

Abstract
The CCAAT/enhancer binding protein δ (C/EBPδ, CRP3, CELF, NF-IL6β) regulates gene expression and plays functional roles in many tissues, such as in acute phase response to inflammatory stimuli, adipocyte differentiation, and mammary epithelial cell growth control. In this study, we examined the expression of human C/EBPδ (NF-IL6β) gene by epidermal growth factor (EGF) stimulation in human epidermoid carcinoma A431 cells. NF-IL6β was an immediate-early gene activated by the EGF-induced signaling pathways in cells. By using 5′-serial deletion reporter analysis, we showed that the region comprising the –347 to +9 base pairs was required for EGF response of the NF-IL6β promoter. This region contains putative consensus binding sequences of Sp1 and cAMP response element-binding protein (CREB). The NF-IL6β promoter activity induced by EGF was abolished by mutating the sequence of cAMP response element or Sp1 sites in the –347/+9 base pairs region. Both in vitro and in vivo DNA binding assay revealed that the CREB binding activity was low in EGF-starved cells, whereas it was induced within 30 min after EGF treatment of A431 cells. However, no change in Sp1 binding activity was found by EGF treatment. Moreover, the phosphatidylinositol 3 (PI3)-kinase inhibitor (wortmannin) and p38MAPK inhibitor (SB203580) inhibited the EGF-induced CREB phosphorylation and the expression of NF-IL6β gene in cells. We also demonstrated that CREB was involved in regulating the NF-IL6β gene transcriptional activity mediated by p38MAPK. Our results suggested that PI3-kinase/p38MAPK/CREB pathway contributed to the EGF activation of NF-IL6β gene expression.
INTRODUCTION
C/EBPδ belongs to the CCAAT/enhancer binding protein family that is involved in tissue differentiation, liver regeneration, metabolism, healing, and immune response (Ramji and Foka, 2002). All of the members of the C/EBP family have a C-terminal leucine zipper domain for dimerization and a basic domain for DNA binding. Recently, six distinct C/EBP isoforms have been identified: C/EBPα, C/EBPβ (also known as NF-IL6, LAP, AGP/EBP, IL-6DBP, or NF-M), C/EBPγ (immunoglobulin [Ig]/EBP or GPE1BP), C/EBPδ, C/EBPε (CRP1), and CHOP (gadd153) (Lekstrom-Mines and Xanthopoulos, 1998). The majority of the family members recognize similar DNA sequences in their target genes, where they bind either as homodimers or heterodimers with other C/EBP family members or with other leucine zipper factors (Hsu et al., 1994).
C/EBPδ has been implicated in the control of adipogenesis and in mediating the acute phase response to inflammatory stimuli (Wedel and Ziegler-Heibrock, 1995; Mandrup and Lane, 1997; Tanaka et al., 1997). Studies of the expression of mouse C/EBPδ show that it is typically undetectable in most cell types and tissue but that it is rapidly induced by stimulators, such as interleukin (IL)-1 (Okazaki et al., 2002), lipopolysaccharide (LPS) (Kravchenko et al., 2003; Liu et al., 2003), interferon (IFN)-α, IFN-γ (Tengku-Muhammad et al., 2000), IL-6 (Kamaraju et al., 2004), prostacyclin (Belmonte et al., 2001), and tumor necrosis factor-α (Cardinaux et al., 2000). Moreover, it has been reported that C/EBPδ expression is involved in cell cycle control. C/EBPδ mRNA and protein levels are markedly induced in cultured mouse mammary epithelial cells during G0 growth arrest and apoptosis initiated by serum and growth factor withdrawal (O'Rourke et al., 1997). It also plays an important role in inducing growth arrest of mammary epithelium cells by oncostatin M and in promoting prostate epithelial cell growth arrest and/or apoptosis after androgen withdrawal (Yang et al., 2001; Hutt and DeWille, 2002). In mouse embryonic fibroblasts, the lacking of C/EBPδ results in genomic instability and centrosome amplification in vitro. These results suggest that C/EBPδ may play a substantial role in tumor suppression in vivo (Hung et al., 2004).
Studies on the signaling pathways that regulate transcription of C/EBPδ are still limited. Species-specific autoregulation has been proposed for the regulation mechanism of the C/EBPδ gene. For example, the autoregulation of the rat C/EBPδ is through two downstream binding sites at +3350 and +3700 of the C/EBPδ gene (Yamada et al., 1998). In contrast, the 5′ ends of the mouse and the ovine C/EBPδ gene are sufficient for autoactivation (O'Rourke et al., 1999; Davies et al., 2000). STAT3 and Sp1 mediated the IL-6–induced mouse C/EBPδ gene expression in hepatoma cells (Cantwell et al., 1998). STAT3 also is involved in the regulation of C/EBPδ gene expression in G0 growth-arrested mouse mammary epithelial cells (Hutt etal., 2000). In preadipocytes, activation of extracellular signal-activated kinase (ERK) and CREB was shown to increase the expression of mouse C/EBPδ (Belmonte et al., 2001). However, there is no report about the transcription regulation of human C/EBPδ (NF-IL6β).
CREB is a member of the leucine zipper class of cAMP-responsive element binding proteins/activation transcription factor (CREB/ATF). It responds to a variety of external signals and plays important roles in cell proliferation and differentiation (De Cesare et al., 1999; Shaywitz and Greenberg, 1999). CREB requires phosphorylation at Ser133 to become active that is induced by cyclical AMP-elevating agents, mitogens, or exposure to cellular stresses (De Cesare et al., 1999; Shaywitz and Greenberg, 1999). When activated by mitogenic stimuli, such as the isoforms of mitogen-activated protein kinase (MAPK)-activated protein kinase 1 (MAPKAP-K1, also called RSK) and stress-activated protein kinase phosphorylate CREB at Ser133 in vitro (Deak et al., 1998; Caivano and Cohen, 2000; Wiggin et al., 2002). Recent evidence indicates that p90rsk may be responsible for CREB phosphorylation at Ser133, both in vitro and in vivo, in response to growth factor stimulation (Böhm et al., 1995; Xing et al., 1996; Monaco and Sassone-Corsi, 1997). Also, the stress-induced phosphorylation of CREB is prevented by SB203580, an inhibitor of another MAPK family member including stress-activated protein kinase 2 (SAPK2) and p38MAPK, which is a component of a distinct signal transduction pathway (Deak et al., 1998).
The epidermal growth factor (EGF) receptor (EGFR) is a 170-kDa transmembrane glycoprotein with intrinsic tyrosine kinase activity (Carpenter, 1987; Hunter and Cooper, 1985). On ligand binding, the EGFR undergoes autophosphorylation and initiates multiple intracellular signaling cascades, leading to the induction of cell growth (Hill and Treisman, 1995; Treisman, 1996). EGFR activation also induces other signaling pathways that turn off EGFR signaling through endocytosis. Attenuation of the signaling is important for the control of EGFR mitogenic properties (Wiley et al., 1991; Sorkin and Waters, 1993). Many effector molecules were reported to be involved in the EGF signal cascades, including phospholipase C-γ1, Ras-mitogen-activated protein kinase kinase (MEK)-MAPK, phosphatidylinositol 3 (PI3)-kinase, Akt, Src, and STATs (Olayioye et al., 2000; Yarden and Sliwkowski, 2001). Whereas EGF is a potent mitogen, it paradoxically induces apoptosis in cells that overexpress EGFR such as the human epidermoid carcinoma A431 cells (Haigler et al., 1978). Its growth in monolayer culture had been shown to exhibit a biphasic response to EGF. A431 cells are weakly stimulated by picomolar concentrations of ligand but exhibit a marked inhibition of proliferation in the presence of nanomolar concentration of EGF (Barnes, 1982; Kawamoto et al., 1984).
In this study, we demonstrated that EGF up-regulated the transcriptional activity of NF-IL6β gene under growth arrest condition in A431 cells. We further showed that CREB and Sp1 bound to the cAMP response element (CRE) and Sp1 sites of NF-IL6β gene promoter region, respectively. EGF induced the phosphorylation of p38MAPK in A431 cells. The activation of p38MAPK and CREB was involved in EGF-activated PI3-kinase signaling pathway. Pretreatment with p38 inhibitor SB203580 reduced both CREB phosphorylation in Ser133 and the transcriptional product of NF-IL6β gene after EGF stimulation. These results suggested that the EGF-induced NF-IL6β gene expression was mediated through the p38MAPK signaling pathway and CREB activation.
MATERIALS AND METHODS
Materials
Human EGF was purchased from Peprotech (Rocky Hill, NJ). All the chemical inhibitors, wortmannin, SB203580, PD98059, SP200126, anisomycin, and cycloheximide were obtained from Calbiochem (San Diego, CA). Antibodies against CREB, Sp1, Sp3, COX-1, NF-IL6β, and p38 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phosphorylated CREB and p38 antibodies were purchased from Cell Signaling Technology (Beverly, MA). Lipofectamine 2000, Dulbecco's modified Eagle's medium, and Opti-MEM medium were obtained from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Hyclone Laboratories (Logan, UT). All other reagents used were of the highest purity obtainable.
