

ABSTRACT
Messenger ribonucleic acid (mRNA) therapeutics are attracting attention as promising tools in cancer immunotherapy due to their ability to leverage the in vivo expression of all known protein sequences. Even small amounts of mRNA can have a powerful effect on cancer vaccines by promoting the synthesis of tumor‐specific antigens (TSA) or tumor‐associated antigens (TAA) by antigen‐presenting cells (APC). These antigens are then presented to T cells, eliciting strong antitumor immune stimulation. The potential of mRNA can be further enhanced by expressing immunomodulatory agents, such as cytokines, antibodies, and chimeric antigen receptors (CAR), enhancing tumor immunity. Recent research also explores mRNA‐encoded tumor death inducers or tumor microenvironment (TME) modulators. Despite its promise, the clinical translation of mRNA‐based anticancer strategies faces challenges, including inefficient targeted delivery in vivo, failure of endosomal escape, and inadequate intracellular mRNA release, resulting in poor transfection efficiencies. Inspired by the approval of lipid nanoparticle‐loaded mRNA vaccines against coronavirus disease 2019 (COVID‐19) and the encouraging outcomes of mRNA‐based cancer therapies in trials, innovative nonviral nanotechnology delivery systems have been engineered. These aim to advance mRNA‐based cancer immunotherapies from research to clinical application. This review summarizes recent preclinical and clinical progress in lipid and polymeric nanomedicines for delivering mRNA‐encoded antitumor therapeutics, including cytokines and antibody‐based immunotherapies, cancer vaccines, and CAR therapies. It also addresses advanced delivery systems for direct oncolysis or TME reprogramming and highlights key challenges in translating these therapies to clinical use, exploring future perspectives, including the role of artificial intelligence and machine learning in their development.
Keywords: cancer immunotherapy, messenger RNA (mRNA) therapeutics, mRNA‐based delivery systems, nanotechnology
mRNA‐loaded nanocarrier engineering and cancer immunotherapy applications. By adjusting mRNA and nanocarrier properties, each mRNA‐encoded immunomodulator can be delivered and produced with the precise spatiotemporal control desired for its therapeutic effect. APC: antigen‐presenting cell; CAR: chimeric antigen receptor; M: macrophage; mRNA: messenger ribonucleic acid; NK: natural killer cell; TCR: T‐cell receptor; UTR: untranslated region.

1. Introduction
Cancer is a life‐threatening disease characterized by uncontrolled cell growth that can disseminate throughout the body when untreated. Treating cancer is challenging, as conventional therapies including surgery, radiotherapy, and chemotherapy are commonly inefficient in abolishing tumor cells, which skillfully evade immune surveillance and repolarize neighboring immune cell function into a pro‐tumoral state (Mortezaee 2020). Cancer immunotherapy has emerged as a powerful strategy to treat cancer by using the body's own immune system to target, fight, and eradicate cancer cells without destroying healthy cells (Riley et al. 2019). In contrast to conventional therapeutic approaches, immunotherapy triggers innate and adaptive tumor‐specific immune responses resulting in immunological memory, reduced tumor burden, and prolonged overall survival rates (Conniot et al. 2019; Del Paggio 2018). In 2020, the successful and worldwide approval for clinical purposes of the two rapid‐response Pfizer‐BioNTech (Comirnaty, BNT162b2) and Moderna (Spikevax, messenger ribonucleic acid (mRNA)‐1273) vaccines against coronavirus disease 2019 (COVID‐19) highlighted the massive potential of mRNA technology, sparking unprecedented interest in the development of mRNA‐based therapeutics as a groundbreaking tool for cancer immunotherapy (Acúrcio et al. 2024; Barbier et al. 2022; Florindo et al. 2020; Morris and Kopetz 2022; Xie, Chen, and Wong 2021). mRNA is a polyanionic single‐stranded RNA molecule that bridges the delivery of genetic information from a deoxyribonucleic acid (DNA) template with its translation into essential cell‐functioning proteins (Cobb 2015).
Nowadays, synthetic pure mRNA molecules are easily and promptly produced on a large scale with minimal batch‐to‐batch variation due to great improvements in mRNA manufacturing and in vitro transcription (IVT) techniques (Ingels et al. 2022). From a linear DNA template, the IVT method guarantees, with high fidelity, the production of synthetic single‐stranded mRNA similar to naturally derived mature transcripts, composed of an open‐reading fragment (ORF) (coding sequence) flanked by a five‐prime (5′) and three‐prime (3′) untranslated regions (UTR), a 5′ cap structure and a 3′ poly(A) tail. Under the guidance of the exogenous mRNA molecules released in the cytoplasm, translation occurs through the internal cell machinery (e.g., ribosomes, enzymes, amino acids) resulting in the biosynthesis of encoded proteins indistinguishable from protein translated from endogenous mRNA (Beck et al. 2021).
Among the several cancer immunotherapeutic approaches, including monoclonal antibodies (mAbs), immune checkpoint inhibitors (ICIs), cytokines, and chimeric antigen receptor (CAR) therapy, mRNA has been generally employed as a therapeutic cancer vaccine by taking advantage of both genetic delivery and immunostimulatory abilities. Unlike DNA, mRNA as a cancer vaccine results in rapid translation into therapeutic proteins without the need to cross the nuclear membrane (Jahanafrooz et al. 2020). After translation, mRNA can be degraded by ribonucleases (RNases) and does not integrate into the host genome, avoiding continuous cell reprogramming (Pelechano, Wei, and Steinmetz 2015). The protein is eventually processed, and peptides are presented through major histocompatibility complex (MHC) molecules. In addition, minimal doses of mRNA can induce safe and potent immune responses against tumors without affecting nonmalignant cells. Exogenous mRNA also displays both inherent immunogenic and adjuvant properties, making it an interesting advantage in treating diseases where the immune system plays a key role, such as cancers and infections (Kranz et al. 2016; Miao, Zhang, and Huang 2021). In parallel to the translation, mRNA displays a strong adjuvanticity by triggering the release of type I interferon (IFN) and pro‐inflammatory cytokines through its binding to endosomal toll‐like receptors (TLR) 3, 7, and 8, or retinoic acid‐inducible gene 1 and melanoma differentiation‐associated protein 5 in the cytoplasm (Pastor et al. 2018). Although mRNA holds a fragile non‐stable structure, mRNA‐based therapeutics have shown to be highly effective during the rapid‐response mRNA vaccine development to solve the COVID‐19 pandemic. Indeed, COVID‐19 mRNA vaccines were rapidly developed under good manufacturing practices (GMP) conditions for easy scale‐up and presented great activity at relatively low dose ranges (Anderson et al. 2020; Polack et al. 2020; Walsh et al. 2020).
These key features have raised hope to advance from the bench to the clinical implementation of mRNA‐based cancer medicines, such as Moderna's personalized mRNA vaccine against melanoma under clinical evaluation (ClinicalTrials.gov identifier NCT03897881) (J. S. Weber et al. 2024). Despite its promising potential, the translation of mRNA‐based cancer immunotherapeutics to the clinic is still challenging due to several limitations including: (1) mRNA repels the negatively charged cell membrane's lipid bilayer which avoids its translocation, (2) RNases in vivo degrade mRNA, and (3) mRNA can undergo preliminary phagocytosis by the mononuclear phagocytic system before attaining the target. These hurdles have driven the design and development of proper advances for efficient mRNA delivery and improved therapeutic effectiveness (Zeng et al. 2022).
Current improvements in mRNA chemical modification strategies and delivery systems have addressed these challenges. For instance, several structural modifications of the mRNA backbone, such as nucleoside replacement/modification (Bornewasser, Domnick, and Kath‐Schorr 2022); the remolding of 5′ cap, poly(A) tail, and UTR (K. Lee et al. 2020; Linares‐Fernández et al. 2020); and the codon optimization of mRNA ORF region (Presnyak et al. 2015; T. Yang et al. 2020); have been developed to enhance the mRNA stability and translation efficiency as well as to regulate its overpowering adjuvant and immunogenic effects.
Furthermore, Pfizer–BioNTech and Moderna vaccines also highlighted the impact of nanotechnology‐based systems as critical players in defeating the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection and overcoming the challenging mRNA delivery. Since then, a renewed and deep research interest in mRNA nanomedicines has been booming, fueled by the promising clinical translation of lipid nanoparticle (LNP) platforms (Horejs 2021; X. Huang et al. 2022). For cancer immunotherapy, delivery systems can be properly modified to ensure a selective and targeted mRNA delivery to specific tissues and cells (e.g., tumor or antigen‐presenting cells (APC)) and an efficient endosomal escape to the cytoplasm, while promoting an enhanced transfection efficiency to trigger strong antitumor immune responses and reducing the off‐target and side effects (Kim, Seo, and Park 2022; J. Shi, Huang, et al. 2022b). In addition, several mRNA molecules encoding multiple antigens (Sahin et al. 2017) and/or immunostimulatory proteins (Van Lint et al. 2016) can be co‐delivered by the same nanoplatform to boost precise and powerful immunotherapeutic effects against tumors, as is currently ongoing in trials for triple‐negative breast cancer (NCT02316457), melanoma (NCT02410733), or other advanced malignancies (NCT03739931 and NCT03871348).
This review focuses on the recent advances in designed nanotechnology‐based approaches for enhancing mRNA's biological and pharmacological potential in cancer immunotherapy. A concise, yet comprehensive overview of the preclinical and clinical landscape of LNP and polymeric nanoparticles (PNP)‐based medicines developed to deliver mRNA‐encoded cancer immunotherapeutic strategies, including cytokines and antibody‐based immunotherapies, cancer vaccines, and CAR therapies, as well as to modulate the tumor site to promote a direct tumor killing and/or tumor microenvironment (TME) reprogramming against immunosuppression, is disclosed (Figure 1). As mRNA immunotherapy has emerged as a transformative strategy in cancer treatment, future perspectives of mRNA therapeutics as novel therapeutic modalities in cancer immunotherapy and the challenges anticipated for future research are also discussed.
FIGURE 1.
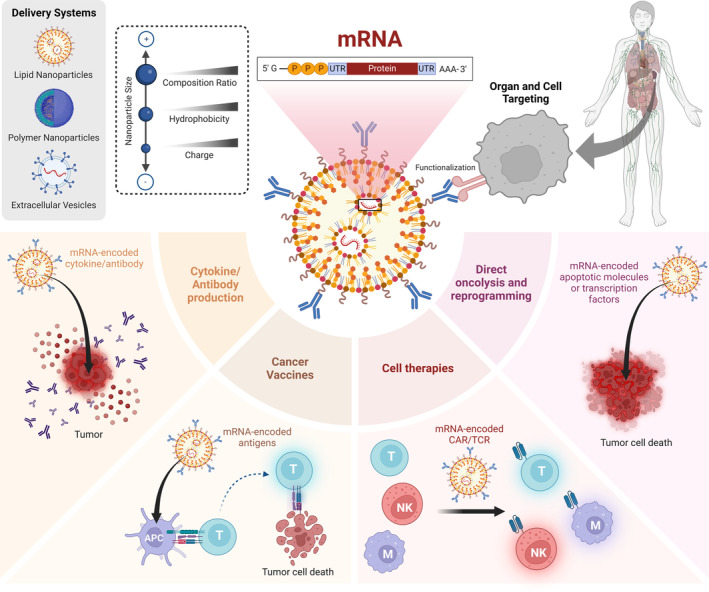
mRNA‐loaded nanocarrier engineering and cancer immunotherapy applications. By adjusting mRNA and nanocarrier properties, each mRNA‐encoded immunomodulator can be delivered and produced with the precise spatiotemporal control desired for its therapeutic effect. APC: antigen‐presenting cell; CAR: chimeric antigen receptor; M: macrophage; mRNA: messenger ribonucleic acid; NK: natural killer cell; TCR: T‐cell receptor; UTR: untranslated region.
2. Advancing mRNA Technology for Cytokine and Antibody‐Based Immunotherapies—Multiple Cell Targeting Approaches
2.1. Cytokine Delivery: Systemic vs. Intratumoral vs. Immune Cell Targeting
Cytokines are powerful and soluble immunomodulatory proteins that play a key role in homeostasis and communication between the innate and adaptive systems (Holder et al. 2022). The application of pro‐inflammatory cytokines was among the first immunotherapeutic strategies approved by the U.S. Food and Drug Administration (FDA), with IFN‐α approved for treating hairy cell leukemia in 1986 and interleukin (IL)‐2 for metastatic renal cell cancer in 1992 (Holder et al. 2022). Systemically administered cytokines can activate and boost the survival of effector immune cells, induce IFN‐γ production, stimulate antigen presentation, and modulate the TME (Beck et al. 2024; Luke et al. 2015). However, the high doses required, the narrow therapeutic window, poor accumulation in tumor tissues, short half‐life, severe adverse effects, and complex, expensive, and time‐consuming production processes have restricted the therapeutic success of recombinant cytokines via the systemic route. As an alternative, intratumoral administration strategies have been studied (Jiang et al. 2024; J.‐Q. Liu et al. 2022; Neshat et al. 2023; vom Berg et al. 2013). Compared to systemic injection, local administration of cytokines enables smaller doses that are spatially restricted to the tumor tissue, reducing systemic exposure while potentiating immune responses against tumor cells. Thus, several therapies based on IL‐2, IL‐12, IFN‐γ, and tumor necrosis factor (TNF)‐α have been developed with intratumoral injection, leading to improved response rates, higher tolerability, and better recruitment of lymphocytes to the tumor tissue (Figure 2) (Hewitt et al. 2020; Hotz et al. 2021; H. Shin et al. 2023).
FIGURE 2.
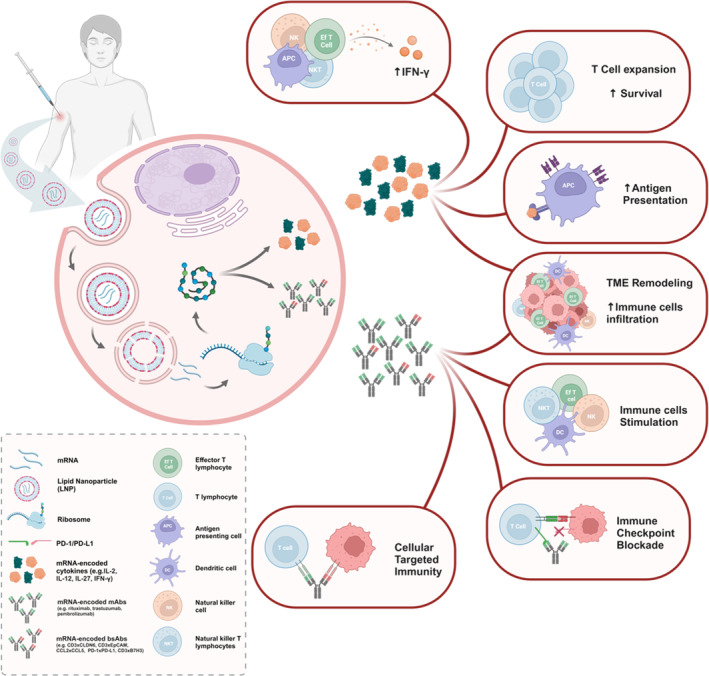
In vivo antitumoral responses induced by NP‐delivered mRNA‐encoded cytokines and antibodies. mRNA is delivered into target cells via NP endocytosis. During internalization, the acidic pH triggers the endosomal escape effect of the ionizable lipids by fusing the NP with the endosome membrane, releasing the mRNA into the cell cytoplasm. Once in the cytoplasm, mRNA is translated into the encoded proteins, which undergo posttranslation modifications to acquire the proper folding and chemical structure. Following their expression, mRNA‐encoded cytokines can induce potent antitumoral responses in both injected and non‐injected lesions through several immune mechanisms, including IFN‐γ level increase, T‐cell expansion and survival enhancement, stimulation of APC antigen presentation through immune cell activation or increased MHC molecule expression, and TME remodeling toward a TH1 phenotype with higher immune infiltration ratios. Similarly, in vivo synthesized monoclonal antibodies (mAbs) and bispecific antibodies (bsAbs) may also generate pronounced antitumor responses. These responses include promoting an M1 proinflammatory TME, improving immune cell infiltration into the tumor, activating different immune cell subsets, inhibiting immune checkpoint interaction with their ligands, or bringing immune cells like T cells closer to cancer cells, thereby inducing targeted T cell‐dependent cytotoxicity. APC: antigen‐presenting cell; B7H3: B7 homolog 3 protein; bsAb: bispecific antibody; CCL: chemokine (C–C motif) ligand; CLDN6: claudin 6; DC: dendritic cell; EpCAM: epithelial cellular adhesion molecule; IFN‐γ: interferon‐γ; IL: interleukin; LNP: lipid nanoparticle; mAb: monoclonal antibody; NK: natural killer cell; NKT: natural killer T cell; PD‐1: programmed cell death protein‐1; PD‐L1: programmed cell death‐ligand 1; TME: tumor microenvironment.
