

Abstract
Background
Cyclocarya paliurus is a high-value tree, and it contains a variety of bioactive secondary metabolites which have broad application prospects in medicine, food and health care. Triterpenoids can improve the bioactive function of C. paliurus health tea and also improve the efficacy of health care tea.
Results
The results of this study showed that there were 69 kinds were terpenoids, and triterpenoids accounted for more than 80%. We excavated 5 kinds of triterpenoid metabolites with high content and significant difference dynamics, namely, corosolic acid, asiatic acid, maslinic acid, ursolic acid and oleanolic acid. The co-expression analysis identified CYP71D8 and CYP716A15 co-expressed with β-AS may generate oleanane type triterpenoids by modifying β-amyrin, while CYP71AN24 and CYP98A2 co-expressed with LUS may play a key role in lupine type triterpenoids biosynthesis. MYB,Whirly,WRKY and bHLH families, which showed strong correlation with function genes, may play an important role in the regulation of P450 and OSC expression. A total of 20 modules were identified by WGCNA analysis, and CYP71AU50 and CYP716A15 in tan and orange modules may play a major role in the synthesis of oleanolic acid, ursolic acid and asiatic acid, while CYP82D47 in lightcyan 1 module may be the hub gene for the biosynthesis of corosolic acid and maslinic acid.
Conclusions
Our findings mined candidate genes closely related to triterpenoid synthesis in C. paliurus. The results of this paper can provide scientific reference for breeding high-content triterpenoid varieties of C. paliurus.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-024-11125-0.
Keywords: Cyclocarya paliurus, Triterpenoid biosynthesis, Metabolomics, Transcriptomics, WGCNA
Background
Cyclocarya paliurus (Batal.) Iljinsk., also known as Cash Cow tree, belongs to the single genus of Cyclocurus in the walnut family and is a rare tree species unique to China, known as “the giant panda in the plant world” and “the third tree in the medical world” [1]. C. paliurus leaves are rich in terpenoids, flavonoids, polysaccharides, phenolic acids, steroids and other functional active components as well as trace elements such as manganese, iron, copper, zinc, selenium, chromium, alum and germanium, making them good natural health food resources [2]. Pharmacological experiments have shown that these active ingredients can make C. paliurus have physiological activities such as lowering blood pressure and sugar, antioxidant and lowering blood lipids, and have a good effect on the prevention and treatment of hypertension, diabetes and other diseases [3–5]. C. paliurus wood has high strength, slightly thin structure, light soft and shiny, straight grain, not obvious heartwood, easy cutting and smooth section, and is an excellent wood for furniture. With beautiful appearance and fruit resembling copper coins, C. paliurus can be used as an excellent ornamental greening tree and afforestation tree. Therefore, C. paliurus is a precious tree with many values such as medicine, health care, material and ornamental use.
Terpenoids are a class of important secondary metabolites in C. paliurus, including monoterpenoids, sesquiterpenoids, triterpenoids and other types of terpenoids, which have anti-inflammatory, anti-tumor, antibacterial, antiviral, hypoglycemic and other biological activities [6]. The terpenoids are mainly extracted from the leaves of C. paliurus, but they are difficult to meet the market demand due to the problems such as low yield, large environmental impact, long harvest cycle and inability to produce on a large scale. The terpenoids isolated from C. paliurus include asiatic acid, 2α-Hydroxyursolic acid, pterocaryoside B, α-Amyrenone, 11-Keto-ursolic acid, cyclocarioside K, cyclocaric acid A, β-Amyrenone, pomolic acid, cyclocaric acid B, cyclocarioside A, cyclocarioside F, cyclocarioside H, corosolic acid and ursolic acid. The wide range of physiological and pharmacological activities of terpenoids has endowed them with high commercial value, and the research on the synthesis of specific components of terpenoids has been increasingly in-depth. The main synthetic pathways of terpenoids include methyl-light-valerate (MVA) pathway and plastids (MEP) pathway. The precursors of all terpenoids, isopentylene pyrate-pyruvate (IPP) and dimethylallylpyrophosphate (DMAPP), are synthesized from acetyl-coenzyme A, pyruvate and phosphoglyceric acid at different sites [1].
In the previous research, we conducted whole genome sequencing of C. paliurus, and assembled the genome of C. paliurus with a size of 634.90 Mb (2n = 4x = 64) and an N50 of 30.98 Mb. 46,292 encoded protein genes were predicted, and ncRNAs, repeats, transcription factors were predicted. After homologous comparison and functional annotation of KEGG pathway, 16 genes can be annotated to the known triterpenoid saponin synthesis pathway. Including acetyl-CoA acyltransferase (ACAT), 3-hydroxy-3-methylglutaryl-CoA synthetase (HMGS), 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), mevalonate kinase (MVK), 5-pyrophosphate mevalonate decarboxylase (MVD), 1-deoxyd-xylulose-5-phosphate synthetase (DXS), 1-deoxyd-xylose-5-phosphate reduction isomerase (DXR), Cytidine 2-C-methyl-D-erythritol kinase (CMK), Cytidine 2-C-methyl-D-erythritol synthetase (CMS), 2-C-methyl-D-erythritol 2, 4-cyclodiphosphate synthase (ISPF), (E) -4-hydroxy-3-methyl-2-butyl-pyrophosphate synthase (HDS), (E) -4-hydroxy-3-methyl-2-butyl-pyrophosphate reductase (HDR), isopentenylpyrophosphate isomerase (IDI), Farnesyl pyrophosphate synthase (FPS), squalene synthase (SS), squalene oxidase (SE) [1].
But at present, the research about the breeding, cultivation and production technology of C. paliurus health tea are mainly focused on. However, the potential hub genes involved in the downstream pathway of triterpenoid biosynthesis have not been discovered. In view of this, this study will analyze triterpene synthesis pathway from the perspective of transcriptome and metabolome, explore the hub genes involved in the downstream triterpene synthesis pathway and highly related metabolic pathways, and identify the related gene families. The results can provide a scientific basis for the breeding of high content triterpenoid C. paliurus varieties.
Results
Determination of total triterpenoids in C. paliurus
The result of determination of total secondary metabolites in C. paliurus detected by Ultra Performance Liquid Chromatography-Mass Spectrometry (UPLC-MS/MS) was 751, which belonged to amino acids and their derivatives, phenolic acids, nucleotides and their derivatives, flavonoids, lignans and coumarins, tannins, alkaloids, terpenes, organic acids, lipids and other metabolites, respectively (Fig. 1A). Among these, 69 metabolites were identified as terpenoids, of which 60 were triterpenoids, making up more than 80% of the total terpenoid content (Table S1). The total triterpenoid content in the leaves of C. paliurus from August to November is illustrated in Fig. 1B. A highly significant difference in triterpenoid content was observed between August and November (p < 0.01). However, no significant difference was found between September and November (p < 0.05). August triterpenoid content was significantly different from that of September, October, and November. The highest triterpenoid accumulation was recorded in August and November, while it was lower in September and November. Overall, triterpenoid accumulation exhibited a decreasing trend from August to November. The transition from summer to autumn, particularly during the fruiting period from August to September, likely influenced the distribution and transport of metabolites, contributing to the reduction in triterpenoid content. In October, cooler temperatures and environmental stress led to an increase in terpenoid accumulation in the leaves. By November, during the leaf yellowing and senescence phase, the total triterpenoid accumulation reached its lowest point.
Fig. 1.

Characteristics of Secondary Metabolites and Triterpenoid Content in C. paliurus.A: Breakdown of the categories and proportions of secondary metabolites in C. paliurus, highlighting the dominance of flavonoids and phenolic acids. B: Seasonal variation in total triterpenoid content in C. paliurus leaves from August to November. Statistically significant differences between months are denoted by different letters (p < 0.05). C: Composition and relative proportions of different terpenoid subclasses, including triterpenoids, diterpenoids, triterpenoid saponins, and sesquiterpenoids, across the months. D: Pearson correlation coefficients of the samples within the same month, indicating strong biological repeatability, as demonstrated by values close to 1
Seasonal variation of total triterpenoid content and triterpenoid compositional profile in C. paliurus
The comprehensive analysis of secondary metabolites in C. paliurus leaves, depicted in Fig. 1A, identified 751 compounds across several classes, with phenolic acids (18%), flavonoids (19%), lipids (12%), and organic acids (11%) representing the major groups. Notably, terpenoids comprised 9% of the metabolite profile, with triterpenoids accounting for over 80% of this class, highlighting their prominent role in the plants secondary metabolism and potential biological activity. The seasonal fluctuation in total triterpenoid content, as shown in Fig. 1B, reveals that triterpenoid levels peak in October, reaching approximately 8%, while August shows a slightly lower content at around 6%. This content notably declines in September and November to approximately 4% and 3%, respectively. These variations suggest that external factors, such as seasonal temperature changes and environmental stresses, may influence triterpenoid biosynthesis. The pronounced rise in triterpenoid levels in October likely reflects an adaptive response to cooler weather, which can trigger secondary metabolite production. Additionally, Table S1 confirms the significant presence of 69 individual terpenoid compounds, predominantly triterpenoids, aligning with the trends observed in Fig. 1A.
