

Abstract
The number of chemicals requiring risk evaluation exceeds our capacity to provide the underlying data using traditional methodology. This has led to an increased focus on the development of novel approach methodologies. This work aimed to expand the panel of gene expression-based biomarkers to include responses to estrogens, to identify training strategies that maximize the range of applicable concentrations, and to evaluate the potential for two classes of small non-coding RNAs (sncRNAs), microRNA (miRNA) and piwi-interacting RNA (piRNA), as biomarkers. To this end larval Pimephales promelas (96 hpf +/− 1h) were exposed to 5 concentrations of 17α-ethinylestradiol (0.12, 1.25, 2.5, 5.0, 10.0 ng/L) for 48 h. For mRNA-based biomarker development, RNA-seq was conducted across all concentrations. For sncRNA biomarkers, small RNA libraries were prepared only for the control and 10.0 ng/L EE2 treatment. In order to develop an mRNA classifier that remained accurate over the range of exposure concentrations, three different training strategies were employed that focused on 10 ng/L, 2.5 ng/L or a combination of both. Classifiers were tested against an independent test set of individuals exposed to the same concentrations used in training and subsequently against concentrations not included in model training. Both random forest (RF) and logistic regression with elastic net regularizations (glmnet) models trained on 10 ng/L EE2 performed poorly when applied to lower concentrations. RF models trained with either the 2.5 ng/L or combination (2.5 + 10 ng/L) treatments were able to accurately discriminate exposed vs. non-exposed across all but the lowest concentrations. glmnet models were unable to accurately classify below 5 ng/L. With the exception of the 10 ng/L treatment, few mRNA differentially expressed genes (DEG) were observed, however, there was marked overlap of DEGs across treatments. Overlapping DEGs have well established linkages to estrogen and several of the 81 DEGs identified in the 10 ng/L treatment have been previously utilized as estrogenic biomarkers (vitellogenin, estrogen receptor-β). Following multiple test correction, no sncRNAs were found to be differentially expressed, however, both miRNA and piRNA classifiers were able to accurately discriminate control and 10 ng/L exposed organisms with AUCs of 0.83 and 1.0 respectively. We have developed a highly discriminative estrogenic mRNA biomarker that is accurate over a range of concentrations likely to be found in real-world exposures. We have demonstrated that both miRNA and piRNA are responsive to estrogenic exposure, suggesting the need to further investigate their regulatory roles in the estrogenic response.
Keywords: epigenetics, miRNA, estrogens, piwiRNA, RNA-seq, Fathead minnow
1. Introduction
The number of chemicals requiring risk evaluation exceeds our capacity to provide the underlying data using traditional methodology. This realization has driven research focused on developing new methods that are higher throughput, less resource intensive, and that can facilitate chemical read across (Kavlock et al., 2018). Among the primary focal points of these research efforts is the development of mechanistic-based grouping of chemicals (Kienzler et al., 2017). Experimental platforms, such as microarrays and more recently RNA-seq, are able to reproducibly characterize large portions of the transcriptome, making them amenable to mechanistic grouping (Gibb, 2008; Lamb et al., 2006). Thus far, transcriptomic measures and tools have proven highly effective in chemical grouping and in the characterization of chemical MOA (Biales et al., 2016; De Abrew et al., 2016; Rees et al., 2016; Wu et al., 2017). The primary application of these approaches has been to human health and relied largely on mammalian model species or in vitro systems. Our group and others have demonstrated that these transcriptional approaches can also be successfully applied to non-mammalian species (Biales et al., 2016; Wang et al., 2016).
Recently, transcriptomic studies have shifted away from the use of microarrays toward next generation sequencing (NGS)-based technologies. NGS has allowed not only mRNA to be evaluated, but also non-coding RNAs (ncRNAs). ncRNA are a broad group that are distinguished based largely on size, function, biogenesis, structural features and interacting proteins (Farazi et al., 2008). Examples of ncRNA include, but are not limited to, microRNAs (miRNA), P-Element Induced Wimpy Testis (PIWI)-interacting RNA (piRNA), small interfering RNA (siRNA) and long non-coding RNA (lncRNA). These ncRNA act to silence gene expression either pre- or post-transcriptionally through diverse mechanisms that range from regulation of open chromatin states and DNA methylation status of promoters to direct targeting of mRNA species for enzymatic degradation to translational repression (Esteller, 2011). It has become clear that many key biological processes are regulated through ncRNA, which has led to an increased interest in their use for biomarker development (Vrijens et al., 2015). Several factors suggest the potential of ncRNA as biomarkers. Collectively the ncRNA comprise 60–70% of the total transcriptome and their aberrant expression has been associated with etiology of diverse diseases (Esteller, 2011). They are thought to regulate up to 60% of genes and their expression and activity has been shown to be responsive to a broad array of environmental stressors including chemical exposure (Friedman et al., 2009; Nilsson et al., 2018; Vrijens et al., 2015). Many ncRNAs have been found extracellularly in plasma, as well as in urine (Mall et al., 2013), saliva (Bahn et al., 2015), and semen (Barcelo et al., 2018), thus they can be sampled non-invasively. Extracellular ncRNAs have been shown to be stable in accessible fluids (Mall et al., 2013; Yang et al., 2015) and are often encapsulated in extracellular vesicles which further protect them from degradation (Mall et al., 2013; Valadi et al., 2007), suggesting that ncRNA-based biomarkers may act as a longer lasting signal relative to mRNA biomarkers. Lastly, because as a group they rely upon such a broad array of mechanisms for gene silencing and can have very different cellular targets, if evaluated together in a single assay, they may provide a systemic picture of physiological status that may not be possible using just mRNA.
Of the ncRNA, miRNAs are the most well-characterized and several diagnostic miRNA biomarkers on have been developed and are currently available for clinical use (Bonneau et al., 2019). A single miRNA can regulate multiple genes and conversely multiple miRNAs can regulate the same gene (Enright et al., 2003). Gene regulation by miRNAs is mediated through the ribonucleoprotein miRNA/argonaut RNA-Induced Silencing Complex (RISC). The miRNA functions to target the complex to mRNA based on complementary base pairing. Once bound, the RISC can degrade the target mRNA through its endonuclease activity or, if there is limited complementarity, can destabilize the target mRNA through removal of the poly-A tail or through translational repression (Jo et al., 2015; O’Brien et al., 2018). Another class of ncRNAs, piRNA, has also been used in biomarker development. piRNAs were previously thought to only be expressed in the germ line where they protect the integrity of the genome by suppressing expression of transposable element (TE) mRNA. More recently, piRNA expression has been observed in the soma (Yan et al., 2011) and they have been shown to be differentially expressed in response to chemical insult, tissue injury, and in a numerous cancer types independent of TE mRNA (Rojas-Rios and Simonelig, 2018).
Using larval Pimephales promelas (fathead minnow; FHM) as a model organism, we have previously demonstrated that mRNA signatures are able to identify organisms exposed to chemicals with similar MOA at concentrations below those that elicit acute responses (Biales et al., 2016; Kostich et al., 2019). For example, an mRNA biomarker trained on the pyrethroid bifenthrin was able to accurately classify larvae exposed to other pyrethroids, demonstrating that the domain of applicability spanned chemicals with the same molecular target (Biales et al., 2016). This same biomarker also successfully discriminated control and organophosphate pesticide exposed organisms. The OP pesticides also target neuronal signaling, but at a different point in the signaling cascade, suggesting that the domain of applicability of the bifenthrin biomarker may be extended to include additional neurotoxicological responses (Kostich et al., 2019). The current work aims to expand the number of existing transcriptomic biomarkers to include estrogenic compounds and to evaluate training and testing strategies to maximize the range of concentrations-where the biomarker remains accurate. Estrogenic compounds are structurally diverse (Blair et al., 2000) and can cause effects in both estrogen receptor (ER) dependent and independent mechanisms (Marino et al., 2006). This potentially complicates the identification of putative estrogens using either in silico approaches or receptor-based assays. A third aim of this study was to evaluate the biomarker potential of small ncRNAs (sncRNA; <200 nucleotides). This study focused on miRNA and piRNA, both of which are responsive to chemical exposure but utilize different mechanisms for gene silencing. Because miRNA and piRNA are similarly sized, they can be isolated together opening the possibility of a including both in the development of biomarkers.