Alongside local administration, cell‐targeting concepts have also been applied to cytokine‐based therapies, enabling the generation of chimeric proteins with increased specificity. Immunocytokines are a type of chimeric protein formed by fusing cytokines with immunomodulatory activity and antibodies with specificity for a target antigen, thereby triggering robust and targeted antitumor responses (Holder et al. 2022). The momentum gained from the approval of T‐VEC in 2015 for melanoma treatment and more recently from COVID‐19 vaccines has facilitated the introduction of mRNA‐based strategies as a simple and efficient method to trigger cytokine production in vivo (Barbier et al. 2022). Despite the first use of mRNA as a therapeutic tool in the 1980s, several optimizations over the years have fine‐tuned its structure to decrease its immunogenicity and boost expression rates, achieving viable in vivo protein production (J. Han et al. 2023). Consequently, mRNA‐encoded cytokines have overcome several challenges associated with previous DNA plasmid and viral vector strategies, resulting in higher, rapid, cytoplasmic, and transient expression rates, with no risk of genome integration, and safer profiles. mRNA‐translated cytokines also offer advantages over recombinant proteins, including straightforward production processes, longer serum half‐life, and a lower rate of aberrant posttranslational modifications during in vivo production (Barbier et al. 2022).
Considering the added value of mRNA for cytokine‐based therapies, several studies demonstrated the potential of this strategy. Hewitt et al. showed that a single intratumoral dose of mRNA‐encoded IL‐12 was sufficient to induce IFN‐γ and CD8+ T cell‐dependent rejection of both treated and untreated lesions in several murine models (Hewitt et al. 2020). Hotz et al. proved that intratumoral administration of a saline mixture of mRNAs for multiple cytokines, including IL‐12, IL‐15, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), and IFN‐α, resulted in a synergistic strategy against B16F10 tumors with good tolerability (Hotz et al. 2021). Saline mRNA strategies have also been studied to encode immunocytokines in vivo, with higher therapeutic indices and safer profiles in a target‐dependent manner. Cirella et al. designed an intratumoral mRNA encoding IL‐12 fused with transforming growth factor (TGF)‐β and CD137 antibodies, leading to potent and synergistic antitumoral responses in B16OVA melanoma models and increased cytotoxic T lymphocyte (CTL) infiltration in treated tumors (Cirella et al. 2023). Similarly, Di Trani et al. fused mRNA encoding colony‐stimulating factor 1 receptor (CSF1R) and avelumab (anti‐programmed cell death‐ligand 1 (PD‐L1)) antibodies with mRNA‐encoded IL‐12, preserving the robust antitumoral effect of non‐chimeric IL‐12, achieving lower systemic exposure, and remodeling the TME toward higher immune cell infiltration (Di Trani et al. 2023).
To potentiate antitumor responses induced by mRNA‐based cytokine therapies, several groups have explored combination therapies. Beck et al. combined the administration of mRNA‐encoded IL‐2 with the anti‐programmed cell death protein‐1 (PD‐1) antibody to treat B16F10 and MC38‐bearing mice, resulting in complete tumor rejection and long‐term survival rates of up to 60% (Beck et al. 2024). Despite the promising therapeutic value of mRNA‐encoded cytokines, the lack of efficient delivery methods remains a significant challenge (Barbier et al. 2022).
Although promising, there is still room for improvement, as the susceptibility of mRNA to enzymatic degradation and modest in vivo protein expression efficiency continue to hinder the success of mRNA‐encoded cytokines. Circular RNA (cRNA) and self‐amplifying RNA (saRNA) could address these challenges. As an alternative nucleic acid structure, cRNA features covalently linked 5′ and 3′ ends, protecting it from blood RNases, thereby enhancing RNA stability and sustaining protein expression over time (Hwang and Kim 2024; J. Yang et al. 2022). saRNA is another promising structure that has gained increasing attention, particularly after the approval of the first saRNA vaccine, ARCT‐154 (Hồ et al. 2024). Unlike its non‐replicative counterpart, saRNA not only encodes the intended protein but also takes advantage of the host cell machinery to express a replicase, increasing protein expression efficiency while requiring fewer administration cycles and lower doses (Papukashvili et al. 2022). As such, cRNA and saRNA will be pivotal in the development of the next generations of mRNA‐encoded cytokines.
2.2. Antibody Delivery: Systemic vs. Intratumoral vs. Immune Cell Targeting
Antibodies are macromolecules composed of two polypeptide regions and are currently the standard of biological treatment for various diseases, including cancer (Deal, Carfi, and Plante 2021). The potential of antibodies as anti‐cancer passive immunotherapies was first realized in 1997 with the FDA approval of rituximab for chronic lymphocytic leukemia and non‐Hodgkin's lymphoma (Beck et al. 2021). Since then, the production of recombinant mAbs with decreasing murine content has led to several approvals (Van Hoecke and Roose 2019). The success of recombinant antibodies as cancer therapies lies in their ability to activate APC, repolarize the TME toward a pro‐inflammatory macrophages type‐1 (M1) phenotype, stimulate CD8+ T cells, natural killer (NK), and natural killer T (NKT) cells, and increase CTL infiltration into the tumor tissue to induce oncolysis. Despite their potential as immunotherapy, conventional antibody production in mammalian cells involves complex, customized, and expensive processes, posing challenges for intravenous, intramuscular, or subcutaneous administration of high doses (Zhao et al. 2023). This production process is also time‐consuming, introduces a high analytical burden, and can lead to aberrant posttranslational modifications that impact biological activity (Van Hoecke and Roose 2019). Additionally, issues such as low storage stability, susceptibility to enzymatic degradation, and safety problems related to T‐cell overstimulation have been reported (Zhao et al. 2023). While mAbs have demonstrated antitumor activity and therapeutic success, these limitations have prompted the search for better alternatives (J. Han et al. 2023).
Similarly to cytokines, intratumoral administration of recombinant antibodies has emerged as a viable option to increase tumor bioavailability, minimize systemic off‐target effects, and achieve superior antitumor efficacy with smaller, localized doses. In addition, the intratumoral combination of recombinant antibodies with cytokines, other antibodies, or dendritic cells (DCs) generates synergistic effects with low systemic exposure and abscopal antitumor responses (A. Huang et al. 2020; Melero et al. 2021). However, direct injection of antibodies into tumor tissue can be limited by rapid endocytic clearance and poor tumor retention (A. Huang et al. 2020). To address this, targeting antibody‐based therapies to immune cells has been explored in several studies (Melero et al. 2021). Bispecific antibodies (bsAbs) have been developed to recognize two epitopes: one on a tumor cell (e.g., epidermal growth factor receptor (EGFR), CD19, IMCgp100) and one on an immune cell (e.g., CD3 or CD16), thereby bringing T or NK lymphocytes and cancer cells together (C. Huang et al. 2023; Wei et al. 2022). This approach underpins the antitumor activity of bispecific T cell engagers (BiTES), a type of bsAbs that recognize CD3 on T cells, promoting targeted cytotoxic activity and effector cells with lower dose requirements, thus generating a strong antitumor response and improving the safety profile (Stadler et al. 2017; Wei et al. 2022). The use of therapeutic combinations with ICI or cytokines can also enhance the safety and therapeutic index of BiTES (Suurs et al. 2019).
To overcome possible challenges with BiTES, new targeting approaches and strategies based on polyethylene glycol (PEG), plasma proteins, and antibody fragment crystallizable (Fc) regions have been investigated to increase serum circulation time and enable cellular antitumor responses without activating regulatory T (Treg) cells (Suurs et al. 2019; Wei et al. 2022). The discovery of mRNA's potential to encode antibodies in 2008, along with advances in in vitro transcription technology, has enabled the in vivo production of mAbs and bsAbs as an elegant immunotherapeutic strategy to treat cancer (Figure 2) (C. Huang et al. 2023; Van Hoecke and Roose 2019). Administering mRNA systemically and intratumorally helps to circumvent several dilemmas associated with recombinant antibodies (Golubovskaya, Sienkiewicz, Sun, Huang, et al. 2023a; Zhao et al. 2023). The production process for mRNA does not require mammalian cell lines, making it universal for various antibodies, easily adaptable, and less expensive (Rybakova et al. 2019; Sanz and Álvarez‐Vallina 2021; Van Hoecke and Roose 2019). Moreover, the translation of the mRNA up to 72 h after administration and naturally occurring posttranslational modifications increase the stability and half‐life of the antibodies (L. Wu et al. 2022). Milligram doses in a single injection can generate robust antitumor responses, given that mRNA‐encoded antibodies achieve higher or equivalent expression rates compared to recombinant antibodies (e.g., trastuzumab) (C. Huang et al. 2023; Sanz and Álvarez‐Vallina 2021). Lastly, mRNA‐encoded antibodies present an optimized pharmacokinetic profile, with lower volumes of distribution, rapid expression, and prolonged therapeutic serum concentrations (Y. Wang et al. 2021; Zhao et al. 2023).
However, despite the promising preclinical performance of mRNA‐encoded antibodies, the efficient delivery of mRNA remains a significant challenge (Van Hoecke and Roose 2019; Zhao et al. 2023). To address this, several researchers have explored the potential of nanoparticle‐based systems to deliver mRNA as a replacement for clinically approved antibodies. Wu et al. developed a full‐size mRNA‐encoded pembrolizumab systemically delivered within LNP, which was well‐tolerated and showed tumoral growth inhibition rates 18%–21% higher than those achieved by the recombinant antibody (L. Wu et al. 2022). Similar antitumor responses were found in receptor tyrosine‐protein kinase erbB‐2 (HER2)+ breast cancer‐bearing mice treated with LNP‐containing mRNA‐encoded trastuzumab, which revealed plasma concentrations 64% higher than recombinant trastuzumab (Rybakova et al. 2019). Thran et al. and Duy et al. also reported significant tumor growth reduction after administering mRNA‐encoded rituximab‐loaded LNP in lymphoma mouse models and mRNA‐encoded bevacizumab‐loaded polymeric nanoparticles (PNP) in non‐small cell lung cancer (NSCLC) mouse models, compared to the respective recombinant antibodies (Le et al. 2024; Thran et al. 2017). mRNA has also been used to express bsAbs in LNPs formulations, achieving tumor rejection by targeting CD3 × claudin 6 (CLDN6), CD3 × epithelial cell‐attached molecules (EpCAM), chemokine (C–C motif) ligand 2 CCL2 × chemokine (C–C motif) ligand 5 (CCL5), PD‐1 × PD‐L1, and CD3 × B7 homolog 3 protein (B7H3), both as monotherapy and in combination with cell therapies or ICI, in murine tumor models (C. Huang et al. 2023; Stadler et al. 2017; Y. Wang et al. 2021; L. Wu et al. 2022).
To evade unnatural posttranslational modifications and concerns related to short half‐lives, new frontiers in antibody immunotherapy emerged with the development of polyspecific antibodies and proteolysis‐targeting antibodies (PROTABs). Following the clinical approval of blinatumomab (BiTE CD19 × CD3), a new generation of polyspecific antibodies has begun to appear, exploring their inherent binding specificity while extending circulation times. Consequently, research on trispecific antibodies is gaining momentum, providing preliminary evidence of their safety and pronounced efficacy at picomolar doses (Austin et al. 2021; Passariello et al. 2022). Considering the promising results of mRNA‐encoded bsAbs, mRNA holds enormous potential as a delivery platform for trispecific antibodies (Sanz and Álvarez‐Vallina 2021). Considerable potencies in the nanomolar range have also been observed for biological proteolysis‐targeting chimeras (BioPROTACs), which are bifunctional proteins resulting from the fusion of an E3 ligase with targeted proteins, including antibodies (Lim et al. 2020). These structures can combine the selectivity of antibodies with the catalytic effect of E3 ligases to trigger targeted protein degradation via the ubiquitin‐proteosome pathway. Currently, proof‐of‐concept studies have already demonstrated the incorporation of mRNA‐encoded BioPROTACS into LNPs, achieving rapid and noticeable protein degradation, and laying the groundwork for future advances (Chan et al. 2024; Chang et al. 2022).
2.3. mRNA Expression as a Tool to Advance Cell‐Specific Targeted Production of Therapeutic Proteins
Recently, mRNA has garnered significant attention as a viable alternative for inducing protein expression in vivo, potentially replacing recombinant cytokines and antibodies (Beck et al. 2021). However, while it is possible to transfect cells with naked mRNA, its negative charge and high molecular weight hinder its permeability across biological membranes, reducing the rate of protein expression (Di Trani et al. 2023; Van Hoecke and Roose 2019). Consequently, nanoparticulate delivery systems are essential to ensure the intracellular delivery of mRNA, enhancing protein expression by decreasing mRNA immunogenicity, providing sustained release, and minimizing enzymatic degradation (Clemente et al. 2023; Melero et al. 2021).
With the advent of COVID‐19 vaccines, LNP have gained visibility and are now the most widely used vehicle in mRNA‐based therapeutics (Barot et al. 2023; Neshat et al. 2023). Generally, LNP are composed of four components: (1) ionizable lipids crucial for mRNA encapsulation and endosomal escape; (2) cholesterol to regulate membrane fluidity; (3) auxiliary lipids that impact membrane rigidity; and (4) PEG‐modified lipids to enhance NP circulation time (J. Han et al. 2023; Jiang et al. 2024). Although expression rates are high, mRNA‐encoded cytokines and antibodies may require targeted delivery to specific tissues and cells to avoid systemic adverse events and achieve higher expression efficiencies (Deshpande, Biswas, and Torchilin 2013). However, hepatic accumulation of LNP, due to the dense and leaky hepatic vessel network and their interaction with Apolipoprotein E, remains a significant challenge (C. Huang et al. 2023; B. Kong et al. 2023).
To target mRNA to extrahepatic tissues, both passive and active targeting approaches have been developed (B. Kong et al. 2023; Veiga et al. 2018). Passive targeting of LNP exploits the enhanced permeability and retention effect in tumors, where the permeability of tumor capillaries and reduced tumor lymphatic drainage increase NP retention (Deshpande, Biswas, and Torchilin 2013; Rybakova et al. 2019). To improve LNP delivery performance, factors such as size, morphology, charge, and circulation time must be finely optimized (Guo et al. 2020). In addition, developing new ionizable lipids (e.g., IC8, 113‐O12B, and cKK‐E12) with optimized targeting abilities, modulating the ratio of lipid constituents, replacing PEG‐linked (PEGylated) lipids with polymers that have a better safety profile (e.g., polysarcosine), and using protamine to condense mRNA and facilitate encapsulation have all been explored as valuable strategies (Beck et al. 2021; Fenton et al. 2017; C. Huang et al. 2023; B. Kong et al. 2023; Lei et al. 2020; Nogueira et al. 2020; Rybakova et al. 2019; Shimosakai et al. 2022).
Rybakova et al. demonstrated the assembly of LNP using the novel ionizable lipid cKK‐E12 to deliver mRNA‐encoded trastuzumab (Rybakova et al. 2019). Intravenous administration of cKK‐E12‐LNP showed hepatic tropism with high levels of protein expression and inhibition rates compared to recombinant antibody administration in breast cancer mouse models. To achieve higher delivery efficiencies than conventional LNP, Liu et al. used ionizable lipids with diamino groups (J.‐Q. Liu et al. 2022). This approach, combined with intratumoral administration, triggered targeted expression of mRNA‐encoded IL‐12 and IL‐27 in tumor cells and B lymphocytes, inducing potent CTL and NK infiltration and long‐lasting reduction of melanoma growth.
Active targeting relies on ligand‐receptor interactions, where LNP are decorated/coated with small molecules, peptide sequences, or antibodies (Clemente et al. 2023; Deshpande, Biswas, and Torchilin 2013). C‐type lectin receptors (e.g., mannose receptors), TLR (e.g., TLR2), CD proteins (e.g., CD3), and other surface proteins (e.g., Ly6c) can be used for the active targeting of mRNA‐LNP (Clemente et al. 2023; Yuan et al. 2023). For example, mRNA‐encoded reporter protein encapsulated in anti‐CD3‐coated LNP accumulated in the spleen and showed expression levels in T lymphocytes dependent on the anti‐CD3 amount (Kheirolomoom et al. 2022). The choice of the most appropriate route of administration, considering the properties of LNP and the desired response, also plays a major role in the biodistribution of NP and the rate of mRNA translation (Hou et al. 2021).
In addition to LNP, other strategies such as extracellular vesicles, PNP, and inorganic NP have been studied for in vivo expression of antibodies and cytokines, offering high delivery efficiency, better distribution, and improved safety profiles (Dong et al. 2023; Le et al. 2024; Neshat et al. 2023; H. Shin et al. 2023). For example, taking advantage of the affinity of antibodies against CD71 highly expressed in glioblastoma, Dong et al. designed extracellular vesicles from fibroblasts conjugated with anti‐CD71 and anti‐PD‐L1 antibodies to target glioblastoma (Dong et al. 2023). These vesicles, after systemic administration, crossed the blood–brain barrier and induced in vivo translation of mRNA‐encoded INF‐γ, significantly inhibiting glioma cell growth in GL261 and SB28 mouse models (Dong et al. 2023). Extracellular vesicles derived from exosomes were also exploited for translating mRNA‐encoded IL‐12 after intratumoral administration in lung cancer models, resulting in efficient tumor growth deceleration (M. Liu et al. 2024).