The detailed terpenoid composition from August to November, as shown in Fig. 1C and Table S2, highlights a consistent predominance of triterpenoids within the total terpenoid profile. In August, triterpenoids represent 91.14% of the terpenoid content, with a minor decrease to 84.35% in October. Conversely, September and November exhibit lower proportions, with triterpenoids comprising 79.28% and 81.27%, respectively. This seasonal fluctuation suggests that environmental or physiological factors could regulate triterpenoid biosynthesis. Interestingly, triterpenoid saponins showed the highest concentrations in September and November (13.51% and 13.78%, respectively), indicating a possible compensatory relationship between triterpenoids and their saponin derivatives. While diterpenoids and sesquiterpenoids were present at lower levels, they also displayed subtle seasonal variations, with slightly higher concentrations in October. The data in Table S2 corroborates these observations, illustrating individual terpenoid fluctuations, including notable compounds such as Madasiatic acid and Corosolic acid. Madasiatic acid reached its peak in November (22.96%), while Corosolic acid exhibited its highest content also in November (4.67%). Overall, these findings emphasize the dynamic regulation of triterpenoids and their derivatives throughout the year, underscoring the unique seasonal profiles of specific compounds in C. paliurus.
Correlation and cluster analysis of terpenoid metabolites across sampling months
By calculating the Pearson correlation coefficient, which serves as an indicator of biological duplication within samples, a value closer to 1 indicates stronger similarity between the samples. As shown in Fig. 1D, the Pearson correlation coefficient for samples collected within the same month was near 1, demonstrating excellent biological repeatability. In contrast, the Pearson correlation coefficients between samples from different months ranged from 0.2 to 0.6, reflecting substantial differences in their biological characteristics. Although the Pearson correlation coefficient and PCA utilize different algorithms, both methods consistently highlight the significant variation in terpenoid composition from August to November.
Differential analysis of terpenoid metabolites and identification of key triterpenoids
To accurately identify triterpenoids with high content and significant dynamic differences, a combination of p-values and fold change values was used in univariate analysis. The screening criteria for differential terpenoid metabolites were set as fold change ≥ 2 and VIP ≥ 1 (Fig. S1). In the comparison of 8 M-ML vs. 9 M-ML, 8 metabolites were significantly upregulated, 18 were significantly downregulated, and 43 showed no significant changes. The top five upregulated metabolites included isololiolide, progesterone, betulin, cyclocalsalic acid A, and 12,13-dihydroursolic acid, while downregulated metabolites were 6,9-dihydroxy-7-megastigmen-3-one, gentiopicroside, geniposide, 2-hydroxyoleanolic acid, and corosolic acid (Table S3).
In the comparison of 8 M-ML vs. 10 M-ML, 16 metabolites were significantly upregulated, 14 downregulated, and 39 remained unchanged. The top upregulated metabolites were isololiolide, genipin, progesterone, swertiamarin, and lupenone, while 6,9-dihydroxy-7-megastigmen-3-one, geniposide, epikatonic acid, oleanolic acid, and corosolic acid were downregulated (Table S4). Similarly, in the 8 M-ML vs. 11 M-ML comparison, 8 terpenoid metabolites were upregulated, 7 were downregulated, and 54 had no significant change. Notably, progesterone, 24,30-dihydroxy-12(13)-enolupinol, betulinic acid, ursolic acid, and ivy saponin were upregulated, while gentiopsin, arbutin, β-boswellic acid, α-boswellic acid, and 3,11-dioxo-19α-hydroxyurs-12-en-28-oic acid were downregulated (Table S5). Further comparisons, such as 9 M-ML vs. 10 M-ML and 9 M-ML vs. 11 M-ML, revealed significant dynamic differences in terpenoid metabolites. Key metabolites such as genipin, lupenone, betulin, and asiatic acid displayed notable shifts, confirming the temporal regulation of terpenoid biosynthesis (Tables S6, S7). In the final comparison, 10 M-ML vs. 11 M-ML, 17 terpenoids were upregulated, while 17 were downregulated. The most significantly upregulated metabolites included betulin, epikatonic acid, corosolic acid, and genipin, among others (Table S8).
Through this screening process, five key triterpenoids—corosolic acid, asiatic acid, maslinic acid, ursolic acid, and oleanolic acid—were identified as having high content and significant dynamic differences (Fig. S2). In comparisons such as 8 M-ML vs. 9 M-ML and 9 M-ML vs. 10 M-ML, these metabolites were generally downregulated. However, in comparisons involving later months (e.g., 10 M-ML vs. 11 M-ML), they were upregulated, indicating their role in the seasonal accumulation patterns of C. paliurus triterpenoids (Fig. S2).
K-means clustering and venn diagram analysis of terpenoid metabolites
To further investigate the relative content trends of differential terpenoid metabolites, we conducted k-means clustering analysis. As shown in Fig. 2A, the differential metabolites were grouped into 10 clusters. Among these, 28 terpenoids exhibited higher content in 10 M-ML compared to 8 M-ML, 9 M-ML, and 11 M-ML, while 22 terpenoids had lower content in 10 M-ML. Subclass 1, which contained 11 terpenoid metabolites, demonstrated higher species and quantities compared to the other subclasses, following a trend where 11 M-ML was significantly higher than 8 M-ML, 9 M-ML, and 10 M-ML. Key terpenoids in this group included 6,9-dihydroxy-7-megastigmen-3-one, progesterone, betulinic acid, ursolic acid, and oleanolic acid-3-O-glucosyl(1→2)glucoside. Subclass 3 and 4, containing fewer terpenoids, displayed similar but lower trends, with metabolites such as arjunic acid and asiatic acid. Triterpenoids with high content and significant differences, including corosolic acid, asiatic acid, maslinic acid, ursolic acid, and oleanolic acid, were mainly found in subclasses 1, 3, and 5, showing distinct clustering patterns.
Fig. 2.

K-means Clustering and Upset Plot Analysis of Differential Terpenoid Metabolites. A: K-means clustering analysis of differential terpenoid metabolites. The sub-classes represent groups of metabolites with similar change trends across the sampling months. The x-axis shows the sample groups, and the y-axis represents the standardized relative content of terpenoid differential metabolites. B: Upset plot and bar diagram illustrating the intersection and unique elements of differential terpenoid metabolites between sample groups. The upper portion shows the unique and common metabolites across different groups, while the lower part visualizes the intersection classification with connecting lines indicating shared elements. The bar chart on the left shows the number of unique metabolites in each group
To illustrate the relationships between terpenoid metabolites across the six comparison groups, Venn diagrams and petal diagrams were generated using Venn analysis (Fig. 2B). The petal diagrams revealed five unique terpenoid differential metabolites across the comparisons, such as cyclocarioside I, pterocaryoside B, ambolic acid, and ursolaldehyde. Interestingly, no common terpenoid metabolites were identified across all six comparison groups. However, the intersection diagram showed that maslinic acid was common in all groups except 8ML-ML vs. 11 M-ML, while corosolic acid and asiatic acid were common in all groups except for 8ML-ML vs. 11 M-ML and 9ML-ML vs. 10 M-ML.