2. Materials and methods
2.1. Exposure organisms
Larval FHM were obtained from the on-site culture at the U.S. EPA Andrew W. Breidenbach Environmental Research Center in Cincinnati, OH as described in (Office of Water, 2002). However, to ensure that larvae were closely synchronized in their development, spawning tiles were placed in breeding tanks in the morning and were removed after one hour. Eggs and larvae were maintained in an incubator at 25°C in dechlorinated tap water supplemented with CaCO3 to a hardness of 180 mg/L.
2.2. Test chemicals and exposure water
All exposures were performed in moderately hard reconstituted water (MHRW; (Office of Water, 2002). A master stock solution of EE2 was prepared in DMSO at a concentration of 1.0 μg/L. A 10 ng EE2/L exposure solution was prepared by adding 40 μl of the stock solution to 4.0 L MHRW. This solution was then serially diluted to produce 5.0, 2.5, 1.2, and 0.12 ng/L solutions. The concentration of DMSO was kept at 0.002% in all exposure solutions, which were prepared each morning.
2.3. Exposures
mRNA: Three identical replicate exposure experiments were performed over a three-week period. For each experiment, larvae that had hatched at the start of the experiment (96 hpf /- 1 h) were used in exposures. Thirteen larvae were placed in replicate 150 mL beakers containing 130 mL exposure solution, for a loading rate of 1 mg/10 ml (0.1 g/L). Larvae were maintained at 25°C under a 16:8 h light:dark cycle. The control, 1.25, and 10 ng/L treatments each had 5 replicate beakers (n = 65 total per treatment), while the remaining treatments had three (n = 39 total per treatment). Mortality was assessed at 6 and 24h and dead larvae were removed. Overall, mortality was 2.7% with no difference between treatments. Exposures were conducted for 48 h, with a renewal of approximately 90% of the exposure solution at 24 h. At 48 h, five individuals from each exposure vessel were randomly selected, placed into separate 1.5 ml microcentrifuge tubes containing a 3.2 mm stainless steel bead (n = 10 per treatment X 3 exposures) and snap frozen in liquid nitrogen separately. Samples were subsequently stored at −80°C until used. sncRNA: For the sncRNA, a fourth exposure was conducted for the control and 10 ng/L EE2 treatment only (3 beakers per treatment; n = 39 total per treatment). The increased mass needed for small RNA library preparation necessitated that two larvae be pooled and treated as a biological replicate (n = 4 pools of two larvae per beaker – 12 per treatment total for exposure). Collection and storage were as described above.
2.4. Analytical water chemistry
Water samples were collected for analysis at initiation of each of the four exposure and after 24 h. Initial volumes were one liter and final volumes ranged from approximately 360–960 ml, depending upon the number of beakers used per treatment. All samples collected for water chemistry were stored in 1 L silanized amber bottles at 4°C until extraction and analysis.
Concentration of EE2 in the exposures was verified by liquid chromatography tandem mass spectrometry (LC-MS/MS) using a Quattro micro API Tandem Quadrupole System (Waters Corporation).
Methanol was added to each water sample prior to analysis to achieve a concentration of 1% (v/v). Samples were filtered through a 1.2 μm glass microfiber filter and then passed through a C-18 solid phase extraction (SPE) column. The column was extracted with acetone, concentrated, and derivatized with dansyl chloride. Equilin, equilenin, and testosterone-[d5] were used as surrogate analytes. Ethynylestradiol-[d4] was used as the internal standard.
The reporting limit for EE2 analysis was 0.1 ng/L. Average recovery of surrogate standards for all analyzed water samples was 75% for equilin, 76% for equilenin, and 100% for testosterone-[d5].
2.5. RNA isolation, library preparation, and sequencing
RNA was extracted from larvae using MagMAX™−96 Total RNA Isolation Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. Samples were removed from −80°C and immediately placed on ice. MagMax lysing/binding buffer (100 μl) was added to each tube. Samples were homogenized using a Bullet Blender Storm 24 homogenizer (Next Advance, Troy, NY). RNA was quantified spectrophotometrically on a Synergy™ HTX Multi-Mode Microplate Reader using a Take3 Micro-Volume Plate (Biotek). RNA quality was assessed using a 4200 TapeStation (Agilent).
For gene expression profiling, mRNA libraries were prepared from 30 samples in the control, 1.2, and 10 ng EE2/L treatment groups, and 18 samples from the 0.12, 2.5 and 5 ng EE2/L groups. Each treatment group contained an equal number of samples from each replicate exposure experiment. Libraries were prepared from 250 ng RNA using the SENSE mRNA-Seq Library Prep Kit V2 (Lexogen, Greenland, NH) according to the supplied protocol. The concentration of each library was determined using the Qubit™ dsDNA HS Assay with a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA). Equimolar amounts of libraries from 16 samples were pooled and loaded onto one lane of an Illumina HiSeq 4000 flow cell and sequenced in the 1 × 50bp single read format using HiSeq 4000 SBS reagents (Illumina, San Diego, CA). Base calling was done by Illumina Real Time Analysis (RTA) v2.7.7 and output of RTA was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v2.19.1. 16 individual libraries were pooled. onto one lane of an Illumina HiSeq 4000 flow cell and sequenced in the 1 × 50bp single read format using HiSeq 4000 SBS reagents. Base calling was done by Illumina Real Time Analysis (RTA) v2.7.7 and output of RTA was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v2.19.1.
For small RNA sequencing, 1000 ng of total RNA was used as input for library preparation using the TruSeq Small RNA Library preparation kit (Illumina) following manufacturer’s protocol (n=10 per treatment). Sequencing was performed as described above with the exception that these 20 samples were split evenly into 2 pools for sequencing in two different lanes. Treatments were split across sequencing lanes to avoid batch effects.
2.6. mRNA mapping
The 50 base long reads in the RNA-Seq fastq(.gz) file for each sample were mapped to indexed transcripts from each of the 47,578 gene models (which include isoforms) from the newly updated FHM genome (WIOS00000000; BioSample SAMN12875914; (Martinson et al., 2021) using the STAR alignment software (version2.7.1a)(Dobin et al., 2013). Following STAR mapping, sam output was filtered to only preserve primary mappings in the reverse strand, and only keeping target_id and any XS or AS tags. A feature X sample matrix was generated from filtered sam files and a target fasta file
Count matrices were filtered to so that only those features for which at least three samples had at least 10 counts were included. All analyses were based on the reverse strand count matrix. An initial QC assessment was done at this stage involving generation of density plots, a box plot, and MDS plots for each toxicant/control combination. Density and box plots showed no unusual count distributions, but MDS plots revealed three outlier samples which were removed from analysis.