Methods: Cell Culture and Transfection
Human epidermoid carcinoma cell line, A431 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 units/ml penicillin. Transient transfection into A431 cells was carried out using Lipofectamine 2000 transfection reagent. For experiments with chemical inhibitors, the following concentrations and compounds were used: 0.1 μM wortmannin, 10 μM SB203580, 20 μM PD98059, 50 nM SP200126, 50 ng/ml anisomycin, and 10 μg/ml cycloheximide. All inhibitors were added 30 min before the 50 ng/ml EGF treatment of cells.
Western Blotting
An analytical 10% SDS-polyacrylamide slab gel electrophoresis was performed. The cell lysates were prepared from control and EGF-treated cells, and 70 μg of protein of each was analyzed. For immunoblotting, proteins in the SDS gels were transferred to a polyvinylidene difluoride membrane by an electroblot apparatus. Immunoblot analysis was carried out with goat or rabbit IgG antibody coupled to horseradish peroxidase. An enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ) was used for detection.
Reporter Plasmids and Luciferase Assay
Cells were transfected with plasmids by Lipofectamine 2000 according to the manufacturer's instruction. The longest promoter fragment was cloned by PCR of genomic DNA from strain 129 mice or A431 cells, and inserted into the TA cloning vector. The primers used are as follows: C/EBPδ-2015, 5′-CCAAGTCTGACAGTGCTCTCTG; C/EBPδ+14; 5′-GGGAAGCTTCCTGGCGTCCA AGTTGGCTG-3′; NF-IL6β-1717; 5′-GGGGTACCTTGGTGTTCCGACGCAGATC-3′; and NF-IL6β+9; 5′-GGGAAGCTTGGCTGTCACCTCGCTGGGCC-3′. The luciferase reporter plasmids containing various fragments of mouse C/EBPδ promoter (C/EBPδ-1680/+14, C/EBPδ-1268/+14, C/EBPδ-1065/+14, and C/EBPδ-275/+14) were generated from the C/EBPδ-2015 by digestion with various restriction enzymes, StuI, PmlI, SspI, or SmaI. The shorter fragment of NF-IL6β promoter, NF-IL6β-347/+9 was generated by PCR with primers NF-IL6β+9 and NF-IL6β-347, 5′-GGGGTACCGAGGAGGTTCCAAGCCCAC-3′, and digested with KpnI and HindIII. These treated fragments were subcloned into the multicloning sites of the promoterless vector pGL2-Basic (Promega, Madison, WI). The numbers in parentheses indicate the nucleotide position with respect to the transcriptional initiation site. Other site-directed mutagenesis reporters of the NF-IL6β promoter (NF-IL6β-347/+9mC, NF-IL6β-347/+9mSp1-1 NF-IL6β-347/+9mSp1-2, NF-IL6β-347/+9mCmSp1-1, NF-IL6β-347/+9mdSp1, NF-IL6β-347/+9mCmSp1-2, NF-IL6β-347/+9mCmdSp1) were derived from NF-IL6β-347/+9. Plasmid pGL2-NF-IL6β1xCRE was derived by inserting one copy of the DNA fragment containing CRE site (sequence as below NF-IL6βCRE) into the SmaI site of the pGL2-Promoter vector (Promega). Transfected cells were seeded in growth medium with or without EGF. Sixteen hours after transfection, cells were harvested and assayed for luciferase activity. Luciferase activities were normalized with the amount of protein in the cell lysate.
Reverse Transcription (RT)-PCR
A431 cells were maintained for 6 h in serum-free medium and restimulated with EGF under various time courses and chemical compounds treatment. Total RNA was isolated using the TRIzol RNA extraction kit, and 1 μg of RNA was subjected to RT-PCR with SuperScriptII. The NF-IL6β–specific primers 5′-AGCGCAACAACATCGCCGTG-3′ and 5′-GTCGGGTCTGAGGTATGGGTC-3′ were used for analysis. The β-actin primers sense strain, 5′-CCCAAGGCCAACCGCGAGAAG-3′ and antisense strain, 5′-TCTTCATTGTGCTGGGTGCCA-3′ were used as controls. The PCR products were separated by 1% agarose-gel electrophoresis and visualized with ethidium bromide staining.
Nuclear Extract and Gel Shift Assays
Gel shift assays were carried out essentially as described previously (Kawamoto et al., 1984). Briefly, the 32P-labeled oligonucleotide probes (–0.2 to 0.5 ng) containing the CRE, Sp1-1, or Sp1-2 site were incubated with 8 μg of nuclear extracts or 1 μl of in vitro-translated CREB in the specific binding buffer, as described below, containing 1 μg of poly(dI-dC). After 20 min of incubation at room temperature, the reaction mixtures were resolved in a 5% native polyacrylamide gel (acrylamide/bisacrylamide ratio, 30:1) at 4°C, and the specific protein complexes were visualized by autoradiography. The CRE binding buffer contained 10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM dithiothreitol (DTT), 1 mM EDTA, and 10% (vol/vol) glycerol. The Sp1 binding buffer contained 20 mM HEPES, pH 7.9, 0.1 mM KCl, 2 mM MgCl2, 15 mM NaCl, 0.2 mM EDTA, 5 mM DTT, 10% (vol/vol) glycerol, and 2% (wt/vol) polyvinyl alcohol. For antibody supershifting experiments, 1 μg of various indicated antibodies, such as α-CREB, α-Sp1, and α-Sp3 or control rabbit IgG was included in the binding reaction mixture. For competition experiments, a 100-fold molar excess of unlabeled wild-type or mutant oligonucleotides was included in the binding reaction mixture. The sense strand sequences of various oligonucleotides used are as follows: NF-IL6βCRE (hCRE), 5′-GGGGCGTGCACGTCAGCCGGG-3′; NF-IL6βmCRE (muthCRE), 5′-GGGGCGTGGATCCCAGCCGGG-3′; NF-IL6βSp1-1, 5′-AAGGCTCGGGGCGGCTCCGGGG-3′; NF-IL6βSp1-2, 5′-CCGGAGTCGGGGCGGGGCGTGC-3′; mouse C/EBPδCRE (mCRE), 5′-GGGGCGTGCGCGTCAGCTGGG-3′; and consensus Sp1 oligonucleotide (cSp1), 5′-ATTC GATCGGGGCGGGGCGAGC-3′.
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation (ChIP) assay was carried out essentially as described previously (Wang et al., 2003) with a minor modification. Briefly, A431 cells, with or without prior stimulation with EGF, were treated with 1% formaldehyde for 15 min. The cross-linked chromatin was then prepared and sonicated to an average size of 300–400 base pairs before be immunoprecipitated with antibody specific to CREB, pCREB, and Sp1 or control rabbit IgG at 4°C for overnight. After reversal of cross-linking, the immunoprecipitated chromatin was amplified by PCR with various sets of primers as indicated. The amplified DNA products were resolved by agarose gel electrophoresis and confirmed by sequencing. For PCR amplification of specific regions of the NF-IL6β genomic locus, the NF-IL6β-347 and NF-IL6β+9 were used.
RESULTS
Induction of NF-IL6β Expression by EGF Treatment
Treatment of A431 cells with EGF resulted in a rapid increase in the steady-state level of NF-IL6β mRNA without effect on β-actin mRNA level (Figure 1A). NF-IL6β mRNA induction was detectable in 30 min after EGF treatment and was maximal after 2 h with an increase of four- to fivefold. To assess whether NF-IL6β protein expression also was correlated with transcriptional regulation by EGF, Western blotting with NF-IL6β–specific antibodies was performed. The EGF-induced NF-IL6β protein expression was elevated in 30 min and sustained up to 2 h (Figure 1B). The induction of NF-IL6β protein expression was correlated with that of NF-IL6β mRNA increasing as shown in Figure 1A. From Figure 1C, treatment with cycloheximide did not affect the induction of NF-IL6β mRNA by EGF. It indicated that de novo protein synthesis was not required for the EGF response.
Figure 1.
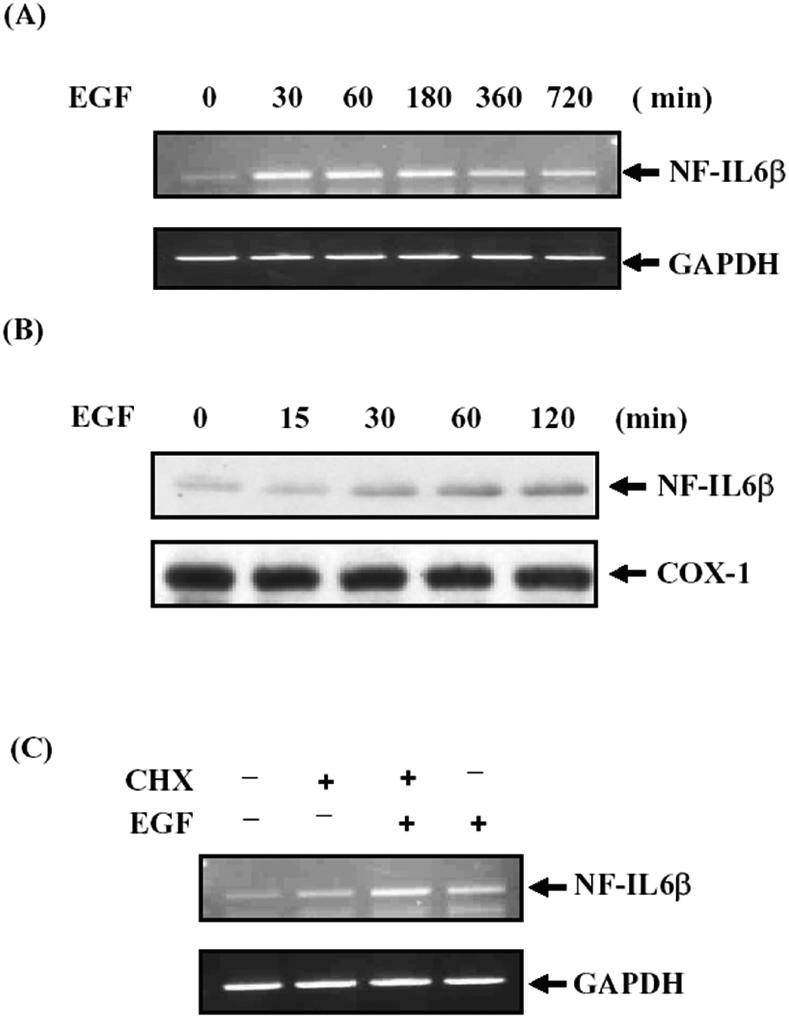
NF-IL6β production was increased by EGF in A431 cells. (A and B) A431 cells were starved for 6 h and then restimulated with 50 ng/ml EGF. Total RNA or cell lysates were harvested for different time points as indicated for examining by RT-PCR and Western blotting analysis of NF-IL6β mRNA and protein level, respectively. (C) Effect of cycloheximide treatment on the transcription of NF-IL6β mRNAs. The A431 cells were starved for 6 h and then pretreated with cycloheximide for 20 min before addition of EGF. Total RNA was isolated after 1 h of EGF treatment for RT-PCR assay.