KEGG functional annotation and enrichment analysis of terpenoid metabolites
KEGG annotation was performed on the terpenoid differential metabolites identified in the six comparison groups, and the results are shown in Fig. S3. Across the comparisons, including 8 M-ML vs. 9 M-ML, 8 M-ML vs. 11 M-ML, 9 M-ML vs. 10 M-ML, and 10 M-ML vs. 11 M-ML, gentiopicroside was the only metabolite annotated for both monoterpenoid biosynthesis and secondary metabolite biosynthesis pathways. The enrichment analysis revealed that gentiopicroside was significantly enriched in all four terpenoid groups. This metabolite was downregulated in 8 M-ML vs. 9 M-ML, 8 M-ML vs. 11 M-ML, and 10 M-ML vs. 11 M-ML, while it was upregulated in 9 M-ML vs. 10 M-ML, indicating its dynamic role in terpenoid biosynthesis across different months. This analysis suggests that gentiopicroside plays a key role in the metabolic changes observed between different stages of plant growth, particularly in its involvement in monoterpenoid and secondary metabolite biosynthesis pathways. Further studies on its regulatory mechanisms could provide deeper insights into the functional roles of terpenoids in C. paliurus.
Transcriptomic analysis
Transcriptomic data quality assessment and alignment analysis
After quality control of the sequencing data, a total of 162.73 Gb of clean data were generated from the transcriptome sequencing, as detailed in Table S9. The number of clean reads per sample ranged from 40,403,964 to 52,318,574. The quality scores for the sequencing data were high, with Q20 exceeding 96.13% and Q30 surpassing 89.48%, indicating a base recognition accuracy rate of 99.9%. The GC content fluctuated by around 5%, with values lower than 42.65%. These results demonstrate that the RNA-seq data are of high quality and provide a reliable foundation for subsequent transcriptomic analysis.
When compared to the reference genome, the mapping results, shown in Fig. S4, indicated that mapped reads ranged from 9.47 to 53.35%. Unique mapped reads varied from 8.84 to 49.68%, while multiple mapped reads ranged between 0.63% and 4.90%. Reads mapping to the positive strand (‘+’) ranged from 4.46 to 25.00%, and reads mapping to the negative strand (‘-‘) ranged from 4.62 to 26.00%. The overall mapping efficiency ranged from 9.47 to 53.56%, confirming that the selected reference genome was generally appropriate for the analysis requirements.
Gene expression analysis and principal component analysis
To measure gene expression levels, FPKM values were calculated, and genes with low expression (FPKM < 1) were filtered out. The overall gene expression profile was visualized using box plots and FPKM density distribution maps, as shown in Fig. S5. The box plots reveal that the upper quartile, lower quartile, and median gene expression levels are consistent across all samples except for 10M2, suggesting minimal variation and sampling error in most samples. The FPKM density distribution also shows that, aside from 10M2, the curves for other samples overlap, indicating good repeatability and low error across samples.
PCA analysis was performed using the FactoMineR package in R to assess gene expression patterns. The first three principal components, PC1 (22.92%), PC2 (20.01%), and PC3 (13.02%), explained 55.95% of the total variation in the data (Fig. S6). Biological replicates for each month clustered closely, indicating good repeatability, while samples from different months were clearly separated, demonstrating significant differences in gene expression. These variations suggest that genes involved in terpenoid synthesis pathways may also show distinct expression patterns across different months.
Gene expression correlation analysis results are displayed in Fig. S7. Notably, there is a strong correlation (~ 0.8) between 11 M and 8 M, 9 M, and 10 M, suggesting similar expression patterns among these months. However, the correlation between 9 M and both 11 M and 8 M is lower (~ 0.6), indicating some divergence in expression patterns. Biological replicates for each month showed high correlations (0.9–1.0), confirming good repeatability within each group. However, while correlation cluster analysis provides an overview of gene expression patterns across different months, it does not specifically reflect the expression of genes involved in the biosynthesis of key triterpenoids, warranting further investigation.
Comprehensive gene annotation across multiple databases
All genes were annotated using nine major databases: NR, Swiss-Prot, COG, KOG, GO, KEGG, TrEMBL, eggNOG, and Pfam, with the results illustrated in Fig. 3A. A total of 21,411 genes were successfully annotated, accounting for 84.27% of all identified genes. The NR database annotated 21,313 genes, representing 99.54% of the total annotations. In the eggNOG database, 18,139 genes were annotated, making up 84.72% of the total. The TrEMBL database provided annotations for 21,343 genes, covering 99.68% of the total annotation count. Swiss-Prot annotated 15,067 genes (70.37%), while 16,626 genes (77.65%) were annotated in the Pfam database. The KOG database annotated 11,800 genes (55.11%), and the KEGG database provided annotations for 14,938 genes (69.77%). The GO database contributed annotations for 17,461 genes, representing 81.55% of the total. Finally, the COG database annotated 6,654 genes, accounting for 31.08% of all gene annotations.
Fig. 3.

Gene Annotation and KEGG/GO Enrichment Analysis of DEGs. A: Pie chart representing the proportion of transcriptome gene annotations across nine databases, including GO, KEGG, SwissProt, Pfam, and others. B: KEGG enrichment clustering of DEGs. The color gradient from red to green indicates the relative expression levels, with red representing higher expression and green representing lower expression. The number in parentheses next to each term indicates the number of significantly different genes associated with that pathway. C: GO enrichment analysis of DEGs, categorized by biological process, cellular component, and molecular function. The x-axis represents the logarithm of the enrichment significance (-log10 p-value), and the y-axis lists the corresponding GO terms
Identification and functional annotation of differentially expressed genes (DEGs)
Gene expression was compared across samples collected from August (8 M) to November (11 M), with each month serving as both a control and experimental group in six different comparison sets. DEGs were identified using the screening criteria of FDR < 0.05 and |log2(fold change)| > 1. The statistical analysis of DEGs is presented in Table S10, showing a total of 15,951 DEGs. The results indicate that the number of DEGs decreases progressively over time. Additionally, in all six comparison groups, the number of downregulated genes exceeded the number of upregulated ones, suggesting that environmental and internal factors are likely influencing gene downregulation. These downregulated genes could play a critical role in the biosynthesis of triterpenoids.
KEGG annotation of these DEGs identified a total of 3,247 genes, categorized into 118 metabolic pathways. A cluster analysis of the top 30 genes with the highest expression and most significant differences in metabolic pathways is illustrated in Fig. 3B. Notably, four of these genes were involved in the citrate cycle (TCA cycle), which showed the highest expression levels across all 12 samples, significantly outpacing other pathways. This suggests that the TCA cycle plays an important regulatory role in triterpenoid biosynthesis in C. paliurus. KEGG enrichment analysis further highlighted the involvement of pathways such as Flavonoid biosynthesis, Terpenoid backbone biosynthesis, Pyruvate metabolism, and Aminoacyl-tRNA biosynthesis. The consistent expression patterns of these pathways suggest that the terpenoid skeleton biosynthesis pathway is closely linked to other metabolic processes, potentially regulating terpenoid production. Interestingly, pathways like N-glycan biosynthesis and cyanoamino acid metabolism showed elevated expression levels in November but lower expression in earlier months (August, September, and October). Conversely, isoquinoline alkaloid biosynthesis and limonene and pinene degradation pathways were more active in August and September but less so in October and November. Among the annotated genes, nine pathways related to terpenoid biosynthesis were identified, as listed in Table S11. The largest number of differential genes was associated with pathways such as terpenoid skeleton biosynthesis, carotenoid biosynthesis, and ubiquinone and other terpenoid-quinone biosynthesis. In contrast, pathways like zein biosynthesis and steroid biosynthesis had fewer DEGs, while monoterpene biosynthesis had the fewest.
GO analysis was performed to observe the distribution of DEGs across the 12 samples, with gene annotations classified into biological processes, cellular components, and molecular functions, as shown in Fig. 3C. GO enrichment analysis revealed that most genes were involved in cellular components, followed by biological processes and molecular functions. Within the molecular function category, the most prominent activities were microtubule binding, hydrolase activity, and protein kinase activity. Cellular components were largely related to the cell wall, extracellular region, and plasma membrane. Key biological processes included cell wall organization, carbohydrate metabolic processes, and pectin catabolism. These results suggest that genes involved in these functional groups likely play pivotal roles in the regulation and biosynthesis of triterpenoids in C. paliurus.
Analysis of the Triterpenoid Biosynthesis Pathway in C. paliurus
Based on transcriptome analysis and GO and KEGG annotations, we examined DEGs involved in terpenoid skeleton biosynthesis as well as sesquiterpene and triterpene biosynthesis across six comparison groups. Volcano plot analysis of these differential genes, illustrated in Fig. S8, revealed that downregulated genes were more prevalent than upregulated genes, suggesting that the downregulated genes in these pathways may contribute significantly to the accumulation of terpenoids in C. paliurus. Notably, no significant differences were found between the 8 M vs. 10 M and 9 M vs. 10 M groups. However, one gene in the 10 M vs. 11 M group showed substantial upregulation, indicating its potential involvement in triterpenoid biosynthesis during October and November. Comparative analysis across other groups—8 M vs. 9 M, 8 M vs. 10 M, and 9 M vs. 11 M—showed both upregulated and downregulated genes, with downregulated genes outnumbering upregulated ones in most cases. For example, the 8 M vs. 10 M comparison revealed one significantly upregulated gene and two significantly downregulated genes, while the 9 M vs. 11 M group had two upregulated genes and three downregulated genes.
