

Abstract
Objectives
To report a case of adult-onset non-dystrophic myotonia complicated by recurrent episodes of laryngospasm.
Methods
The patient is a 35-year-old man who was admitted to our hospital for recurrent episodes of apnea requiring endotracheal intubation with mechanical ventilation. He underwent extensive evaluation, including EMG, laryngoscopy, muscle biopsy, and genetic testing, which revealed a diagnosis of non-dystrophic myotonia.
Results
His myotonic disorder was due to the synergistic effects of a pathogenic CLCN1 variant and a newly reported SCN4A variant. His muscle biopsy demonstrated myofibrillar disorganization with Z-band streaming, which may reflect the severity of his clinical and electrographic myotonia. Treatment with mexiletine resulted in resolution of his episodes of laryngospasm and symptoms of myotonia in the extremities.
Discussion
Our case adds to the literature on the potentiating effects of chloride channelopathies on sodium channel myotonia. This is the first reported case of an adult-onset sodium channelopathy manifesting with respiratory failure due to laryngospasm. In addition, we present muscle biopsy findings that have not been described in the recent literature. This case also highlights that a myotonic disorder should be considered in the differential diagnosis for recurrent episodes of mixed hypoxic and hypercarbic respiratory failure.
Introduction
The non-dystrophic myotonias are muscle channelopathies caused by pathogenic variants in the skeletal muscle sodium (SCN4A) or chloride (CLCN1) channel genes.1 Classically, variants in the SCN4A gene are associated with paramyotonia congenita and sodium channel myotonia while variants in the CLCN1 gene are associated with myotonia congenita.1 Myotonia is delayed relaxation of skeletal muscle, which patients experience as stiffness.1 Paramyotonia congenita is an autosomal dominant disorder characterized by myotonia that worsens with exercise and is typically cold induced.1 By contrast, myotonia congenita, which can have either an autosomal dominant or a recessive mode of inheritance, presents with the warm-up phenomenon, myotonia that improves with repetitive muscle activation.1 In actuality, variants in the genes encoding these ion channels have been shown to result in a spectrum of clinical presentations with complex genotype-phenotype correlations.1 Furthermore, patients with SCN4A and CLCN1 variants can have features of both archetypal channelopathies.2-5
Case Report
In this article, we report the case of a 35-year-old man with a history of an elevated left hemidiaphragm, tracheal diverticulum, and learning disability, who was found to have severe myotonia. He was born at 39 weeks of gestation through forceps-assisted vaginal delivery after an uncomplicated pregnancy. He had feeding difficulties as an infant, with frequent emesis and trouble gaining weight. He sat independently at 9–10 months and started to walk at 14 months. In high school, he played basketball and ran cross-country without limitations. He developed muscle stiffness at age 32. He described this as leg tightness on arising and taking the first few steps, which would gradually resolve as he walked. He also experienced grip myotonia and reported painless muscle stiffness provoked by cold weather. In recent years, he required recurrent hospital admissions for respiratory failure characterized by abrupt-onset severe hypoxia and hypercarbia from suspected laryngospasm, which would immediately resolve on endotracheal intubation with mechanical ventilation.
He presented to our hospital after one such apneic episode. Laryngoscopy performed shortly after endotracheal extubation did not reveal vocal fold paralysis but did show paradoxical vocal fold motion (Figure 1), which could be explained by myotonia or irritation from the endotracheal tube. On examination, he had striking percussion myotonia of the bilateral finger extensors and grip myotonia. On confrontational motor testing, he demonstrated full strength in the limbs. He also had normal bulk and tone in his limbs with mild, symmetric hyporeflexia in the upper and lower extremities. EMG showed dense electrographic myotonia in the 6 muscles tested, including the right deltoid muscle.
Figure 1. Laryngoscopy Findings.