Identification of the EGF Responsive Element in NF-IL6β Promoter Region
By comparing with the proximal region sequences of human, mouse, and rat C/EBPδ promoters, several conserved transcription factor binding sites, including Sp1 and CRE motifs were found. However, no acute phase response element (APRE) exists in the human NF-IL6β promoter (Figure 2A). As shown in Figure 2B, transient transfection with full-length mouse C/EBPδ promoter, –1680/+14 base pairs, or human NF-IL6β promoter, –1717/+9 base pairs, resulted in an average of 4.5-fold increase in luciferase activity upon EGF treatment. These results demonstrated that the 5′-flanking regions of the NF-IL6β gene ranging from –1717 to +9 base pairs and the C/EBPδ gene range from –1680 to +14 base pairs provided inducibility for EGF response. To further determine the EGF responsive element in them, the reporters containing 5′-serial deletion of NF-IL6β and C/EBPδ promoter were performed. Both 5′-serial deletion of promoter regions of mouse C/EBPδ, –275/+14 base pairs, and human NF-IL6β, –347/+9 base pairs showed the same EGF inducibility as the individual full-length reporter. It suggested that the proximal regions near the transcriptional initiation sites were important for EGF activity in A431 cells.
Figure 2.

Mapping of promoter elements required for EGF activation of the NF-IL6β reporter gene. (A) Putative conserved sites alignment of promoter sequences of human, mouse and rat C/EBPδ genes. The consensus sequences of the Sp1/APRE, CRE, and TATA sites are blocked. The mutated sequence in each individual construct is shown in italics and bold. (B) Activation of the C/EBPδ and NF-IL6β promoter by EGF in A431 cells. A431 cells transfected with various luciferase reporters as indicated were stimulated with or without EGF. The average fold inductions by EGF for each construct are shown. The minimal EGF responsive regions of C/EBPδ and NF-IL6β gene were represented by bold. (C and D) Characterization of heterologous reporters containing APRE or CRE elements contributed to EGF response. The heterologous reporters were transiently transfected to A431 cells and treated with or without EGF for 15 h. (E) Functional roles of binding sites for Sp1- or CREB-related transcriptional factors in the NF-IL6β promoter activity. A431 cells transfected with various luciferase reporters as indicated were stimulated with or without EGF, and the lysates were analyzed for luciferase activity (left). Similar results were obtained from three independent experiments, and the data shown here were from one representative assay. The average fold induction (mean ± SD, n = 3) by EGF for each construct was analyzed (right).
It was previously reported that IL-6-induced mouse C/EBPδ transcription is through the APRE and Sp1 motifs (Okazaki et al., 2002). To clarify the function of Sp1/APRE region in human NF-IL6β promoter, DNA gel-shift assay using the predicted APRE-like oligonucleotide 5′-GGCTCCGGGGGGCTCCCAGGGCG-3′ of human NF-IL6β promoter was performed. However, no slow mobility shifting pattern was observed (our unpublished data). An APRE-like oligonucleotide was inserted into the pGL2 Promoter vector containing a simian virus 40 (SV40) promoter. The luciferase reporter results indicated that the human APRE-like site and mouse APRE site did not confer EGF activity on the SV40 promoter in A431 cells (Figure 2C). However, EGF could induce the promoter activity of the heterologous reporter containing CRE site (Figure 2D). These results suggested that the CRE site, but not the APRE site, contributed to the EGF function in A431 cells.
To identify the responsive motifs involved in EGF activation of the NF-IL6β promoter, a series of reporters with mutations as illustrated in Figure 2E were constructed. NF-IL6βmSp1-1 and NF-IL6βmSp1-2 with the individually mutated Sp1 site resulted in a more significant attenuation of the basal promoter activity than NF-IL6βmCRE with the mutated CRE site. It suggested that the Sp1 sites were critical for the basal promoter activity. Furthermore, the single site mutants of Sp1-1, Sp1-2, or CRE site (NF-IL6β-347/+9mSp1-1, NF-IL6β-347/+9mSp1-2, or NF–IL6β-347/+9mCRE) diminished the EGF induction of the promoter activity by 30–40% respectively, whereas the triple mutant, NF-IL6β-347/+9mC/dSp1 with two Sp1 sites and a CRE site, resulted in a complete elimination of EGF response. These results indicated that the Sp1-1, the Sp1-2, and the CRE motifs indeed played important roles in the EGF induction and the basal activity of NF-IL6β promoter.
Binding of CREB to NF-IL6β Promoter
To further identify the transcription factors bound to the CRE site of NF-IL6β promoter, gel shift assays with nuclear extracts prepared from EGF-treated A431 cells were performed. As shown in Figure 3A, CRE binding activity was low in cells deprived of EGF (lane 1) and was rapidly induced within 30 min after EGF stimulation (lanes 2 and 3) and decreased in 60 min (lane 4). The CRE motif of mouse C/EBPδ promoter (mCRE), specific for CREB/ATF-1 binding (Belmonte et al., 2001), was used as a competitor. The retarded band was competed out by 100-fold mCRE (lane 5), suggesting CREB/ATF-1 could be the binding protein of CRE motif. To determine the possible CRE-binding protein, the EGF-induced CRE-binding complex was examined by antibodies recognized CREB or ATF-2 in gel-shift assay. The α-CREB antibodies completely shifted and blocked the EGF-induced CRE-binding complex (lane 8), but α-ATF-2 antibodies did not (lane 9). Additionally, the CREB protein, synthesized in vitro by the TNT-coupled reticulocyte lysate system (Promega), bound specifically to the CRE probe (Figure 3B, lane 2). Moreover, addition of the CREB-specific antibodies, but not the control rabbit IgG shifted the specific CREB/CRE-binding complex to a higher molecular weight region (Figure 3B, compare lane 3 with lane 4). Excess human NF-IL6β CRE (hCRE, Figure 3B, lane 5) and mCRE (Figure 3B, lane 6) oligonucleotides competed with CREB for the formation of CREB/CRE binding complex, but mutant hCRE (muthCRE) oligonucleotide did not (Figure 3B, lane 7). These results indicated that the CREB protein bound to the CRE binding element in the human NF-IL6β promoter.
Figure 3.

CREB was one component of the CRE-binding complex and directly bound to NF-IL6β promoter in vitro. (A) In lanes 1–4, nuclear extracts from A431 cells deprived of serum and restimulated with EGF for various time courses were incubated with the 32P-labeled CRE element of NF-IL6β gene. In lane 5, competition analysis was carried out by the addition of an 100-fold molar excess of unlabeled mCRE oligonucleotides. In lane 6, no antibody was present in the binding reaction. In lanes 7, 8 and 9, binding assays were performed with control, CREB, or ATF2 specific antibody, respectively. The solid arrow indicates the specific DNA-protein complex, and the “SS” arrow indicates the band supershifted by the antibody. (B) The CREB protein synthesized in vitro by the TNT-coupled reticulocyte lysate system was allowed to bind to the CRE probe (lane 2). Incubation with a control rabbit IgG (lane 3), purified rabbit anti-CREB antibodies (lane 4), excess unlabeled mCRE (lane 5), hCRE (wt, lane 6) or hCRE-mutant (muthCRE, lane 7), were performed respectively, and then resolved on the nondenaturing gel. “CREB” represents the CREB retard shifting, and “SS” indicates the supershifted band by antibodies.
