs were negative for this marker throughout the expansion period (our unpublished data).
Figure 3.

Stem cell marker expression. (A) CD34 and Sca-1 immunophenotyping after expansion. Total percentage of cells expressing CD34 is as follows: 0–50 PDs (85 ± 18; n = 9), 51–100 (81 ± 22; n = 12), 101–150 (78 ± 21%; n = 11), 151–200 (94 ± 7; n = 8), 201–300 (65 ± 22; n = 10), and >300 (37 ± 22; n = 5). Total percentage of cells expressing Sca-1 is as follows: 0–50 PDs (78 ± 15; n = 9), 51–100 (84 ± 12; n = 12), 101–150 (83 ± 9; n = 11), 151–200 (71 ± 17; n = 8), 201–300 (71 ± 23; n = 10), and >300 (76 ± 18; n = 5). The double-positive cell fraction (CD34+/Sca-1+) remained large for the first 200 PDs but significantly decreased in size after 200 PDs. There was no significant difference in the double-negative cell fraction (CD34–/Sca-1–) over time (ANOVA). (B) Flow cytometry dot plots from one representative expansion for 12 different time points throughout the expansion process.
Myogenic Behavior
Myogenic Markers. Less than 20% of the MDSC population expressed the myogenic marker desmin upon analysis shortly after isolation (20 PDs), and the early MDSC population was negative for Pax-7 and m-cadherin expression, two markers of satellite cells (Irintchev et al., 1994; Seale et al., 2000). After expansion, we observed a slight increase in desmin expression by MDSCs expanded beyond 200 PDs (19 ± 9%) compared with desmin expression by MDSCs evaluated at 61–200 PDs (range 4.5–7%; p < 0.05); however, there was no significant difference between desmin expression by MDSCs at 200 PDs, and desmin expression by MDSCs expanded for <60 PDs (7 ± 5%; Figure 4A). Immunocytochemical staining illustrates that only a few cells expressing desmin were present among a much larger number of desmin-negative cells (Figure 4B). MDSCs did not express Pax-7 or m-cadherin at any point during the expansion period (our unpublished data).
Figure 4.

Immunochemical analysis of myogenic markers and differentiation. (A) Desmin expression by MDSCs expanded for various numbers of PDs (mean ± SD) is as follows: 0–30 PDs (6.5 ± 2%; n = 3), 31–60 (7.4 ± 7%; n = 4), 61–100 (4.5 ± 7%; n = 8), 101–150 (4.8 ± 5%; n = 5), 151–200 (6.8 ± 4%; n = 6), and 201–300 (19.2 ± 9%; n = 6). (B) Immunochemical staining of MDSCs at the different expansion levels. Desmin, red, Hoechst nuclei, blue (200×). (C) In vitro myogenic differentiation values (as percent myosin heavy chain positive) are as follows: 0–50 PDs (65 ± 4%; n = 4), 51–100 (44 ± 26%; n = 5), 101–150 (40 ± 20%; n = 5), 151–200 (40 ± 7%; n = 5), 201–300 (24 ± 15%; n = 4), and >300 (15 ± 8%; n = 4). ANOVA revealed a significant decrease between both the 201–300 PD time point and the >300 PDs time point compared with all earlier time points (ANOVA; p < 0.05). (D) Immunocytochemical staining for myogenic differentiation into myotubes expressing myosin heavy chain after incubation for 7 d in differentiation-inducing conditions. Myosin heavy chain, red; Hoechst nuclei, blue (200×).
Myogenic Differentiation. The extent of myogenic differentiation, i.e., myotube formation, declined with increased passaging of MDSCs. Cells at low doublings (<30 PDs) exhibited a high degree of differentiation, as indicated by the formation of multinucleated myotubes with 65% of the nuclei positive for myosin heavy chain (fast MyHC) after culturing the MDSCs under low-serum and high-density conditions for 7 d (Figure 4C). Generally, the MDSCs began to fuse on day 3 or 4 under such differentiation-inducing conditions, before reaching 100% confluence in culture. A significantly smaller percentage of MDSCs underwent fusion or differentiation to form myotubes on day 7 at the 200 PD level (24 ± 15%; p < 0.05) and the 300 PD level (15 ± 8%; p < 0.05; Figure 4D) than at the <50 PD level (65 ± 4%).