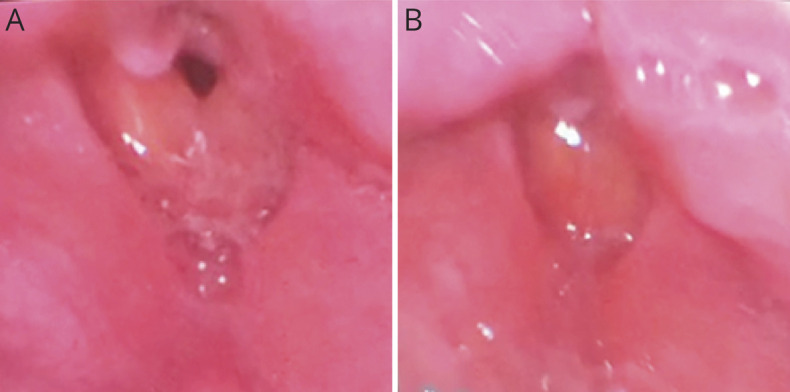
(A) Bedside laryngoscopy performed shortly after endotracheal extubation revealed laryngeal edema, likely due to intubation, and normal abduction of the vocal folds. (B) Quiet breathing was interrupted by episodes of inappropriate adduction of the vocal folds. Prolonged laryngospasm, induced and/or perpetuated by myotonia, was believed to be responsible for this patient's apneic episodes and respiratory failure.
A left deltoid muscle biopsy, performed while awaiting the return of genetic testing results, revealed multiple histologic and ultrastructural myopathic abnormalities (Figure 2). Of interest, electron microscopy showed prominent myofibrillar disorganization with Z-band streaming, a distinctive feature that is not usually seen in non-dystrophic myotonias but has been previously reported in an archival case series of patients with severe myotonia congenita.6
Figure 2. Left Deltoid Biopsy Findings.

(A and B) H&E-stained cryosections showed moderate fiber size variation (fiber size range, 5–120 µm), mild endomysial fibrosis without fatty infiltration, and moderate increase in internal nucleation without subsarcolemmal nuclear aggregates or nuclear chains. Small clear irregular regions of the sarcoplasm were notable on high magnification (arrowheads in B). (C) The NADH-TR stain demonstrated moderate abnormalities of the myofibrillar matrix, with mini cores/moth-eaten change in type 1 muscle fibers and linearized internal architecture in type 2 muscle fibers. (D) The esterase stain showed scattered esterase-positive sarcolemmal patches (arrowheads). (E) Acid phosphatase stain was unremarkable, with no enlarged lysosomes or sarcoplasmic puncta (F and G). Electron microscopy showed disruption of the myofibrillar architecture with Z-band streaming (F) and occasional spheroid bodies (G), but no autophagic vacuoles, no significant increase in glycogen, no mitochondrial abnormalities, and no tubular aggregates. Scale bars: A, C, D, and E, 50 µm; B, 25 µm; F and G, 1 µm.
There were no repeat expansions of the dystrophia myotonica protein kinase or zinc finger protein 9 genes, and acid alpha glucosidase enzyme activity was normal. Genetic testing using a multigene test panel revealed that he had a pathogenic missense variant in CLCN1 c.1478C>A and a deletion in SCN4A c.5107_5109del of uncertain pathogenicity.
Genetic testing of his family revealed that the CLCN1 variant was inherited from his father, who does not have clinical or electrographic myotonia (Figure 3). The SCN4A variant was inherited from his mother, who has subtle percussion myotonia of the bilateral finger extensors and mild electrographic myotonia (Figure 3). The patient's younger brother is asymptomatic and does not have either variant (Figure 3).
Figure 3. Pedigree.
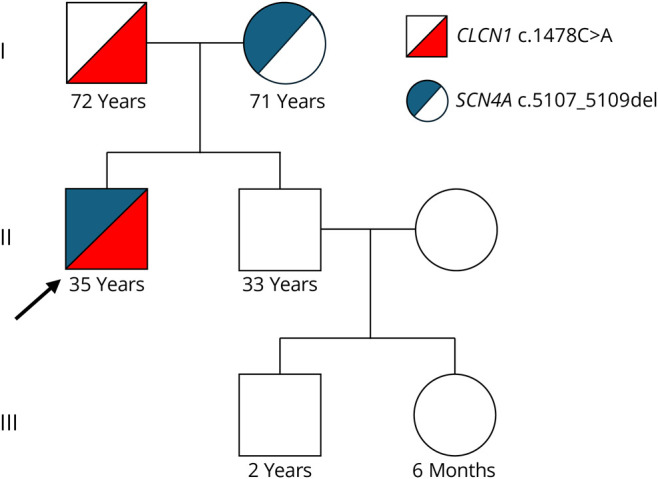
The patient inherited a pathogenic variant in the CLCN1 gene from his asymptomatic father and a variant of uncertain significance in the SCN4A gene from his mother, who has milder clinical and electrographic myotonia. The patient's brother, niece, and nephew are unaffected.