A predicted pathway for triterpenoid biosynthesis was constructed based on these transcriptomic findings and gene differences within the terpenoid biosynthesis pathways (Fig. 4). A total of 33 genes were found to participate in terpene skeleton, sesquiterpene, and triterpene biosynthesis. In the upstream pathway, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) are generated via the MVA and MEP pathways, followed by the formation of farnesyl pyrophosphate (FPP) by geranyl pyrophosphate synthase. Further downstream, squalene is produced by two squalene synthases (SQS1, SQS2), which undergoes epoxidation by squalene epoxidase to yield 2,3-oxidosqualene. Gene expression patterns across different months reveal that the MEP pathway plays a prominent role in November, with significant upregulation of key genes like DXS,ISPE,ISPG, ISPH, and ISPF compared to other months. Meanwhile, the MVA pathway is more active in August and September, as indicated by the upregulation of HGMR,MVAK2, and ACAT during these months. Additionally, isopentenyl pyrophosphate isomerase (IDI) shows increased expression in September and November, suggesting that the MEP and MVA pathways may work in tandem to regulate triterpenoid biosynthesis. Moreover, two genes encoding squalene synthase (SQS1, and SQS2) in the intermediate pathway showed upregulation in November, implying that the intermediate and MEP pathways collectively regulate triterpenoid biosynthesis in C. paliurus at this time.
Fig. 4.

Upstream Pathway of Triterpenoid Synthesis in C. paliurus. Solid lines indicate pathways where differential genes have been identified in the transcriptome analysis, while dotted lines represent pathways without identified differential genes. The colored blocks next to each enzyme represent the FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values of the corresponding genes, with expression levels shown from August to November. The color gradient moves from blue (lower expression) to red (higher expression), illustrating the dynamic expression of genes involved in the triterpenoid biosynthesis pathway. Gene name abbreviation: acetyl-CoA acyltransferase (ACAT), 3-hydroxy-3-methylglutaryl-CoA synthetase (HMGS), 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), mevalonate kinase (MVK), phosphomevalonate kinase (MVAK2), 5-pyrophosphate mevalonate decarboxylase (MVD), 1-deoxyd-xylulose-5-phosphate synthetase (DXS), 1-deoxyd-xylose-5-phosphate reduction isomerase (DXR), 2-C-methyl-D-erythritol 2, 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase (ISPE), 4-cyclodiphosphate synthase (ISPF), (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (ISPG), 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase (ISPH), isopentenyl pyrophosphate isomerase (IDI), farnesyl diphosphate synthase (FDPS), (2Z,6Z)-farnesyl diphosphate synthase (ZFPS), squalene synthase 1 (SQS1), squalene synthase 2 (SQS2)
We conducted a Pfam database annotation to identify genes containing conserved P450 domains and performed homology analysis using Arabidopsis thaliana P450 sequences. This allowed us to identify 193 P450 genes in the transcriptome of C. paliurus. These P450 genes were classified into 34 families, based on amino acid sequence homology, with ≥ 40% homology classifying them into the same family and ≥ 55% into the same subfamily. The families CYP71,CYP72,CYP85, and CYP86 were found to form large multi-family clusters. Among the P450 families, CYP707,CYP71,CYP75, and CYP82 had the largest number of members, followed by CYP705,CYP78,CYP87,CYP714, and CYP716. The functional annotation and distribution of these P450 family members are presented in Fig. S9 and Fig. S10. GO enrichment analysis of these P450 genes revealed significant involvement in iron binding, monooxygenase activity, and integral membrane components, with biological processes primarily enriched in methylation. We also performed transcription factor (TF) prediction using the PlantTFDB database and identified 1,219 transcription factors (TFs) in the leaves of C. paliurus, which were classified into 33 families (Fig. S11). The most abundant TF families were BHLH,NAC,WRKY,MYB-related, and FAR1, containing 170, 133, 108, 104, and 100 members, respectively. An analysis of transcription factor expression across different months showed that most TFs, such as bZIP,CAMTA,HSF,MYB-related, NF-X1,NF-YB,NF-YC,WRKY, and Nin-like, exhibited higher expression levels in August and November. In contrast, CPP,Dof,GeBP,WRKY, and HSF showed lower expression levels in September and October. These TFs, with varying expression levels, likely play essential roles in regulating triterpenoid biosynthesis in C. paliurus. Cluster analysis of all DEGs revealed that β-amyrin synthase (β-AS) and lupine synthase (LUS), two key functional genes, were located in the same gene cluster containing 570 genes, indicating similar expression patterns (Fig. S12). Additionally, CYP716A15, known for its involvement in triterpenoid skeleton modification, was also present in this cluster. Another form of CYP716A15 displayed a distinct expression pattern from these genes. Further mining of genes in this cluster revealed high Pearson correlation coefficients between CYP71D8 and β-AS (R = 0.83), and between CYP71AN24,CYP98A2, and LUS (R > 0.99), suggesting these genes are co-expressed and may function together in triterpenoid biosynthesis. Similarly, CYP51G1 was highly correlated with CYP716A15 (R > 0.99), while transcription factors from the MYB,WRKY,Whirly, and bHLH families were strongly correlated with these functional genes, indicating their regulatory role in downstream triterpenoid biosynthesis pathways (Fig. 5A).
Fig. 5.

P450 Enzymes and Transcription Factors Correlated with β-AS,LUS, and CYP716A15 in the Downstream Triterpenoid Synthesis Pathway in C. paliurus. A: Heatmap showing the expression profiles (FPKM values) of P450 enzymes and transcription factors that exhibit high correlation (Pearson > 0.7) with key functional genes in triterpenoid biosynthesis, including β-AS,LUS, and CYP716A15. The left panel lists the gene IDs and corresponding gene names, while the heatmap in the middle presents the normalized gene expression levels across different months. The right panel highlights highly correlated P450 enzymes and transcription factors. B: Schematic representation of the downstream pathway of triterpenoid synthesis. Solid lines indicate steps where differential genes have been identified in the transcriptome, while dashed lines represent potential unknown steps or genes not yet identified. The colored blocks represent FPKM values for differentially expressed genes (DEGs) from August to November, with the color gradient indicating changes in expression levels
In the downstream triterpenoid synthesis pathway (Fig. 5B), five genes encoding 2,3-epoxy squalene cyclase (OSC) were identified, alongside one gene encoding β-AS, which was upregulated in August and November, closely mirroring the accumulation pattern of oleanolic acid triterpenoids. Four genes encoding LUS were upregulated or downregulated in different months, paralleling the content changes in lupinoid triterpenoids. The P450 family, responsible for modifying the triterpenoid skeleton, showed significant increases in gene expression in November, highlighting their role in this biosynthetic process. Co-expression analysis suggests that genes like CYP71D8 and CYP716A15, co-expressed with β-AS, are involved in the modification of β-amyrin to produce oleanolic acid triterpenoids, while CYP71AN24 and CYP98A2, co-expressed with LUS, may be key regulators in lupinoid triterpenoid biosynthesis.
Integrated transcriptome and metabolome analysis of triterpenoid biosynthesis pathways
To refine our gene selection, we conducted Mfuzz clustering analysis of DEGs and 69 terpenoid metabolites, resulting in 10 clusters, as shown in Fig. S13 and Fig. S14. Key terpenoids, such as asiatic acid, corosolic acid, maslinic acid, and oleanolic acid, were primarily located in DAM Cluster 1, while ursolic acid was mostly in DAM Cluster 7. Comparisons of DAM clusters with DEG clusters revealed that DAM Clusters 1 and 7 displayed expression patterns similar to DEG Clusters 3 and 1, respectively. DEG Cluster 3 showed higher gene expression levels in August and November, while DEG Cluster 1 had elevated expression in November. We focused on the 8 M vs. 9 M and 9 M vs. 11 M groups in DEG Cluster 3 and the 8 M vs. 11 M and 9 M vs. 11 M groups in DEG Cluster 1, as they demonstrated the largest expression differences.