Involvement of Sp1 in EGF Stimulation of NF-IL6β Gene Transcription through the Sp1 Motifs
To determine whether Sp1 proteins bound to both of the Sp1 sites, gel-shift assay was carried out with probes containing individual Sp1 site and nuclear extracts from cells treated with EGF as indicated (Figure 4A). The binding pattern of the Sp1 sites was different from that of the CRE sites. Both of the Sp1-1 and Sp1-2 binding activities were already near maximum in serum starvation cells and were not further induced by EGF treatment, as shown in Figure 4A (lanes 1–4 and lanes 9–12). To examine the binding specificity of Sp1 on Sp1-1 and Sp1-2 motifs, the excess oligonucleotides competition assays were performed using commercially available consensus Sp1 (cSp1) and wild-type (Wt) oligonucleotides. Both Sp1-1– and Sp1-2–binding complexes were completely abolished by the cSp1 oligonucleotides (Figure 4A, lanes 5 and 13) or the Wt oligonucleotides (Figure 4A, lanes 6 and 14). A supershift pattern also was observed when antibodies against human Sp1 were added to the mixture of Sp1 probes and nuclear extracts (Figure 4A, lanes 7 and 15); however, the α-Sp3 antibodies did not (Figure 4A, lanes 8 and 16). These results suggested that the Sp1 sites were specifically for Sp1 binding but not for Sp3. To study the functional role of Sp1 with or without EGF treatment, reporter gene assay was performed. Cells were cotransfected with NF-IL6β reporter genes together with either a control vector, pCDNA3, or a vector expressing Sp1 as shown in Figure 4B. Overexpression of Sp1 could transactivate the reporter activity of NF-IL6β-347/+9 but not NF-IL6β-347/+9mdSp1 without EGF stimulation (lanes 3 and 7). Although the reporter activities of both constructs were enhanced under EGF stimulation (compare lanes 1 and 2 with lanes 5 and 6, respectively). A further increase in reporter activity due to the overexpression of Sp1 was observed only in cells transfected with NF-IL6β-347/+9 reporter (compare lane 2 with lane 4), but not in cells transfected with NF-IL6β-347/+9mdSp1 reporter (compare lane 6 with lane 8). These results suggested that the two Sp1 motifs on the essential promoter region played important roles in regulating EGF-induced gene expression of NF-IL6β.
Figure 4.

Sp1 was one component of the Sp1 binding complexes and played a role in the EGF stimulation of the NF-IL6β reporter. (A) Left and right, respectively, show that DNA binding pattern of Sp1-1 and Sp1-2 oligonucleotides. Lanes 1–4 and lanes 9–10 indicate the time course lysates extracted from EGF-restimulated A431 cells. The commercialized consensus Sp1 (cSp1, lanes 5 and 13) and wild-type oligonucleotides (Sp1-1 or Sp1-2, lanes 6 or 14) were, respectively, preincubated with nuclear extract for competition assay. The retard probe signals were determined with specific α-Sp1 (lanes 7 and 15) and α-Sp-3 (lanes 8 and 16) antibodies, and “SS” represents the supershifting band. (B) A431 cells were transfected with NF-IL6β-347/+9 (lanes 1–4) or containing Sp1 sites mutagenic reporter, NF-IL6β-347/+9mdSp1 (lanes 5–8), with a control or Sp1 expression vector, as indicated. The transfectants were stimulated with or without EGF for 12 h before the cell lysates were prepared and analyzed for luciferase activity.
Binding of CREB and Sp1 to NF-IL6β Promoter In Vivo
To confirm the results of the in vitro-DNA binding assay, binding of CREB and Sp1 to the NF-IL6β gene promoter in vivo was examined by using the ChIP assay. The primers NF-IL6β-347 and NF-IL6β+9 were used to specifically amplify the promoter region containing CRE, Sp1-1 and Sp1-2 motifs of the NF-IL6β gene locus as illustrated in Figure 5A. The predicted size of PCR fragment was confirmed by agarose gel electrophoresis, which was further characterized by DNA sequencing. As shown in Figure 5B, CREB and Sp1 bound to the promoter region of NF-IL6β gene in control cells (lanes 10 and 13). On EGF treatment, antibodies recognizing the active form of the CREB (pCREB) or the CREB protein specifically coprecipitated with the fragment of NF-IL6β promoter in an EGF inducible manner (lanes 8 and 11). This inducible binding pattern of CREB was also observed in the IL-3–stimulated transcriptional regulation of mcl-1 gene (Wang et al., 2003). Moreover, Sp1 constitutively bound to the promoter region of NF-IL6β gene. These results were consistent with the above-mentioned observation of gel-shift assay (Figure 4A).
Figure 5.

CREB and Sp1 bound to the NF-IL6β gene promoter in vivo. (A) Schematic representation of the NF-IL6β genomic locus spanning the promoter region. Primer NF-IL6β+9 and primer NF-IL6β-347 were generated for PCR reaction. (B) ChIP analysis of CREB or Sp1 binding to the NF-IL6β gene locus. Sheared formaldehyde cross-linked chromatin was immunoprecipitated with antibodies as indicated and processed for PCR amplification. As a positive control, PCR amplification also was carried out with input DNA from chromatin before the immunoprecipitation step (lanes 1, 2, and 3). The chromatin was isolated from cells with (lanes 5, 8, 11, and 14) and without (lanes 4, 7, 10, and 13) EGF treatments, or anisomycin treatments (lanes 6, 9, 12, and 15) and the immunoprecipitation (IP) step was performed with various antibodies (IP antibody), as indicated. The “–347/+9” indicates the PCR products after specific primers amplification with purified templates from specific antibody-IP step.
Role of p38MAPK and PI3-Kinase Signal Transduction Pathway in NF-IL6β Regulation
EGF interacts with EGFR to result in receptor autophosphorylation and initiates multiple intracellular signaling cascades, leading to the induction of cell growth (Hill and Treisman, 1995; Treisman, 1996). PI3-kinase and MAPKs pathways have been reported to play an important role in EGF signaling in A431 cells (Soltoff et al., 1994; Matthew and Jan, 2001; Chen et al., 2002). To determine the possible involvement of signaling pathways in EGF induction of NF-IL6β mRNA, the pharmacological inhibitors of signal transduction components were used to study the EGF-induced NF-IL6β regulation. The effect of various specific MAPK inhibitors, SP600125 (JNK inhibitor), SB203580 (p38 inhibitor), and PD98059 (MEK1 inhibitor), on NF-IL6β mRNA expression was studied. SB203580, a specific inhibitor of p38MAPKα- and β-isoforms, apparently inhibited EGF-induced NF-IL6β transcriptional activity (Figure 6A, compare lane 2 with lane 4); however, no effects were found upon SP600125 (lane 3) or PD98059 (lane 5) treatment.
Figure 6.

Wortmannin and SB203580 attenuated EGF-inducted NF-IL6β mRNA and protein expression. (A and B) Cells were pretreated with various MAPK inhibitors as indicated for 30 min before restimulation with EGF for 90 min. After stimulation, total RNA was prepared from these cells and analyzed by RT-PCR with specific NF-IL6β primers for detection of the NF-IL6β mRNA. (C) Cell were treated with compounds as indicated for 30 min before restimulation with EGF for 4 h. Lysates from various treatment were analyzed by Western blot with α-NF-IL6β or α-cyclooxygenase (COX)-1 antibodies. The relative density of NF-IL6β protein expression was normalized with COX-1 protein in densitometer. The pharmacological inhibitors were dissolved in dimethyl sulfoxide (DMSO), and the final 0.5% of DMSO was used in incubation.
Overexpression of p110CAAX, a constitutively activated PI3-kinase, as well as insulin induces mRNA expression and nuclear expression of C/EBPβ and C/EBPδ in vascular smooth muscle cells (Sekine et al., 2002). To clarify the possible contribution of PI3-kinase to the human NF-IL6β gene regulation in A431 cells, the effect of PI3-kinase inhibitor wortmannin was studied. Pretreatment of cells with 100 nM wortmannin attenuated the effect on EGF induction of NF-IL6β mRNA (Figure 6B, compare lane 2 with lane 3). These results indicated that PI3-kinase and p38MAPK were involved in EGF-induced NF-IL6β transcription. For investigating the signal pathway transduced by these two kinases on NF-IL6β transcription, combined treatment with wortmannin and SB203580 were performed, and it did not result in any synergistic inhibition of EGF-induced transcription of NF-IL6β mRNA (Figure 6B, compare lane 4 and lane 5). A similar inhibition phenomenon was observed at the NF-IL6β protein level (Figure 6C). These results strongly suggested that PI3-kinase and p38MAPK activation was on the same signaling pathway to regulate NF-IL6β transcription under EGF-stimulation in A431 cells.
In rat osteosarcoma UMR cells, EGF activates members of the MAPK family, including p38MAPK and ERKs. Treatment of cells with either SB203580 or PD98059 prevents phosphorylation of CREB at Ser133 induced by EGF (Swarthout et al., 2002). Because CREB bound to NF-IL6β promoter was demonstrated in our system, whether the p38MAPK phosphorylated the CREB was conducted. By detecting with antibodies against phosphorylated Ser133 CREB, a rapid induction of the phosphorylation of Ser133 on CREB in A431 cells was found (Figure 7A, lanes 1–4). Under similar experimental condition, the EGF-induced phosphorylation of CREB was prevented by pretreatment of SB203580 (Figure 7A, lanes 5 and 6). It suggested that p38MAPK was an activator mediating the EGF-induced CREB activation. To further confirm this observation, treatment with anisomycin, p38 MAPK activator, in A431 cells was carried out. Induction of Ser-133 phosphorylation of CREB by anisomycin was observed (our unpublished data). Together, these results suggested that p38MAPK played a functional role in CREB activation in EGF-stimulated A431 cells.