Le et al. used poly(β‐amino esters) (PBAE) for lung‐targeted delivery of mRNA‐encoded bevacizumab, achieving selective expression in the pulmonary endothelium and superior antitumor efficacy compared to recombinant antibody in murine NSCLC models (Le et al. 2024). Neshat et al. explored the same polymer to prepare mRNA‐encoded IL‐12 PNP, which, when locally administered ensured good safety profiles and induced tumor rejection with enhanced immune memory in breast and colon cancer mouse models (Neshat et al. 2023).
Silica NP have also been reported as an effective intratumoral delivery vehicle for mRNA‐encoded IL‐2, resulting in robust IL‐2 expression and inhibition of both ICI‐treated and untreated tumors (H. Shin et al. 2023). To further improve tumor accumulation of NP and extend shelf life, poly(lactic‐co‐glycolic acid) (PLGA) gelling copolymers, oxidants, and stabilizers have been added as adjuvants (Barot et al. 2023; Neshat et al. 2023).
Currently, 12 immunotherapies are undergoing clinical evaluation, taking advantage of the promising preclinical results of mRNA‐encoded cytokines and antibodies (Table 1). LNP are the preferential nanostructured vehicles, being selected in 10 of the 12 clinical trials (Table 1). In the remaining two clinical trials, naked mRNA is administered directly via internodal or intratumoral injections. Although these mRNA‐based cytokine and antibody immunotherapies have advanced to the clinical phase, their safety and tolerability are still in an early stage of development.
TABLE 1.
Clinical trials on mRNA‐encoded cytokines and antibodies for cancer immunotherapy.
| Name | Sponsor | Cancer type | Target | Route/nanodelivery system | Therapy | Phase | Status | National clinical trial identifier |
|---|---|---|---|---|---|---|---|---|
| mRNA‐2752 | Laura Esserman (University of California, San Francisco) | Ductal carcinoma in situ | Human OX40L, IL‐23, and IL‐36γ | Intratumoral/LNP | Monotherapy and combined with anti‐PD‐1 pembrolizumab | I | Recruiting | NCT02872025 |
| ECI‐006 | eTheRNA immunotherapies | Melanoma | 5 TAA, caTLR4, CD40L, and CD70 | Intranodal/Naked mRNA | Monotherapy | I | Terminated | NCT03394937 |
| mRNA‐2752 | Laura Esserman | High‐risk ductal carcinoma | Human OX40L, IL‐23, and IL‐36γ | Intratumoral/LNP | Combined with anti‐PD‐1 pembrolizumab | I | Recruiting | NCT02872025 |
| ModernaTX Inc. | Relapsed/refractory solid tumor malignancies or lymphoma | Monotherapy and combined with anti‐PD‐L1 durvalumab | I | Active, not recruiting | NCT03739931 | |||
| mRNA‐2416 | Human OX40L | I/II | Terminated | NCT03323398 | ||||
| SAR441000 | Sanofi | Advanced solid tumors | IL‐12, IFN‐α 2b, GM‐CSF, and IL‐15sushi | Intratumoral/Saline mixture | Monotherapy and combined with anti‐PD‐1 cemiplimab | I | Terminated | NCT03871348 |
| MEDI1191 | MedImmune LLC | Advanced solid tumors | IL‐12 | Intratumoral/LNP | Monotherapy and combined with anti‐PD‐L1 durvalumab | I | Completed | NCT03946800 |
| BNT151 | BioNTech SE | Solid tumors | IL‐2 | Intravenous/LNP | Monotherapy and combined with the respective SoC | I/IIa | Terminated | NCT04455620 |
| BNT152 and BNT153 | IL‐7 and IL‐2 | Monotherapy | I | Recruiting | NCT04710043 | |||
| BNT141 | CLDN18.2‐positive solid tumors | mAb anti‐CLDN18.2 | Monotherapy and combined with paclitaxel and gemcitabine | I/II | Terminated | NCT04683939 | ||
| BNT142 | CLDN6‐positive solid tumors | BiTE (CLDN6 × CD3) | Monotherapy | I/IIa | Recruiting | NCT05262530 | ||
| ABOD2011 | Cancer Institute and Hospital, Chinese Academy of Medical Sciences | Advanced solid tumors | IL‐12 | Intratumoral/LNP | Monotherapy | I | Recruiting | NCT05392699 |
Abbreviations: BiTE: bispecific T cell engagers; CLDN18.2: claudin‐18.2 protein; CLDN6: claudin‐6 protein; GM‐CSF: granulocyte‐macrophage colony‐stimulating factor; IFN: interferon; IL: interleukin; LAMP: lysosome‐associated membrane glycoproteins; LNP: lipid nanoparticle; mRNA: messenger RNA; NA: not available; NCT: national clinical trial identifier; OX40L: OX40 ligand; PD‐L1: programmed cell dead‐ligand 1; SoC: standard of care; TAA: tumor‐associated antigen.
3. mRNA as a Tool to Advance Cancer Vaccines
Cancer vaccines have been under development for decades, holding the potential to revolutionize cancer treatment by offering a more targeted and personalized therapeutic strategy with fewer side effects. However, therapeutic cancer vaccines have historically demonstrated limited clinical efficacy. To date, only Sipuleucel‐T (Provenge) has been approved by the FDA to treat prostate cancer, providing minimal survival benefits of approximately 4 months (Kantoff et al. 2010).
Eliciting antitumor responses is challenging because the immune system eliminates self‐reactive immune cells to prevent toxic, autoreactive immune responses. Despite these challenges, exciting data shows that cancer vaccines can be designed to target specific tumor antigens, triggering strong in vivo antitumor T and B cell responses (Lang et al. 2022; Matos et al. 2023; Peres et al. 2024). Among the various vaccination platforms, mRNA‐based technology has emerged as a particularly attractive platform for inducing and strengthening antitumor responses and controlling tumor regression.
Several preclinical and clinical studies have demonstrated the feasibility and effectiveness of mRNA cancer vaccines. mRNA vaccines offer many advantages over conventional vaccine platforms, including rapid production and a broad range of applications achievable through simple modification of the mRNA sequence. While significant progress has been made, much remains to be learned about the mechanisms of mRNA‐based therapeutics, their interactions with the human body, and how to optimize their efficacy, safety, and accessibility for all patients. These challenges are particularly evident in mRNA cancer vaccines, which face high manufacturing, handling, and storage costs (Uddin and Roni 2021; Wadhwa et al. 2020). The requirement for a cold chain—including production, distribution, ultra‐cold storage, and administration—presents significant logistical hurdles that must be carefully considered when selecting mRNA technology. Moreover, several key questions remain regarding the design of mRNA cancer vaccines: which antigens, delivery platforms, and administration routes can trigger effective and sustained antitumor immunity with clinical relevance? This overview addresses these questions and provides insights into the use of mRNA in cancer vaccines.
3.1. mRNA Encoding Tumor Antigens
The most critical vaccine component is antigens binding to an antigen receptor, either an antibody or a T‐cell receptor (TCR). Tumor antigens are generally divided into two groups: tumor‐associated antigens (TAA) and tumor‐specific antigens (TSA). Typically, TAA are overexpressed in tumor cells but are also present in normal tissues, which means they have weak tumor specificity and immunogenicity. In contrast, TSA, also known as neoantigens, are “non‐self” antigens derived from mutations in tumor cells. They possess high tumor specificity and immunogenicity without affecting central immune tolerance.
Several personalized vaccines encoding multiple TSA have been developed and are currently under preclinical and clinical evaluation. Notably, two independent efforts using tailor‐made mRNA vaccines have reported initial success in melanoma (mRNA‐4157/V940) and pancreatic cancer (BNT122/cevumeran), revitalizing the field of therapeutic cancer vaccines (Rojas et al. 2023; J. S. Weber et al. 2024). Moderna and Merck have announced that 3‐year data for mRNA‐4157 in combination with KEYTRUDA demonstrated sustained improvement in recurrence‐free survival and distant metastasis‐free survival in patients with high‐risk stage III/IV melanoma (NCT03897881). Next‐generation sequencing and epitope‐selection algorithms have been instrumental in advancing personalized cancer vaccines (Figure 3). However, these vaccines still face several limitations. Reducing the cost and manufacturing time will be instrumental in ensuring timely and equitable access to patient‐specific therapeutics. In addition, as the clinical use of mRNA in vaccines is relatively new, long‐term safety needs to be assessed. Although TSA‐based vaccines represent the most attractive strategy, TAA‐based vaccines have been the most used.
FIGURE 3.
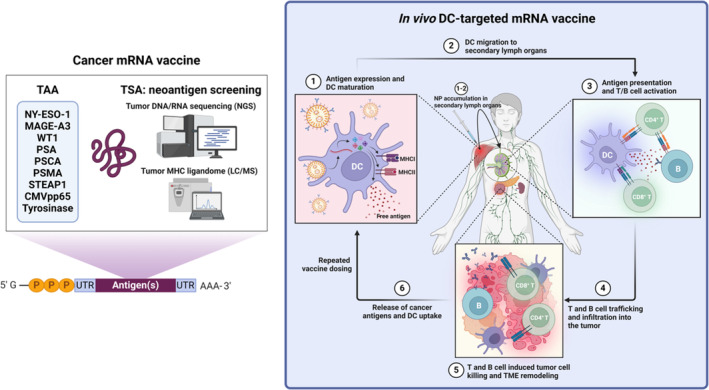
Cancer mRNA vaccine antigen composition and in vivo DC‐targeted mRNA vaccine‐induced antitumor response mechanisms. Each mRNA‐loaded nanocarrier vaccine can encode one or multiple TAA and/or TSA based on the antigen landscape of the patient's tumor. Due to the versatility of mRNA‐loaded nanocarriers, personalized cancer vaccines can be tailored using tumor neoantigen screening techniques such as NGS and LC/MS coupled with antigen selection by immunogenicity prediction algorithms. Upon administration, DC‐targeted mRNA‐loaded nanocarriers are taken up by local DC and accumulate in secondary lymph organs. Following mRNA cytoplasmic release and translation, DC mature and present the mRNA‐encoded antigens via MHC class I, MHC class II, and free antigen secretion. These DC activate T and B cells in secondary lymph organs to induce a broad adaptive immune response. The activated immune cells subsequently traffic to and infiltrate the tumor, inducing tumor cell death and potentially initiating the antitumor immunity cycle through TME remodeling and epitope spreading. CD: cluster of differentiation; CMVpp65: cytomegalovirus matrix protein pp65; DC: dendritic cell; LC/MS: liquid chromatography‐mass spectrometry; MAGE‐A3: melanoma‐associated antigen 3; MHC: major histocompatibility complex; NGS: next‐generation sequencing; NP: nanoparticle; NY‐ESO‐1: New York esophageal squamous cell carcinoma 1; PSA: prostate‐specific antigen; PSCA: prostate stem cell antigen; PSMA: prostate‐specific membrane antigen; STEAP1: six transmembrane epithelial antigen of the prostate 1; TAA: tumor‐associated antigen; TME: tumor microenvironment; TSA: tumor‐specific antigen; UTR: untranslated region; WT1: Wilm's tumor 1.
3.2. Ex Vivo vs. In Vivo mRNA Vaccination
DC are critical for establishing effective and prolonged antitumor immunity. They achieve this through MHC‐mediated antigen presentation, expression of T‐cell co‐stimulatory molecules, secretion of T‐cell activating cytokines, cell migration, and memory T‐cell activation to control tumor relapse (Palucka and Banchereau 2013; Perez and De Palma 2019). Therefore, DC are an ideal and frequently used delivery vehicle for mRNA cancer vaccines.
Patient‐derived DC have been transfected with mRNA encoding TAA following the successful electroporation of DC with mRNA encoding ovalbumin (Boczkowski et al. 1996). DC vaccines have been developed for multiple cancers, including leukemia, melanoma, and pancreatic cancer (Conniot et al. 2019; Matos et al. 2019; Peres et al. 2021). Clinical studies have demonstrated that DC electroporated with mRNA encoding various TAA (WT1, PRAME, and CMVpp65) are safe and feasible for treating patients with acute myeloma leukemia (Lichtenegger et al. 2020). In addition, whole tumor RNA extracted from tumor tissue has been investigated in cancer vaccine design to avoid TAA selection. Clinical trials have shown that this approach is safe and feasible for melanoma, pancreatic cancer, neuroblastoma, and glioma (NCT01983748, NCT04157127, NCT04837547). However, most patients do not respond to DC vaccines, as DC generated outside the body often fail to sufficiently re‐activate host immunity to trigger effective T‐cell activation against tumors (Darvin et al. 2018; Havel, Chowell, and Chan 2019). High DC numbers and multiple doses are often required to ensure enough DC reach the secondary lymphoid organs, promoting to the formation of specific cytotoxic T cells capable of infiltrating the tumor mass. Moreover, these therapies are costly and involve numerous technical challenges related to complex production procedures (e.g., leukocyte isolation, ex vivo DC generation, DC loading, and activation with tumor antigens, followed by reinjection into patients who are often severely immunosuppressed). Given these limitations, direct RNA delivery systems have been considered as an alternative to DC mRNA vaccines. mRNA delivery systems include lipids, lipid‐like materials, polymers, and protein derivatives (Hou et al. 2021; Kowalski et al. 2019; B. Li, Zhang, and Dong 2019; Meng et al. 2021). These systems incorporate mRNA through electrostatic interactions, preserving mRNA stability and preventing degradation by extracellular RNases. They also facilitate cellular endosomal uptake and subsequent escape through membrane disruption, allowing for mRNA cytosolic release.
Among mRNA delivery systems, LNP have dominated the vaccine landscape, with several preclinical and clinical studies ongoing (Table 2). A liposome‐protamine mRNA vaccine encoding four TAA (PSA, PSCA, PSMA, and STEAP1) in advanced prostate cancer patients demonstrated inconsistent immune responses over time (Koch et al. 2014; Kübler et al. 2015). Lipo‐MERIT (BNT111), a lipid‐protected mRNA vaccine encoding TAA (NY‐ESO‐1, MAGE‐A3, TPTET, and tyrosinase), is currently being evaluated in an ongoing clinical trial against melanoma (NCT02410733). Notably, Lipo‐MERIT has shown strong and durable antigen‐specific CD4+ and CD8+ T cell responses (Kranz et al. 2016; Sahin et al. 2020). Furthermore, studies have demonstrated that LNP provide intrinsic adjuvant signals by activating the NLR family pyrin domain containing 3 (NLRP3) and the stimulator of interferon genes (STING) pathways (Miao et al. 2019; Tahtinen et al. 2022).
TABLE 2.
Clinical trials using lipidic nanodelivery systems for mRNA‐based vaccines.