2.7. Differential expression analysis and feature selection
Differential expression analysis was performed using the R limma package (version 3.40.6; (Ritchie et al., 2015). Between sample normalization was conducted using the trimmed mean of M-values (Robinson and Oshlack, 2010) method implemented by the calcNormFactors function from the R edgeR package (version 3.26.8) (Robinson et al., 2010). Precision weights (Law et al., 2014) were calculated using the voom function from the limma package. The limma lmFit function was used to fit a generalized linear model with a term for chemical treatment effects and a term for experimental batches. Moderated t-statistics (Phipson et al., 2016) were calculated for chemical treatment effects using the limma eBayes function. The limma topTable function was used to sort features (i.e. genes) based on their nominal p-values. For comparison of the top 100 features across dose, all samples were used for generation of rankings (n = 29, 28, 17, 18, 30, 17 for the control, 10, 5.0, 2.5, 1.25, and 0.125 ng/L EE2 treatments respectively) and the Benjamini-Hochberg (BH) procedure was used to correct raw p values for multiple testing (Hochberg and Benjamini, 1990).
The majority of the top 100 genes were annotated based on (Martinson et al., 2021). Sequences for the remaining genes were compared to the NCBInt database using the megablast algorithm and annotations were taken directly from the top scoring alignment. Heatmaps were generated for the 30 transcripts with the lowest adjusted p values (Metsalu and Vilo, 2015)
2.8. Classifier tuning, training, and testing
The paradigm employed for feature selection was based on the discriminative power of the features and did not consider known mechanistic links to the chemical class of interest (Kostich et al., 2019). No p-value or FDR cutoff was employed for feature selection for use in classification since the number of top features used during classification was treated as an empirically tunable parameter as described below. Feature counts were quantile normalized and batch-adjusted. Subsequently, features were analyzed for correlations – features with correlation coefficients greater than or equal to 0.9 were removed. Binary classifiers (control vs. treatment) were developed using both random forest (RF, version 4.6–14, (Breiman, 2001)) and logistic regression with elastic net regularizations (glmnet, version 2.0–18, (Friedman et al., 2000)).
Classifiers were developed to discriminate EE2-exposed from control organisms. In order to maximize the concentration domain of applicability for the classifier, three different training strategies were evaluated: 1) 10 ng/L EE2 vs. control, 2) 2.5 ng/L vs. control, or 3) 2.5 + 10.0 ng/L (pooled) vs. control. Prior to training, samples were split at a 75/25 ratio into training and test sets respectively using the caTools R package. Training was conducted using a five-fold cross validation (CV) approach.
Classifiers were empirically tuned using a grid search approach. Sixty points in parameter space are chosen using a uniform random sample either on the raw scale or, if indicated, on a log scale. For feature selection and classification, a subset of the most significantly differentially expressed genes, i.e., features with the lowest adjusted p-value, is defined by randomly selecting the number of top features on a log scale in the interval [10, 1000]. Additional details of parameter tuning, including the specific parameters tuned for each algorithm, can be found in (Kostich, et al., in preparation).
Best performing classifiers from CV were then evaluated against the 25% test set (samples not included in training) which were exposed to the same concentrations of EE2 as the respective training set. Classifier performance was evaluated using the area under the receiver operator curve (AUC) and the Brier score. Classifiers were then evaluated for their ability to correctly discriminate EE2 treated from control fish at each of the concentrations not used in training.
2.9. miRNA identification
FHM microRNAs (miRNA) were identified previously using the command-line version of miRDeep*, MDS_command_line_V37(An et al., 2013) as described in Martinson et al. (Martinson et al., 2021). Using the miRDeep* log-odds probability score for being a genuine miRNA precursor, 620 miRNAs resulted after applying a score filter of 100 (only 2 of these received “No hits” when compared to the miRBase.org mature miRNA database). Furthermore, this list included 118 of 374 zebrafish miRNA reported at mirBase.org.
2.10. miRNA mapping, feature counts and classification
FHM small RNA (smRNA) reads were mapped against the 620 FHM miRNAs using Bowtie (bowtie-1.2.3-linux-x86_64)(Langmead et al., 2009). Counts of alignments for features proceeded as described above for FHM mRNA. Likewise, RF classifications of EE2 miRNA features proceeded as for FHM mRNA with the exception that classification was not evaluated across additional concentrations. Since both DNA strands have been shown to be used in miRNA transcription (Song et al., 2020), feature counts and classifications were analyzed for each strand separately. Feature lists were ultimately generated from those ranked features from the 75% training split that were included in the random forest classifier model.
2.11. piRNA mapping, feature counts and classification
Unlike for the miRNAs, at present a high quality FHM piRNA reference set does not exist, therefore to putatively identify FHM piRNAs and to create count matrices, all smRNA reads were mapped against the existing zebrafish piRNA reference set which has approximately 1.33 M piRNAs (zebrafish piRNA v2 library in piRBase) (Wang et al., 2019b; Zhang et al., 2014) using Bowtie. Approximately 10% of FHM smRNA mapped to published zebrafish piRNA sequences (3% of reads with at least one reported alignment). Count matrices were generated as described for mRNA. Random Forest classification was conducted as described for miRNA. As was the case with miRNA, piRNAs may be transcribed in the sense or antisense orientation (Choudhuri, 2011; Ozata et al., 2019) thus all analyses were conducted using each strand separately. Feature lists were ultimately generated from those features that were included in the classifier model.
3. Results
3.1. Exposure concentrations
Except for the lowest exposure concentration (0.12 ng/L) measured EE2 values were similar to nominal (Table 1). There were no appreciable losses of EE2 following 24 hours of exposure, suggesting more or less constant exposure levels over the exposure. For the fourth exposure experiment, measured values were slightly below the nominal.
Table 1.
Measured values of EE2. Water was renewed after 24 h of exposure. Samples for water chemistry at Time 0, just prior to renewal, just after renewal, and at the completion of the study.
| Target conc. | Time 0 | 24 h | Time 0 renewal | 24 h | Average conc. |
|---|---|---|---|---|---|
| 0 | 0.03 | 0.06 | 0.00 | 0.00 | 0.02 |
| 0.125* | 0.09 | 0.00 | 0.15 | 0.07 | 0.08 |
| 1.25 | 1.37 | 1.15 | 1.40 | 1.49 | 1.35 |
| 2.5 | 2.73 | 2.46 | 2.77 | 2.40 | 2.59 |
| 5 | 5.28 | 5.11 | 5.89 | 5.15 | 5.36 |
| 10 | 10.67 | 9.43 | 11.87 | 11.50 | 10.87 |
| Target Conc | Time 0 | 24 h | Time 0 renewal | 24 h | Average conc. |
| 0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 10 | 8.51 | 8.91 | 10.47 | 8.31 | 9.05 |
Units are ng/L EE2.
indicates at minimum reporting limit.
3.2. EE2 exposures–FHM mRNA analyses
Accurate classification over a broad range of concentrations broadens the utility of biomarkers for most applications. We sought to develop a binary classifier able to distinguish EE2-exposed from non-exposed fish that remained accurate over all or most concentrations tested. To this end, three different training strategies were evaluated to determine which produced a classifier with the broadest concentration domain of applicability. Training sets included organisms from: 1) high exposure (10 ng/L EE2), 2) low exposure (2.5 ng/L EE2), or 3) incorporated both (10 2.5 ng/L). Each of the three resulting classifiers were first tested against a test set, made up of control FHM and organisms exposed to the same concentrations as the training set, but were not used in training. Secondarily, classifiers were tested for their ability to accurately classify organisms exposed to the remaining concentrations. Five concentrations were included in the exposure experiment, which spanned a range of environmental concentrations (0.12, 1.25, 2.5, 5.0 and 10.0 EE2 ng/L).