In Vivo Self-Renewal
The ability of MDSCs to undergo extensive proliferation while preserving their marker profile for at least 200 PDs is indicative of the high self-renewal ability of these cells in vitro. To examine in vivo self-renewal, we used the classic approach of serial transplantation. We were able to reisolate MDSCs that were previously genetically engineered to express neomycin-resistance gene and GFP and were transplanted into the skeletal muscles of dystrophic mice 14 d earlier. We selected the recovered cells in vitro and expanded the cell population for use in a second transplantation. These MDSCs regenerated GFP-positive myofibers within the secondary recipients (Figure 5, GFP-positive fibers), could be reharvested from the secondary recipients (Figure 5, GFP-positive cells in G418 sulfate), and demonstrated the ability to undergo myogenic differentiation in vitro (Figure 5, GFP-positive myotubes). These findings suggest that MDSCs are capable of self-renewal in vivo.
Figure 5.

In vivo self-renewal. MDSCs were isolated and then were transduced with genes encoding for GFP and neomycin resistance. Two weeks after their injection, the labeled cells were reharvested, selected in G418 medium, and transplanted into secondary mdx recipients. Regeneration of muscle fibers was observed by green fluorescence, whereas contralateral muscles were used to reharvest MDSCs from the secondary recipients. Serially transplanted cells regenerated skeletal muscle fibers within the skeletal muscles of secondary recipients and differentiated into myotubes in vitro.
Muscle Regeneration
MDSCs at low passages maintained the ability to regenerate large numbers of dystrophin-positive fibers after transplantation into the gastrocnemius muscles of mdx mice. We analyzed regeneration capacity in terms of the transplanted cells' regeneration index (RI)—the number of dystrophin-positive fibers per 105 donor MDSCs. Fourteen days after transplantation, the average RI for MDSCs expanded for between 0 and 50 PDs was 829 ± 337 dystrophin-positive fibers. MDSCs maintained this high regeneration capacity for up to 195 PDs (RI = 800 ± 170; Figure 6A). Representative dystrophin engraftments are shown in Figure 6A. After 200 PDs, the engraftment efficiency decreased significantly; we observed a quantity of dystrophin-positive fibers comparable with the number of background revertant fibers (200–300 PDs, RI = 32 ± 47; >300 PDs, RI = 3 ± 3, p < 0.001; Figure 6D). Histological examination 14 d after transplantation of MDSCs expanded to 240 PDs revealed a large engraftment of mononuclear cells (Figure 6B). However, small numbers of dystrophin-positive fibers were visible at the graft site (Figure 6B). To confirm delivery of the MDSCs and identify the injection site, we used LacZ to retrovirally label MDSCs expanded for 310 PDs. We verified the site of cell delivery and observed <25 dystrophin-positive fibers in these regions (Figure 6C).
Figure 6.

In vivo regeneration efficiency. (A) Regeneration of dystrophin-positive fibers within the skeletal tissue of mdx mice (harvested at 14 d), as indicated by immunostaining (red), by cells at 45, 60, 140, and 195 PDs (50× magnification in background, 200× magnification in foreground). (B) Eosin staining revealed numerous cells in the host mdx muscle transplanted with MDSCs expanded for 240 PDs. Serial sections contained very few dystrophin-positive fibers (200×). (C) LacZ-positive cells (arrows) enabled confirmation of the cell injection site, which serial sections again revealed to be negative for dystrophin (red, Hoechst blue). Image contrast to reveal fibers was performed on serial sections (400×). (D) Dystrophin-expressing muscle fibers present after cell transplantation were scored as the regeneration index (i.e., the number of dystrophin-positive fibers per 100,000 donor cells). Cells expanded for up to 200 PDs engrafted at a level comparable with that exhibited by newly isolated cells; after this point, however, regeneration efficiency dropped significantly (ANOVA; p < 0.05): 0–50 PDs (829 ± 336; n = 4), 51–100 (610 ± 376; n = 3), 101–150 (457 ± 272; n = 6), 151–200 (800 ± 170; n = 4), 201–300 (32 ± 47; n = 4), and >300 (3 ± 2.8; n = 8).