| Name | Sponsor | Cancer type | Target antigen | Route/nanodelivery system | Therapy | Phase | Status | National clinical trial identifier |
|---|---|---|---|---|---|---|---|---|
| BNT111 | BioNTech SE | Advanced melanoma | NY‐ESO‐1, MAGE‐A3, tyrosinase, and TPTE (TAA) | Intravenous/LPX | Monotherapy | I | Completed | NCT02410733 |
| Monotherapy and combined with anti‐PD‐1 cemiplimab | II | Active, not recruiting | NCT04526899 | |||||
| BNT112 | Prostate cancer | 5 prostate TAA | Monotherapy and combined with anti‐PD‐1 cemiplimab and/or goserelin acetate | I/II | Terminated | NCT04382898 | ||
| BNT113 | University of Southampton | HPV16+ head and neck cancer | HPV16‐ derived oncoproteins E6 and E7 | Intradermal/LPX | Monotherapy | I/II | Completed | NCT03418480 |
| BioNTech SE | Combined with anti‐PD‐1 pembrolizumab | II | Recruiting | NCT04534205 | ||||
| BTN114 (IVAC_W_bre1_uID) | BioNTech SE | TNBC | TNBC TAA, p53, and neoantigens | Intravenous/liposome | Monotherapy | I | Completed | NCT02316457 |
| BNT115 (W_ova1 Vaccine) | University Medical Center Groningen | Ovarian cancer | Ovarian TAA | Intravenous/LPX | Combined with (neo)adjuvant chemotherapy (carboplatin and paclitaxel) | I | Terminated | NCT04163094 |
| BNT116 | BioNTech SE | Advanced NSCLC | 6 TAA | Monotherapy and combined with anti‐PD‐1 cemiplimab or chemotherapy (carboplatin and paclitaxel) | I | Recruiting | NCT05142189 | |
| Regeneron Pharmaceuticals | Combined with anti‐PD‐1 cemiplimab | II | NCT05557591 | |||||
| BNT122 (RO7198457) | Genentech Inc. |
Locally advanced or metastatic solid tumors |
Neoantigens |
Monotherapy and combined with anti‐PD‐L1 atezolizumab | I | Active, not recruiting | NCT03289962 | |
| Untreated advanced melanoma | Combined with anti‐PD‐1 pembrolizumab | II | NCT03815058 | |||||
| Memorial Sloan Kettering Cancer Center | Surgically resected pancreatic cancer | Combined with anti‐PD‐L1 atezolizumab and FOLFIRINOX | I | NCT04161755 | ||||
| BioNTech SE | Surgically resected colorectal cancer | Monotherapy | II | Recruiting | NCT04486378 | |||
| BNT211 | BioNTech Cell & Gene Therapies GmbH | Solid tumors | Claudin 6 | Combined with CLDN6‐specific CAR‐T cells | I | NCT04503278 | ||
| mRNA‐4157 | ModernaTX Inc. | Solid tumors | Neoantigens | Intramuscular/LNP | Monotherapy and combined with anti‐PD‐1 pembrolizumab | I | Recruiting | NCT03313778 |
| High‐risk melanoma | Combined with anti‐PD‐1 pembrolizumab | II | NCT03897881 | |||||
| Merck Sharp & Dohme LLC | High‐risk melanoma | III | NCT05933577 | |||||
| NSCLC | III | NCT06077760 | ||||||
| Cutaneous squamous cell carcinoma | II/III | NCT06295809 | ||||||
| Bladder cancer postradical resection | II | NCT06305767 | ||||||
| Renal cell carcinoma | II | NCT06307431 | ||||||
| mRNA‐2752 | Laura Esserman | High‐risk ductal carcinoma | Human OX40L, IL‐23, and IL‐36γ | Intratumoral/LNP | Combined with anti‐PD‐1 pembrolizumab | I | NCT02872025 | |
| ModernaTX Inc. | Relapsed/refractory solid tumor malignancies or lymphoma | Monotherapy and combined with anti‐PD‐L1 durvalumab | I | Active, not recruiting | NCT03739931 | |||
| mRNA‐2416 | Relapsed/refractory solid tumor malignancies or lymphoma | Human OX40L | I/II | Terminated | NCT03323398 | |||
| mRNA‐5671 | Merck Sharp & Dohme LLC | KRAS‐mutant NSCLC, colorectal cancer, pancreatic adenocarcinoma | KRAS mutations | Intramuscular/LNP | Monotherapy and combined with anti‐PD‐1 pembrolizumab | I | Completed | NCT03948763 |
| MEDI1191 | MedImmune LLC | Solid tumors | IL‐12 | Intratumoral/LNP | Monotherapy and combined with anti‐PD‐L1 durvalumab | I | Completed | NCT03946800 |
| No | University of Florida | Adult glioblastoma | Autologous total tumor and LAMP TAA | Intravenous/Liposome | Monotherapy | I | Recruiting | NCT04573140 |
| No | University of Florida | Advanced melanoma | Autologous total tumor | Intravenous/Liposome | Monotherapy | I | Suspended | NCT05264974 |
Abbreviations: FOLFIRINOX: chemotherapy combination of leucovorin calcium (folinic acid), fluorouracil, irinotecan hydrochloride, and oxaliplatin; HPV: human papillomavirus; IL: interleukin; KRAS: Kirsten rat sarcoma virus; LAMP: lysosome‐associated membrane glycoproteins; LNP: lipid nanoparticle; LPX: lipoplex; MAGE‐A3: melanoma‐associated antigen 3; mRNA: messenger RNA; NCT: national clinical trials; NSCLC: Non‐small cell lung cancer; NY‐ESO‐1: New York esophageal squamous cell carcinoma 1; OX40L: OX40 ligand; PD‐1: programmed cell death protein‐1; PD‐L1: programmed cell dead‐ligand 1; TAA: Tumor‐associated antigen; TNBC: triple‐negative breast cancer; TPTE: transmembrane phosphatase with tensin homology.
3.3. DC‐Targeted mRNA Vaccines
Targeted delivery of mRNA vaccines to tissues rich in immune cells can induce prolonged antitumor immunity and reduce the systemic adverse effects associated with broader administration. In addition, directing tumor antigens specifically to immune cells has proven to be an effective strategy for developing innovative and potent vaccines. In this context, DC, as the most potent APC with a unique ability to prime adaptive immunity, stand out as pivotal candidates for targeted mRNA delivery (Kastenmüller et al. 2014; Figure 3).
Multiple preclinical studies have highlighted the advantages of directly delivering mRNA payloads to APC. For instance, delivering TAA via naked RNA to lymphoid organs has stimulated antitumor immunity in mouse models of melanoma, lymphoma, and leukemia (Diken et al. 2011; Van Lint et al. 2012). For optimal targeted delivery, many NP include active‐targeting ligands to deliver payloads to specific sites. These NP are functionalized with molecules, either antibodies or small molecules, that interact with cellular receptors to enhance their uptake by target cells.
Recently, DC‐targeting virus‐like particles were engineered with a Sindbis‐virus glycoprotein that recognizes the surface proteins on DC (Yin et al. 2024). Among the molecules used to formulate targeted mRNA vaccines, antibodies have been at the forefront in the functionalization of LNP. Typically, antibodies are conjugated to LNP using PEGylated lipids with terminal groups such as maleimide (Ramishetti et al. 2015). However, the chemical conjugation on the LNP surface often lacks control over antibody orientation, resulting in poor interaction with their targets. Consequently, small molecules have been added to LNP via PEG or cholesterol as alternatives (F. Kong et al. 2012). For example, several mannose‐modified LNP have demonstrated targeted mRNA delivery to DC (L. Liu et al. 2018; Perche et al. 2011; Y. Wang et al. 2018). Nevertheless, adding targeting ligands to delivery systems increases complexity, which can hinder the rapid production of mRNA vaccines. Therefore, to direct LNP to immune‐enriched organs without using targeting ligands, LNP have been engineered using lipids that redirect the biodistribution of RNA‐lipoplexes to DC in secondary lymphoid organs and the bone marrow, while avoiding the liver and lungs (Akinc et al. 2010; Chen et al. 2022; Kranz et al. 2016). Details on how LNP composition can guide LNP targeting over DC will be further discussed below.
3.4. LNP Composition and Administration Routes
LNP self‐assemble due to thermodynamically favorable arrangements driven by electrostatic interactions and the amphiphilic nature of lipids (Weng et al. 2020). The first generation of nonviral vectors used for delivering mRNA cancer vaccines were cationic LNP (Hess et al. 2006; Le Moignic et al. 2018; Sahin et al. 2020). Commonly used cationic lipids included dioleoyl‐3‐trimethylammonium propane (DOTAP) and 1,2‐di‐O‐octadecenyl‐3‐trimethylammonium propane (DOTMA) (Conry et al. 1995; Kranz et al. 2016). By adjusting the ratio of cationic lipids, it is possible to specifically target the spleen and lungs (Perche et al. 2011). Additionally, cationic LNP demonstrated high mRNA incorporation and transduction efficiency. However, liver damage was observed when cationic LNP were systemically administered (Landesman‐Milo and Peer 2014; Lv et al. 2006).
To circumvent these limitations, current LNP designs incorporate four key components: an ionizable lipid, cholesterol, a helper phospholipid, and a PEGylated lipid. These components collectively impact biological activity, biodegradability, and structural stability. Ionizable lipids enable neutral or anionic surface charges to make particles inert at physiologic pH, while cholesterol and PEG prevent particle aggregation and maintain LNP size (< 200 nm) for endocytic uptake. Helper lipids prevent mRNA endo‐lysosomal degradation (X. Han et al. 2021; Suk et al. 2016; Y. Zhang et al. 2021). The lipid ratio and properties (e.g., pKa, alkyl chain length, and ester bonds) used in LNP formulation determine the size, surface charge, and composition of LNP, which in turn determine LNP fate (Cornebise et al. 2022). Ionizable lipids, such as DLin‐MC3‐DMA, ALC‐0315, OF‐Deg‐Lin, FTT5, and TT3 exhibited low toxicity and high tissue clearance, reducing the adverse effects observed with cationic lipids. Their increased biocompatibility is attributed to pH‐dependent ionization, which enhances, endosomal escape when LNP use lipids with optimized pKa values or branched tails for stronger protonation at endosomal pH (Hajj et al. 2019).
The other lipid components—cholesterol, helper lipids, and PEGylated lipids—further improve the stability, delivery, tolerance, and biodistribution of LNP. Cholesterol stabilizes and modulates LNP's rigidity, ensuring lipoplex integrity. Helper lipids, such as 1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine (DSPC) or 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE), promote the fusion of NP with cell and endosomal membranes, improving cellular uptake and endosomal release, thus enhancing the NP effect (W. Li and Szoka 2007). Finally, PEGylated lipids increase the LNP half‐time, reduce serum protein adsorption on the NP surface, and decrease NP uptake by reticuloendothelial cells (Jokerst et al. 2011; Knop et al. 2010). However, PEGylated nanomedicine formulations face challenges such as accelerated blood clearance after repeated administrations (Ishida and Kiwada 2008). Subsequent doses can lead to anti‐PEG antibody formation, resulting in particle opsonization and increased particle uptake by the mononuclear phagocytic system (D. Shi, Beasock, et al. 2022a; Q. Yang and Lai 2015).
The delivery route also significantly impacts the magnitude and duration of antigen delivery, influencing the effectiveness of antitumor immunity. Multiple RNA delivery routes are being tested, including local (intramuscular, intradermal, intranodal, intranasal, subcutaneous, and intratumoral) and systemic (intravenous, intraperitoneal, and intratracheal) administration (Phua et al. 2014; Van Lint et al. 2016). Intravenous administration leads to LNP accumulation in lymph nodes, potentially enhancing immune responses to mRNA vaccines (N. Kong et al. 2019; Sayour et al. 2017). However, this method also results in accumulation in the liver. Most advanced‐stage mRNA cancer vaccines are designed for intramuscular administration, as resident APC in the skin and muscle can internalize and process mRNA‐encoded tumor antigens.
4. Cell Therapies—Creating “Living Drugs” With mRNA‐Loaded Nanocarriers
4.1. Chimeric Antigen Receptor (CAR)‐T Cells
CAR‐T cell therapy has revolutionized cancer immunotherapy, demonstrating remarkable efficacies in several B‐cell malignancies (Cappell and Kochenderfer 2023). Inspired by the signaling pathways governing T cell activation and proliferation, the synthetic CAR structure typically comprises an extracellular targeting moiety, a spacer domain, a transmembrane domain, and a CD3ζ intracellular domain, with or without co‐stimulatory domains such as 4‐1BB and CD28. By utilizing different targeting molecules, such as single‐chain variable fragments (scFv), nanobodies, natural ligands, designed ankyrin repeat proteins (DARPins), and aptamers, these synthetic receptors redirect T cell responses toward any cell surface‐expressed targets of interest, independently of the endogenous MHC‐TCR interaction (Figure 4). Moreover, each CAR component can be switched, combined, and optimized to fine‐tune CAR‐T cell activity and phenotype (Jayaraman et al. 2020; Nix and Wiita 2024; Q. Zhang et al. 2024). This extreme versatility unveiled a golden age for synthetic biology, with numerous CAR designs and synthetic biological circuits emerging in the preclinical and clinical pipelines for hematological and solid tumors. Although promising, the expensive, sophisticated, and inflexible CAR‐T cell manufacturing process has significantly hindered its widespread adoption. A critical challenge to clinical implementation has been the spectrum of severe CAR‐T cell‐related adverse effects, including cytokine release syndrome (CRS), immune effector cell‐associated neurotoxicity syndrome (ICANS), and on‐target off‐tumor toxicity (Bailey et al. 2023).
FIGURE 4.
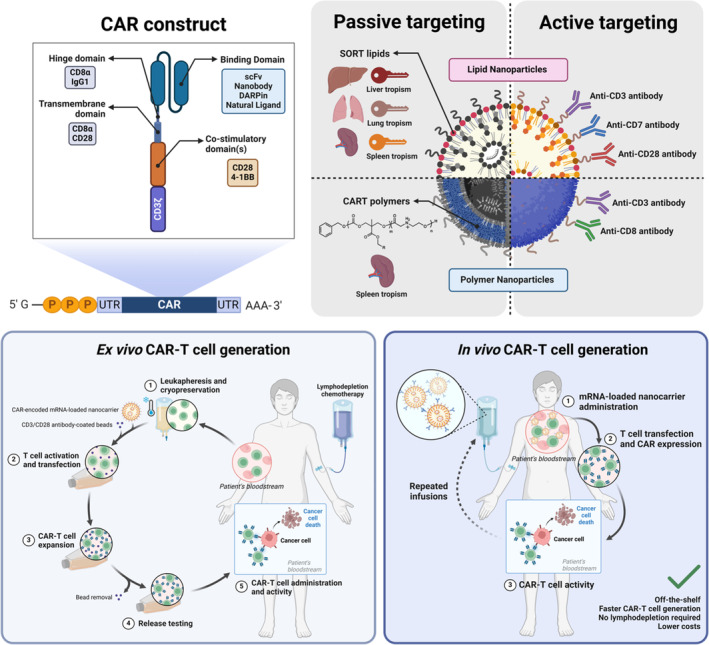
CAR‐encoded mRNA‐loaded nanocarrier engineering and CAR‐T cell generation methods. 4‐1BB: tumor necrosis factor ligand superfamily member 9; CART: charge‐altering releasable transporters; CAR‐T: chimeric antigen receptor‐T cell; CD: cluster of differentiation; DARPin: designed ankyrin repeat proteins; scFv: single‐chain variable fragment; SORT lipid: selective organ‐targeting lipid; UTR: untranslated region.
Currently, all approved CAR‐T cell therapies are generated ex vivo using viral vectors to modify patient‐derived T cells, a process that takes several weeks. Initially, T cells are collected from the patient's blood by leukapheresis and transported to specialized facilities. The harvested T cells are then activated, transduced with γ‐retroviral or lentiviral vectors delivering the CAR transgene, expanded to reach an adequate CAR‐T cell number in the final product, and assessed for their quality attributes (Ayala Ceja et al. 2024). During this time, the patient undergoes a preparatory lymphodepletion regimen, commonly including cyclophosphamide and/or fludarabine to reduce tumor burden and improve CAR‐T cell expansion and persistence (Figure 4) (Lickefett et al. 2023). This manufacturing and treatment workflow costs between $400,000 to over a million dollars per patient, raising significant hurdles to CAR‐T cell affordability and sustainability for healthcare systems (Cliff et al. 2023). Furthermore, the time required to obtain and deliver the CAR‐T cell product compromises its therapeutic utility for patients with a rapidly progressing disease (Caimi et al. 2020).
Although γ‐retroviral and lentiviral vectors reliably yield high T cell transduction efficiencies, their application at a clinical CAR‐T cell scale poses additional issues. First, their nonrandom genome integration profile raises safety concerns regarding insertional oncogenesis, as recently spotlighted by the FDA statement reporting clinical cases of second primary malignancies possibly linked to CAR‐T cell therapies (Banerjee et al. 2024; Cornetta et al. 2023). In addition, CAR‐T cell production requires significant costs and complexity to follow the strict GMP and biosafety regulations, thus representing a key bottleneck in the manufacturing workflow (Ayala Ceja et al. 2024). For these reasons, the technological and logistical challenges of autologous viral vector‐mediated ex vivo CAR‐T cell generation prompted the development of innovative platforms to harness the full potential of CAR‐T cells and improve their accessibility to patients. Among these, nonviral methods for CAR‐T cell production and in vivo (also called in situ) CAR‐T cell generation garnered significant interest in the last years, owing to multiple advancements in nonviral delivery systems engineering and gene editing tools. Notably, the development of these novel approaches was greatly accelerated by the IVT mRNA technology, as will be discussed further (Table 3).
TABLE 3.