Figure 7.

EGF activated the PI3-kinase/p38MAPK/CREB signaling pathway in A431 cells. (A) Inhibition of EGF-induced CREB phosphorylation by SB203580. The A431 cells were pretreated with or without 10 μM SB203580 for 30 min and then stimulated with EGF as indicated. Western blot analysis with α-P-CREB and α-CREB antibodies was performed. (B) Wortmannin attenuated the activation of p38MAPK and CREB after EGF treatment. Cell lysates from A431 cells stimulated with EGF for various times were analyzed by Western blotting with antibodies recognizing the active form p38 (P-p38), CREB (P-CREB), p38, and CREB. The pharmacological inhibitors were dissolved in dimethyl sulfoxide (DMSO), and the final 0.5% of DMSO was used in incubation.
To further determine the relationship between PI3-kinase, P38MAPK, and CREB, the chemical inhibition assay was performed by Western blotting assay. A431 cells were preincubated with or without wortmannin and then stimulated with EGF for various time courses as indicated in Figure 7B. Phosphorylation of p38MAPK and CREB Ser133 was partially inhibited by wortmannin in the same time courses (compare lanes 2 with 5, lanes 3 with 6 and lanes 4 with 7). These results suggested that PI3-kinase was an upstream, but just partial, activator in the EGF-induced activation of p38MAPK and CREB, and p38MAPK was a major CREB Ser133 activator in A431 cells.
To examine whether the –347/+9 fragment of NF-IL6β promoter is required for PI3-kinase and p38MAPK activation, the NF-IL6β-347/+9 was cotransfected with different expression vectors, including wild-type expression vectors of CREB; p38α; two constitutive activation forms of p38MAPK upstream activators, MKK3Ac and MKK6Ac; and p110*, constitutive activation form of PI3-kinase, under the condition without EGF treatment. As shown in Figure 8A, both of the p38-mimic activators MKK3Ac and MKK6Ac increased the transcriptional activity of NF-IL6β-347/+9 (lanes 4 and 5), and p110* also contributed the same effect (lane 6). Furthermore, a dominant negative mutant of p38α (DN-p38α) attenuated the transcriptional activity enhanced by p110* (Figure 8B). These results strongly suggested that PI3-kinase/p38MAPK signaling was involved in the transcriptional activation of human NF-IL6β promoter.
Figure 8.
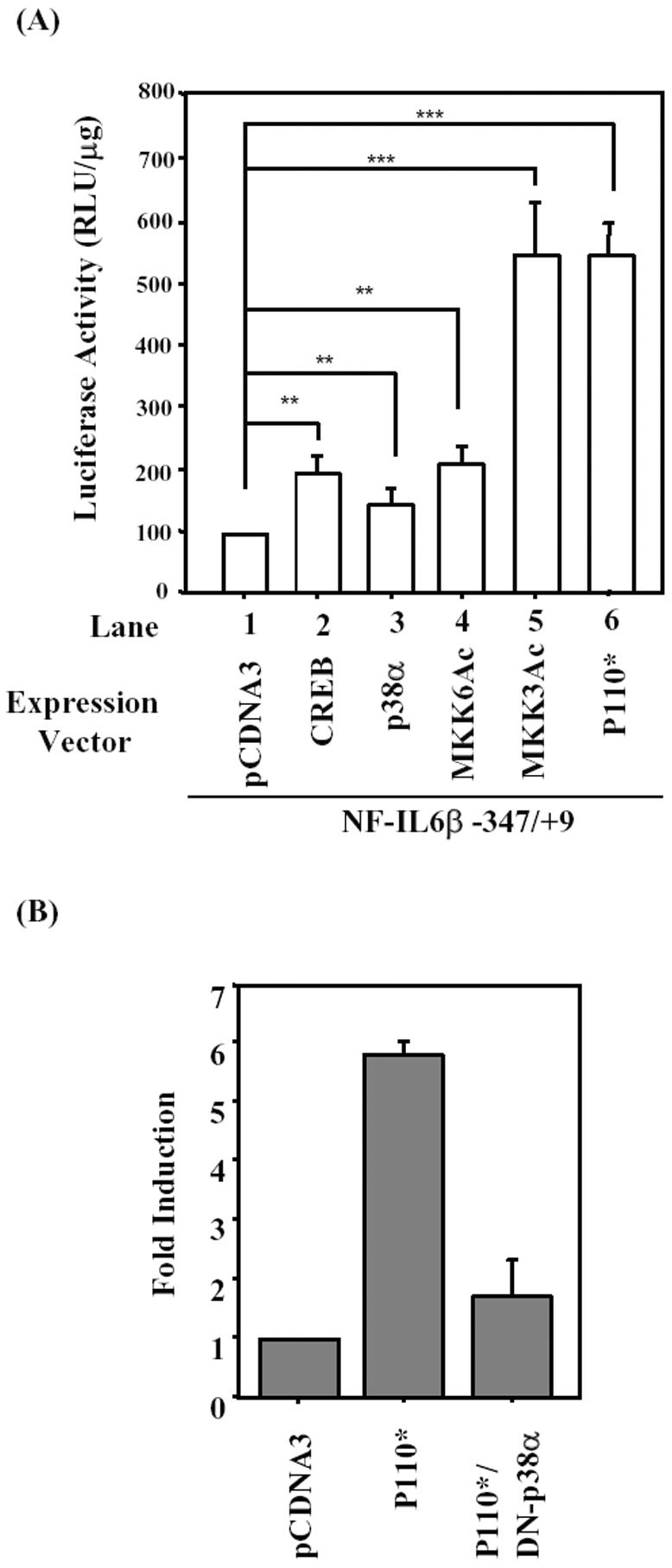
Overexpression of constitutively activated MKK3, MKK6, or p110* up-regulated the promoter activity of NF-IL6β gene. (A) A431 cells were cotransfected with reporter NF-IL6β-347/+9 and various expression vectors as indicated. (B) A431 cells were cotransfected with reporter NF-IL6β-347/+9 and p110* alone or DN-p38α combined with p110*. After growing in serum-free medium for 15 h, the luciferase assay was performed with lysates of those transient transfectants. Statistic analysis was performed by Student's t test.
Mediation of the p38MAPK Signal through CREB in Activation of Human NF-IL6β Promoter Activity
To further confirm whether CREB was a downstream target of p38MAPK and p38MAPK-regulated phosphorylation of CREB Ser133 contributes to NF-IL6β transcription, a dominant negative mutant of CREB, in which serine 133 was replaced by alanine (DN-CREB), was transfected to A431 cells to address this issue. A431 cells transfected with p38α-expressing vector enhanced NF-IL6β-347/+9 reporter activity without EGF treatment (Figure 9A, compare lane 1 with lane 4), and the presence of EGF significantly enhanced reporter activity of NF-IL6β-347/+9 in the presence of p38α expression vector (Figure 9A, compare lane 2 with lane 5). Moreover, overexpressed DN-CREB repressed the reporter activity of NF-IL6β-347/+9 with or without p38αMAPK expression under EGF stimulation (Figure 9A, compare lane 2 with lane 3, and lane 5 with lane 6). For specifically determining whether the CRE motif of the NF-IL6β promoter was a p38α-regulated CREB target site, a heterologous promoter containing CRE motif, pGL2NF-IL6β1xCRE, was constructed and analyzed. Insertion of a CRE site alone was sufficient to confer EGF activity on the SV40 promoter (Figure 9B, compare lane 2 with lane 7). We then examined whether p38α was mediated through phosphorylation of CREB Ser133 to regulate the CRE motif. Cells were cotransfected with p38α expression vectors enhanced the heterologous reporter activity of pGL2NF-IL6β1xCRE under EGF treatment (Figure 9A, compare lane 5 with lane 8). Moreover, similar pattern was observed in the experiment with cotransfection of CREB expression vector (Figure 9B, compare lane 6 with lane 9). We further examined whether the EGF inducibility on the pGL2NF-IL6β/1xCRE could be attenuated by dominant negative forms of p38α or CREB. The results shown in Figure 9C indicated that coexpression of DN-p38α or DN-CREB attenuated EGF inducibility effect on CRE reporter activity (compare lane 1 with lanes 2 or 3). These results suggested that the induction of phosphorylated CREB through activated p38α played a functional role in EGF-induced NF-IL6β transcription.
Figure 9.

Functional role of p38MAPK and CREB in EGF stimulated NF-IL6β promoter activity in A431 cells. (A) A431 cells were transiently transfected with reporter genes as indicated along with a control (pCDNA3) or the pCDNA3/CREBS133A (DN-CREB) expression vectors. After transfection, cells were cultivated in medium with or without EGF. Cell lysates were then prepared and analyzed for luciferase activity. (B) A431 cells transfected with pGL2-promoter or CRE-heterologous reporter genes combined with the control, p38α, or CREB expression vectors, as indicated, were stimulated with or without EGF for 15 h. Cell lysates were then prepared and analyzed for luciferase activity. (C) Dominant negative forms of p38α or CREB were cotransfected with pGL2NF-IL6β1xCRE reporter in A431 cells. For the normalization, the pGL2NF-IL6β1xCRE reporter activity cotransfected with control vector, pCDNA3, was assigned a value of 100. The reporter activities of pGL2NF-IL6β1xCRE combined with DN-p38α or DN-CREB were normalized to the control. The data shown are means ± standard deviations of two independent experiments.