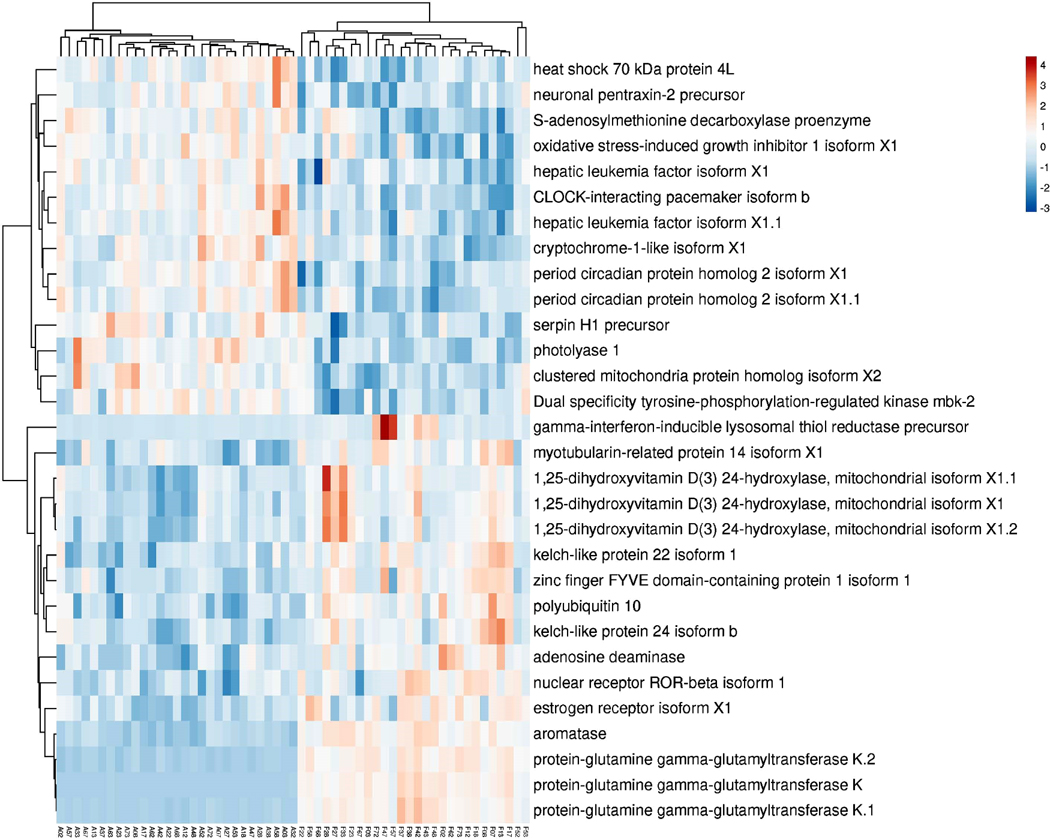
In order to determine the similarity of the transcriptomic response, gene expression was examined at each of the six (5 concentrations plus the 10 and 2.5 EE2 ng/L combination) relative to control fish using all samples. With the exception of 10 EE2 ng/L and the 10 ng + 2.5 ng/L EE2 treatments, which had 81 and 33 differentially expressed genes (DEGs) respectively, few DEGs were identified in any treatment following BH correction (Top 30 DEGs; Table 2). Not surprisingly, of 33 differentially expressed genes (DEGs) in the 10 ng + 2.5 EE2 ng/L treatment, all but one was also identified in the 10 ng/L treatment. Only one gene, which was unique to that treatment, was identified in the lowest treatment level, suggesting that this concentration is at the threshold for a transcriptional response for this duration of exposure. There was significant overlap of DEGs across all treatments, with four transcripts, corresponding to aromatase and isoforms of protein-glutamine gamma-glutamyltransferase K, being identified in all treatments but 0.1 ng EE2 (Fig. 1). The significant overlap among treatments strongly supports the biological relevance of the observed gene expression data.
Table 2.
Top 30 transcripts identified as differentially expressed between the 10 ng/L EE2 and control treatments. Differential expression analysis was conducted using 100% of samples from either the control group or those exposed to 10 ng/L EE2 using the limma R package. p values were adjusted using Benjamini-Hochberg. Repeated gene names represent putative isoforms.
|
Indicates genes were also found in the 5, 2.5, and 1.25 ng/L EE2 treatments.
Fig. 1.

Differentially expressed genes across all treatments. Larval fathead minnow were exposed to one of five concentrations of EE2 for 48 h and RNA-seq was conducted to identify differentially expressed genes (DEGs). DEG analysis was performed using limma (FDR < 0.05)
For classifier development, differential expression analysis was conducted for genes in the training set (75% of samples), all genes were rank ordered by their adjusted p value and those with the lowest values were evaluated as features in classifier training. When feature counts showed correlations greater than or equal to 0.9, only the feature with the highest adjusted p-value for differential expression was retained. Importantly, because no significance cutoff was used, many of the features evaluated were not statistically differentially expressed
Across all concentrations, RF and glmnet performed similarly; however, classification accuracy across concentrations (Fig. 2a–d, Supplementary Fig. 1a–d) and Brier scores were slightly better for RF (Table 3). When evaluated against their corresponding test sets, which were made up of organisms exposed to the same concentrations used in training, all three training strategies proved highly accurate. However, when their ability to accurately classify EE2 exposed vs. non-exposed organisms across concentrations was evaluated, the training strategy that employed the 10 ng EE2/L treatment alone proved much less accurate (for predicting 2.5 and 5 ng EE2 samples) than those that incorporated low-dose exposures into the training. Both the 2.5 ng/L and the 10 + 2.5 ng/L EE2 trained classifier performed similarly, indicating that the addition of the 10 ng samples in training did not add any discriminatory capability. The high degree of overlap of DEGs between the combined and 10 ng/L treatment (32/33) suggested that classification is mostly driven by features identified in the low exposure group.
Fig. 2.

Prediction probabilities of random forest classifiers trained on either 10, 2.5, or a combination of 10 and 2.5 ng/L EE2 fathead minnow larvae. (A) Prediction probabilities of all three training scenarios, (B) 10 ng/L EE2 scenario with error bars, (C) 2.5 ng/L EE2 scenario with error bars or (D) combination training set with error bars.
Table 3.
Evaluation of RF and glmnet algorithms using different training sets against test sets of organisms from the same concentration of EE2.
| Instance | Area Under ROC Curve (AUC) | Brier score |
|---|---|---|
| randomForest; train on 10.0 ng EE2 | 1 | 5.95E-03 |
| randomForest; train on 2.5 ng EE2 | 1 | 1.52E-02 |
| randomForest; train on 2.5/10 ng EE2 combo | 1 | 4.17E-03 |
| glmnet; train on 10.0 ng EE2 | 1 | 4.33E-06 |
| glmnet; train on 2.5 ng EE2 | 1 | 1.42E-02 |
| glmnet; train on 2.5/10 ng EE2 combo | 0.93 | 1.98E-01 |
3.3. EE2 exposures–FHM miRNA analyses
Differential expression of FHM miRNAs was studied using fish that were exposed during a fourth replicate experiment. Though the fish are from the same culture and the same stock solutions were used, fish were from a different hatch, however, exposure conditions were identical. Due to the larger RNA input requirement for the small RNA library preps, replicates were pools of two larvae. Because this was an initial investigation of the potential of ncRNA for biomarker development, only the 10 ng/L EE2 and control treatments were evaluated.
Although 23 miRNAs had unadjusted p-values of less than 0.05, none of them met the false discovery rate criterion for statistical significance (< 0.05). Classification proceeded as described for the mRNA, however, due to its superior performance with mRNA, only the RF algorithm was used. The best performing classifier from cross-validation had 22 (Table 4) and 17 miRNA features from the forward and reverse strand respectively. Classification of the test set was fairly accurate for the forward strand with an AUC of 0.83 (Brier 0.217).