Transformation Analysis
MDSCs at higher doublings demonstrated a heightened ability for anchorage-independent growth (Figure 7A) after 21 d in soft agar. We found that ∼2.3% of MDSCs at 25 PDs initiated colonies, which did not progress beyond the four- or eight-cell stage (∼30 μm in diameter) and that <1% formed colonies >60 μm in diameter (Figure 7B). Above 100 PDs, 12% of the plated cells formed colonies with diameters >30 μm. We detected several intriguing large colonies (>100 μm in diameter and comparable in size to the positive control, the rat 1A 412 transformed cell line) in the MDSCs expanded for 140 PDs (Figure 7B). No significant differences were detected among the five different PD groups expanded for <200 PDs (i.e., 0–30 PDs, 31–60 PDs, 61–100 PDs, 101–150 PDs, and 151–200 PDs; ANOVA; p > 0.05; Figure 7B). However, MDSCs expanded for 295 PDs formed significantly more colonies than did MDSCs expanded for <200 PDs (for MDSCs >295 PDs: 59% [590 ± 93 colonies per 1000 plated cells]; for MDSCs 0–200 PDs: <10%; p < 0.001).
Figure 7.

(A) Anchorage-independent growth on soft agar. Cells were plated at 2000 cells per 9.6-cm2 well, and images were acquired 21 d after cell seeding. The positive control was the rat 1A cell line. Number of colonies formed per 1000 plated cells: 0–50 PDs (23 ± 10; n = 3), 51–100 (82 ± 24; n = 6), 101–150 (61 ± 13; n = 6), 151–200 (125 ± 116; n = 5), 201–300 (122 ± 88; n = 5), and >300 (590 ± 93; n = 5). The Northern Eclipse software package was used to score both large (>60 μm in diameter) and small colonies (<60 μm). (B) Representative images of colony growth (100×). (C) DNA content analysis by flow cytometry revealed a cell cycle distribution that did not differ significantly among the various doubling levels (ANOVA) with the single exception of the percentage of cells in G2/M at 45 PDs (29 ± 2.5%) compared with the percentage of cells in G2/M at 300 PDs (21 ± 0.6%). (D) Metaphase spreads of MDSCs at 15 PDs and 300 PDs (1000×).
We examined DNA content of MDSCs at different passages by performing flow cytometry to detect polyploidy. We observed no difference in the distribution of cells in the different phases of the cell cycle (Figure 7C), with the single exception of the percentage of cells in G2/M at the levels of 45 PDs (29 ± 2.5%) versus 300 PDs (21 ± 0.6%). These findings are consistent with the division time analysis that indicated no significant difference in the cell cycle time of expanded MDSCs (Figure 1E) and suggest normal durations of cell cycle phases (G0/G1: ∼5–7 h, S: ∼2–3 h, and G2/M: ∼3–4 h; Figures 1E and 7C). We also did not detect polyploidy or any increase in populations with >4N DNA content (Figure 7C). These results (Figures 1E and 7C) suggest that DNA content and cell cycle kinetics of MDSCs obtained through extensive expansion are similar to those of freshly isolated stem cells. Analysis of chromosomal number revealed that the cells continued to contain the modal diploid number of 40 chromosomes throughout the expansion process (Figure 7D); however, from one of the three expansions, a population of cells expanded to ∼295–310 PDs contained 76–80 chromosomes. It is interesting to note that the cell cycle analysis did not detect this as tetraploidy.
MDSCs at 15 PDs failed to give rise to tumors in either SCID or syngeneic animals. After implanting MDSCs at 300 PDs into mice, we detected one tumor (∼0.75–1.0 cm in diameter) in a syngeneic mouse but no tumors in the SCID mice (Table 1 and Figure 8). The growth of the tumor in the syngeneic mouse was not detectable by radiography performed 30 or 60 d after injection, but it was slightly visible 150 d after injection; no other mice exhibited abnormal growths detectable by x-ray at any time points (Table 1). Pathological examination of tissue sections revealed neoplastic growth and the presence of reactive cells (Figure 8). The histomorphological appearance of the cells along with the infiltrative nature of the process suggest a neoplasia designation, whereas the appearance of numerous strap-like cells suggests that there are some areas of skeletal muscle differentiation that would indicate a rhabdomyosarcoma-like growth (Klein, personal communication). Although there was no statistical difference between the number of growths found in the two groups (Fisher's exact test; 15 vs. 300 PDs), our analysis of all the findings with the highly expanded cells, together with the incidence of one growth found with MDSC at 300 PDs, suggests that this may not be attributable to chance and should not be disregarded.