DISCUSSION
In this study, we provided several pieces of evidence suggesting that the Sp1 and the CRE sites in the essential promoter region were required for EGF-induced transcription of the human NF-IL6β gene. Several transcription factors, for example, STAT3 (Hutt et al., 2000; Alonzi et al., 2001), CREB/ATF-2 (Belmonte et al., 2001), RunX (McCarthy et al., 2000), and Sp1 (Alonzi et al., 2001), are involved in the transcriptional regulation of C/EBPδ, which are binging in the vicinity of the transcriptional initiation site. The APRE site is important for IL-6–regulated C/EBPδ promoter activity in hepatoma cells (Cantwell et al., 1998). However, the APRE-like sequence does not exist in human NF-IL6β promoter. By comparing the APRE regions on mouse and human promoter sequences, an Sp1, a nuclear factor-κB (NF-κB)–like, and STAT3 binding sites in mouse APRE region were observed (Figure 2A). The mouse Sp1 site corresponds to the human NF-IL6β Sp1-1 site, whereas no homologous sites of NF-κB and STAT3 exist in NF-IL6β. Transient transfection with the heterologous reporter containing the APRE sequence, C/EBPδ 5′-AGCGAGGGCGGGTCGTTCCCAGC-3′ (Sp1 site, underlined; NF-κB site, italics) and NF-IL6β 5′-GGCTCCGGGGGGCTCCCAGGGCG-3′ to A431 cells under EGF treatment was conducted in this study. By comparing with the control reporter construct, there was no induction observed in pGL2NF-IL6β 4xAPRE (Figure 2C, left). However, the increased promoter activity of pGL-2C/EBPδ 4xAPRE might be due to the Sp1 binding element existed in the mouse APRE region (Figure 2C, right). On the other hand, both heterologous reporters containing CRE sequence showed twofold induction under EGF treatment (Figure 2D). These results indicated that NF-IL6β had different promoter usage in A431 cells from mouse C/EBPδ in other systems. The two Sp1 sites and CRE site, but not APRE region, were more important in the basal or in the EGF-induced transcriptional activities of NF-IL6β gene (Figure 2E).
Expression of dominant negative form of CREB dramatically reduces the leukemia inhibitory factor- and prostacyclin-stimulated C/EBPδ expression (Belmonte et al., 2001). In RAW264 cells, C/EBPβ induction seems not to require CREB, because it is not affected by the treatment that abolishes CREB activation (Caivano and Cohen, 2000). Nevertheless, in our study, the same treatments inhibited CREB phosphorylation and also abolished NF-IL6β induction (Figure 9, A and C). The NF-IL6β CRE site, GCACGTCA, has homologous sequence to the ATF/CRE sequence motifs (TGACGTCA) and like as the asymmetric and weak binding sites (Nichols et al., 1992). Transfection of the CRE heterologous reporters of C/EBPδ or NF-IL6β promoter enhanced the EGF induction of transcription activity in A431 cells (Figure 2D). From the results of gel shift and ChIP assays (Figures 3A and 5), it clearly indicated that CREB bound to the NF-IL6β promoter, whereas EGF enhanced the DNA binding activity of CREB through phosphorylation mechanism.
Sp1 is a ubiquitous nuclear factor that plays a key role in maintaining basal transcription of house-keeping genes. Many reports show that the posttranslational modification of Sp1, such as phosphorylation (Banchio et al., 2004) and acetylation (Ryu et al., 2003), is important for its regulation of target genes expression. Mutants of Sp1 sites on NF-IL6β promoter resulted in not only lose of the basal transcription activity but also lose of the EGF-induced activity (Figure 2). Constitutive binding of Sp1 to the NF-IL6β promoter was not affected by the EGF treatment as shown in the gel shift and ChIP assays (Figures 4 and 5). Overexpression of Sp1 also enhanced the basal transcription activity (Figure 4B, lanes 1 and 3), indicating that Sp1 played a major role in the basal transcriptional complex of NF-IL6β promoter. On EGF treatment, the NF-IL6β promoter did not recruit more Sp1 proteins to increase the transcription activity. Instead, the EGF-induced posttranslational modification of Sp1 might account for the elevation of transcription activity.
Several previous reports show that NF-κB pathway is important for transcriptional regulation of mouse C/EBPδ (Tengku-Muhammad et al., 2000; Okazaki et al., 2002), but there was no evidence to show that the direct binding of NF-κB to the promoter region of C/EBPδ gene is crucial. Induction of de novo production biosynthesis through NF-κB might be a possible mechanism to account for the LPS-regulation of mouse C/EBPδ (Caivano et al., 2001). In our hands, NF-IL6β was an immediate-early gene regulated by EGF treatment, and NF-κB could not bind to the APRE-like motif by gel-shifting assay (Wang and Chang, unpublished result). It indicated that NF-κB might not be involved in EGF-induced transcriptional regulation of NF-IL6β gene.
Cycloheximide failed to inhibit the induction of NF-IL6β by EGF, indicating that NF-IL6β is an immediate-early gene (Figure 1C). This result was the same as the immediate-early gene encoding transcription factor c-fos treated with cycloheximide (Morgan and Curran, 1991). NF-IL6β mRNA also is superinduced by cycloheximide (Figure 1C). An unappreciated property of protein translation inhibitors is their ability to activate numerous kinases, such as c-Jun NH2-terminal kinase (JNK), p38MAPK, mTOR, and p70S6 kinase (Barros et al., 1997; Sidhu and Omiecinski, 1998; Khaleghpour et al., 1999). Cycloheximide superinduces glucocorticoid-mediated transcription of a gene encoding the α-epithelial sodium channel protein via a mechanism that can be suppressed by a p38 MAPK inhibitor (Itani et al., 2003). Our results demonstrated that p38MAPK signaling pathway was involved in the EGF-induced NF-IL6β transcription that also might be involved in cycloheximide-induced manner.
Another interesting finding from this study is the demonstration that PI3-kinase was an upstream regulator of p38MAPK. p38MAPK is a JNK-related MAPK that is activated in response to a variety of stimuli, including growth factors, phorbol esters, cytokines, and environmental stress (Minden and Karin, 1997); and the different upstream activators of p38MAPK were reported, such as MKK3/6 (Shuto et al., 2001), Rac (Xu et al., 2003), Src (Daly et al., 1999; Frey et al., 2004), or PI3-kinase (Gibbs et al., 2002; Xu et al., 2003; Gonzalez et al., 2004). IL-4 stimulates Rac and Cdc42, which seem to regulate a protein kinase cascade initiated at the level of PAK and lead to activation of p38MAPK in A431 cells, and are finally able to produce IL-6 (Wery-Zennaro et al., 2000). PI3-kinase coprecipitates with the ErbB3 protein in response to EGF in A431 cells (Soltoff et al., 1994). Using a number of different approaches, several pieces of evidence indicated that p38MAPK contributed to the EGF induction of NF-IL6β. SB203580, which selectively inhibits p38α and p38β2 isoforms but has no effect on JNK and ERK (Cuenda et al., 1997; Kumar et al., 1997), inhibited the EGF-induced NF-IL6β expression (Figure 6). In addition, NF-IL6β promoter activity was inhibited by expression of a dominant-negative p38α mutant and was activated by the overexpression of wild-type p38α (Figure 9). p38MAPK can be phosphorylated and activated by the dual-specific protein kinases MKK3 and MKK6 (Derijard et al., 1995). Overexpression of the constitutively activated p38MAPK-specific kinase MKK6 or MKK3 directly stimulated NF-IL6β promoter activity (Figure 8). Treatment of anisomycin that mimics the p38MAPK activation increased the in vivo-CREB binding activity (Figure 5B) and CREB phosphorylation in A431 cells (Wang and Chang, unpublished result). The evidence concluded that p38 was involved in the signal pathway of EGF-induced NF-IL6β expression. We also investigated the signal transduction cascades that connect EGFR activation to phosphorylation of p38MAPK in A431 cells. Several possible signaling transduction pathways that could be implicated in PI3-kinase–regulated p38MAPK activation have been reported, such as the PI3-kinase/Rca/p38MAPK pathway in Signet-ring cell carcinoma (Xu et al., 2003), the TGFβ1-induced PI3-kinase/p38MAPK/Akt pathway in mesenchymal cells (Horowitz et al., 2004), or the involvement of PI3-kinase/p38MAPK/Akt2 pathway in myogenesis (Gonzalez et al., 2004). The treatment of wortmannin partially inhibited p38MAPK and CREB activation (Figure 7), suggesting that PI3-kinase might not be the only upstream activator.
Based on these observations, a tentative model for regulation mechanism of NF-IL6β gene in A431 cells under EGF treatment was proposed. In human epidermoid carcinoma A431 cells, EGF signal, at least in part, activated the PI3-kinase pathway that led to the phosphorylation of p38MAPK. The phosphorylated p38MAPK in turn induced the CREB phosphorylation and increased its binding to NF-IL6β promoter. Then, the Sp1 cooperated with the phosphorylated CREB to activate the transcription of NF-IL6β gene. In conclusion, these results indicated that induction of PI3-kinase/p38MAPK/CREB pathway plays a functional role in EGF-induced transcription of NF-IL6β gene in A431 cells.