Overview of CAR‐encoded mRNA‐loaded nanocarriers in preclinical development.
| CAR type | CAR targeting domain | CAR target | CAR Intracellular domains | Delivery system | NP composition and other payload | Generation site | NP targeting mechanism | Tumor type and models | References |
|---|---|---|---|---|---|---|---|---|---|
| CAR‐T | scFv | Human CD19 | 4‐1BB/CD3ζ | LNP | DOPE, Cholesterol, DMG‐PEG2000, C14‐4 | Ex vivo | None | Nalm‐6‐Fluc ALL (culture) | (Billingsley et al. 2020, 2022) |
| scFv | Human CD19 | 4‐1BB/CD3ζ | LNP | DOPE and/or DSPC, Cholesterol, DMG‐PEG2000, C14‐4, PD‐1 siRNA | Ex vivo | None | Not evaluated | (Hamilton et al. 2023) | |
| scFv | Human CD19 | 4‐1BB/CD3ζ | LNP | DOPE, Cholesterol, DMG‐PEG2000, DSPE‐PEG2000‐maleimide, C14‐4, CD3, and CD28 Antibody fragments | Ex vivo | Active: anti‐CD3 and anti‐CD28 antibody fragments | Nalm‐6‐Fluc ALL (co‐culture and NSG mice) | (Metzloff et al. 2024) | |
| Undisclosed | LNP | Undisclosed, target‐primed reverse transcriptase mRNA co‐delivery—RNA Gene Writer System | Ex vivo and in vivo | Undisclosed | Undisclosed (humanized and immunodeficient mice, nonhuman primates) | (Magee et al. 2023) | |||
| Undisclosed | Murine CD19 | Undisclosed | LNP | DOPE, Cholesterol, DMG‐PEG2000, DSPE‐PEG2000‐maleimide, C14‐4, anti‐CD3, or anti‐CD7 Antibody fragments | In vivo | Active: anti‐CD3 or anti‐CD7 antibody fragments | Not evaluated (healthy mice only) | (Billingsley et al. 2024) | |
| scFv | Murine CD19 | 4‐1BB or CD28/CD3ζ | LNP | DOPE, Cholesterol, 5A2‐SC8, DMG‐PEG, SORT lipid: 18:1 PA | In vivo | Passive: 18:1 PA—spleen tropism | A20‐Fluc lymphoma (BALB/c mice) | (Álvarez‐Benedicto et al. 2023) | |
| scFv | Human CD19 | 4‐1BB/CD3ζ | PNP | bAC‐CART polymer | Ex vivo | Passive: bAC‐CART—Spleen tropism | Nalm‐6‐GFP&Fluc ALL (culture) | (Z. Li, Amaya, et al. 2023b) | |
| scFv | Human CD19, Human ROR1 | CD28/CD3ζ | PNP | PBAE‐447, PGA‐linked anti‐CD3, or anti‐CD8 antibody fragments | In vivo | Active: anti‐CD3 or anti‐CD8 antibody fragments |
Anti‐CD19 CAR: Raji‐Fluc Burkitt's Lymphoma (co‐culture and NSG mice), Eμ‐ALL01‐Fluc Leukemia, (albino C57BL/6J mice) Anti‐ROR1 CAR: C42‐Fluc prostate adenocarcinoma (orthotopic, NSG mice) |
(Parayath et al. 2020) | |
| CAR‐NK | scFv | Human CD19, Human BCMA |
Anti‐CD19 CAR: CD28/CD3ζ Anti‐BCMA CAR: 4‐1BB/CD3ζ |
LNP | DSPC, cholesterol, SM‐102, and DMG‐PEG2000 | Ex vivo | None |
Anti‐CD19 CAR: Daudi Burkitt's Lymphoma (co‐culture) and Nalm‐6‐EGFP&Fluc ALL (co‐culture and NSG mice) Anti‐BCMA CAR: RPMI 8226 and MM1S multiple myeloma (co‐culture) |
(Golubovskaya, Sienkiewicz, Sun, Zhang, et al. 2023b) |
| scFv | Murine GPC3 | CD28/CD3ζ | LNP | DSPC, cholesterol, DMG‐PEG2000, DOTAP, DLin‐MC3‐DMA | Ex vivo | None | Hepa1c1c7 HCC (co‐culture and orthotopic thioacetamide‐induced liver‐fibrotic BALB/c nude mice) | (H. E. Shin et al. 2024) | |
| scFv | Human or murine CD19 | CD28/CD3ζ | PNP | CART BDK‐O7:N7:A13 polymer | Ex vivo | None | K562 leukemia (co‐culture), Nalm‐6 ALL (co‐culture), Raji Burkitt's Lymphoma (co‐culture) | (Wilk et al. 2020) | |
| CAR‐T and CAR‐M | scFv | Human CD19 | 4‐1BB/CD3ζ | LNP | DOPE, cholesterol, DSPC, DMG‐PEG2000, 76‐O17Se (CAR‐T), or 9322‐O16B (CAR‐M) | Ex vivo | None | Fluc + human B lymphoma (co‐culture) | (Ye et al. 2022) |
| CAR‐M | scFv | Murine GPC3 | CD3ζ only | LNP | DOPE, cholesterol, DMG‐PEG2000, PPZ‐A10, Siglec‐GΔITIM‐encoded mRNA | In vivo | Passive: PPZ‐A10—macrophage specificity | Hepa1‐6 HCC (co‐culture and orthotopic C57BL/6J mice) | (Z. Yang, Liu, et al. 2023b) |
| Undisclosed | Murine CD19 | Undisclosed | LNP | DOPE, Cholesterol, DMG‐PEG2000, C14‐O2 | In vivo | Passive: C14‐O2—macrophage specificity | Not evaluated (healthy mice only) | (Mukalel et al. 2024) | |
Abbreviations: 4‐1BB: tumor necrosis factor ligand superfamily member 9; 18:1 PA: 1,2‐dioleoyl‐sn‐glycero‐3‐phosphate; 9322‐O16B: bis(2‐(dodecyldisulfaneyl)ethyl) 3,3′‐((3‐(2‐methyl‐1H‐imidazol‐1‐yl)propyl)azanediyl)dipropionate; ALL: acute lymphoblastic leukemia; bAC‐CART: charge‐altering releasable transporters with a beta‐amido carbonate backbone; C14‐4: 1,1′‐[[2‐[2‐[4‐[2‐[[2‐[2‐[bis(2‐hydroxytetradecyl)amino]ethoxy]ethyl](2‐hydroxytetradecyl)amino]ethyl]‐1‐piperazinyl]ethoxy]ethyl]imino]bis‐2‐tetradecanol; CAR‐NK: chimeric antigen receptor‐natural killer cell; CAR‐T: chimeric antigen receptor‐T cell; CAR‐M: chimeric antigen receptor‐monocytes/macrophages; CD: cluster of differentiation; DLin‐MC3‐DMA: 4‐(dimethylamino)‐butanoic acid, (10Z,13Z)‐1‐(9Z,12Z)‐9,12‐octadecadien‐1‐yl‐10,13‐nonadecadien‐1‐yl ester; DMG‐PEG2000: 1,2‐dimyristoyl‐rac‐glycero‐3‐methoxypolyethylene glycol‐2000; DOPE: 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine; DOTAP: 1,2‐dioleoyl‐3‐trimethylammonium‐propane; DSPC: 1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine; EGFP: enhanced green fluorescent protein; Fluc: firefly luciferase; GFP: green fluorescent protein; GPC3: glypican 3; HCC: hepatocellular carcinoma; LNP: lipid nanoparticle; mRNA: messenger ribonucleic acid; NP: nanoparticle, PBAE: poly(β‐amino ester); PD‐1: programmed cell death protein‐1; PGA: polyglutamic acid; PNP: polymeric nanoparticle; ROR1: receptor tyrosine kinase‐like orphan receptor 1; scFv: single‐chain variable fragment; Siglec‐CΔITIM: Siglec‐C lacking immunoreceptor tyrosine‐based inhibition motifs; siRNA: small interference ribonucleic acid; SM‐102: 8‐[(2‐hydroxyethyl)[6‐oxo‐6‐(undecyloxy)hexyl]amino]‐octanoic acid, 1‐octylnonyl ester; SORT lipid: selective organ‐targeting lipid.
4.1.1. mRNA‐Loaded NP for Ex Vivo CAR‐T Cell Generation
mRNA technology offers significant advantages for CAR‐T cell generation compared to genome‐integrating vectors and DNA expression systems (Table 4). The transient expression of CAR‐encoding mRNA acts as a built‐in safety feature, preventing major adverse effects of persistent CAR expression, including CRS, ICANS, on‐target off‐tumor toxicity, and anti‐CAR immune reactions (Stock et al. 2023). Additionally, transient CAR expression preserves CAR‐T cell fitness by minimizing T cell exhaustion due to chronic antigen stimulation and tonic signaling (E. W. Weber et al. 2021). As a non‐integrating platform, mRNA‐based CAR‐T cell generation cannot cause insertional oncogenesis, thereby offering a favorable safety profile. Moreover, mRNA‐based CAR‐T cells benefit from the high scalability and flexibility of IVT mRNA manufacturing, with lower costs and fewer production constraints (J. Wu et al. 2024). For these reasons, CAR‐T cell generation with mRNA promises to significantly improve the safety, efficacy, manufacturing, logistical, and accessibility aspects of CAR‐T cell products.
TABLE 4.
Advantages and disadvantages of viral vectors and mRNA‐loaded nanocarriers for CAR‐T cell generation.
| Delivery platform | Advantages | Disadvantages |
|---|---|---|
| Viral vector |
|
|
| mRNA‐loaded nanocarriers |
|
|
Apart from the transfection step, mRNA‐based CAR‐T cell generation follows a similar workflow to that of viral vector‐based CAR‐T cells. However, the CAR‐encoded mRNA must successfully reach the T‐cell cytosol and be efficiently translated by the cell's protein synthesis machinery. To this end, the first and most frequently reported mRNA‐based CAR‐T cells are transfected ex vivo through electroporation, a straightforward and well‐established transfection method that applies an electrical field to temporarily increase cell membrane permeability, allowing mRNA to enter. However, the high cytotoxicity and limited scalability of this technique have driven the development of novel mRNA‐loaded LNP for CAR‐T cell generation (Figure 4). In this approach, the CAR‐encoded mRNA‐loaded LNP are internalized by endocytosis, after which the mRNA payload escapes from the endosome and reaches the cytosol (Kitte et al. 2023). Billingsley et al. synthesized a library of 24 ionizable lipids and identified C14‐4‐based LNP as top‐performing nanocarriers to deliver mRNA to human T cells ex vivo. Importantly, these C14‐4‐LNP‐generated CAR‐T cells exhibited similar CAR expression levels and antileukemic activity, with higher cell viability compared to electroporation‐generated CAR‐T cells (Billingsley et al. 2020). Later, the same research group optimized the previous C14‐4‐based LNP composition using a design of experiments with a library containing different molar ratios of C14‐4, DOPE, cholesterol, and PEG. The best‐identified LNP composition (called B10) achieved even better mRNA transfection efficiencies and lower cytotoxicity than the standard formulation, demonstrating the usefulness of this approach in identifying optimal mRNA‐loaded LNP for T‐cell transfection (Billingsley et al. 2022).
These versatile LNP nanoformulations can be further modified to enhance mRNA‐based CAR‐T cell manufacturability and effector activity. For instance, the previous screening approach was successfully employed to identify and characterize C14‐4‐based LNP formulations co‐delivering CAR‐encoding mRNA and PD‐1‐targeting small interference RNA (siRNA) to overcome PD‐1‐mediated CAR‐T cell resistance (Hamilton et al. 2023). In a recent report, C14‐4‐based LNP were surface‐conjugated with CD3 and CD28 antibody fragments to combine mRNA transfection and T cell activation and expansion in a single step. This method circumvents the need for magnetic beads to activate and expand T cells, reducing the number of steps required to generate a CAR‐T cell product, thereby accelerating and simplifying the CAR‐T cell manufacturing process (Metzloff et al. 2024).
Beyond LNP, other nanomaterials have been employed to generate mRNA‐CAR‐T cells ex vivo (Figure 4). Paul Wender's group developed a lipid‐polymer hybrid material termed charge‐altering releasable transporter (CART). This amphiphilic material comprises a lipophilic block linked to several side‐chain lipids and a polycationic α‐amino ester mRNA‐binding block (McKinlay et al. 2018). Using a CART with a beta‐amido carbonate backbone (bAC‐CART), they formulated CAR‐encoding mRNA‐loaded nanocarriers and demonstrated their enhanced T‐cell transfection ability and minimal cytotoxicity. Remarkably, bAC‐CART‐based nanocarriers delivering luciferase‐encoding mRNA cargo efficiently transfected T cells in vivo and displayed significant spleen tropism, suggesting their application toward generating CAR‐T cells in vivo (Z. Li, Amaya, et al. 2023b). Overall, these nanocarriers' excellent performance, scalability, and versatility can potentially give mRNA‐based CAR‐T cell technology an edge toward better, safer, and cheaper CAR‐T products.
Notwithstanding the advantageous properties of ex vivo CAR‐T cell generation with mRNA‐loaded nanocarriers, this process inherently requires specialized facilities as well as dedicated manufacturing and distribution chains to provide autologous CAR‐T cell products (Ayala Ceja et al. 2024). Due to its transient expression profile, mRNA‐based CAR‐T cell technology requires repeated administration to achieve therapeutic efficacy, greatly increasing the costs and logistical demands. Especially in these settings, a cheap, safe, instantaneous, and widely available (off‐the‐shelf) strategy, such as in vivo CAR‐T cell generation, is extremely desirable.
4.1.2. mRNA‐Loaded NP for In Vivo CAR‐T Cell Generation
When fully realized in a safe and cost‐effective clinical setting, in vivo CAR‐T cell generation will unleash a new era for synthetic immunotherapy. Once a groundbreaking therapy accessible only to a few cancer patients due to its costs and toxicity, CAR‐T cells will become a widely available immunotherapeutic option for a broader range of patients. This technology is recent and still in its early stages of clinical development, made possible by the multiple advances in vector engineering for nucleic acid delivery.
mRNA‐loaded nanocarrier‐mediated in vivo CAR‐T cell generation offers multiple advantages, reducing costs and complexity while improving safety and efficacy (Figure 4). The manufacturing processes for IVT mRNA and the NP are more scalable than those for viral vectors and cell manufacturing, producing batches with well‐defined physicochemical attributes (Short et al. 2024). By directly reprogramming T cells within the patient's body, the need for a lymphodepletion regimen is eliminated, thereby avoiding complications such as opportunistic infections (Lickefett et al. 2023). In this setting, an intact immune system may enhance the ability to mount a broader immune response against non‐CAR‐targeted tumor antigens (epitope spreading), partially overcoming the CAR‐target antigen escape and enhancing therapeutic efficacy (Klampatsa et al. 2020). Although transient mRNA‐CAR expression requires multiple dosing, mRNA‐loaded nanocarrier in vivo administration can rapidly generate CAR‐T cells and allow real‐time pharmacological control of the systemic CAR expression levels. Simple dosing adjustments make it feasible to fine‐tune CAR expression levels, minimizing CAR‐induced CAR‐T cell exhaustion and allowing prompt discontinuation of treatment if toxicity arises (Stock et al. 2023). Additionally, it is theoretically possible to adjust the nanocarrier features to deliver CAR‐encoding mRNA to T cells with distinct phenotypic states, such as tissue‐resident or tumor‐infiltrating T cells. However, the promise of in vivo CAR‐T generation can only be realized if the nanoplatforms used are biodegradable, biocompatible, and capable of specifically and efficiently delivering the mRNA payload to the desired immune cell subsets while avoiding unwanted off‐target transfection in cancer or immunosuppressive cells. Accordingly, multiple NP formulations have been developed with distinct features for T cell‐specific mRNA delivery (Figure 4). The Matthias Stephan Lab's work with PNP was one of the earliest reports demonstrating the feasibility of transfecting T cells in vivo with CAR‐encoding mRNA payload. Specifically, this nanoplatform comprises a biodegradable cationic PBAE‐447 polymer core complexed with mRNA, and an anionic polyglutamic acid (PGA) surface decorated with anti‐CD8 antibody fragments for CD8+ T cell‐targeting. Repeated dosing induced tumor regression at levels comparable to those achieved by repeated administration of ex vivo lentiviral‐transduced CAR‐T cells. Importantly, these nanocarriers were shown not to cause acute systemic toxicity (Parayath et al. 2020). Expanding on this targeting strategy, Billingsley et al. further developed the approach by attaching CD3‐ and CD7‐targeted antibody fragments to the previously discussed B10‐LNP, aiming to assess their ability to induce CAR‐T cell production in vivo. Both CD3‐ and CD7‐targeted B10‐LNP transiently generated significant numbers of CAR‐T cells, resulting in potent B cell depletion in vivo. Furthermore, these formulations exhibited a dose‐dependent effect on CAR expression and cytokine release. Noteworthy, these nanoplatforms have demonstrated efficient in vivo T cell transfection and enhanced delivery to the spleen, with minimal accumulation in the liver (Billingsley et al. 2024). A critical consideration is that NP active targeting requires an additional manufacturing step, limiting scalability. To address this challenge, other research groups have explored alternative approaches for efficient T‐cell delivery of CAR‐encoding mRNA without relying on antibody targeting. For example, Daniel Siegwart's group developed the Selective ORgan Targeting (SORT) nanoplatform, which employs an engineered lipid molecule (SORT lipid) enabling precise tissue‐specific delivery without the need for active targeting ligands. Using a spleen‐targeted SORT lipid termed 18:1 PA, these LNP efficiently induced mRNA‐CAR‐T cell production in vivo, demonstrating potent antitumor activity while not displaying detectable liver tropism (Álvarez‐Benedicto et al. 2023). Together, these nanocarriers greatly extend the capabilities of mRNA‐based CAR‐T cell strategies, paving the way for the next generation of CAR‐T cell therapies with improved efficacy, safety, accessibility, and flexibility.
4.2. Beyond CAR‐T Cells: CAR‐Natural Killer (CAR‐NK) and CAR‐Monocytes/Macrophages (CAR‐M)
The CAR‐T cell‐associated toxicities and the challenge of developing safe, universal allogenic CAR‐T cells prompted the exploration of other CAR‐engineered immune cell populations. In this context, CAR‐NK cells have attracted significant interest due to their unique advantages. While exhibiting a similar cytotoxic ability to CAR‐T cells, CAR‐NK cells do not cause severe CAR‐T cell‐associated adverse effects such as CRS and ICANS. Moreover, CAR‐NK cells do not require genetic modification to prevent graft‐versus‐host disease, making them a suitable source for allogeneic cell therapies (Raftery, Franzén, and Pecher 2023). Most CAR‐NK cells have been developed ex vivo using viral vectors or electroporation. To address the drawbacks of these gene delivery methods, recent reports have demonstrated the potential superiority of CAR‐encoding mRNA nanocarriers for generating CAR‐NK cells. Paul Wender's group found that mRNA‐loaded CART‐based NP achieve better ex vivo NK transfection efficiencies compared to electroporation, without negatively impacting CAR‐NK cell viability, phenotype, or function (Wilk et al. 2020). Similarly, Golubovskaya et al. and Shin et al. demonstrated that mRNA‐loaded LNP efficiently generate CAR‐NK cells ex vivo with potent antitumor activity (Golubovskaya, Sienkiewicz, Sun, Zhang, et al. 2023b; H. E. Shin et al. 2024). Owing to the multiple mRNA‐loaded nanoplatforms engineered for in vivo immune cell reprogramming, several biotech companies are already developing mRNA‐loaded nanocarriers for in vivo CAR‐NK cell generation, with reports expected to be published soon (Diwanji, Getts, and Wang 2024).