Our patient was treated with mexiletine 200 mg 3 times daily, which led to the resolution of symptoms of myotonia and allowed for tracheostomy decannulation. Correspondingly, his clinical and electrographic myotonia also improved with this medication. The patient was pleased with the effects of this treatment.
Discussion
Clinical evaluation and genetic testing of our patient's family members shed light on the etiology of his myotonic disorder. The patient's CLCN1 c.1478C>A variant is a well-characterized pathogenic alanine-to-glutamic acid substitution in exon 14 (p.Ala493Glu). Variants affecting this amino acid, which is positioned adjacent to the chloride ion pathway, are predicted to cause a structural defect in the skeletal muscle chloride channel encoded by the CLCN1 gene.7 This CLCN1 variant has been seen in autosomal dominant and recessive forms of myotonia congenita.7,8 However, in this case, this CLCN1 variant is unlikely to be the primary driver of the patient's clinical phenotype because his father has the same CLCN1 variant but neither clinical nor electrographic myotonia. An alternative explanation is that this CLCN1 variant has variable penetrance.
The patient's SCN4A c.5107_5109del variant has not been previously reported in the literature. It leads to a glutamic acid deletion in exon 24 (p.Glu1703del). This SCN4A deletion, currently classified as a variant of uncertain significance, affects the highly conserved C-terminal tail of the voltage-gated sodium channel, pathogenic variants in which have been shown to impair fast inactivation, thereby leading to clinical and electrical myotonia.9 Because the patient's mother has the same SCN4A variant and exhibits clinical and electrographic myotonia, we believe that our patient's non-dystrophic myotonia is primarily due to his SCN4A variant. His case provides evidence that this newly reported SCN4A variant is pathogenic. Future laboratory expression studies of his SCN4A variant may shed light on its mechanisms of pathogenicity.
The sodium channelopathies have been associated with respiratory distress secondary to severe neonatal episodic laryngospasm.10,11 Analogously, our patient developed recurrent episodes of apnea and laryngospasm, resulting in respiratory failure necessitating mechanical ventilation. Resolution of his respiratory symptoms when he was treated with mexiletine, such that he was able to undergo tracheostomy decannulation, suggests that his elevated hemidiaphragm and tracheal diverticulum are incidental and that his recurrent episodes of laryngospasm were due to myotonia. His clinical presentation establishes that sodium channelopathies can cause respiratory distress secondary to laryngospasm and vocal fold motion abnormalities in adulthood.
We suspect that our patient developed a more severe myotonic disorder than his mother did because of additive effects from the pathogenic CLCN1 variant he inherited from his father. Our patient's myotonia is characterized by the warm-up phenomenon, which is typically associated with CLCN1 variants. However, sodium and chloride channelopathies can have overlapping features.1 Moreover, with chloride channelopathies, patients with the same genetic variant can present with different clinical phenotypes.1 These variable genotype-phenotype correlations have been attributed to genetic modifiers; incomplete penetrance; and epigenetic, hormonal, or environmental factors.1 There are other reports of patients with pathogenic SCN4A and CLCN1 variants acting in combination to affect the clinical presentation.2-5 Thus, our case adds to the growing literature on the potentiating effects of CLCN1 variants on sodium channel myotonia.
In addition, his muscle biopsy results contribute to the limited literature on histopathologic findings in the non-dystrophic myotonias. In myotonia congenita, the severity of the clinical phenotype can correspond to the degree of abnormalities on muscle biopsy.6 We suspect that the striking myofibrillar disorganization with Z-band streaming seen on electron microscopy may reflect the patient's severe clinical and electrographic myotonia.