Acknowledgments
We thank Drs. Wai-Ming Kan, Hsin-Fang Yang-Yen, and Shen K. Yang for critical reviewing and editing of the manuscript. We also thank for Drs. Jiahuai Han for the plasmid expressing DN-p38α, Ming-Zong Lai for the plasmids expressing MKK3Ac and MKK6Ac, Hsin-Fang Yang-Yen for the plasmid p110*, and Jeffrey J.Y. Yen for the plasmids expressing DN-CREB and CREB. This work was supported in part by grant the Ministry of Education Program for Promoting Academic Excellent of University under the grant number 91-B-FA09-1-4 of the Republic of China.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–02–0105) on May 18, 2005.
Abbreviations used: ChIP, chromatin immunoprecipitation; CRE, cAMP response element; CREB, cAMP response element-binding protein; EGF, epidermal growth factor; MAPK, mitogen-activated protein kinase.
References
- Alonzi, T., Maritano, D., Gorgoni, B., Rizzuto, G., Libert, C., and Poli, V. (2001). Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol. Cell. Biol. 2, 1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchio, C., Schang, L. M., and Vance, D. E. (2004). Phosphorylation of Sp1 by cyclin-dependent kinase 2 modulates the role of Sp1 in CTP:phosphocholine cytidylyltransferase alpha regulation during the S phase of the cell cycle. J. Biol. Chem. 279, 40220–40226. [DOI] [PubMed] [Google Scholar]
- Barnes, D. W. (1982). Epidermal growth factor inhibits growth of A431 human epidermoid carcinoma in serum-free cell culture. J. Cell Biol. 93, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros, L. F., Young, M., Saklatvala, J., and Baldwin, S. A. (1997). Evidence of two mechanisms for the activation of the glucose transporter GLUT1 by anisomycin: p38(MAP kinase) activation and protein synthesis inhibition in mammalian cells. J. Physiol. 504, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte, N., et al. (2001). Activation of extracellular signal-regulated kinases and CREB/ATF-1 mediate the expression of CCAAT/enhancer binding proteins β and -δ in preadipocytes. Mol. Endocrinol. 15, 2037–2049. [DOI] [PubMed] [Google Scholar]
- Böhm, M., Moellmann, G., Cheng, E., Alvarez, F. M., Wagner, S., Sassone-Corsi, P., and Halaban, R. (1995). Identification of p90RSK as the probable CREB-Ser133 kinase in human melanocytes. Cell Growth Differ. 6, 291–302. [PubMed] [Google Scholar]
- Cantwell, C. A., Sterneck, E., and Johnson, P. F. (1998). Interleukin-6-specific activation of the C/EBPdelta gene in hepatocytes is mediated by Stat3 and Sp1. Mol. Cell. Biol. 18, 2108–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caivano, M., and Cohen, P. (2000) Role of mitogen-activated protein kinase cascades in mediating lipopolysaccharide-stimulated induction of cyclooxygenase-2 and IL-1 beta in RAW264 macrophages. J. Immunol. 164, 3018–3025. [DOI] [PubMed] [Google Scholar]
- Caivano, M., Gorgoni, B., Cohen, P., and Poli, V. (2001). The induction of cyclooxygenase-2 mRNA in macrophages is biphasic and requires both CCAAT enhancer-binding protein beta (C/EBP β) and C/EBP δ transcription factors. J. Biol. Chem. 276, 48693–48701. [DOI] [PubMed] [Google Scholar]
- Cardinaux, J. R., Allaman, I., and Magistretti, P. J. (2000). Pro-inflammatory cytokines induce the transcription factors C/EBPβ and C/EBPδ in astrocytes. Glia 29, 91–97. [PubMed] [Google Scholar]
- Carpenter, G. (1987). Receptors for epidermal growth factor and other polypeptide mitogens. Annu. Rev. Biochem. 56, 881–914. [DOI] [PubMed] [Google Scholar]
- Chen, B. K., Tsai, T. Y., Huang, H. S., Chen, L. C., Chang, W. C., Tsai, S. B., and Chang, W. C. (2002). Functional role of extracellular signal-regulated kinase activation and c-Jun induction in phorbol ester-induced promoter activation of human 12(S)-lipoxygenase gene. J. Biomed. Sci. 9, 156–165. [DOI] [PubMed] [Google Scholar]
- Cuenda, A., Cohen, P., Buee-Scherrer, V., and Goedert, M. (1997). Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38). EMBO J. 16, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly, J. M., Olayioye, M. A., Wong, A. M., Neve, R., Lane, H. A., Maurer, F. G., and Hynes, N. E. (1999). NDF/heregulin-induced cell cycle changes and apoptosis in breast tumour cells: role of PI3 kinase and p38 MAP kinase pathways. Oncogene 18, 3440–3451. [DOI] [PubMed] [Google Scholar]
- Davies, G. E., Sabatakos, G., Cryer, A., and Ramji, D. P. (2000). The ovine CCAAT-enhancer binding protein delta gene: cloning, characterization, and species-specific autoregulation. Biochem. Biophys. Res. Commun. 271, 346–352. [DOI] [PubMed] [Google Scholar]
- Deak, M., Clifton, A. D., Lucocq, L. M., and Alessi, D. R. (1998). Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare, D., Fimia, G. M., and Sassone-Corsi, P. (1999). Signaling routes to CREM and CREB: plasticity in transcriptional activation. Trends Biochem. Sci. 24, 281–285. [DOI] [PubMed] [Google Scholar]
- Derijard, B., Raingeaud, J., Barrett, T., Wu, I. H., Han, J., Ulevitch, R. J., and Davis, R. J. (1995). Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267, 682–685. [DOI] [PubMed] [Google Scholar]
- Frey, M. R., Golovin, A., and Polk, D. B. (2004). Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. J. Biol. Chem. 279, 44513–44521. [DOI] [PubMed] [Google Scholar]
- Gibbs, B. F., Plath, K. E., Wolff, H. H., and Grabbe, J. (2002). Regulation of mediator secretion in human basophils by p38 mitogen-activated protein kinase: phosphorylation is sensitive to the effects of phosphatidylinositol 3-kinase inhibitors and calcium mobilization. J. Leukoc. Biol. 72, 391–400. [PubMed] [Google Scholar]
- Gonzalez, I., Tripathi, G., Carter, E. J., Cobb, L. J., Salih, D. A., Lovett, F. A., Holding, C., and Pell, J. M. (2004). Akt2, a novel functional link between p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways in myogenesis. Mol. Cell. Biol. 24, 3607–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigler, H., Ash, J. F., Singer, S. J., and Cohen, S. (1978). Visualization by fluorescence of the binding and internalization of epidermal growth factor in human carcinoma cells A-431. Proc. Natl. Acad. Sci. USA 75, 3317–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, C. S., and Treisman, R. (1995). Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell 80, 199–211. [DOI] [PubMed] [Google Scholar]
- Horowitz, J. C., Lee, D. Y., Waghray, M., Keshamouni, V. G., Thomas, P. E., Zhang, H., Cui. Z., and Thannickal, V. J. (2004) Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J. Biol. Chem. 279, 1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu. W., Kerppola, T. K., Chen, P. L., Curran, T., and Chen-Kiang, S. (1994). Fos and Jun repress transcription activation by NF-IL6 through association at the basic zipper region. Mol. Cell. Biol. 14, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung, A., Montagna, C., Sharan, S., Ni, Y., Ried, T., and Sterneck, E. (2004). Loss of CCAAT/enhancer binding protein delta promotes chromosomal instability. Oncogene 23, 1549–1557. [DOI] [PubMed] [Google Scholar]
- Hunter, T., and Cooper, J. A. (1985). Protein-tyrosine kinases. Annu. Rev. Biochem. 54, 897–930. [DOI] [PubMed] [Google Scholar]
- Hutt, J. A., and DeWille, J. D. (2002) Oncostatin M induces growth arrest of mammary epithelium via a CCAAT/enhancer-binding protein delta-dependent pathway. Mol. Cancer Ther. 1, 601–610. [PubMed] [Google Scholar]
- Hutt, J. A., O'Rourke, J. P., and DeWille, J. (2000). Signal transducer and activator of transcription 3 activates CCAAT enhancer-binding protein delta gene transcription in G0 growth-arrested mouse mammary epithelial cells and in involuting mouse mammary gland. J. Biol. Chem. 275, 29123–29131. [DOI] [PubMed] [Google Scholar]
- Itani, O. A., Cornish, K. L., Liu, K. Z., and Thomas, C. P. (2003). Cycloheximide increases glucocorticoid-stimulated α-ENaC mRNA in collecting duct cells by p38 MAPK-dependent pathway. Am. J. Physiol. 284, 778–787. [DOI] [PubMed] [Google Scholar]
- Kamaraju, A. K., Adjalley, S., Zhang, P., Chebath, J., and Revel, M. (2004). C/EBP-delta induction by gp130 signaling. Role in transition to myelin gene expressing phenotype in a melanoma cell line model. J. Biol. Chem. 279, 3852–3861. [DOI] [PubMed] [Google Scholar]
- Kawamoto, T., Mendelsohn, J., Le, A., Sato, G., Lazar, C. S., and Gill, G. N. (1984). Relation of epidermal growth factor receptor concentration to growth of human epidermoid carcinoma A431 cells. J. Biol. Chem. 259, 7761–7766. [PubMed] [Google Scholar]