Table 1.
Abnormal growth in SCID and syngeneic mice
| In vitro cell age | SCID mice (C57BL/6J-Prkdc) | Syngeneic mice (C57BL/6J) |
|---|---|---|
| MDSC-15 PDs | 0/4 | 0/4 |
| MDSC-300 PDs | 0/4 | 1/4 |
MDSCs at 15 PDs or at 300 PDs were subcutaneously injected into both SCID mice (0.3-0.4 × 106 cells, mice sacrificed 130 d after injection) and syngeneic mice (0.75-1.0 × 106 cells, mice sacrificed 150 d after injection) (number of growths observed/total number of injections).
Figure 8.

Histological examination of in vivo growth observed after transplantation of MDSC-300 PDs into syngeneic mice. Visible are cells with “strap”-like appearance, hypercellular areas containing cells of high nuclear-cytoplasm ratio, and extensive areas of connective tissue formation.
DISCUSSION
Although it is theorized that several types of stem cells have extended replicative life spans, few studies have investigated the doubling potential of stem cell populations. MDSCs are postnatal stem cells identified and distinguished from satellite cells by their relatively low level of commitment to the myogenic lineage (Lee et al., 2000; Qu-Petersen et al., 2002). The origin of muscle stem cells isolated from skeletal tissue and their relationship to other stem cells (e.g., muscle satellite cells or blood-derived stem cells) remain unclear, but basic characterization studies have revealed many features of stem cell behavior (Seale et al., 2001; Zammit and Beauchamp, 2001; Deasy and Huard, 2002). Our study presented here reveals that MDSCs have an extended replicative lifetime and a substantial self-renewal capacity that previously has not been demonstrated for postnatal muscle-derived cells.
Freshly isolated MDSC exhibit high expression of CD34 and Sca-1 and low expression of myogenic markers. We were able to maintain this molecular profile for at least 200 PDs. After 200 PDs, MDSCs showed decreased CD34 expression that corresponded with the cells' decreased ability to generate dystrophin-positive muscle fibers after intramuscular transplantation. This finding is consistent with previous reports of CD34-sorted MDSCs, which indicate that, in comparison with CD34-negative populations, sorted CD34-positive populations exhibit a significantly greater ability to regenerate dystrophin-positive muscle fibers (Jankowski et al., 2002).
As with the majority of studies with stem cells, we did not solely use markers to define the MDSC populations; we also examined the behavior of the MDSCs as they differentiated toward the myogenic lineage in vitro and regenerated skeletal tissue in vivo. Changes in MDSCs' expression of molecular markers occurred concomitantly with decreases in the percentage of cells undergoing myogenic differentiation. MDSCs expanded for up to 195 PDs and subsequently transplanted into mdx mice demonstrated a remarkably high level of engraftment, comparable with that exhibited by freshly isolated MDSCs and significantly higher than that displayed by freshly isolated satellite cells (Jankowski et al., 2002; Qu-Petersen et al., 2002).
Few other postnatal stem cell populations have been observed to undergo such extensive expansion (http://www.nih.gov/news/stemcell/scireport.htm; Schwartz et al., 2002). It is interesting to note that the doubling level observed in our study is most similar to the doubling level reported for ES cells (Thomson et al., 1998; Amit et al., 2000; Smith, 2001; Xu et al., 2001). Other postnatal stem cell populations, such as hematopoietic and bone marrow-derived populations, have been expanded only to much lower levels, despite being cultured for several months (Piacibello et al., 1997; Colter et al., 2000; Gilmore et al., 2000; Jiang et al., 2002; Bianchi et al., 2003).