In summary, in this case report, we describe a 35-year-old man with severe non-dystrophic myotonia complicated by episodes of apnea and laryngospasm secondary to pathogenic SCN4A and CLCN1 variants. For 2 years, our patient was undiagnosed, his recurrent episodes of apnea considered idiopathic. His case should prompt providers to consider a myotonic disorder among the possible causes of recurrent mixed hypoxic and hypercarbic respiratory failure.
Acknowledgment
The authors thank Ms. Christine Lin for help with Figure 2.
Author Contributions
M. Tugizova: drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data. M. Margeta: drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. M. Richie: drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. D. Pet: drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. L. Rosow: drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. M. Terrelonge: drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. J.W. Ralph: drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data.
Study Funding
The authors report no targeted funding.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Morales F, Pusch M. An up-to-date overview of the complexity of genotype-phenotype relationships in myotonic channelopathies. Front Neurol. 2019;10:1404. doi: 10.3389/fneur.2019.01404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kato H, Kokunai Y, Dalle C, et al. A case of non-dystrophic myotonia with concomitant mutations in the SCN4A and CLCN1 genes. J Neurol Sci. 2016;369:254-258. doi: 10.1016/j.jns.2016.08.030 [DOI] [PubMed] [Google Scholar]
- 3.Maggi L, Ravaglia S, Farinato A, et al. Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics. 2017;18(4):219-225. doi: 10.1007/s10048-017-0525-5 [DOI] [PubMed] [Google Scholar]
- 4.Furby A, Vicart S, Camdessanche JP, et al. Heterozygous CLCN1 mutations can modulate phenotype in sodium channel myotonia. Neuromuscul Disord. 2014;24(11):953-959. doi: 10.1016/j.nmd.2014.06.439 [DOI] [PubMed] [Google Scholar]
- 5.Vacchiano V, Brugnoni R, Campanale C, et al. Coexistence of SCN4A and CLCN1 mutations in a family with atypical myotonic features: a clinical and functional study. Exp Neurol. 2023;362:114342. doi: 10.1016/j.expneurol.2023.114342 [DOI] [PubMed] [Google Scholar]
- 6.Sokolina NA. Thomsen's myotonia (clinico-morphologic study). Zh Nevropatol Psikhiatr Im S.S. Korsakova 1977;77(3):325-331. [PubMed] [Google Scholar]
- 7.Skálová D, Zídková J, Voháňka S, et al. CLCN1 mutations in Czech patients with myotonia congenita, in silico analysis of novel and known mutations in the human dimeric skeletal muscle chloride channel. PLoS One. 2013;8(12):e82549. doi: 10.1371/journal.pone.0082549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaitán-peñas H, Armand-Ugón M, Macaya A, Estevez R. CLCN1 myotonia congenita mutation with a variable pattern of inheritance suggests a novel mechanism of dominant myotonia: short reports. Muscle Nerve. 2018;58(1):157-160. doi: 10.1002/mus.26098 [DOI] [PubMed] [Google Scholar]
- 9.Horie R, Kubota T, Koh J, et al. EF hand‐like motif mutations of Nav1.4 C‐terminus cause myotonic syndrome by impairing fast inactivation. Muscle Nerve. 2020;61(6):808-814. doi: 10.1002/mus.26849 [DOI] [PubMed] [Google Scholar]
- 10.Pechmann A, Eckenweiler M, Schorling D, Stavropoulou D, Lochmuller H, Kirschner J. De novo variant in SCN4A causes neonatal sodium channel myotonia with general muscle stiffness and respiratory failure. Neuromuscul Disord. 2019;29(11):907-909. doi: 10.1016/j.nmd.2019.09.001 [DOI] [PubMed] [Google Scholar]
- 11.Purkey MR, Valika T. A unique presentation and etiology of neonatal paradoxical vocal fold motion. Int J Pediatr Otorhinolaryngol. 2019;125:199-200. doi: 10.1016/j.ijporl.2019.07.011 [DOI] [PubMed] [Google Scholar]