Despite the remarkable responses of CAR‐T cell therapies in certain hematological malignancies and the promising results obtained with CAR‐NK cells, replicating this success in solid tumors remains challenging. The highly desmoplastic and immunosuppressive TME, along with heterogeneous antigen expression and escape mechanisms, significantly undermines CAR‐T and CAR‐NK cell infiltration, persistence, and effector activity within the solid tumor's environment (Albelda 2024; W. Wang et al. 2024). To surmount these barriers, a recent approach consists of reprogramming monocytes/macrophages with CAR to redirect, leverage, and enhance their ability to infiltrate and reshape the TME, and phagocyte tumor cells, and perform antigen‐presentation (Klichinsky et al. 2020). However, CAR‐M have limited proliferation ability, requiring repeated infusions and increasing manufacturing complexity. Therefore, nanocarrier‐mediated mRNA delivery is particularly suitable for reducing the costs and complexity of CAR‐M therapy. Ye et al. successfully engineered LNP that efficiently generate CAR‐T cells and CAR‐M ex vivo, showing significant antitumor responses in B cell lymphoma models in vitro (Ye et al. 2022). Subsequent reports describe LNP formulations for in vivo CAR‐M generation. For hepatocellular carcinoma (HCC), Yang et al. formulated LNP co‐delivering a glypican 3 (GPC3)‐targeted CAR‐encoding mRNA and a mRNA encoding a version of the inhibitory receptor Siglec‐C lacking Immunoreceptor Tyrosine‐based Inhibition Motifs (ITIM) to act as a decoy receptor. These LNP successfully generated CAR‐M directly in vivo, demonstrating a robust antitumor response enhanced by resistance to tumor‐induced CD24‐mediated immunosuppression (Z. Yang, Liu, et al. 2023b). Mukalel et al. recently developed a novel class of oxidized LNP with inherent tropism toward monocytes/macrophages, identifying LNP formulated with the oxidized ionizable lipid C14‐O2 as efficient mRNA‐loaded nanoplatforms for in vivo CAR‐M generation (Mukalel et al. 2024). Interestingly, none of these nanoplatforms rely on active targeting strategies, simplifying their potential production at a clinical scale. These recent advancements highlight how nanocarrier versatility has enabled the expansion of mRNA‐based CAR technology to novel immunotherapeutic approaches and combinations, which would be otherwise impossible or hardly feasible with traditional mRNA delivery strategies.
5. Nanoparticle Engineering for Direct Oncolysis or Cell Reprogramming
For a long time, mRNA therapeutics were primarily explored as a source of tumor antigens for developing strong cancer vaccines. However, due to the versatility and safety of mRNA compared to plasmid DNA and viral vectors, which can integrate into the host cell genome and cause immunogenicity issues, its application has been extended to novel cancer immunotherapeutic strategies beyond tumor antigen vaccination. In addition to encoding cytokines, immune checkpoint mAbs, and CAR, mRNA has recently been explored as a trigger for direct tumor cell death and as a modulator of the dense network of malignant and nonmalignant immunosuppressive cells to reprogram the TME (Figure 5).
FIGURE 5.
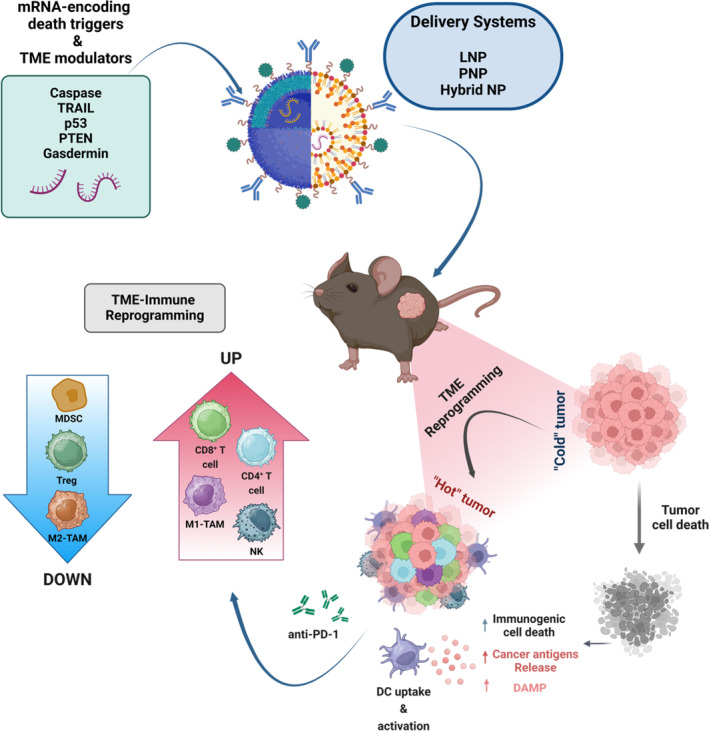
Schematic representation of mRNA‐loaded NP that prompts the synthesis of toxic intracellular proteins, forcing tumor cells to self‐destruction. Moreover, autophagy‐mediated tumor cell death can also trigger ICD, which promotes the release of cytokines and the secretion of TSA and DAMP, resulting in immune cell recruitment that contributes to TME reprogramming. The polarization of a “cold” and immunosuppressive TME to an immunogenic “hot” is mediated by (1) increased levels of tumor‐infiltrating antitumor cells, including CD8+ and CD4+ T, and NK cells; (2) TAM polarization toward the M1‐type antitumor phenotype; (3) enhanced expression of proinflammatory cytokines (TNF–α and IFN‐γ); and (4) downregulation of immunosuppression‐related Treg and MDSC levels. DAMP: damage‐associated molecular patterns; ICD: immunogenic cell death; IFN: interferon; MDSC: myeloid‐derived suppressor cells; mRNA: messenger ribonucleic acid; NK: natural killer; NP: nanoparticle; TAM: tumor‐associated macrophages; TME: tumor microenvironment; TNF: tumor necrosis factor; Treg: regulatory T cells; TSA: tumor‐specific antigens.
5.1. NP‐Based mRNA Delivery‐Mediated Direct Oncolysis
Despite the effective and stable transduction rate of commonly used viral gene transfer methods, recent improvements have highlighted the advantages of safe, poorly immunogenic, and low‐cost cationic and ionizable LNP as promising nonviral systems for mRNA delivery (Guevara, Persano, and Persano 2020). These mRNA‐loaded LNP have been used to induce tumor cells to self‐destruction by triggering the synthesis of toxic intracellular proteins. For instance, Nakashima et al. demonstrated the strong antitumoral activity of inducible caspase‐9 (iC9) mRNA‐loaded cationic LNP with chemical inducers of dimerization (CID) in HER2+ SKBR3 and triple‐negative breast cancer MDA‐MB231 breast cancer cells in vitro (Nakashima et al. 2022). iC9, a suicide gene, is activated through CID‐induced dimerization of the caspase‐9 protein fused with a modified human FK‐binding protein (Stasi et al. 2011). Efficient delivery and translation of the encapsulated iC9 mRNA led to the expression of apoptosis‐related genes, low levels of antiapoptotic protein B cell lymphoma 2 (Bcl‐2), high Bcl‐2‐associated X protein (BAX)/Bcl‐2 ratios, and the activation of downstream caspase‐9 and caspase‐3/7, resulting in cell apoptosis. This makes cancer gene therapy a promising alternative for treating breast cancers (Nakashima et al. 2022). However, the authors did not evaluate the safety and efficacy of the iC9‐LNP treatment in vivo.
Building on knowledge acquired during the COVID‐19 pandemic regarding SARS‐CoV‐2's binding affinity to angiotensin‐converting enzyme 2 (ACE2) receptors, Gu et al. developed an LNP system to deliver mRNA encoding TNF‐related apoptosis‐inducing ligand (TRAIL) combined with the SARS‐CoV‐2 spike receptor‐binding domain (RBD) to colon cancer tissues with high ACE2 receptor expression levels (Gu et al. 2024). TRAIL ligand binds to death receptors 4 and 5, triggering caspase activation that leads to programmed cell death. By combining the delivery of RBD for specific tumor cell targeting via ACE2 receptors with the apoptosis‐inducing TRAIL, the intratumoral injection of RBD‐TRAIL mRNA‐loaded LNP significantly induced targeted apoptosis of ACE2‐overexpressing cancer cells, resulting in a robust tumor growth inhibition in a colon cancer patient‐derived xenograft mouse model (Gu et al. 2024). Although promising, the RBD‐TRAIL therapy raises concerns related to potential human immunity against SARS‐CoV‐2 constituents and the need for models that mimic complex human antitumor immunity, thus highlighting the need for further clinical evaluation of its safety and efficacy. Following a similar strategy, da Silva et al. explored combining physical TME normalization with improved delivery of TRAIL mRNA to induce targeted tumor cell apoptosis (da Silva et al. 2024). Combining intratumoral administration of ionizable LNP developed to deliver TRAIL mRNA (LNP‐TRAIL) with TME normalization using Losartan or angiotensin 1–7 to increase vascular perfusion and reduce extracellular matrix deposition, resulted in increased levels of tumor‐infiltrating CD4+ and CD8+ T cell levels and caspase‐3‐based tumor cell death. This approach demonstrated strong inhibition of colon cancer growth in humanized mice (da Silva et al. 2024). Intratumoral mRNA delivery allows local treatment with high doses and low systemic exposure, potentially resulting in advanced responses in tumors poorly responsive to systemic ICI, as explored in an ongoing phase 1 trial (NCT03739931) (Hewitt et al. 2019).
5.2. NP‐Based mRNA Delivery‐Mediated TME Reprogramming and Its Combination With Direct Tumor Cell Death
Beyond cancer cells, stromal and immune cells that constitute the TME complex network are crucial modulators of the immunosuppressive environment, contributing to tumor progression, angiogenesis, and metastasis. Targeting these cells for TME reprogramming toward an antitumor phenotype has been explored as an added value for cancer treatment, as described below (Figure 5). Generally, the dysregulation of apoptotic mechanisms, including the upregulation of antiapoptotic proteins and inactivation of tumor suppressors, is associated with poor anticancer responses or treatment resistance. For instance, the p53 tumor suppressor gene, a strong regulator of apoptosis, also impacts the tumor‐immune cell interactions within the TME. It promotes antitumor immunity through the activation of APC and NK cells, in addition to suppressing protumorigenic M2‐type tumor‐associated macrophages (TAM) (L. Li et al. 2015; Sharma et al. 2018). However, p53 is one of the most frequently altered genes in several human cancers. Loss of p53 function is associated with tumorigenesis, immunosuppression, and immunotherapy resistance, and has been reported in approximately 34% of HCC cases (Ng et al. 2022) and 56% of NSCLC cases according to The Cancer Genome Atlas database in the cBio Cancer Genomics Portal (http://cbioportal.org).
Kong et al. demonstrated the importance of using p53‐encoding mRNA‐loaded redox‐responsive lipid‐polymer hybrid NP to restore p53 expression. This not only induced apoptosis‐mediated tumor growth inhibition but also sensitized p53‐deficient HCC and NSCLC to the mammalian target of rapamycin (mTOR) inhibitor, everolimus, which had previously failed in clinical trials for these advanced tumors (N. Kong et al. 2019). Subsequently, the same research group, led by Jinjun Shi, optimized a CXCR4‐targeted lipid‐polymer hybrid NP designed to restore p53 expression in p53‐null murine HCC models and reprogram the TME (Xiao et al. 2022). When combined with PD‐1 checkpoint blockade, the p53 mRNA‐loaded nanoplatform, functionalized with the CTCE ligand for CXCR4‐expressing HCC targeting, strongly induced global reprogramming of the immune cellular and molecular elements within the TME. This resulted in effective HCC growth inhibition and prolonged survival of mice bearing p53‐deficient intrahepatic/ectopic HCC (Xiao et al. 2022). This combinatorial strategy offers hope for patients suffering from the most prevalent and lethal liver cancer, who generally do not respond to immune checkpoint blockade treatment alone or in combination with other therapies, including anti‐vascular‐endothelial growth factor therapy.
In another approach, Zhang et al. designed targeted polymeric NP comprising PBAE, PGA, and di‐mannose moieties loaded with mRNAs encoding for the co‐expression of interferon regulatory factor 5 (IRF5) and its activating kinase IKKβ (IRF5/IKKβ‐encoding NP). This reprogrammed pro‐tumoral TAM function toward a potent pro‐inflammatory and cytotoxic M1 phenotype (F. Zhang, Parayath, et al. 2019b). The therapeutic efficacy of IRF5/IKKβ‐encoding NP was confirmed through immunosuppression reversion, TAM polarization to an antitumor phenotype, and reduction of tumor progression or clearance in metastatic melanoma, glioblastoma, and ovarian cancer advanced‐stage models, without triggering systemic toxic effects that disturb immune homeostasis (F. Zhang, Parayath, et al. 2019b). In addition, a phase 1 clinical trial of this nano‐based technology as an intraperitoneal monotherapy is expected to treat patients with ovarian cancer at Fred Hutchinson Cancer Research Center (F. Zhang, Parayath, et al. 2019b).
Furthermore, the delivery of mRNA encoding factors that combine both tumor cell death and TME reprogramming has also been described. Jinjun Shi's group evidenced that recovering the expression of the tumor suppressor gene phosphatase and tensin homolog (PTEN) using a hybrid nanoplatform for systemic PTEN mRNA delivery resulted in remarkably apoptotic‐mediated prostate cancer cell growth inhibition both in vitro and in vivo (Islam et al. 2018). PTEN expression was shown to trigger autophagy, whereas its deletion or function loss in cancer cells is related to the inhibition of cell death and resultant development and progression of different cancer types, including glioblastomas, melanomas, endometrial, prostate, colon, bladder, lung, and breast cancers (Aquila et al. 2020). PTEN genetic alterations also promote TME remodeling, correlating with an immunosuppressive network and a poor response to ICI therapy (Aquila et al. 2020). Autophagy‐mediated tumor cell death can promote the secretion of damage‐associated molecular patterns (DAMP), which act as strong adjuvants that trigger antitumor immune activation. Shi and co‐workers also explored the ability of PTEN restoration to trigger cell death‐related immune activation via DAMP release. The systemic administration of PTEN mRNA‐loaded PEG‐PLGA‐based NP (mPTEN@NP) in B16F10‐bearing mice not only reactivated the PTEN expression and induced tumor cell death but also triggered the DAMP secretion. This contributed to TME reprogramming by promoting higher levels of tumor‐infiltrating CD8+ T cells, enhancing the expression of proinflammatory cytokines (IL‐12, TNF–α, and IFN‐γ), and downregulating immunosuppression‐related Treg and myeloid‐derived suppressor cell in vivo compared to saline‐ and empty NP‐treated groups (Lin et al. 2021). PTEN restoration by the mPTEN@NP sensitized Pten‐mutated melanoma B16F10 and Pten‐null prostate tumors to anti‐PD‐1 immunotherapy, demonstrating remarkable tumor growth control and improved survival compared with monotherapies (Lin et al. 2021).
Li et al. developed ionizable cationic LNP loaded with gasdermin N‐terminal domain‐encoding mRNA (GSDMBNT mRNA@LNP) for cancer therapy by inducing tumor cell pyroptosis (F. Li, Zhang, et al. 2023a). Pyroptosis, described as gasdermin protein cleavage‐mediated programmed cell death, can also trigger immunogenic cell death (ICD), which induces the release of cytokines and immune cell recruitment, polarizing the cold and immunosuppressive TME to an immunogenic hot state. The expression of gasdermin is suppressed or even silenced in different cancers (Z. Zhang et al. 2020; Zhou et al. 2020). GSDMBNT mRNA@LNP administered intratumorally exerted antitumor effects by controlling tumor growth and prolonging overall survival in highly aggressive and poorly immunogenic 4T1‐ and B16F10‐bearing mice models (F. Li, Zhang, et al. 2023a). Even with low levels of cancer cell pyroptosis, GSDMBNT mRNA@LNP triggered the secretion of proinflammatory cytokines (including, TNF‐α, IFN‐γ, IL‐1β, and IL‐18) and improved tumor‐infiltrating CD4+, CD8+, and NKT cells, creating a positive feedback loop to endorse robust antitumor immune responses. This antitumoral effect of pyroptosis‐triggering GSDMBNT mRNA@LNP was clearly enhanced when combined with anti‐PD‐1 immunotherapy, leading to the eradication of large melanomas and inhibiting the growth of distant B16F10 tumors (F. Li, Zhang, et al. 2023a). Compared to chemotherapy and thermotherapy, this clinical setting can take advantage of the strong antitumor effect with low levels of gasdermin‐mediated pyroptosis, allowing for decreased high‐dose toxicity and preventing off‐target side effects. Wang et al. demonstrated the abolishment of 4T1 mammary tumor grafts with less than 15% of tumor cell pyroptosis‐triggering GSDMA3(N + C)‐loaded NP (Q. Wang et al. 2020).