KEGG annotation of differential genes highlighted key metabolic pathways involved in triterpenoid biosynthesis (Fig. S15-16). In DEG Cluster 1, upregulated genes in 8 M vs. 11 M and 9 M vs. 11 M were enriched in plant-pathogen interaction, circadian rhythm, amino acid metabolism, and terpenoid backbone biosynthesis pathways. This alignment between gene upregulation and terpenoid biosynthesis suggests these pathways may contribute to ursolic acid accumulation. In DEG Cluster 3, genes were downregulated in the 8 M vs. 9 M comparison and upregulated in the 9 M vs. 11 M comparison, potentially explaining the fluctuating levels of asiatic acid, corosolic acid, maslinic acid, and oleanolic acid.
Further analysis showed distinct pathways in DEG Cluster 3, including basal transcription factors, inositol phosphate metabolism, oxidative phosphorylation, and ubiquitin-mediated proteolysis, which were not present in DEG Cluster 1. These pathways may account for the unique accumulation patterns of several triterpenoid metabolites compared to ursolic acid. Mfuzz clustering was also applied to transcription factors (TFs) predicted by the PlantTFDB database, dividing them into 10 clusters (Fig. S17-S18). Notably, TFs clusters 6 and 10 shared expression patterns with DAM Clusters 1 and 7. TFs in Cluster 6 were primarily bHLH and FAR1, followed by AGTA,GRAS,HSF,NAC,NF-YB, and MYB-related. In Cluster 10, bHLH,bZIP, and WRKY were the most prominent families, with WRKY being unique to this cluster, indicating it may play a pivotal role in triterpenoid regulation. Additionally, NF-YB,TCP, and S1Fa-like TFs were unique to TFs Cluster 6, suggesting these TFs may also contribute to the regulation of triterpenoid biosynthesis in C. paliurus.
Correlation between seasonal triterpenoid content and gene expression in C. paliurus
In this study, WGCNA was utilized to correlate key biosynthetic genes with the monthly variation in triterpenoid content in C. paliurus, identifying clusters of genes with expression patterns aligned to seasonal shifts in compound accumulation. The analysis began with filtering for genes with FPKM ≥ 1 and removing outliers to ensure robust network construction. An optimal soft threshold of β = 19 (R² = 0.821) was applied to construct a scale-free network, resulting in 20 gene modules, each representing groups of genes with similar expression patterns (Fig. S19, Fig. 6A).
Fig. 6.

Gene Clustering Tree and Heatmap of Correlations with Triterpenoid Metabolites. A: Gene clustering tree showing module division. Different branches represent clusters of co-expressed genes, with each module marked by a distinct color. B: Heatmap representing the correlation between gene modules and the relative content of triterpenoid metabolites. The color gradient represents the correlation values, with red indicating positive correlations and blue indicating negative correlations between gene expression modules and triterpenoid metabolite levels
To investigate the relationship between gene modules and triterpenoid levels, five primary triterpenoid metabolites—asiatic acid, corosolic acid, maslinic acid, oleanolic acid, and ursolic acid—were mapped to these modules, revealing significant correlations (Fig. 6B). The selection of these five triterpenoids for WGCNA analysis is based on their high concentrations and significant variation across the sampling periods. And these compounds represent the most biologically relevant triterpenoids in C. paliurus and have established roles in the plants medicinal properties. The lightcyan 1 module showed a positive correlation with maslinic acid, corosolic acid, and oleanolic acid, while the tan module was strongly correlated with ursolic acid. Similarly, the orange module was associated with asiatic acid. These correlations reflect distinct accumulation patterns, as shown in Fig. 1B, with ursolic acid exhibiting unique trends compared to other triterpenoids, suggesting that gene expression in these modules influences triterpenoid biosynthesis differently across months. KEGG pathway enrichment analysis of the tan, lightcyan 1, and orange modules provided insights into the metabolic processes linked to triterpenoid biosynthesis (Fig. 7A, C and E). In the tan module, enriched pathways included terpenoid backbone biosynthesis, sesquiterpenoid and triterpenoid biosynthesis, and the MEP pathway, which appear to regulate ursolic acid synthesis, especially during periods of elevated accumulation in November. Similarly, genes in the lightcyan1 module were enriched in pathways like starch and sucrose metabolism and oxidative phosphorylation, which may contribute to the biosynthesis of asiatic acid, corosolic acid, and oleanolic acid. The orange module, associated with asiatic acid, was enriched in photosynthesis and carbon fixation pathways, suggesting a unique regulatory role in asiatic acid accumulation, which is higher in August and November.
Fig. 7.

KEGG Enrichment Histograms and Genetic Interaction Network Diagrams. A: KEGG enrichment histogram for the tan module, showing the distribution of genes across various metabolic and biological pathways. B: Genetic interaction network diagram representing the top 30 genes with the highest connectivity in the lightCyan 1 module. C: KEGG enrichment histogram for the lightCyan1 module, illustrating the most significantly enriched pathways. D: Genetic interaction network diagram for the top 30 genes with the highest connectivity in the orange module. E: KEGG enrichment histogram for the tan module, highlighting the enriched KEGG pathways. F: Genetic interaction network diagram for the top 30 genes with the highest connectivity in the tan module
These modules contained critical enzymes and transcription factors influencing triterpenoid biosynthesis (Fig. 7B, D and F, Table S12, Table S13, and Table S14). Key genes, such as CYP761A15 (a C-28 oxidase), are involved in ursolic acid formation [7], while CYP82D47 in the lightcyan 1 module, co-expressed with ERF109, appears to regulate the synthesis of corosolic acid, maslinic acid, and oleanolic acid. The expression patterns and high correlations of these genes with specific triterpenoids underscore the genetic regulation underlying seasonal shifts in triterpenoid levels. This study establishes a strong association between the seasonal variation in triterpenoid content and gene expression in C. paliurus, offering insights into the molecular mechanisms that drive triterpenoid biosynthesis across different months.
Quantitative real-time qPCR validation for DEGs
To validate the identified DEGs, we selected 8 candidate genes (CpACAT,CpFDPS1,CpAFDPS2,CpLUP4,Cpβ-AS,CpDXS,CpHMGS,CpWRKY17) for qRT–PCR analysis to validate the identified DEGs. The qRT–PCR results were consistent with the expression levels of DEGs, indicating that the results of this transcriptome analysis had high reliability (Fig. S20).
Discussion
In this study, we examined the dynamic changes in total triterpenoid content in C. paliurus leaves from August to November. The results demonstrated that total triterpenoid content was significantly higher in August and October, with lower levels observed in September and November. The distinct seasonal variations in triterpenoid accumulation can be attributed to the plants growth rhythm, metabolic processes, and seasonal redistribution of metabolites [8]. Our findings suggest that environmental factors and internal metabolic regulation both influence triterpenoid biosynthesis and accumulation during different stages of plant growth [9].
Triterpenoid biosynthesis processes of many plants have been discovered and reported, such as gardenia [10], coltwinter flower [11], and plum [12]. To further explore these variations, we performed metabolomics analysis on C. paliurus leaves across the same months, identifying 751 secondary metabolites. These metabolites include amino acids and their derivatives, phenolic acids, nucleotides, flavonoids, and, most significantly, 69 terpene metabolites. Among the terpenes, triterpenoids were the most abundant, followed by saponins, diterpenoids, and sesquiterpenoids, indicating the crucial role of triterpenoids in the plants response to environmental stimuli. The metabolomics data confirmed that triterpenoid levels were highest in August and November, closely aligning with the total triterpenoid content results. These findings align with previous studies on the ecological functions of terpenoids in plant growth and defense mechanisms [13, 14]. This further highlights the need for additional research on the role of triterpenoid metabolites in C. paliurus under varying environmental conditions.
Principal component analysis (PCA) of the metabolomic data revealed clear separation of samples based on their collection months, indicating significant changes in the terpenoid profile across the seasons. The clustering and K-means analysis showed that certain triterpenoid metabolites, such as ursolic acid, oleanolic acid, and their derivatives, were differentially expressed. For instance, ursolic acid exhibited upregulation in August and November, while metabolites like asiatic acid and corosolic acid showed different patterns of up- and down-regulation. These changes suggest that specific terpenoid biosynthesis pathways are activated at different times during the plants growth cycle, with notable regulatory shifts occurring from August to November. The results further emphasize the importance of investigating these metabolite dynamics for understanding C. paliurus adaptive responses to environmental stressors.