Table 4.
10 ng EE2 miRNA Features from forward strand orientation. miRNA features were selected by RF classifier. t = t statistic, p.value is the unadjusted p value, adj.P.Value is the p value following BH correction. Sequences were compared to miRBase using Blastn to identify homologous miRNAs. E values are the expected value given a database of that size.
| FHM miRNA name | t | P.Value | adj.P.Val | Blastn of sequence vs. mirbase.org database | E value |
|---|---|---|---|---|---|
| novelMiR_1551 | 4.04 | 9.29E-04 | 0.49 | Drosophila melanogaster miR-305 | 27 |
| novelMiR_10582 | 2.68 | 0.02 | 0.82 | Monodelphis domestica miR-759 | 17 |
| novelMiR_16250 | 2.66 | 0.02 | 0.82 | Homo sapiens miR-27a | 20 |
| novelMiR_13155 | 2.52 | 0.02 | 0.82 | Homo sapiens miR-25 | 12 |
| novelMiR_14487 | 2.47 | 0.02 | 0.82 | Schistosoma mansoni miR-8467 | 10 |
| novelMiR_13707 | 2.41 | 0.03 | 0.82 | Mus musculus miR-182 | 9.00E-04 |
| novelMiR_39194 | 2.38 | 0.03 | 0.82 | Mus musculus miR-204 | 9.00E-04 |
| novelMiR_16920 | 2.36 | 0.03 | 0.82 | Sorghum bicolor miR6227 | 7 |
| novelMiR_23779 | -2.31 | 0.03 | 0.82 | Epstein Barr virus miR-BART3 | 8.5 |
| novelMiR_35382 | 2.29 | 0.04 | 0.82 | Echinococcus granulosus miR-10240 | 3.2 |
| novelMiR_15023 | −2.25 | 0.04 | 0.82 | Gallus gallus miR-210b | 8.5 |
| novelMiR_13066 | −2.24 | 0.04 | 0.82 | Danio rerio miR-146a | 8.00E-04 |
| novelMiR_1190 | −2.20 | 0.04 | 0.82 | Bos taurus miR-2346 | 11 |
| novelMiR_44859 | −2.19 | 0.04 | 0.82 | Bombyx mori miR-3257 | 57 |
| novelMiR_37380 | 2.17 | 0.05 | 0.82 | Columba livia miR-1641 | 7 |
| novelMiR_9594 | −2.17 | 0.05 | 0.82 | Danio rerio miR-202 | 0.002 |
| novelMiR_35457 | −2.14 | 0.05 | 0.82 | Danio rerio miR-1788 | 0.002 |
| novelMiR_2139 | −2.13 | 0.05 | 0.82 | Glycine max miR5033 | 11 |
| novelMiR_36470 | −2.10 | 0.05 | 0.82 | Brugia malayi miR-5879a | 3.6 |
| novelMiR_34701 | −2.05 | 0.06 | 0.82 | Danio rerio miR-148 | 0.002 |
| novelMiR_37665 | −2.04 | 0.06 | 0.82 | Danio rerio miR-15b | 9.00E-04 |
| novelMiR_34702 | 2.01 | 0.06 | 0.82 | Danio rerio miR-10d | 9.00E-04 |
3.4. EE2 exposures–analyses of FHM piwi-interacting RNA
Similar to miRNAs, no statistical differences in piRNA was observed following post-hoc correction. Classifiers were developed for the piRNAs in both forward and reverse strand orientation with RF as described for the miRNAs, which resulted in 13 (forward) and 12 (reverse) features (Table 5). Classification conducted resulted in a highly accurate prediction of controls and 10 ng EE2 test set using the reverse strand orientation AUC of 1.00 (Brier 0.060; forward strand AUC = 0.5).
Table 5.
Features included in the RF-based piRNA classifier based on the reverse strand orientation. In order to identify FHM piRNA small RNA sequences were compared to the zebrafish piRNA reference library. Best scoring matches were used as identity.
| piRNA name | t | P.Value | adj.P.Val |
|---|---|---|---|
| piR-dre-6663 | −4.09 | 7.68E-04 | 0.37 |
| piR-dre-66662 | −3.69 | 1.84E-03 | 0.37 |
| piR-dre-29844 | −3.67 | 1.91E-03 | 0.37 |
| piR-dre-1081409 | 3.24 | 4.84E-03 | 0.53 |
| piR-dre-29926 | −3.20 | 5.32E-03 | 0.53 |
| piR-dre-16862 | −3.12 | 6.25E-03 | 0.53 |
| piR-dre-10207 | −3.03 | 7.69E-03 | 0.53 |
| piR-dre-47571 | −2.97 | 8.57E-03 | 0.53 |
| piR-dre-453260 | −2.96 | 8.77E-03 | 0.53 |
| piR-dre-24682 | −2.95 | 9.08E-03 | 0.53 |
| piR-dre-31286 | −2.88 | 1.04E-02 | 0.55 |
| piR-dre-49631 | 2.56 | 2.03E-02 | 0.80 |
4. Discussion
Omics-based biomarkers have demonstrated their utility for chemical and phenotypic read-across (Brum et al., 2015; Lv et al., 2017; Wang et al., 2016). Much of the progress in the development of omics-based biomarkers has focused on protecting human health and has employed mammalian model species or in vitro systems. However, these biomarkers may have limited applicability for the protection of ecological health given the diversity of taxa and exposure routes found in an ecosystem. Recently, our group has successfully developed omics-based biomarkers using FHM larvae, a commonly used ecotoxicological model species. Though still early in their development, these biomarkers have demonstrated increased sensitivity relative to apical toxicological endpoints, such as mortality (Biales et al., 2016), and are accurate at levels of regulatory concern (Kostich et al., 2019). Additionally, in initial and albeit limited studies, we have demonstrated that omics-based biomarkers are capable of accurately identifying organisms exposed to chemicals mechanistically related to those used in biomarker development (Biales et al., 2016; Kostich et al., 2019). One objective of the current study is to build upon these previous studies to further characterize the performance of omics-based biomarkers. Specifically, we sought to identify the concentration domain of applicability of newly developed biomarkers and to identify model training strategies that would maximize the range of concentrations where the biomarker remains accurate.
Three approaches were used to train classifiers that differed in that they utilized organisms exposed to relatively low levels of EE2 (2.5 ng/L), high levels (10 ng/L) or a combination of both. When tested on holdout sets that were exposed to the same concentration used in the training set, all three strategies performed well, with near perfect accuracy (Fig. 2). However, when tested at other concentrations, the model trained only on the 10 ng/L concentrations was able to classify 5 ng/L EE2 but performed poorly when applied to low concentrations (Fig. 2b). Using the RF algorithm for prediction, both the low and combination training strategies correctly predicted EE2 exposure against all concentrations tested with the exception of the lowest concentration (0.12 ng/L). The average measured concentration of the nominal 0.12 ng/L treatment was 0.08 ng/L (Table 1), suggesting that it was below the threshold for biological activity for an exposure of this duration. However, it is worth noting that though the mean probability of class membership using the glmnet classifier was well below the 0.5 threshold, three biological replicates were identified as exposed with probabilities of exposure of 0.98, 0.64, and 0.82 respectively. It is unclear whether this is biologically significant or just random noise, however, to our knowledge, no studies have observed biological effects at EE2 concentrations this low. Several studies observed EE2 dependent biological responses at low ng to high pg concentrations, however, none employed concentrations as low as 0.08 ng/L (Lange et al., 2001; Parrott and Blunt, 2005). Further, even with full life-cycle exposures, low-level effects were not evident until later points in the life cycle. Together, this supports the idea that 0.08 ng/L EE2 is below the biologically active concentration for EE2. The variability in the probability of class membership increased at the 1.25 ng/L in both strategies, but overall, the RF classifier was able to determine that these organisms had been exposed. Given that the classifiers trained on either the 2.5 ng/L and the 10 + 2.5 ng/L EE2 performed similarly, inclusion of features only present in the 10 ng/L EE2 exposure added no discriminative ability. It is also worth noting that though only five DEGs were identified in the 2.5 ng/L treatment, the classifier contained 14 features, suggesting that the use of a hard DEG cutoff for feature selection may be too conservative. Our data suggest that training classifiers on lower concentrations results in a broader range of applicability than training on a relatively high concentration, though this assertion is based on a single evaluation. Further work will be conducted to substantiate or invalidate this observation.