The MDSCs' high replicative potential, coupled with the ability to maintain their high regeneration capacity, provides evidence that these cells are capable of long-term self-renewal with preservation of the undifferentiated phenotype. However, the failure of MDSCs to engraft after expansion for >240 PDs (the first doubling level at which we observed a decline) indicates that their potential for extended in vitro self-renewal under the conditions used in our study is, in fact, limited. Several other groups of researchers have reported the loss of multipotency by expanded populations of postnatal stem cells. In comparison with unexpanded HSCs isolated from mice, HSCs expanded for only 2–7 d exhibited a decreased ability to perform hematopoietic reconstitution (Knobel et al., 1994; Peters et al., 1995; Traycoff et al., 1996). In another study, murine MSCs cultured for 24 or 48 h exhibited a decreased ability to home to the bone marrow and spleen (Rombouts and Ploemacher, 2003). Other researchers have reported that human MSC populations expanded for 15 or 22 PDs have a decreased ability to differentiate toward osteogenic, chondrogenic, and adipogenic lineages (Digirolamo et al., 1999; Muraglia et al., 2000). By comparison, the MDSCs investigated in our study retained their phenotype and their ability to efficiently promote skeletal regeneration in vivo for up to 200 PDs, a remarkable property of these cells.
We also investigated whether the extended expansion process resulted in transformation of MDSCs, which is a concern for using stem cells in cell and gene therapy, and would be expected for culture-expanded populations (Figure 9). In comparison with earlier populations of MDSCs, the highly expanded MDSC population showed changes in morphology, a heightened ability to grow on soft agar, a decreased ability to exit the cell cycle, a loss of contact inhibition, and, in one case, the capacity to cause a neoplastic growth in vivo. After 200 PDs, MDSCs continued to proliferate even when cultured at high confluence within a reduced cytokine environment, conditions that normally stimulate MDSCs to fuse and form terminally differentiated multinucleated myotubes. Our assessment of MDSCs expanded for >200 PDs revealed dramatic increases in terms of cell growth on agar, with 59% of the cells able to grow on soft agar and form colonies. However, a region of mononuclear cells showing minimal to no cell fusion was visible 2 wk after the transplantation of these populations into the skeletal muscle of mdx mice. Furthermore, one of the eight mice injected subcutaneously with the highly expanded MDSC population developed a neoplastic growth. Together, these findings demonstrate a change in the phenotype of the MDSC population after extended expansion for 200 or more PDs. Although not investigated in our study, the development of structural chromosomal abnormalities due to excessive culturing of the MDSCs—perhaps by receiving multiple genetic hits—would not be surprising (Knudson, 2001).
Figure 9.

In vitro cell aging and clinical expansion of stem cells. Expansion of stem cells will involve generating quantities that are sufficient for basic investigation as well as future therapeutic applications. Identifying and controlling the appropriate expansion and selfrenewal conditions represent a major focus in stem cell biology. It is necessary to know the limit of the expansion as it approaches cell transformation. In the study presented here, MDSC lost their stem cell phenotype after 200 PDs. Stimulated expansion, e.g., cytokine stimulation, can be expected to increase the rate at which a clinical dose is reached and also the rate at which cells may reach their limit.
These results also are particularly intriguing in light of recent studies indicating that stem cells share certain characteristics with cancer cells. Recent studies have investigated the hypothesis that cancer may be driven by cancer stem cells. Small populations of stem cells that share properties with normal cells but also have the ability to form cancer have been identified in cases of leukemia (Bonnet and Dick, 1997; Cobaleda et al., 2000; Miyamoto et al., 2000), breast cancer (Al-Hajj et al., 2003; Dick, 2003), and brain tumors (Hemmati et al., 2003; Singh et al., 2003). The obvious similarity between cancer cells and stem cells is their capacity for self-renewal. Our finding that MDSCs expanded for up to 200 PDs retain their stem cell properties in vitro and exhibit sustained in vivo engraftment efficiency suggests that MDSCs do not undergo transformation unless subjected to very extensive cell replication. It remains to be determined whether the loss of stem cell phenotype and apparent transformation observed here occurred due to chromosomal abnormalities resulting from cell culturing or to the selection of a small fraction of cancer-like stem cells within the MDSC population. Future sorting experiments to identify subpopulations within the expanded populations may help to answer this question.