Gene expression analysis across the same time period revealed 15,951 DEGs, with downregulated genes playing a pivotal role in the regulation of triterpenoid biosynthesis. Notably, the MEP pathway was more active in November, while the MVA pathway dominated in August and September. The upregulation of β-amyrin synthase (β-AS) in August and November highlights its role in triterpenoid biosynthesis during these months. Similar trends have been observed in other species, such as Gardenia jasminoides and Tripterygium wilfordii, where temporal regulation of OSCs and P450s is crucial for triterpenoid production [10, 15]. The number of P450s family is large, the substrate specificity is strong, and the sequence similarity is low [16], and the P450 identified to be involved in triterpenoid nucleus modification is still less, and there are also differences in OSC, P450 and transcription factors in different plants. Zhou G L et al. believe that CYP71AU subfamily members play a role of hydroxylation in diterpenoid synthesis [17], which is similar to our annotation results on CYP71AU50. However, the variation in gene expression patterns across months suggests that the regulation of triterpenoid biosynthesis in C. paliurus is uniquely adapted to its seasonal growth cycle.
The co-expression network analysis identified several transcription factors, including MYB,WRKY, and bHLH, which exhibited strong correlations with key biosynthetic genes such as CYP716A15 and β-AS. These transcription factors likely play significant roles in modulating the expression of P450s and OSCs, which are essential for triterpenoid backbone modification [18, 19]. For instance, the correlation between ERF109 and CYP82D47 in the lightcyan1 module suggests a regulatory mechanism for the biosynthesis of corosolic acid and maslinic acid, similar to findings in other plant species [20]. Additionally, WRKY and ERF transcription factors, present in the tan and orange modules, may also regulate the biosynthesis of triterpenoids such as ursolic acid and asiatic acid [21]. while Whirly is different from our results. Whirly may regulate P450 associated with triterpene synthesis and participate in biological and abiotic stress and allelopathy [22, 23].
The observed seasonal variations in triterpenoid content in C. paliurus reflect the plant adaptive response, with peaks in October and lower levels in September and November. WGCNA analysis revealed gene modules that correlate strongly with these patterns, linking triterpenoid fluctuations to the expression of key biosynthetic genes. The lightcyan1 module, associated with maslinic acid, corosolic acid, and oleanolic acid, showed heightened gene activity in August and October, suggesting a response to environmental shifts that trigger triterpenoid biosynthesis. Similarly, the tan module correlated closely with ursolic acid content, showing increased expression in November. Genes within this module were enriched in the MEP pathway, contrasting with the MVA pathway, which dominated in August. This shift implies that specific biosynthetic routes are activated according to seasonal changes, potentially enhancing C. paliurus protective responses. The correlation between triterpenoid content and gene expression highlights the plant finely tuned response to external cues, suggesting that triterpenoid biosynthesis is both dynamic and adaptable to environmental factors, particularly during stress or seasonal transitions.
This study provides valuable insights into the biosynthesis and regulation of triterpenoids in C. paliurus leaves. The integration of metabolomics and transcriptomics analyses revealed critical metabolic pathways and gene networks involved in triterpenoid accumulation. Notably, the differential expression of key biosynthetic genes and the identification of transcription factors that regulate these pathways highlight potential targets for future research. These findings offer a foundation for the development of C. paliurus varieties with enhanced triterpenoid content, which could have significant implications for medicinal and industrial applications.
Conclusions
The leaf of C. paliurus provide ideal materials for studying the regulatory mechanisms of triterpenoid biosynthesis. By metabolomic analyses, we identified a total of 69 kinds terpenoids, in which there were 5 kinds of triterpenoid metabolites with high content and significant difference dynamics, namely, corosolic acid, asiatic acid, maslinic acid, ursolic acid and oleanolic acid. Integrative analysis of the metabolome and transcriptome showed that the 570 gene clusters with similar expression patterns to the downstream functional genes (β-AS,LUS,CYP176A15) identified by co-expression analysis. In the downstream pathway, CYP71D8 and CYP716A15 co-expressed with β-AS may generate oleanane type triterpenoids by modifying βamyrin, while CYP71AN24 and CYP98A2 co-expressed with LUS may play a key role in lupine type triterpenoids biosynthesis. Notably, MYB,Whirly,WRKY and bHLH families, which showed strong correlation with function genes, may play an important role in the regulation of P450 and OSC expression. A total of 20 modules were identified by WGCNA analysis, and CYP71AU50 and CYP716A15 in tan and orange modules may play a major role in the synthesis of oleanolic acid, ursolic acid and asiatic acid, while CYP82D47 in lightcyan1 module may be the hub gene for the biosynthesis of corosolic acid and maslinic acid. The results of this paper can provide scientific reference for breeding high-content triterpenoids varieties.
Materials and methods
Plant material
The plant material was identified by Professor Binghua Chen, College of Life Sciences, Fujian Normal University according to the voucher specimen of C. paliurus (Code number: CBH01302) stored in the Herbarium of Zhejiang University. Plant materials were collected from C. paliurus tree in Anxi Taoyuan organic tea farm, Taozhou Township of Anxi County. Sampling was permitted by Anxi Taoyuan organic tea farm Co., Ltd.
The C. paliurus leaves were collected from a managed plantation located in Taozhou Township, Anxi County, Fujian Province, China (E117°45′42′, N25°22′48′). This region is situated in a mountainous area renowned for its suitability for tea cultivation, characterized by a subtropical monsoon climate. The climate features hot and humid summers with temperatures averaging around 25–33 °C, and mild, dry winters with temperatures averaging around 5–15 °C, providing optimal conditions for agricultural practices. The soil in this region is well-drained, slightly acidic, and rich in organic matter, which supports the healthy growth of trees. The C. paliurus trees were grown in a managed plantation, where they were cultivated alongside tea plants. The plantation follows standard agricultural practices for the cultivation of both species, with the plants being at 7 years old in vegetative stage at the time of leaf collection. Healthy leaves were collected from 8 to 10 plants of similar height and morphology. Midsections (6–9 cm) from 10 to 30 leaves per plant were harvested and pooled for analysis. The leaf samples were frozen in liquid nitrogen and stored in an ultra-low temperature refrigerator. Leaf samples from August, September, October and November were divided into three biological replicates (8 M-ML1/2/3, 9 M-ML1/2/3, 10 M-ML1/2/3, 11 M-ML1/2/3) for the determination of total triterpenoid content, metabolomics and transcriptomics analysis.
Determination of total triterpenoids in leaves of C. paliurus
Samples of C. paliurus leaves were freeze-dried in vacuum for 3 days, then ground in a grinder. Take no less than 200 g representative sample, smash it with a sample grinder, pass a 0.425 mm standard mesh screen, and put the sample in a sealed container, stored at 0 ℃~20 ℃ for use. Weigh and remove 0.5 g (accurate to 0.0001 g) sample into 250 mL corked conical bottle, accurately add 50 mL anhydrous ethanol, cover the stopper tightly, cast evenly, and place the sample in ultrasonic extraction instrument for 1 h ultrasonic extraction, during which the sample is often cast, and mix evenly after extraction. Centrifuge an appropriate volume of 8 000 r/min for 10 min, and take the supernatant as the sample extraction solution for use. The extracted liquid was filtered with 0.22 μm filter membrane. And then dry the solvent methanol in a water bath at 60 ℃, add 5% vanillin-glacial acetic acid (0.1 mL) and perchloric acid (0.8mL), and react in a water bath at 60 ℃ for 20 min. Ice water bath for 3–5 min, quickly add 5 mL of ice acetic acid, mix well, stand for 10 min, and determine the absorbance at 550 nm wavelength. The principle of this method is that triterpenes in plants react with vanillin under acidic conditions to produce blue-purple molecules, which have maximum absorption at 550 nm wavelength, and the light absorption value is proportional to the total triterpene content. We use oleanolic acid to make the standard curve because it is a well-characterized and widely studied triterpenoid with a stable structure, which makes it ideal for use as a reference compound. Oleanolic acid is one of the most prevalent triterpenoids found in many medicinal plants, including C. paliurus. Its availability and well-documented properties allow for consistent and reliable quantification in spectrophotometric methods [24, 25].