Finally, a dual‐targeted mRNA‐based inhaled advanced strategy was also recently explored to promote the expression of target proteins in the lung to treat lung‐related diseases, including cancer (Tang et al. 2023). After inhalation, these stable cationic lipid (1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine (DSPE)‐PEG‐Mannose) and hyaluronic acid–based NP accumulated in both lung tumor cells and inflammatory macrophages by targeting CD44 proteins and glucose transporters, effectively transfecting p53, EGFR and imaging (luciferase and green fluorescence protein) proteins into lung tissues in vivo (Tang et al. 2023). Considering the lung as a common metastatic site for several cancers, this dual‐targeted approach can also be used to tackle pulmonary cancer metastases.
These results reinforce the promising potential of mRNA‐loaded nano‐based therapies with excellent targeting capabilities to trigger an efficient transfection of proteins involved in direct tumor cell death and TME polarization from a cold to a hot state. A recent approach by Asgard Therapeutics aims to reprogram tumor cells into type 1 conventional DC (DC1) through the delivery of several transcription factors. Specifically, the transcription factors PU.1, IRF8, and BATF3 successfully reprogrammed tumor cells into cDC1 in vitro, eliminating their tumorigenicity. These tumor‐derived cDC1 exerted potent in vivo activity when delivered intratumorally, underscoring their potential to induce TME polarization and long‐term antitumor immune responses (Zimmermannova et al. 2023). Using nanocarriers delivering mRNA encoding PU.1, IRF8, and BATF3, these cells could be directly generated in vivo, making this strategy readily deployable at a clinical scale to greatly improve the efficacy of current immunotherapies (Zimmermannova, Ferreira, and Pereira 2024).
6. Conclusion
mRNA technology has dramatically expanded the frontiers of protein‐based therapies. With its remarkable versatility to encode any protein of interest for extracellular or intracellular targeting, novel proteins can be designed and manufactured programmatically. However, the clinical potential of mRNA was historically hindered by its low in vivo stability, poor cellular uptake, high immunogenicity, and a limited range of available transfection methods. The emergence of innovative nanoplatforms solving these issues and the convergence between these technologies has enabled the delivery of customizable mRNA molecules to specific organs and cells. This flexibility has thus positioned mRNA‐loaded nanocarriers as biotechnological Swiss Army knives for any imaginable therapeutic protein expression application. With the historic approval and widespread rollout of clinically safe and effective mRNA vaccines against SARS‐CoV‐2, this technology is poised to become a novel platform for a broad range of indications in the foreseeable future.
In this overview, we discussed how mRNA‐loaded nanocarriers have been successfully employed in cancer immunotherapy to deliver various immunomodulators, including cytokines, antibodies, cancer antigens, synthetic receptors, transcription factors, apoptotic proteins, and oncolytic RNA viral genomes. With mRNA‐loaded nanocarriers, cytokines, antibodies, and oncolytic modulators can be expressed for a prolonged time and/or produced intratumorally to avoid their systemic effects while precisely remodeling the TME. In cancer vaccination, mRNA can simultaneously encode personalized TAA and TSA and be specifically delivered in vivo by APC‐targeted nanocarriers to enhance antigen presentation and T‐cell responses. Synthetic biology approaches like CAR‐engineered immune cells benefit from CAR‐encoding mRNA‐loaded nanocarriers, mediating ex vivo and in vivo immune cell reprogramming with lower costs, reduced manufacturing constraints, and better control over safety and efficacy profiles compared to traditional methods. From a broader perspective, it becomes evident that the versatility of mRNA‐loaded nanocarriers provides significant advantages in surmounting various limitations of each immunotherapeutic approach. By precisely controlling the location and duration of mRNA expression and the degree of immunogenicity required for each application, formulations can be screened and adjusted for their NP composition, hydrophobicity, size, charge, and targeting moieties (e.g., mannose receptor targeting for APC delivery, CD3 targeting for T cell delivery).
mRNA‐based nanoformulations can be further integrated with conventional treatments such as chemotherapy and radiotherapy to optimize clinical outcomes. In the context of cancer vaccination, the cytotoxic effects of these regimens temporarily reduce the tumor burden, thereby prolonging the time available for the vaccine to elicit a potent antitumor response. Notably, chemotherapy and radiotherapy can also synergize with immunotherapies by exerting several immunostimulatory mechanisms, including immunogenic cell death, depletion of immunosuppressive cell subsets (e.g., MDSC, M2‐type TAM, and Treg cells), DC maturation, and effector T‐cell activation and proliferation (Hader et al. 2020; Kerr et al. 2021). Despite the immunomodulatory benefits observed when combined with non‐mRNA vaccines, mRNA vaccine‐based combinations with chemotherapy and radiotherapy have not consistently demonstrated clear benefits compared to immunotherapy alone in clinical trials. These mixed outcomes highlight design differences between clinical trials (e.g., vaccination, chemotherapy and radiotherapy schedules, and tumor types), and underscore the need to carefully balance the immunostimulatory benefits of chemotherapy and radiotherapy with their cytotoxicity‐induced immunosuppression to achieve synergistic responses (Sayour et al. 2024). To enhance the effectiveness and safety profile of chemotherapy and radiotherapy, a promising strategy involves co‐delivering cytotoxic agents within mRNA‐loaded nanocarriers. This approach harnesses the antitumor activity of mRNA‐encoded immunotherapeutic molecules to overcome therapy resistance, while the tumor‐targeted nanocarrier enhances tumor accumulation and minimizes toxicity to healthy tissues. For instance, Zhang et al. developed a synergistic combination of chemotherapy and direct oncolysis using LNP containing paclitaxel‐conjugated amino lipids and mRNA‐encoded P53. These nanoparticles achieved superior in vitro and in vivo antitumor efficacy in TNBC models and demonstrated a higher paclitaxel loading capacity than clinically used formulations such as Abraxane and Lipusu (C. Zhang, Zhang, et al. 2019a).
Despite the rapid pace of mRNA‐based nanoimmunotherapy development, several clinical translation challenges persist. While the self‐adjuvant effect of mRNA and NP can enhance vaccination‐elicited responses, it may negatively impact the performance and safety of mRNA‐encoded cytokines, antibodies, and synthetic receptors, particularly after repeated infusions (Rohner et al. 2022). Regardless of the application, it is crucial to understand the immunological effects of mRNA‐loaded nanocarriers, their route of administration, and to properly balance immune stimulation without potentially triggering adverse effects such as immunoglobulin E‐mediated allergy, complement‐associated pseudoallergy, and autoimmune reactions (Y. Lee et al. 2023). Despite the improvements achieved in preclinical models for extrahepatic NP delivery, our knowledge of the optimal NP properties for specific human organ/cell targeting remains very limited. Therefore, it is advisable to diversify the mRNA‐based nanoplatform development pipeline with various nanomaterials and properties while thoroughly assessing their pharmacokinetics, biodistribution, and metabolic and immunological effects. Concerning their clinical efficacy, mRNA‐based therapies are yet to overcome solid tumor immunosuppression and heterogeneity. Accordingly, the recent progress with personalized mRNA‐LNP neoantigen vaccines underscores the need to actively consider intertumoral and intratumoral antigen expression heterogeneity for more effective immunotherapies. Importantly, achieving groundbreaking efficacies in solid tumors will require combining multiple immunotherapeutic strategies aimed at tackling immunosuppression and re‐engaging the entire antitumor immunity cycle, as demonstrated by the combination of mRNA‐LNP neoantigen vaccines with ICI, and claudin 6‐specific CAR‐T cells with claudin 6‐encoding mRNA‐LNP vaccines (Mackensen et al. 2023; Sayour et al. 2024). While mRNA‐based formulations can be rapidly and cheaply produced, most require a cold distribution chain that substantially increases costs and logistical demands. Solving this issue involves prioritizing shelf‐life stability during the development cycle by selecting the most stable formulations under optimal storage conditions or employing lyophilization to improve stability at higher temperatures.
Considering the progressive development of optimal mRNA‐loaded nanocarrier properties for cancer treatment advances, we envision that future mRNA‐based immunotherapeutic strategies will be able to outpace tumor evolution and immune escape more effectively. Advances in artificial intelligence and machine learning, particularly in silico receptor and de novo antibody structure prediction algorithms, might expand personalized neoantigen‐based approaches to develop neoantigen‐targeted synthetic receptors, as showcased by the first‐in‐human trial of personalized, computationally predicted neoantigen‐specific TCR therapy (Foy et al. 2023; X. Yang, Nishimiya, et al. 2023a). Compared to personalized mRNA‐LNP neoantigen vaccines, CAR‐encoded mRNA‐loaded nanocarriers could engineer immune cells with personalized CAR constructs and combinations, creating truly personalized cell therapies. The transient CAR expression and pharmacological control from in vivo nanocarrier‐mediated CAR engineering could reduce the risks of using untested in silico‐predicted CAR constructs in clinical settings.
Another promising topic for established extrahepatic delivery nanoplatforms is in vivo mRNA‐encoded immunomodulator delivery to other cell populations involved in tumor progression and survival, especially B cells, innate‐like T cells—γδ‐T cells, invariant NKT (iNKT) cells, and mucosal‐associated invariant T (MAIT) cells, and nonimmune cells (cancer‐associated fibroblasts (CAF), endothelial cells (EC) and mesenchymal stem cells). For instance, CAR‐engineered iNKT, γδ‐T cells, and MAIT cells have already been shown to improve solid antitumor responses and could be directly modified in vivo (Neo et al. 2023). Noteworthy, engineering B cells with synthetic constructs like CAR and antibodies has recently garnered significant attention for its ability to harness their innate and adaptive functions, including antigen presentation, antibody production, and differentiation into long‐lived memory B cells. In this context, mRNA‐based CAR‐B cells could exhibit superior features for inducing both short‐term and long‐term antitumor responses by stimulating direct but transient cytotoxic activity, neoantigen presentation, and B cell activation and proliferation for antibody production and tertiary lymphoid structure formation within tumors (Boyle et al. 2024; Pesch et al. 2019). Although B cells have traditionally been challenging to transfect, novel mRNA‐loaded nonviral gene delivery vectors have achieved efficient in vivo B cell transfection rates, laying the groundwork for mRNA‐based in vivo B cell engineering applications (Cheng et al. 2020). CAF‐ and EC‐targeted NP initially developed for chemotherapeutic drug and siRNA delivery could be reengineered to carry mRNA‐encoding immunomodulators for tumor stroma remodeling and tumor vasculature normalization (Q. Huang et al. 2024; G. W. Liu et al. 2023). To conclude, since mRNA does not require nuclear delivery, mRNA‐loaded nanocarriers can co‐deliver mRNAs encoding transgenes and/or gene editors for long‐term gene expression applications. For example, mRNA‐encoded target‐primed retrotransposon systems in LNP were shown to successfully generate CAR‐T cells without relying on DNA templates (Magee et al. 2023). Even when using transgene‐encoded DNA templates, co‐delivering mRNA‐encoded gene editors like transposases and nucleases can reduce genotoxicity by temporally restricting their expression to the necessary timeframe for their function (Popovitz et al. 2023).
Author Contributions
Henrique M. B. Carvalho: conceptualization (lead), data curation (lead), formal analysis (lead), writing – original draft (lead), writing – review and editing (equal). Tiago A. S. Fidalgo: data curation (supporting), formal analysis (supporting), writing – original draft (supporting). Rita C. Acúrcio: data curation (supporting), formal analysis (supporting), writing – original draft (supporting), writing – review and editing (supporting). Ana I. Matos: conceptualization (lead), data curation (lead), formal analysis (lead), supervision (equal), writing – original draft (lead), writing – review and editing (lead). Ronit Satchi‐Fainaro: conceptualization (lead), funding acquisition (lead), validation (lead), writing – review and editing (lead). Helena F. Florindo: conceptualization (lead), data curation (lead), funding acquisition (lead), project administration (lead), supervision (lead), validation (lead), writing – review and editing (lead).
Conflicts of Interest
Ronit Satchi‐Fainaro is a Board Director at Teva Pharmaceutical Industries Ltd. All the other authors declare no conflicts of interest.
Related Wires Articles
Funding: Ronit Satchi‐Fainaro and Helena F. Florindo thank the following funding agencies for their generous support: The Fundação para a Ciência e Tecnologia‐Ministério da Ciência, Tecnologia e Ensino Superior (FCT‐MCTES) under the framework of the projects UIDB/04138/2020, UIDP/04138/2020; “la Caixa” Foundation under the framework of the Healthcare Research call 2019 (LCF/PR/HR19/52160021; NanoPanther), 2022 (LCF/PR/HR22/52420016; MultiNano@BBM), and 2024 (LCF/PR/HR24/00968; PINT); and CaixaImpulse LCF/TR/CD20/52700005 (Co‐Vax). In addition, Ronit Satchi‐Fainaro thanks to the generous financial support from the European Research Council (ERC), PoC Grant Agreement no. 591187—ImmuNovation and ERC Advanced Grant Agreement no. (835227)—3DBrainStrom, the Israel Science Foundation (3706/24), the Melanoma Research Alliance (Established Investigator Award no. 615808), the Israel Cancer Research Fund (ICRF) Professorship award (no. PROF‐18‐682), the Morris Kahn Foundation.
Associate Editor: Olivia Merkel
Co‐Editor‐in‐Chief: Fabiana Quaglia
Contributor Information
Ana I. Matos, Email: aimatos@ff.ulisboa.pt.
Ronit Satchi‐Fainaro, Email: ronitsf@tauex.tau.ac.il.
Helena F. Florindo, Email: hflorindo@ff.ulisboa.pt.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- Acúrcio, R. C. , Kleiner R., Vaskovich‐Koubi D., et al. 2024. “Intranasal Multiepitope PD‐L1‐siRNA‐Based Nanovaccine: The Next‐Gen COVID‐19 Immunotherapy.” Advanced Science 11: 2404159. 10.1002/advs.202404159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc, A. , Querbes W., De S., et al. 2010. “Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand‐Based Mechanisms.” Molecular Therapy 18, no. 7: 1357–1364. 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albelda, S. M. 2024. “CAR T Cell Therapy for Patients With Solid Tumours: Key Lessons to Learn and Unlearn.” Nature Reviews Clinical Oncology 21, no. 1: 47–66. 10.1038/s41571-023-00832-4. [DOI] [PubMed] [Google Scholar]
- Álvarez‐Benedicto, E. , Tian Z., Chatterjee S., et al. 2023. “Spleen SORT LNP Generated In Situ CAR T Cells Extend Survival in a Mouse Model of Lymphoreplete B Cell Lymphoma.” Angewandte Chemie International Edition 62, no. 44: e202310395. 10.1002/anie.202310395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, E. J. , Rouphael N. G., Widge A. T., et al. 2020. “Safety and Immunogenicity of SARS‐CoV‐2 mRNA‐1273 Vaccine in Older Adults.” New England Journal of Medicine 383, no. 25: 2427–2438. 10.1056/NEJMoa2028436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquila, S. , Santoro M., Caputo A., Panno M. L., Pezzi V., and De Amicis F.. 2020. “The Tumor Suppressor PTEN as Molecular Switch Node Regulating Cell Metabolism and Autophagy. Implications in Immune System and Tumor Microenvironment.” Cells 9, no. 7: 7. 10.3390/cells9071725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin, R. J. , Lemon B. D., Aaron W. H., et al. 2021. “TriTACs: A Novel Class of T‐Cell–Engaging Protein Constructs Designed for the Treatment of Solid Tumors.” Molecular Cancer Therapeutics 20, no. 1: 109–120. 10.1158/1535-7163.MCT-20-0061. [DOI] [PubMed] [Google Scholar]
- Ayala Ceja, M. , Khericha M., Harris C. M., Puig‐Saus C., and Chen Y. Y.. 2024. “CAR‐T Cell Manufacturing: Major Process Parameters and Next‐Generation Strategies.” Journal of Experimental Medicine 221, no. 2: e20230903. 10.1084/jem.20230903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, S. R. , Berger T. R., Graham C., Larson R. C., and Maus M. V.. 2023. “Four Challenges to CAR T Cells Breaking the Glass Ceiling.” European Journal of Immunology 53, no. 11: 2250039. 10.1002/eji.202250039. [DOI] [PubMed] [Google Scholar]
- Banerjee, R. , Poh C., Hirayama A. V., et al. 2024. “Answering the “Doctor, Can CAR‐T Therapy Cause Cancer?” Question in Clinic.” Blood Advances 8, no. 4: 895–898. 10.1182/bloodadvances.2023012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier, A. J. , Jiang A. Y., Zhang P., Wooster R., and Anderson D. G.. 2022. “The Clinical Progress of mRNA Vaccines and Immunotherapies.” Nature Biotechnology 40, no. 6: 840–854. 10.1038/s41587-022-01294-2. [DOI] [PubMed] [Google Scholar]
- Barot, S. , Patel H., Yadav A., and Ban I.. 2023. “Recent Advancement in Targeted Therapy and Role of Emerging Technologies to Treat Cancer.” Medical Oncology 40, no. 11: 324. 10.1007/s12032-023-02184-6. [DOI] [PubMed] [Google Scholar]
- Beck, J. D. , Diken M., Suchan M., et al. 2024. “Long‐Lasting mRNA‐Encoded Interleukin‐2 Restores CD8+ T Cell Neoantigen Immunity in MHC Class I‐Deficient Cancers.” Cancer Cell 42, no. 4: 568–582.e11. 10.1016/j.ccell.2024.02.013. [DOI] [PubMed] [Google Scholar]
- Beck, J. D. , Reidenbach D., Salomon N., et al. 2021. “mRNA Therapeutics in Cancer Immunotherapy.” Molecular Cancer 20, no. 1: 69. 10.1186/s12943-021-01348-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley, M. M. , Gong N., Mukalel A. J., et al. 2024. “In Vivo mRNA CAR T Cell Engineering via Targeted Ionizable Lipid Nanoparticles With Extrahepatic Tropism.” Small 20, no. 11: 2304378. 10.1002/smll.202304378. [DOI] [PubMed] [Google Scholar]
- Billingsley, M. M. , Hamilton A. G., Mai D., et al. 2022. “Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for mRNA Engineering of CAR T Cells.” Nano Letters 22, no. 1: 533–542. 10.1021/acs.nanolett.1c02503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley, M. M. , Singh N., Ravikumar P., Zhang R., June C. H., and Mitchell M. J.. 2020. “Ionizable Lipid Nanoparticle‐Mediated mRNA Delivery for Human CAR T Cell Engineering.” Nano Letters 20, no. 3: 1578–1589. 10.1021/acs.nanolett.9b04246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczkowski, D. , Nair S. K., Snyder D., and Gilboa E.. 1996. “Dendritic Cells Pulsed With RNA Are Potent Antigen‐Presenting Cells In Vitro and In Vivo.” Journal of Experimental Medicine 184, no. 2: 465–472. 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornewasser, L. , Domnick C., and Kath‐Schorr S.. 2022. “Stronger Together for In‐Cell Translation: Natural and Unnatural Base Modified mRNA.” Chemical Science 13, no. 17: 4753–4761. 10.1039/D2SC00670G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle, K. , Park H., Kothakota S., Selby M., Brennan T., and Williams L. T.. 2024. “Modified B Cells and Methods of Use Thereof (United States Patent US11913023B2).” https://patents.google.com/patent/US11913023B2/en.