Our study shows that expansion of MDSCs for up to 200 PDs will yield a large number of phenotypically comparable cells, which would theoretically be more than enough for any clinical application. The use of these cells may represent an alternative solution to the need of immortalization of human myogenic cells to obtain enough cells for transplantation (Berghella et al., 1999). In fact, the large yield suggests that one allogeneic donor could provide a sufficient number of cells for many recipients. Traditionally, whether the cells are being brought closer to senescence has been a central question of in vitro cytokine-stimulated expansion studies. Because we show that the “natural progression” of MDSCs cultured in 20% serum conditions for up to 200 PDs (>4 mo in culture) is to maintain their stem cell characteristics and regeneration capacity, we now can focus our efforts on reducing the initial lag period during MDSC expansion, in essence by decreasing the PDT (Figure 9).
This study clearly demonstrates that MDSCs at high expansion levels maintain their ability to efficiently regenerate skeletal muscle. Indeed, our study suggests that MDSCs' remarkable capacity for self-renewal and long-term expansion is comparable with that exhibited by ES cells. Our results also highlight the need to conduct more intensive investigations into basic stem cell behavior and its potential link to transformation before proceeding with the translation of cell therapies to clinical settings. Nevertheless, this is the first study to demonstrate that MDSCs exhibit a notable capacity for long-term proliferation and self-renewal.
Acknowledgments
We thank Drs. Ed Klein (Division of Laboratory Animal Resources University of Pittsburgh, Pittsburgh, PA) and Aaron Pollett (Mount Sinai Hospital, Toronto, Ontario, Canada) for histomorphological assessment of abnormal growth, Michael Mentzer and Marcelle Huard for technical assistance, Jim Cummins and Bruno Péault for scientific discussions, and Ryan Sauder for editorial assistance. This work was supported by the Muscular Dystrophy Association, National Institutes of Health Grants 1R01 AR49684-01 and P01 AR45925-05), the William F. and Jean W. Donaldson Chair at Children's Hospital of Pittsburgh, and the Henry J. Mankin Chair at the University of Pittsburgh. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant No. C06 RR-14489 from the National Center for Research Resources, National Institutes of Health.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–02–0169) on May 4, 2005.
References
- Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., and Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 100, 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit, M., Carpenter, M. K., Inokuma, M. S., Chiu, C. P., Harris, C. P., Waknitz, M. A., Itskovitz-Eldor, J., and Thomson, J. A. (2000). Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev. Biol. 227, 271–278. [DOI] [PubMed] [Google Scholar]
- Berghella, L., et al. (1999). Reversible immortalization of human myogenic cells by site-specific excision of a retrovirally transferred oncogene. Hum. Gene Ther. 10, 1607–1617. [DOI] [PubMed] [Google Scholar]
- Bhardwaj, G., Murdoch, B., Wu, D., Baker, D. P., Williams, K. P., Chadwick, K., Ling, L. E., Karanu, F. N., and Bhatia, M. (2001). Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat. Immunol. 2, 172–180. [DOI] [PubMed] [Google Scholar]
- Bianchi, G., Banfi, A., Mastrogiacomo, M., Notaro, R., Luzzatto, L., Cancedda, R., and Quarto, R. (2003). Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Exp. Cell Res. 287, 98–105. [DOI] [PubMed] [Google Scholar]
- Bonnet, D., and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737. [DOI] [PubMed] [Google Scholar]
- Cobaleda, C., Gutierrez-Cianca, N., Perez-Losada, J., Flores, T., Garcia-Sanz, R., Gonzalez, M., and Sanchez-Garcia, I. (2000). A primitive hematopoietic cell is the target for the leukemic transformation in human Philadelphia-positive acute lymphoblastic leukemia. Blood 95, 1007–1013. [PubMed] [Google Scholar]