The standard curve was obtained with the content of oleanolic acid as y and absorbance as x: y = 337.29x-15.31 (R2 = 0.9948). The total triterpenoid content is calculated as follows:
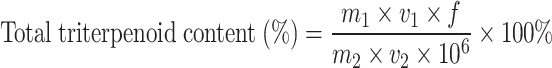 |
m1: The mass of oleanolic acid (µg) was obtained according to the standard curve; v1: extraction volume (mL) by adding methanol; f: dilution ratio of the extraction solution when measured; m2: quality of extracted leaf sample (g); v2: Volume (mL) of the solution to be measured during colorimetric determination [26].
UPLC-MS /MS was used to detect the terpenoids of C. paliurus
To analyze the dynamic changes in terpenoids of C. paliurus from August to October, metabolome analysis was conducted. Freeze-dried leaves were ground using a Retsch MM 400 grinder at 30 Hz for 1.5 min. A 100 mg sample was mixed with 1.2 mL of 70% methanol, vortexed for 30 s, and left overnight at 4 °C. After centrifugation and filtration, the supernatant was analyzed via UPLC/MS/MS. Quantification was done via multiple reaction monitoring on a triple quadrupole mass spectrometer. Relative substance concentrations were inferred from peak areas. MetWare’s database was used for metabolite identification. Statistical analyses, including PCA, HCA, and OPLS-DA, were performed in R, followed by KEGG database enrichment to identify significant metabolites [27].
Screening and analysis of terpenoid differential metabolites
The MetaboAnalyst R package was installed in R, and the VIP value of the OPLS-DA model was obtained through calculation and setting the confidence interval of OPLS-DA. The screening criteria were metabolites with fold change ≥ 2, fold change ≤ 0.5 and VIP ≥ 1 [28]. The upsetR chart is drawn using tbtools. Select upset plot (up to any set), set 6 groups of data, and click Start. K-means clustering analysis refers to the method of Jia et al. [29]. Refer to the annotation method [30].
Transcriptomics analysis
Total RNA was prepared separately from leaf samples of C. paliurus using TRIzol reagent (Invitrogen, California, USA). mRNAs and noncoding RNAs (rRNA-depleted RNAs) were enriched by removing rRNAs from total RNA with a Ribo-Zero™ rRNA Removal Kit (Illumina, United States). Libraries were constructed using the TruSeq Stranded Total RNA Prep kit (Illumina, San Diego, USA) and sequenced on the Illumina NextSeq 500 platform. After removal of adapters, low quality reads were removed by FastQC software with more than 5% unscored bases, or with 50% of the bases with low quality score (PHRED scor 5) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmed sequences were aligned to the reference genome using HISAT2 [31]. The expression profiles (FPKM values and read counts) of mRNAs were calculated using StringTie (version 1.3.1) [32]. Differential expression analysis of mRNA was performed by screening based on an absolute fold change ≥ 2.0, p-value < 0.05 and FDR < 0.05. Co-expression analysis was performed based on the Pearson Correlation Coefficient (PCC ≥ 0.8, p-value < 0.05).
Functional annotation of genes and identification of differentially expressed genes
KEGG annotation of sequencing data was performed using ClusterProfile in R software [33], with the functional pathways and classifications retrieved from the KEGG database (http://www.genome.jp/kegg/). For gene functional annotation, we used several databases, including eggNOG (http://eggnogdb.embl.de/), GO (http://geneontology.org/), and COG (http://www.ncbi.nlm.nih.gov/COG/). The similarity threshold for gene annotation was set to 90% identity for significant matches in all BLAST-based searches. The fasta sequence file of the longest transcript from C. paliurus was uploaded to the Plant Transcription Factor Database, where plant transcription factors from Arabidopsis thaliana were selected for classification predictions. The prediction score cutoff for including significant transcription factors was set to 0.8, meaning only predictions with a score ≥ 0.8 were considered in the analysis. DEGs were identified using the DESeq2 package, comparing paired samples from different months. DEGs were determined based on the following thresholds: False Discovery Rate (FDR) < 0.05 and |log2 fold change| > 1. The versions of the databases used for annotation and functional classification are as follows: eggNOG database v6.0, release date: May 2022, KEGG database v105, release date: January 2023, COG database v2.0, release date: January 2021 [34], Plant Transcription Factor Database v5.0, release date: December 2022.
WGCNA analysis
All the differential gene expression (FPKM), the relative contents of 5 triterpenes and the expression of transcription factors (FPKM) matrix files were imported into R, and the Mfuzz package was used to analyze DEGs. After pre-processing the missing or outlier values, the random seed number was set to 123 and the cluster number to 10. The standardized data was extracted to extract the clusters to which all the differential genes belonged, and finally the clustering information was derived. The WGCNA package [35] in R software was used to filter the gene expression matrix of 12 samples, remove more than 90% of the data with FPKM < 1, and delete the outliers. Filter the soft threshold and set the threshold of R2 to 0.8. The threshold of module merging was set at 0.5, the genes in the same module had the same expression pattern, and the number of genes in the module was ≥ 30. By calculating the eigenvalues (ME) of 5 triterpenoids and modules, the phenotypes were associated with modules. The module network was visualized by Cytoscape software, and the scores of each node were calculated by cytoHubba plug-in in Cytoscape software. In cytoHubba plug-in, MCC was selected to calculate the top 30 genes in each module’s connectivity, order them, and annotate the gene functions of hub genes in the top ranking of connectivity, so as to accurately discover the hub genes related to 5 major triterpenoids.
Quantitative real-time qPCR validation
To validate the identified key genes, we selected 8 genes encoding DEGs for quantitative PCR analysis. Sequence-specific primers of all selected genes for RT-qPCR analysis (Table S15) were designed using Primer 5.0. The mRNA level of GAPDH was used as the internal reference. Total RNA from leaves of C. paliurus was extracted using Rapid Plant RNA Extraction Kit (HERUIBIO, Fujian, China). RNA concentration was determined with DS-11 Spectrophotometer (DeNovix, USA), The first Strand cDNA was synthesized using HRbio™ III 1st Strand cDNA Synthesis SuperMix for qPCR (OneStep gDNA Removal) one-step genomic DNA synthesis kit. Quantitative analysis was performed using the CFX96TM Real-Time System (Bio-Rad Laboratories, USA). The qPCR amplification procedure of these genes was as follows: 95 ℃ for 5 min, 1 cycle; 95 ℃ 10 s, 60 ℃ 30 s, 39 cycles. Data were calculated using 2 −△△Ct, and at least 3 repeated experiments were performed for each gene.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Not applicable.
Author contributions
TX and DC designed the research. DC performed the data analysis and wrote the manuscript. XC collected the experimental materials and performed the statistical analysis. XZ performed the data analysis. JZ collected the experimental materials and performed the statistical analysis. TX performed the data analysis and wrote the manuscript.All authors read and approved the final manuscript.
Funding
This work was supported by Special Sci-tech Team Commissioner of Fujian province (Grant No. T202005007).
Data availability
Raw sequencing data for RNA-seq have been deposited and is available in the BIG Sub system under BioProject accession number CRA004539 (https://ngdc.cncb.ac.cn/gsa/s/7qFh666G).
Declarations
Ethics approval and consent to participate
Plant materials were collected from C. paliurus tree in Anxi Taoyuan organic tea farm, Taozhou Township of Anxi County (E117.45’42", N25.22’48"). Sampling was permitted by Anxi Taoyuan organic tea farm Co., Ltd.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Duo Chen, Email: chenduo@fjnu.edu.cn.
Ting Xue, Email: xueting@fjnu.edu.cn.