- Khaleghpour, K., Pyronnet, S., Gingras, A. C., and Sonenberg, N. (1999). Translational homeostasis: eukaryotic translation initiation factor 4E control of 4E-binding protein 1 and p70 S6 kinase activities. Mol. Cell. Biol. 19, 4302–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravchenko, V. V., Mathison, J. C., Schwamborn, K., Mercurio, F., and Ulevitch, R. J. (2003). IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J. Biol. Chem. 278, 26612–26619. [DOI] [PubMed] [Google Scholar]
- Kumar, S., McDonnell, P. C., Gum, R. J., Hand, A. T., Lee, J. C., and Young, P. R. (1997). Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem. Biophys. Res. Commun. 235, 533–538. [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes, J., and Xanthopoulos, K. G. (1998). Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J. Biol. Chem. 273, 28545–28548. [DOI] [PubMed] [Google Scholar]
- Liu, Y. W., Tseng, H. P., Chen, L. C., Chen, B. K., and Chang, W. C. (2003). Functional cooperation of simian virus 40 promoter factor 1 and CCAAT/enhancer-binding protein beta and delta in lipopolysaccharide-induced gene activation of IL-10 in mouse macrophages. J. Immunol. 171, 821–828. [DOI] [PubMed] [Google Scholar]
- Mandrup, S., and Lane, M. D. (1997). Regulating adipogenesis. J. Biol. Chem. 272, 5367–5370. [DOI] [PubMed] [Google Scholar]
- Matthew, W., and Jan, D. (2001). Recruitment of the class II phosphoinositide 3-kinase C2beta to the epidermal growth factor receptor: role of Grb2. Mol. Cell. Biol. 21, 6660–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, T. L., Ji, C., Chen, Y., Kim, K. K., Imagawa, M., Ito, Y., and Centrella, M. (2000). Runt domain factor (Runx)-dependent effects on CCAAT/enhancer-binding protein delta expression and activity in osteoblasts. J. Biol. Chem. 275, 21746–21753. [DOI] [PubMed] [Google Scholar]
- Minden, A., and Karin, M. (1997). Regulation and function of the JNK subgroup of MAP kinases. Biochim. Biophys. Acta 1333, 85–104. [DOI] [PubMed] [Google Scholar]
- Monaco, L., and Sassone-Corsi, P. (1997). Cross-talk in signal transduction: Ras-dependent induction of cAMP-responsive transcriptional repressor ICER by nerve growth factor. Oncogene 15, 2493–2500. [DOI] [PubMed] [Google Scholar]
- Morgan, J. I., and Curran, T. (1991). Proto-oncogene transcription factors and epilepsy. Trends Pharmacol. Sci. 12, 343–349. [DOI] [PubMed] [Google Scholar]
- Nichols, M., Weih, F., Schmid, W., DeVack, C., Kowenz-Leutz, E., Luckow, B., Boshart, M., and Schutz, G. (1992). Phosphorylation of CREB affects its binding to high and low affinity sites: implications for cAMP induced gene transcription. EMBO J. 11, 3337–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki, K., Li, J., Yu, H., Fukui, N., and Sandell, L. J. (2002). CCAAT/enhancer-binding proteins beta and delta mediate the repression of gene transcription of cartilage-derived retinoic acid-sensitive protein induced by interleukin-1 beta. J. Biol. Chem. 277, 31526–31533. [DOI] [PubMed] [Google Scholar]
- Olayioye, M. A., Neve, R. M., Lane, H. A., and Hynes, N. E. (2000). The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19, 3159–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke, J. P., Hutt, J. A., and DeWille, J. (1999). Transcriptional regulation of C/EBPδ in G(0) growth-arrested mouse mammary epithelial cells. Biochem. Biophys. Res. Commun. 262, 696–701. [DOI] [PubMed] [Google Scholar]
- O'Rourke, J. P., Yuan, R., and DeWille, J. (1997). CCAAT/enhancer-binding protein-delta (C/EBP-delta) is induced in growth-arrested mouse mammary epithelial cells. J. Biol. Chem. 272, 6291–6296. [DOI] [PubMed] [Google Scholar]
- Ramji, D. P., and Foka, P. (2002). CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem. J. 365, 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, H., Lee, J., Olofsson, B. A., Mwidau, A., Deodeoglu, A., Escudero, M., Flemington, E., Azizkhan-Clifford, J., Ferrante, R. J., and Ratan, R. R. (2003). Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc. Natl. Acad. Sci. USA 100, 4281–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine, O., Nishio, Y., Egawa, K., Nakamura, T., Maegawa, H., and Kashiwagi, A. (2002). Insulin activates CCAAT/enhancer binding proteins and proinflammatory gene expression through the phosphatidylinositol 3-kinase pathway in vascular smooth muscle cells. J. Biol. Chem. 277, 36631–36639. [DOI] [PubMed] [Google Scholar]
- Shaywitz, A. J., and Greenberg, M. E. (1999). CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861. [DOI] [PubMed] [Google Scholar]
- Shuto, T., Xu, H., Wang, B., Han, J., Kai, H., Gu, X. X., Murphy, T. F., Lim, D. J., and Li, J. D. (2001). Activation of NF-κB by nontypeable Hemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK α/β-IκBα and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc. Natl. Acad. Sci. USA 98, 8774–8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu, J. S., and Omiecinski, C. J. (1998). Protein synthesis inhibitors exhibit a nonspecific effect on phenobarbital-inducible cytochrome P450 gene expression in primary rat hepatocytes. J. Biol. Chem. 273, 4769–4775. [DOI] [PubMed] [Google Scholar]
- Soltoff, S. P., Carraway, K. L. 3rd., Prigent, S. A., Gullick, W. G., and Cantley, L. C. (1994) ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell. Biol. 14, 3550–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin, A., and Waters, C. M. (1993). Endocytosis of growth factor receptors. Bioessays 15, 375–382. [DOI] [PubMed] [Google Scholar]
- Swarthout, J. T., Tyson, D. R., Jefcoat, S. C., Jr., Partridge, N. C., and Efcoat, S. C., Jr. (2002). Induction of transcriptional activity of the cyclic adenosine monophosphate response element binding protein by parathyroid hormone and epidermal growth factor in osteoblastic cells. J. Bone Miner. Res. 17, 1401–1407. [DOI] [PubMed] [Google Scholar]
- Tanaka, T., Yoshida, N., Kishimoto, T., and Akira, S. (1997). Defective adipocyte differentiation in mice lacking the C/EBPβ and/or C/EBPδ gene. EMBO J. 16, 7432–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengku-Muhammad, T. S., Hughes, T. R., Ranki, H., Cryer, A., and Ramji, D. P. (2000). Differential regulation of macrophage CCAAT-enhancer binding protein isoforms by lipopolysaccharide and cytokines. Cytokine 12, 1430–1436. [DOI] [PubMed] [Google Scholar]
- Treisman, R. (1996). Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell Biol. 8, 205–215. [DOI] [PubMed] [Google Scholar]
- Wang, J. M., Lai, M. Z., and Yang-Yen, H. F. (2003). Interleukin-3 stimulation of mcl-1 gene transcription involves activation of the PU. 1 transcription factor through a p38 mitogen-activated protein kinase-dependent pathway. Mol. Cell. Biol. 23, 1896–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel, A., and Ziegler-Heibrock, H.W.L. (1995). The C/EBP family of transcription factors. Immunology 193, 171–185. [DOI] [PubMed] [Google Scholar]
- Wery-Zennaro, S., Zugaza, J. L., Letourneur, M., Bertoglio, J., and Pierre, J. (2000). IL-4 regulation of IL-6 production involves Rac/Cdc42- and p38 MAPK-dependent pathways in keratinocytes. Oncogene 19, 1596–1604. [DOI] [PubMed] [Google Scholar]
- Wiggin, G. R., Soloaga, A., Foster, J. M., Murray-Tait, V., Cohen, P., and Arthur, J. S. (2002). MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol. Cell. Biol. 22, 2871–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, H. S., Herbst, J. J., Walsh, B. J., Lauffenburger, D. A., Rosenfeld, M. G., and Gill, G. N. (1991). The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 266, 11083–11094. [PubMed] [Google Scholar]
- Xing, J., Ginty, D. D., and Greenberg, M. E. (1996). Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273, 959–963. [DOI] [PubMed] [Google Scholar]
- Xu, Q., Karouji, Y., Kobayashi, M., Ihara, S., Konishi, H., and Fukui, Y. (2003). The PI 3-kinase-Rac-p38 MAP kinase pathway is involved in the formation of signet-ring cell carcinoma. Oncogene 22, 5537–5544. [DOI] [PubMed] [Google Scholar]
- Yamada, T., Tsuchiya, T., Osada, S., Nishihara, T., and Imagawa, M. (1998). CCAAT/enhancer-binding protein δ gene expression is mediated by autoregulation through downstream binding sites. Biochem. Biophys. Res. Commun. 242, 88–92. [DOI] [PubMed] [Google Scholar]
- Yang, G., Gregory, C. W., Shan, Q., O'Brien, D. A., and Zhang, Y. L. (2001). Differential expression of CCAAT/enhancer-binding protein-delta (c/EBPδ) in rat androgen-dependent tissues and human prostate cancer. J. Androl. 22, 471–480. [PubMed] [Google Scholar]
- Yarden, Y., and Sliwkowski, M. X. (2001). Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2, 127–137. [DOI] [PubMed] [Google Scholar]