- Colter, D. C., Class, R., DiGirolamo, C. M., and Prockop, D. J. (2000). Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proc. Natl. Acad. Sci. USA 97, 3213–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deasy, B. M., and Huard, J. (2002). Gene therapy and tissue engineering based on muscle-derived stem cells. Curr. Opin. Mol. Ther. 4, 382–389. [PubMed] [Google Scholar]
- Deasy, B. M., Jankowski, R. J., Payne, T. R., Cao, B., Goff, J. P., Greenberger, J. S., and Huard, J. (2003). Modeling stem cell population growth: incorporating terms for proliferative heterogeneity. Stem Cells 21, 536–545. [DOI] [PubMed] [Google Scholar]
- Dick, J. E. (2003). Breast cancer stem cells revealed. Proc. Natl. Acad. Sci. USA 100, 3547–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digirolamo, C. M., Stokes, D., Colter, D., Phinney, D. G., Class, R., and Prockop, D. J. (1999). Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br. J. Haematol. 107, 275–281. [DOI] [PubMed] [Google Scholar]
- Gilmore, G. L., DePasquale, D. K., Lister, J., and Shadduck, R. K. (2000). Ex vivo expansion of human umbilical cord blood and peripheral blood CD34(+) hematopoietic stem cells. Exp. Hematol. 28, 1297–1305. [DOI] [PubMed] [Google Scholar]
- Hayflick, L., and Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. [DOI] [PubMed] [Google Scholar]
- Hemmati, H. D., Nakano, I., Lazareff, J. A., Masterman-Smith, M., Geschwind, D. H., Bronner-Fraser, M., and Kornblum, H. I. (2003). Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 100, 15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irintchev, A., Zeschnigk, M., Starzinski-Powitz, A., and Wernig, A. (1994). Expression pattern of M-cadherin in normal, denervated, and regenerating mouse muscles. Dev. Dyn. 199, 326–337. [DOI] [PubMed] [Google Scholar]
- Jankowski, R. J., Deasy, B. M., Cao, B., Gates, C., and Huard, J. (2002). The role of CD34 expression and cellular fusion in the regeneration capacity of myogenic progenitor cells. J. Cell Sci. 115, 4361–4374. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., Henderson, D., Blackstad, M., Chen, A., Miller, R. F., and Verfaillie, C. M. (2003). Neuroectodermal differentiation from mouse multipotent adult progenitor cells. Proc. Natl. Acad. Sci. USA 100 (suppl 1), 11854–11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., et al. (2002). Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418, 41–49. [DOI] [PubMed] [Google Scholar]
- Karanu, F. N., Murdoch, B., Gallacher, L., Wu, D. M., Koremoto, M., Sakano, S., and Bhatia, M. (2000). The notch ligand jagged-1 represents a novel growth factor of human hematopoietic stem cells. J. Exp. Med. 192, 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobel, K. M., McNally, M. A., Berson, A. E., Rood, D., Chen, K., Kilinski, L., Tran, K., Okarma, T. B., and Lebkowski, J. S. (1994). Long-term reconstitution of mice after ex vivo expansion of bone marrow cells: differential activity of cultured bone marrow and enriched stem cell populations. Exp. Hematol. 22, 1227–1235. [PubMed] [Google Scholar]
- Knudson, A. G. (2001). Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 1, 157–162. [DOI] [PubMed] [Google Scholar]
- Lee, J. Y., et al. (2000). Clonal isolation of muscle-derived cells capable of enhancing muscle regeneration and bone healing. J. Cell Biol. 150, 1085–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, T., Weissman, I. L., and Akashi, K. (2000). AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc. Natl. Acad. Sci. USA 97, 7521–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky, A. V., Pardal, R., and Morrison, S. J. (2004). Diverse mechanisms regulate stem cell self-renewal. Curr. Opin. Cell Biol. 16, 700–707. [DOI] [PubMed] [Google Scholar]
- Muraglia, A., Cancedda, R., and Quarto, R. (2000). Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J. Cell Sci. 113, 1161–1166. [DOI] [PubMed] [Google Scholar]