References
- 1.Zheng X, Xiao H, Su J, Chen D, Chen J, Chen B, et al. Insights into the evolution and hypoglycemic metabolite biosynthesis of autotetraploid Cyclocarya paliurus by combining genomic, transcriptomic and metabolomic analyses. Ind Crop Prod. 2021;173:114154. 10.1016/j.indcrop.2021.114154. [Google Scholar]
- 2.Zheng X, Xiao H, Chen J, Zhu J, Fu Y, Ouyang S, et al. Metabolome and whole-transcriptome analyses reveal the molecular mechanisms underlying hypoglycemic nutrient metabolites biosynthesis in Cyclocarya paliurus leaves during different harvest stages. Front Nutr. 2022;9:851569. 10.3389/fnut.2022.851569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mo J, Tong Y, Ma J, Wang K, Feng Y, Wang L, et al. The mechanism of flavonoids from Cyclocarya paliurus on inhibiting liver cancer based on in vitro experiments and network pharmacology. Front Pharmacol. 2023;14:1049953. 10.3389/fphar.2023.1049953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lou D, Zhang X, Jiang C, Zhang F, Xu C, Fang S, et al. 3β, 23-Dihydroxy-12-ene-28-ursolic acid isolated from cyclocarya paliurus alleviates NLRP3 inflammasome-mediated gout via PI3K-AKT-mTOR-Dependent autophagy. Evid-Based Compl ALT. 2022;2022(5541232). 10.1155/2022/5541232. [DOI] [PMC free article] [PubMed]
- 5.Shen Y, Peng Y, Zhu X, Li H, Zhang L, Kong F, et al. The phytochemicals and health benefits of Cyclocarya paliurus (Batalin) Iljinskaja. Front Nutr. 2023;10:1158158. 10.3389/fnut.2023.1158158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khouya T, Ramchoun M, Elbouny H, Hmidani A, Bouhlali E, Alem C. Loquat (Eriobotrya japonica (Thunb) Lindl.): evaluation of nutritional value, polyphenol composition, antidiabetic effect, and toxicity of leaf aqueous extract. J Ethnopharmacol. 2022;296:115473. 10.1016/j.jep.2022.115473. [DOI] [PubMed] [Google Scholar]
- 7.Fukushima EO, Seki H, Ohyama K, et al. CYP716A Subfamily members are multifunctional oxidases in Triterpenoid Biosynthesis. Plant Cell Physiol. 2011;52(12):2050–61. 10.1093/pcp/pcr146. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, He Y, Sun J, Zhang J, Zhan Y. Effects of nitrogen deficiency on physiology and growth of Fraxinus mandshurica. Pak J Bot. 2018;50(1):179–87. [Google Scholar]
- 9.Sun C, Shang X, Ding H, Cao Y, Fang S. Natural variations in flavonoids and triterpenoids of Cyclocarya paliurus leaves. J for Res. 2021;32(2):805–14. 10.1007/s11676-020-01139-1. [Google Scholar]
- 10.Pan Y, Chen D, Li L. 2020. Analysis of Transcriptonme and Excavation Iridoid Biosynthesis Pathway Key Genes in Gardenia jasminoides Ellis. Molecular Plant Breeding, 2020, 18(12):3923–3931. 10.13271/j.mpb.018.003923
- 11.He R, Wang X, Han Y, Liu J, Du C, Wang L. 2020. Analysis of key genes and their expression characteristics related to terpenoid biosynthesis in Tussilago farfara based on transcriptome sequencing. Chinese Traditional and Herbal Drugs, 2020, 51(20): 5302–5310.
- 12.Zheng X, Luo X, Xu H, Zhan R, Chen W. Transcriptomic Analysis and systematic mining of genes involved in Biosynthetic Pathway of Triterpenoid Saponins in Ilex Asprella. Modernization Traditional Chin Med Materia Medica-World Sci Technol. 2014;16(7):1505–12. [Google Scholar]
- 13.Alwattar MT, Yaqub HM. Terpenoids as natural allelopathic compounds in plants. Rafidain J Sci. 2023;32(4):106–16. 10.33899/rjs.2023.181268. [Google Scholar]
- 14.Shi R, Xiong B, He S, Liu C, Ben-Asher J, Horowitz AR, et al. Comparative metabolic profiling of root, leaf, fruit, and stem tissues of Panax notoginseng. Int J Food Prop. 2022;25(1):1132–45. 10.1080/10942912.2022.2071294. [Google Scholar]
- 15.Hansen NL, Kjaerulff L, Heck QK. TripterWilfordiifordii cytochrome P450s catalyze the methyl shift and epoxidations in the biosynthesis of triptonide. Nat Commun. 2022;13(1):5011. 10.1038/s41467-022-32667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuo H, Huang H, Lin YCD, et al. Enzyme activity of Natural products on Cytochrome P450. Molecules. 2022;27(2):515–33. 10.3390/molecules27020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou GL, Li Y, Pei F, et al. Chromosome-scale genome assembly of Rhododendron molle provides insights into its evolution and terpenoid biosynthesis. BMC Plant Biol. 2022;22(1):1–17. 10.1186/s12870-022-03720-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Niu Y, Xu J, et al. Discovery of WRKY transcription factors through transcriptome analysis and characterization of a novel methyl jasmonate-inducible PqWRKY1 gene from Panax quinquefolius. Plant Cell Tiss Organ Cult. 2013;114:269–77. 10.1007/s11240-013-0323-1. [Google Scholar]
- 19.Mertens J, Pollier J, Vanden Bossche R, et al. The bHLH transcription factors TSAR1 and TSAR2 regulate triterpene saponin biosynthesis in Medicago truncatula. Plant Physiol. 2016;170:194–210. 10.1104/pp.15.01645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu YY, Zhu CQ, Xu CJ, et al. Integration of Metabolite Profiling and Transcriptome Analysis reveals genes related to volatile terpenoid metabolism in Finger Citron(C. Medica var. Sarcodactylis). Molecules. 2019;24(14):2564–85. 10.3390/molecules24142564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zu KL, Dong SB, Li JX, Xu SJ, Zhao LC. Differentially expressed genes analysis of terpenoid biosynthesis related to aril development in Celastrus orbiculatus Thunb. Plant Sci J. 2017;35(2):276–82. [Google Scholar]
- 22.Yan Y, Liu W, Wei Y, et al. MeCIPK23 interacts with whirly transcription factors to activate abscisic acid biosynthesis and regulate drought resistance in Cassava. Plant Biotechnol J. 2020;18(7):1504–6. 10.1111/pbi.13321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y, Liu Z, Ye Z, Luo R, Pu J, Zhang H. 2021. Identification of Mango Whirly Gene and Its Expression Analysis in the Pathogen Infection. Acta Bot. Boreal, Occident. Sin., 2021, 41(1): 37–45. 10.7606/j.issn.1000-4025.2021.01.0037
- 24.Hiai S, Oura H, Nakajima T. Color reaction of some sapogenins and saponins with vanillin and sulfuric acid. Planta Med. 1976;29(2):116–22. 10.1055/s-0028-1097639. [DOI] [PubMed] [Google Scholar]
- 25.Siddiqui BS, Afshan F, Gulzar T, Hanif M. Tetracyclic triterpenoids from the leaves of Azadirachta indica. Phytochemistry. 2004;65(16):2363–7. 10.1016/j.phytochem.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 26.Huang W, Xue A, Niu H, Jia Z, Wang J. Optimised ultrasonic-assisted extraction of flavonoids from Folium eucommiae and evaluation of antioxidant activity in multi-test systems in vitro. Food Chem. 2009;114(3):1147–54. 10.1016/j.foodchem.2008.10.079. [Google Scholar]
- 27.Shang X, Huang D, Wang Y, Xiao L, Ming R, Zeng W, et al. Identification of nutritional ingredients and medicinal components of Pueraria lobata and its varieties using UPLC-MS/MS-based metabolomics. Molecules. 2021;26(21):6587. 10.3390/molecules26216587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li SF, Guo YJ, Li JR, Zhang DX, Wang BX, Li N, Deng CL, Gao WJ. The landscape of transposable elements and satellite DNAs in the genome of a dioecious plant spinach (Spinacia oleracea L). Mol DNA. 2019;10:3. 10.1186/s13100-019-0147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia K, Zhang X, Meng Y, Liu S, Liu X, Yang T, et al. Metabolomics and transcriptomics provide insights into the flavonoid biosynthesis pathway in the roots of developing Aster tataricus. J Plant Res. 2023;136(1):139–56. 10.1007/s10265-022-01426-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun C, Fang S, Shang X. Triterpenoids biosynthesis regulation for leaf coloring of wheel wingnut (Cyclocarya paliurus). Forests. 2021a;12(12):1733. 10.3390/f12121733. [Google Scholar]
- 31.Daehwan K, Ben L, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–67. 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–80. 10.1093/nar/gkh063. PMID: 14681412; PMCID: PMC308797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res., 28(1): 33 – 6. 10.1093/nar/28.1.33. PMID: 10592175; PMCID: PMC102395. [DOI] [PMC free article] [PubMed]
- 35.Peter L, Steve H. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(559):1–13. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data for RNA-seq have been deposited and is available in the BIG Sub system under BioProject accession number CRA004539 (https://ngdc.cncb.ac.cn/gsa/s/7qFh666G).