This degree of sensitivity of the classifier appears to be within the range of EE2 concentrations that induce reproductive effects after prolonged exposure (Rutherford et al., 2020), suggesting that the biomarker may have utility as a relatively rapid (48h vs. 21d) screening tool for estrogenic compounds. The sensitivity of this biomarker also compares favorably to other commonly used molecular biomarkers, such as vitellogenin (vtg) mRNA expression in adult fish (Flick et al., 2014). The use of larvae as opposed to adults offers practical advantages, as they require considerably less resources both in terms of needing to rear test organisms to adulthood and the amount of exposure media needed to meet recommended loading rate for aquatic toxicity testing (Office of Water, 2002), making them more amenable to high throughput screening.
Feature selection is data driven, based on the discriminative ability and consistency of the potential features, and established mechanistic links to the condition of interest (e.g. estrogenic activity) are not considered in selection. However, once the biomarker is established, evaluation of the features may help identify relevant toxicity pathways including those not previously associated with the condition. Additionally, evaluating overlap among DEG lists generated from related treatments (across concentrations or experiments) may also be used as orthogonal evidence of the biological relevance of included features. In the current work, with the exception of the 10 ng/L EE2 treatment (81 DEGs), few genes were identified as differentially expressed in lower concentrations (maximum of five in the 2.5 ng/L treatment), however, DEG lists among treatments were markedly consistent. The same four transcripts were expressed in all treatments with the exception of 0.12 ng/L. These transcripts correspond to aromatase and paralogues of protein-glutamine gamma-glutamyltransferase K, both of which have previously been shown to be responsive to estrogens (Cleuren et al., 2010; Martyniuk et al., 2006). We subjected the 100 genes with the lowest FDR (maximum FDR = 0.076) to enrichment analysis using Ingenuity Pathway Analysis. Few canonical pathways were enriched, which likely results from the pooling of multiple tissues, however, circadian rhythm (CR) signaling was found to be enriched (data not shown). Estrogens play a role in controlling CR and early exposure to estrogens or androgens may disrupt development programming, resulting in altered CR during adulthood (reviewed in (Hatcher et al., 2020)). Variation in the levels of circulating hormones has been previously related to CR in fish (Lamba et al., 1983). Recent work has demonstrated that exogenous exposure to steroid hormones alters the expression of circadian rhythm related genes in fish (Liang et al., 2019). As CR play important roles in all cyclic behavioral activities (e.g. feeding, reproduction, etc) this has potentially important implications for later life effects resulting from developmental exposure to xenoestrogens.
The differential expression of the well-known estrogenic biomarker, vtg, was only observed in the 10 ng/L treatment. This is somewhat surprising as previous studies observed up-regulation at lower concentrations (Biales et al., 2007; Flick et al., 2014). The lack of observed up-regulation in the current study may have been a consequence of the technological approach. The majority of published studies that examined vtg transcription utilized PCR-based technologies and though RNA-seq is highly sensitive and its ability to identify DEGs is comparable to that of PCR (Li et al., 2014), the underlying technology and statistical approaches differ significantly. The current study also used very early life stage (96 hpf) organisms of indeterminate sex, whereas vtg is generally considered a reliable estrogenic biomarker when up-regulated in the livers of adult male fish. Though the use of whole larvae offers some practical advantages over adults, it requires that the entire larvae be sampled which results in tissue specific transcriptional profiles being averaged across all tissues. The ability to identify differentially expressed transcripts will be a function of the tissue specific magnitude of expression and the relative mass of the target tissue. Though this averaging effect somewhat confounds interpretation of the biological response, we have demonstrated both in the current study, as well as previously, that the sensitivity of the larval derived biomarker is comparable to biomarkers developed in the target tissue (Kostich et al., 2019). Further, in both this and previous studies, we and others have observed the up-regulation of genes known to be responsive to the exposure chemical (Kostich et al., 2019; Liao et al., 2009).
Despite the importance of small ncRNAs in regulating a huge array of biological processes (Esteller, 2011) including many with toxicological relevance (Tsuchiya et al., 2006), there are relatively few studies characterizing small ncRNA responses in fish species and even fewer that have developed and evaluated sncRNAs as potential biomarkers of exposure. Several studies have identified the differential expression of sncRNAs in fish following exposure to chemical toxicants (Kure et al., 2013; Wang et al., 2019a; Wang et al., 2013). In the current study, no sncRNAs were identified as differentially expressed following multiple test correction. This was somewhat surprising as miRNAs have been shown to be responsive to estrogens in several model systems including fish (Cohen and Smith, 2014; Klinge, 2012). Despite the lack of statistical significance, both miRNA- (AUC = 0.83) and piRNA-(AUC = 1.0) biomarkers accurately discriminated exposed from control larvae. Given that the ability to accurately discriminate two conditions requires that they have different expression profiles, classification can be considered a functional test of differential expression. This suggests that the cutoff employed was too stringent. The lack of observed statistical significance may be an artifact of averaging across tissues, as sncRNAs expression is known to be tissue specific (Volinia et al., 2006). Further supporting the biological relevance of sncRNA features, several of the miRNAs that were identified (Table 3) have been previously linked to estrogens (Bailey et al., 2015; Ferraro et al., 2012; Liu and Li, 2015; Nothnick and Healy, 2010). Though piRNAs have also been shown to be differentially regulated by estrogens (Oner et al., 2016), their role is not as well characterized and interpretation of mechanistic relevance in the estrogenic piRNA response is complicated by the lack of a high quality FHM piRNA reference set. However, that both miRNAs and piRNAs were able to accurately discriminate EE2-exposed larvae from control, suggests that they are involved in the organismal response to estrogens and further study into their mechanistic relevance is warranted.
5. Conclusions
We have developed an mRNA-based biomarker for estrogen exposure that remains accurate at both low and high concentrations of EE2 and displays similar or increased sensitivity compared to existing methods. As opposed to in vitro or cell-free systems, the current method utilizes a whole organism and can account for metabolic processes that may alter the toxicity of the parent compound. The use of whole larvae should also be able to identify estrogens that work though both the genomic (i.e. binding to the ER-a nuclear receptor) and non-genomic estrogenic pathways which is not possible using current in vitro systems (Klinge, 2015). Further, whole larvae incorporate all toxicity pathways and is not limited by a priori considerations of target tissue or MOA. As a single biomarker it may be considered expensive or resource intensive, but the long-term intent is to incorporate this biomarker into a larger panel of omics-based biomarkers targeting other MOA all developed within the same model system. This would provide an unbiased, assumption-free means to screen multiple MOA simultaneously, thus reducing the per assay cost. We have also evaluated the biomarker potential of miRNAs and piRNAs. Biomarkers based on either were able to accurately classify organisms exposed to 10 ng/L EE2. Both of the sncRNAs species utilized are known to be responsive to estrogens and are associated with the etiology of breast cancer, suggesting that their mechanistic role in the estrogen response be further investigated.