- Peters, S. O., Kittler, E. L., Ramshaw, H. S., and Quesenberry, P. J. (1995). Murine marrow cells expanded in culture with IL-3, IL-6, IL-11, and SCF acquire an engraftment defect in normal hosts. Exp. Hematol. 23, 461–469. [PubMed] [Google Scholar]
- Piacibello, W., Sanavio, F., Garetto, L., Severino, A., Bergandi, D., Ferrario, J., Fagioli, F., Berger, M., and Aglietta, M. (1997). Extensive amplification and self-renewal of human primitive hematopoietic stem cells from cord blood. Blood 89, 2644–2653. [PubMed] [Google Scholar]
- Potten, C. S., and Morris, R. J. (1988). Epithelial stem cells in vivo. J. Cell Sci. (suppl) 10, 45–62. [DOI] [PubMed] [Google Scholar]
- Qu-Petersen, et al. (2002). Identification of a novel population of muscle stem cells in mice: potential for muscle regeneration. J. Cell Biol. 157, 851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya, T., Morrison, S. J., Clarke, M. F., and Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. [DOI] [PubMed] [Google Scholar]
- Reyes, M., Lund, T., Lenvik, T., Aguiar, D., Koodie, L., and Verfaillie, C. M. (2001). Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood 98, 2615–2625. [DOI] [PubMed] [Google Scholar]
- Rombouts, W. J., and Ploemacher, R. E. (2003). Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 17, 160–170. [DOI] [PubMed] [Google Scholar]
- Rubin, H. (1997). Cell aging in vivo and in vitro. Mech. Ageing Dev. 98, 1–35. [DOI] [PubMed] [Google Scholar]
- Rubin, H. (2002). The disparity between human cell senescence in vitro and lifelong replication in vivo. Nat. Biotechnol. 20, 675–681. [DOI] [PubMed] [Google Scholar]
- Seale, P., Asakura, A., and Rudnicki, M. A. (2001). The potential of muscle stem cells. Dev. Cell. 1, 333–342. [DOI] [PubMed] [Google Scholar]
- Seale, P., Sabourin, L. A., Girgis-Gabardo, A., Mansouri, A., Gruss, P., and Rudnicki, M. A. (2000). Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786. [DOI] [PubMed] [Google Scholar]
- Sherley, J. L., Stadler, P. B., and Stadler, J. S. (1995). A quantitative method for the analysis of mammalian cell proliferation in culture in terms of dividing and non-dividing cells. Cell Prolif. 28, 137–144. [DOI] [PubMed] [Google Scholar]
- Singh, S. K., Clarke, I. D., Terasaki, M., Bonn, V. E., Hawkins, C., Squire, J., and Dirks, P. B. (2003). Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828. [PubMed] [Google Scholar]
- Smith, A. G. (2001). Embryo-derived stem cells: of mice and men. Annu. Rev. Cell. Dev. Biol. 17, 435–462. [DOI] [PubMed] [Google Scholar]
- Stiles, B., Groszer, M., Wang, S., Jiao, J., and Wu, H. (2004). PTENless means more. Dev. Biol. 273, 175–184. [DOI] [PubMed] [Google Scholar]
- Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., and Jones, J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Traycoff, C. M., Cornetta, K., Yoder, M. C., Davidson, A., and Srour, E. F. (1996). Ex vivo expansion of murine hematopoietic progenitor cells generates classes of expanded cells possessing different levels of bone marrow repopulating potential. Exp. Hematol. 24, 299–306. [PubMed] [Google Scholar]
- van Noort, M., and Clevers, H. (2002). TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev. Biol. 244, 1–8. [DOI] [PubMed] [Google Scholar]
- Varnum-Finney, B., Xu, L., Brashem-Stein, C., Nourigat, C., Flowers, D., Bakkour, S., Pear, W. S., and Bernstein, I. D. (2000). Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat. Med. 6, 1278–1281. [DOI] [PubMed] [Google Scholar]
- Xu, C., Inokuma, M. S., Denham, J., Golds, K., Kundu, P., Gold, J. D., and Carpenter, M. K. (2001). Feeder-free growth of undifferentiated human embryonic stem cells. Nat. Biotechnol. 19, 971–974. [DOI] [PubMed] [Google Scholar]
- Zammit, P., and Beauchamp, J. (2001). The skeletal muscle satellite cell: stem cell or son of stem cell? Differentiation 68, 193–204. [DOI] [PubMed] [Google Scholar]
- Zipori, D. (2004). The nature of stem cells: state rather than entity. Nat. Rev. Genet. 5, 873–878. [DOI] [PubMed] [Google Scholar]