Supplementary Material
Acknowledgment
The views expressed in this [article/presentation/poster] are those of the author(s) and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Sequencing was performed at the Research Technology Support Facility (RTSF) Genomics Core at Michigan State University.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.aquatox.2021.105807.
References
- An J, Lai J, Lehman ML, Nelson CC, 2013. miRDeep*: an integrated application tool for miRNA identification from RNA sequencing data. Nucl. Acids Res 41, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, Xiao X, 2015. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin. Chem 61, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey ST, Westerling T, Brown M, 2015. Loss of estrogen-regulated microRNA expression increases HER2 signaling and is prognostic of poor outcome in luminal breast cancer. Cancer Res. 75, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelo M, Mata A, Bassas L, Larriba S, 2018. Exosomal microRNAs in seminal plasma are markers of the origin of azoospermia and can predict the presence of sperm in testicular tissue. Hum. Reprod 33, 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biales AD, Bencic DC, Flick RW, Lazorchak J, Lattier DL, 2007. Quantification and associated variability of induced vitellogenin gene transcripts in fathead minnow (Pimephales promelas) by quantitative real-time polymerase chain reaction assay. Environ. Toxicol. Chem 26, 287–296. [DOI] [PubMed] [Google Scholar]
- Biales AD, Kostich MS, Batt AL, See MJ, Flick RW, Gordon DA, Lazorchak JM, Bencic DC, 2016. Initial development of a multigene ‘omics-based exposure biomarker for pyrethroid pesticides. Aquat. Toxicol 179, 27–35. [DOI] [PubMed] [Google Scholar]
- Blair RM, Fang H, Branham WS, Hass BS, Dial SL, Moland CL, Tong W, Shi L, Perkins R, Sheehan DM, 2000. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: structural diversity of ligands. Toxicol. Sci 54, 138–153. [DOI] [PubMed] [Google Scholar]
- Bonneau E, Neveu B, Kostantin E, Tsongalis GJ, De Guire V, 2019. How close are miRNAs from clinical practice? A perspective on the diagnostic and therapeutic market. EJIFCC 30, 114–127. [PMC free article] [PubMed] [Google Scholar]
- Breiman L, 2001. Random forests. Mach. Learn 45, 5–32. [Google Scholar]
- Brum AM, van de Peppel J, van der Leije CS, Schreuders-Koedam M, Eijken M, van der Eerden BC, van Leeuwen JP, 2015. Connectivity map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc. Natl. Acad. Sci. U. S. A 112, 12711–12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri S, 2011. Epigenetic regulation of gene and genome expression. In: Gupta RC (Ed.), Reproductive and Developmental Toxicology. Academic Press, pp. 801–813. [Google Scholar]
- Cleuren AC, Van der Linden IK, De Visser YP, Wagenaar GT, Reitsma PH, Van Vlijmen BJ, 2010. 17alpha-Ethinylestradiol rapidly alters transcript levels of murine coagulation genes via estrogen receptor alpha. J. Thromb. Haemost 8, 1838–1846. [DOI] [PubMed] [Google Scholar]
- Cohen A, Smith Y, 2014. Estrogen regulation of microRNAs, target genes, and microRNA expression associated with vitellogenesis in the zebrafish. Zebrafish 11, 462–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Abrew KN, Kainkaryam RM, Shan YK, Overmann GJ, Settivari RS, Wang X, Xu J, Adams RL, Tiesman JP, Carney EW, Naciff JM, Daston GP, 2016. Grouping 34 chemicals based on mode of action using connectivity mapping. Toxicol. Sci 151, 447–461. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS, 2003. MicroRNA targets in Drosophila. Genome Biol. 5, R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, 2011. Non-coding RNAs in human disease. Nat. Rev. Genet 12, 861–874. [DOI] [PubMed] [Google Scholar]
- Farazi TA, Juranek SA, Tuschl T, 2008. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development 135, 1201–1214. [DOI] [PubMed] [Google Scholar]
- Ferraro L, Ravo M, Nassa G, Tarallo R, De Filippo MR, Giurato G, Cirillo F, Stellato C, Silvestro S, Cantarella C, Rizzo F, Cimino D, Friard O, Biglia N, De Bortoli M, Cicatiello L, Nola E, Weisz A, 2012. Effects of oestrogen on microRNA expression in hormone-responsive breast cancer cells. Horm. Cancer 3, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick RW, Bencic DC, See MJ, Biales AD, 2014. Sensitivity of the vitellogenin assay to diagnose exposure of fathead minnows to 17alpha-ethynylestradiol. Aquat. Toxicol 152, 353–360. [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R, 2000. Additive logistic regression: a statistical view of boosting. Ann. Statist 28 (2), 337–407. [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP, 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb S, 2008. Toxicity testing in the 21st century: a vision and a strategy. Reprod. Toxicol 25, 136–138. [DOI] [PubMed] [Google Scholar]
- Hatcher KM, Royston SE, Mahoney MM, 2020. Modulation of circadian rhythms through estrogen receptor signaling. Eur. J. Neurosci 51, 217–228. [DOI] [PubMed] [Google Scholar]
- Hochberg Y, Benjamini Y, 1990. More powerful procedures for multiple significance testing. Stat. Med 9, 811–818. [DOI] [PubMed] [Google Scholar]
- Jo MH, Shin S, Jung SR, Kim E, Song JJ, Hohng S, 2015. Human Argonaute 2 has diverse reaction pathways on target RNAs. Mol. Cell 59, 117–124. [DOI] [PubMed] [Google Scholar]
- Kavlock RJ, Bahadori T, Barton-Maclaren TS, Gwinn MR, Rasenberg M, Thomas RS, 2018. Accelerating the pace of chemical risk assessment. Chem. Res. Toxicol 31, 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienzler A, Barron MG, Belanger SE, Beasley A, Embry MR, 2017. Mode of action (MOA) assignment classifications for ecotoxicology: an evaluation of approaches. Environ. Sci. Technol 51, 10203–10211. [DOI] [PubMed] [Google Scholar]
- Klinge CM, 2012. miRNAs and estrogen action. Trends Endocrinol. Metab 23, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge CM, 2015. miRNAs regulated by estrogens, tamoxifen, and endocrine disruptors and their downstream gene targets. Mol. Cell. Endocrinol 418 (Pt 3), 273–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostich MS, Bencic DC, Batt AL, See MJ, Flick RW, Gordon DA, Lazorchak JM, Biales AD, 2019. Multigene biomarkers of pyrethroid exposure: exploratory experiments. Environ. Toxicol. Chem 38, 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kure EH, Saebo M, Stangeland AM, Hamfjord J, Hytterod S, Heggenes J, Lydersen E, 2013. Molecular responses to toxicological stressors: profiling microRNAs in wild Atlantic salmon (Salmo salar) exposed to acidic aluminum-rich water. Aquat. Toxicol 138–139, 98–104. [DOI] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR, 2006. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Lamba VJ, Goswami SV, Sundararaj BI, 1983. Circannual and circadian variations in plasma levels of steroids (cortisol, estradiol-17 beta, estrone, and testosterone) correlated with the annual gonadal cycle in the catfish, Heteropneustes fossilis (Bloch). Gen. Comp. Endocrinol 50, 205–225. [DOI] [PubMed] [Google Scholar]
- Lange R, Hutchinson TH, Croudace CP, Siegmund F, Schweinfurth H, Hampe P, Panter GH, Sumpter JP, 2001. Effects of the synthetic estrogen 17 alpha-ethinylestradiol on the life-cycle of the fathead minnow (Pimephales promelas). Environ. Toxicol. Chem 20, 1216–1227. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL, 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK, 2014. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tighe SW, Nicolet CM, Grove D, Levy S, Farmerie W, Viale A, Wright C, Schweitzer PA, Gao Y, Kim D, Boland J, Hicks B, Kim R, Chhangawala S, Jafari N, Raghavachari N, Gandara J, Garcia-Reyero N, Hendrickson C, Roberson D, Rosenfeld J, Smith T, Underwood JG, Wang M, Zumbo P, Baldwin DA, Grills GS, Mason CE, 2014. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat. Biotechnol 32, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YQ, Huang GY, Zhen Z, Tian F, Hou L, Lin Z, Ying GG, 2019. The effects of binary mixtures of estradiol and progesterone on transcriptional expression profiles of genes involved in hypothalamic-pituitary-gonadal axis and circadian rhythm signaling in embryonic zebrafish (Danio rerio). Ecotoxicol. Environ. Saf 174, 540–548. [DOI] [PubMed] [Google Scholar]
- Liao T, Guo QL, Jin SW, Cheng W, Xu Y, 2009. Comparative responses in rare minnow exposed to 17beta-estradiol during different life stages. Fish Physiol. Biochem 35, 341–349. [DOI] [PubMed] [Google Scholar]
- Liu J, Li Y, 2015. Trichostatin A and Tamoxifen inhibit breast cancer cell growth by miR-204 and ERalpha reducing AKT/mTOR pathway. Biochem. Biophys. Res. Commun 467, 242–247. [DOI] [PubMed] [Google Scholar]
- Lv C, Wu X, Wang X, Su J, Zeng H, Zhao J, Lin S, Liu R, Li H, Li X, Zhang W, 2017. The gene expression profiles in response to 102 traditional Chinese medicine (TCM) components: a general template for research on TCMs. Sci. Rep 7, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall C, Rocke DM, Durbin-Johnson B, Weiss RH, 2013. Stability of miRNA in human urine supports its biomarker potential. Biomark. Med 7, 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M, Galluzzo P, Ascenzi P, 2006. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom 7, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Office of Water, U.S. E.P.A. 2002. Short-term methods for estimating the chronic toxicity of effluents and receiving water to freshwater organisms. Fourth edition. EPA-821-R-02–013. [Google Scholar]
- Martinson J, Bencic DC, Toth GP, Kostich M, Flick RW, See MJ, Lattier DL, Biales AD, Huang W, 2021. De novo assembly and annotation of a highly contiguous reference genome of the fathead minnow (Pimephales promelas) reveals an AT-rich repetitive genome with compact gene structure. bioRxiv. doi: 10.1101/2021.02.24.432777. [DOI] [Google Scholar]
- Martyniuk CJ, Xiong H, Crump K, Chiu S, Sardana R, Nadler A, Gerrie ER, Xia X, Trudeau VL, 2006. Gene expression profiling in the neuroendocrine brain of male goldfish (Carassius auratus) exposed to 17alpha-ethinylestradiol. Physiol. Genom 27, 328–336. [DOI] [PubMed] [Google Scholar]
- Metsalu T, Vilo J, 2015. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucl. Acids Res 43, W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson E, Klukovich R, Sadler-Riggleman I, Beck D, Xie Y, Yan W, Skinner MK, 2018. Environmental toxicant induced epigenetic transgenerational inheritance of ovarian pathology and granulosa cell epigenome and transcriptome alterations: ancestral origins of polycystic ovarian syndrome and primary ovarian insufiency. Epigenetics 13, 875–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothnick WB, Healy C, 2010. Estrogen induces distinct patterns of microRNA expression within the mouse uterus. Reprod. Sci 17, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J, Hayder H, Zayed Y, Peng C, 2018. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol 9, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Office of Water, U.S.E.P.A., 2002. Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms. EPA-821-R-01-012, Washington, D.C. [Google Scholar]
- Oner C, Turgut Cosan D, Colak E, 2016. Estrogen and androgen hormone levels modulate the expression of PIWI interacting RNA in prostate and breast cancer. PLoS ONE 11, e0159044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A, O’Carroll D, Zamore PD, 2019. PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet 20, 89–108. [DOI] [PubMed] [Google Scholar]
- Parrott JL, Blunt BR, 2005. Life-cycle exposure of fathead minnows (Pimephales promelas) to an ethinylestradiol concentration below 1 ng/L reduces egg fertilization success and demasculinizes males. Environ. Toxicol 20, 131–141. [DOI] [PubMed] [Google Scholar]
- Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK, 2016. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat 10, 946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, Soule CK, Alexander B, Li A, Montgomery P, Kotz JD, Hon CS, Munoz B, Liefeld T, Dancik V, Haber DA, Clish CB, Bittker JA, Palmer M, Wagner BK, Clemons PA, Shamji AF, Schreiber SL, 2016. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol 12, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A, 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Rios P, Simonelig M, 2018. piRNAs and PIWI proteins: regulators of gene expression in development and stem cells. Development 145, 1–13. [DOI] [PubMed] [Google Scholar]
- Rutherford R, Lister A, Bosker T, Blewett T, Gillio Meina E, Chehade I, Kanagasabesan T, MacLatchy D, 2020. Mummichog (Fundulus heteroclitus) are less sensitive to 17alpha-ethinylestradiol (EE2) than other common model teleosts: a comparative review of reproductive effects. Gen. Comp. Endocrinol 289, 113378. [DOI] [PubMed] [Google Scholar]
- Song Y, Li L, Yang W, Fu Q, Chen W, Fang Z, Li W, Gu N, Zhang R, 2020. Sense–antisense miRNA pairs constitute an elaborate reciprocal regulatory circuit. Genome Res. 30, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y, Nakajima M, Takagi S, Taniya T, Yokoi T, 2006. MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res. 66, 9090–9098. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO, 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM, 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U. S. A 103, 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrijens K, Bollati V, Nawrot TS, 2015. MicroRNAs as potential signatures of environmental exposure or effect: a systematic review. Environ. Health Perspect 123, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Liu F, Chen W, 2019a. Exposure to triclosan changes the expression of microRNA in male juvenile zebrafish (Danio rerio). Chemosphere 214, 651–658. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang P, Lu Y, Li Y, Zheng Y, Kan Y, Chen R, He S, 2019b. piRBase: a comprehensive database of piRNA sequences. Nucl. Acids Res 47, D175–D180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Bammler TK, Beyer RP, Gallagher EP, 2013. Copper-induced deregulation of microRNA expression in the zebrafish olfactory system. Environ. Sci. Technol 47, 7466–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RL, Biales AD, Garcia-Reyero N, Perkins EJ, Villeneuve DL, Ankley GT, Bencic DC, 2016. Fish connectivity mapping: linking chemical stressors by their mechanisms of action-driven transcriptomic profiles. BMC Genom. 17, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Huang J, Zhong Y, Huang Q, 2017. DrugSig: a resource for computational drug repositioning utilizing gene expression signatures. PLoS ONE 12, e0177743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Hu HY, Jiang X, Maierhofer V, Neb E, He L, Hu Y, Hu H, Li N, Chen W, Khaitovich P, 2011. Widespread expression of piRNA-like molecules in somatic tissues. Nucl. Acids Res 39, 6596–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Cheng Y, Lu Q, Wei J, Yang H, Gu M, 2015. Detection of stably expressed piRNAs in human blood. Int. J. Clin. Exp. Med 8, 13353–13358. [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Si X, Skogerbo G, Wang J, Cui D, Li Y, Sun X, Liu L, Sun B, Chen R, He S, Huang DW, 2014. piRBase: a web resource assisting piRNA functional study. Database (Oxford). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.