- Caimi, P. , Reese J. S., Otegbeye F., et al. 2020. “On Site Manufacture of AntiCD19 CAR‐T Cells. Responses in Subjects With Rapidly Progressive Refractory Lymphomas.” Biology of Blood and Marrow Transplantation 26, no. 3: S234–S235. 10.1016/j.bbmt.2019.12.478. [DOI] [Google Scholar]
- Cappell, K. M. , and Kochenderfer J. N.. 2023. “Long‐Term Outcomes Following CAR T Cell Therapy: What We Know So Far.” Nature Reviews Clinical Oncology 20, no. 6: 359–371. 10.1038/s41571-023-00754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, A. , Haley R. M., Najar M. A., et al. 2024. “Lipid‐Mediated Intracellular Delivery of Recombinant bioPROTACs for the Rapid Degradation of Undruggable Proteins.” Nature Communications 15, no. 1: 5808. 10.1038/s41467-024-50235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, S. C. , Gopal P., Lim S., et al. 2022. “Targeted Degradation of PCNA Outperforms Stoichiometric Inhibition to Result in Programed Cell Death.” Cell Chemical Biology 29, no. 11: 1601–1615.e7. 10.1016/j.chembiol.2022.10.005. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Ye Z., Huang C., et al. 2022. “Lipid Nanoparticle‐Mediated Lymph Node–Targeting Delivery of mRNA Cancer Vaccine Elicits Robust CD8+ T Cell Response.” Proceedings of the National Academy of Sciences 119, no. 34: e2207841119. 10.1073/pnas.2207841119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Q. , Wei T., Farbiak L., Johnson L. T., Dilliard S. A., and Siegwart D. J.. 2020. “Selective Organ Targeting (SORT) Nanoparticles for Tissue‐Specific mRNA Delivery and CRISPR–Cas Gene Editing.” Nature Nanotechnology 15, no. 4: 313–320. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirella, A. , Bolaños E., Luri‐Rey C., et al. 2023. “Intratumoral Immunotherapy With mRNAs Encoding Chimeric Protein Constructs Encompassing IL‐12, CD137 Agonists, and TGF‐β Antagonists.” Molecular Therapy ‐ Nucleic Acids 33: 668–682. 10.1016/j.omtn.2023.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente, B. , Denis M., Silveira C. P., Schiavetti F., Brazzoli M., and Stranges D.. 2023. “Straight to the Point: Targeted mRNA‐Delivery to Immune Cells for Improved Vaccine Design.” Frontiers in Immunology 14: 1294929. 10.3389/fimmu.2023.1294929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff, E. R. S. , Kelkar A. H., Russler‐Germain D. A., et al. 2023. “High Cost of Chimeric Antigen Receptor T‐Cells: Challenges and Solutions.” American Society of Clinical Oncology Educational Book 43: e397912. 10.1200/EDBK_397912. [DOI] [PubMed] [Google Scholar]
- Cobb, M. 2015. “Who Discovered Messenger RNA?” Current Biology 25, no. 13: R526–R532. 10.1016/j.cub.2015.05.032. [DOI] [PubMed] [Google Scholar]
- Conniot, J. , Scomparin A., Peres C., et al. 2019. “Immunization With Mannosylated Nanovaccines and Inhibition of the Immune‐Suppressing Microenvironment Sensitizes Melanoma to Immune Checkpoint Modulators.” Nature Nanotechnology 14, no. 9: 891–901. 10.1038/s41565-019-0512-0. [DOI] [PubMed] [Google Scholar]
- Conry, R. M. , LoBuglio A. F., Wright M., et al. 1995. “Characterization of a Messenger RNA Polynucleotide Vaccine Vector.” Cancer Research 55, no. 7: 1397–1400. [PubMed] [Google Scholar]
- Cornebise, M. , Narayanan E., Xia Y., et al. 2022. “Discovery of a Novel Amino Lipid That Improves Lipid Nanoparticle Performance Through Specific Interactions With mRNA.” Advanced Functional Materials 32, no. 8: 2106727. 10.1002/adfm.202106727. [DOI] [Google Scholar]
- Cornetta, K. , Lin T.‐Y., Pellin D., and Kohn D. B.. 2023. “Meeting FDA Guidance Recommendations for Replication‐Competent Virus and Insertional Oncogenesis Testing.” Molecular Therapy ‐ Methods & Clinical Development 28: 28–39. 10.1016/j.omtm.2022.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva, W. N. , Costa P. A. C., Júnior S. R. A. S., et al. 2024. “Ionizable Lipid Nanoparticle‐Mediated TRAIL mRNA Delivery in the Tumor Microenvironment to Inhibit Colon Cancer Progression.” International Journal of Nanomedicine 19: 2655–2673. 10.2147/IJN.S452896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvin, P. , Toor S. M., Sasidharan Nair V., and Elkord E.. 2018. “Immune Checkpoint Inhibitors: Recent Progress and Potential Biomarkers.” Experimental & Molecular Medicine 50, no. 12: 1–11. 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal, C. E. , Carfi A., and Plante O. J.. 2021. “Advancements in mRNA Encoded Antibodies for Passive Immunotherapy.” Vaccine 9, no. 2: 2. 10.3390/vaccines9020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Paggio, J. 2018. “Cancer Immunotherapy and the Value of Cure.” Nature Reviews Clinical Oncology 15, no. 5: 268–270. 10.1038/nrclinonc.2018.27. [DOI] [PubMed] [Google Scholar]
- Deshpande, P. P. , Biswas S., and Torchilin V. P.. 2013. “Current Trends in the Use of Liposomes for Tumor Targeting.” Nanomedicine 8, no. 9: 1509–1528. 10.2217/nnm.13.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Trani, C. A. , Cirella A., Arrizabalaga L., et al. 2023. “Intratumoral Injection of IL‐12‐Encoding mRNA Targeted to CSF1R and PD‐L1 Exerts Potent Anti‐Tumor Effects Without Substantial Systemic Exposure.” Molecular Therapy ‐ Nucleic Acids 33: 599–616. 10.1016/j.omtn.2023.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diken, M. , Kreiter S., Selmi A., et al. 2011. “Selective Uptake of Naked Vaccine RNA by Dendritic Cells Is Driven by Macropinocytosis and Abrogated Upon DC Maturation.” Gene Therapy 18, no. 7: 702–708. 10.1038/gt.2011.17. [DOI] [PubMed] [Google Scholar]
- Diwanji, N. , Getts D., and Wang Y.. 2024. “Chimeric Antigen Cytotoxic Receptors for In Vivo Engineering of Tumor‐Targeting NK Cells.” ImmunoHorizons 8, no. 1: 97–105. 10.4049/immunohorizons.2300099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, S. , Liu X., Bi Y., et al. 2023. “Adaptive Design of mRNA‐Loaded Extracellular Vesicles for Targeted Immunotherapy of Cancer.” Nature Communications 14, no. 1: 6610. 10.1038/s41467-023-42365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton, O. S. , Kauffman K. J., Kaczmarek J. C., et al. 2017. “Synthesis and Biological Evaluation of Ionizable Lipid Materials for the In Vivo Delivery of Messenger RNA to B Lymphocytes.” Advanced Materials 29, no. 33: 1606944. 10.1002/adma.201606944. [DOI] [PubMed] [Google Scholar]
- Florindo, H. F. , Kleiner R., Vaskovich‐Koubi D., et al. 2020. “Immune‐Mediated Approaches Against COVID‐19.” Nature Nanotechnology 15, no. 8: 630–645. 10.1038/s41565-020-0732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy, S. P. , Jacoby K., Bota D. A., et al. 2023. “Non‐Viral Precision T Cell Receptor Replacement for Personalized Cell Therapy.” Nature 615, no. 7953: 687–696. 10.1038/s41586-022-05531-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya, V. , Sienkiewicz J., Sun J., et al. 2023a. “mRNA‐Lipid Nanoparticle (LNP) Delivery of Humanized EpCAM‐CD3 Bispecific Antibody Significantly Blocks Colorectal Cancer Tumor Growth.” Cancers 15, no. 10: 2860. 10.3390/cancers15102860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya, V. , Sienkiewicz J., Sun J., et al. 2023b. “CAR‐NK Cells Generated With mRNA‐LNPs Kill Tumor Target Cells In Vitro and In Vivo.” International Journal of Molecular Sciences 24, no. 17: 17. 10.3390/ijms241713364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, T. , Wang M., Fu X., et al. 2024. “Intratumoural Delivery of TRAIL mRNA Induces Colon Cancer Cell Apoptosis.” Biomedicine & Pharmacotherapy 174: 116603. 10.1016/j.biopha.2024.116603. [DOI] [PubMed] [Google Scholar]
- Guevara, M. L. , Persano F., and Persano S.. 2020. “Advances in Lipid Nanoparticles for mRNA‐Based Cancer Immunotherapy.” Frontiers in Chemistry 8: 589959. 10.3389/fchem.2020.589959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, S. , Xu C., Yin H., Hill J., Pi F., and Guo P.. 2020. “Tuning the Size, Shape and Structure of RNA Nanoparticles for Favorable Cancer Targeting and Immunostimulation.” WIREs Nanomedicine and Nanobiotechnology 12, no. 1: e1582. 10.1002/wnan.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hader, M. , Frey B., Fietkau R., Hecht M., and Gaipl U. S.. 2020. “Immune Biological Rationales for the Design of Combined Radio‐ and Immunotherapies.” Cancer Immunology, Immunotherapy 69, no. 2: 293–306. 10.1007/s00262-019-02460-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajj, K. A. , Ball R. L., Deluty S. B., et al. 2019. “Branched‐Tail Lipid Nanoparticles Potently Deliver mRNA in Vivo due to Enhanced Ionization at Endosomal pH.” Small 15, no. 6: 1805097. 10.1002/smll.201805097. [DOI] [PubMed] [Google Scholar]
- Hamilton, A. G. , Swingle K. L., Joseph R. A., et al. 2023. “Ionizable Lipid Nanoparticles With Integrated Immune Checkpoint Inhibition for mRNA CAR T Cell Engineering.” Advanced Healthcare Materials 12, no. 30: 2301515. 10.1002/adhm.202301515. [DOI] [PubMed] [Google Scholar]
- Han, J. , Lim J., Wang C.‐P. J., et al. 2023. “Lipid Nanoparticle‐Based mRNA Delivery Systems for Cancer Immunotherapy.” Nano Convergence 10, no. 1: 36. 10.1186/s40580-023-00385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, X. , Zhang H., Butowska K., et al. 2021. “An Ionizable Lipid Toolbox for RNA Delivery.” Nature Communications 12, no. 1: 7233. 10.1038/s41467-021-27493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel, J. J. , Chowell D., and Chan T. A.. 2019. “The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy.” Nature Reviews Cancer 19, no. 3: 133–150. 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, P. R. , Boczkowski D., Nair S. K., Snyder D., and Gilboa E.. 2006. “Vaccination With mRNAs Encoding Tumor‐Associated Antigens and Granulocyte‐Macrophage Colony‐Stimulating Factor Efficiently Primes CTL Responses, but Is Insufficient to Overcome Tolerance to a Model Tumor/Self Antigen.” Cancer Immunology, Immunotherapy 55, no. 6: 672–683. 10.1007/s00262-005-0064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt, S. L. , Bai A., Bailey D., et al. 2019. “Durable Anticancer Immunity From Intratumoral Administration of IL‐23, IL‐36γ, and OX40L mRNAs.” Science Translational Medicine 11, no. 477: eaat9143. 10.1126/scitranslmed.aat9143. [DOI] [PubMed] [Google Scholar]
- Hewitt, S. L. , Bailey D., Zielinski J., et al. 2020. “Intratumoral IL12 mRNA Therapy Promotes TH1 Transformation of the Tumor Microenvironment.” Clinical Cancer Research 26, no. 23: 6284–6298. 10.1158/1078-0432.CCR-20-0472. [DOI] [PubMed] [Google Scholar]
- Hồ, N. T. , Hughes S. G., Ta V. T., et al. 2024. “Safety, Immunogenicity and Efficacy of the Self‐Amplifying mRNA ARCT‐154 COVID‐19 Vaccine: Pooled Phase 1, 2, 3a and 3b Randomized, Controlled Trials.” Nature Communications 15, no. 1: 4081. 10.1038/s41467-024-47905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder, P. G. , Lim S. A., Huang C. S., et al. 2022. “Engineering Interferons and Interleukins for Cancer Immunotherapy.” Advanced Drug Delivery Reviews 182: 114112. 10.1016/j.addr.2022.114112. [DOI] [PubMed] [Google Scholar]
- Horejs, C. 2021. “From Lipids to Lipid Nanoparticles to mRNA Vaccines.” Nature Reviews Materials 6, no. 12: 1075–1076. 10.1038/s41578-021-00379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotz, C. , Wagenaar T. R., Gieseke F., et al. 2021. “Local Delivery of mRNA‐Encoded Cytokines Promotes Antitumor Immunity and Tumor Eradication Across Multiple Preclinical Tumor Models.” Science Translational Medicine 13, no. 610: eabc7804. 10.1126/scitranslmed.abc7804. [DOI] [PubMed] [Google Scholar]
- Hou, X. , Zaks T., Langer R., and Dong Y.. 2021. “Lipid Nanoparticles for mRNA Delivery.” Nature Reviews Materials 6, no. 12: 1078–1094. 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A. , Pressnall M. M., Lu R., et al. 2020. “Human Intratumoral Therapy: Linking Drug Properties and Tumor Transport of Drugs in Clinical Trials.” Journal of Controlled Release 326: 203–221. 10.1016/j.jconrel.2020.06.029. [DOI] [PubMed] [Google Scholar]
- Huang, C. , Duan X., Wang J., et al. 2023. “Lipid Nanoparticle Delivery System for mRNA Encoding B7H3‐Redirected Bispecific Antibody Displays Potent Antitumor Effects on Malignant Tumors.” Advanced Science 10, no. 3: 2205532. 10.1002/advs.202205532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Q. , Ge Y., He Y., et al. 2024. “The Application of Nanoparticles Targeting Cancer‐Associated Fibroblasts.” International Journal of Nanomedicine 19: 3333–3365. 10.2147/IJN.S447350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Kong N., Zhang X., Cao Y., Langer R., and Tao W.. 2022. “The Landscape of mRNA Nanomedicine.” Nature Medicine 28, no. 11: 2273–2287. 10.1038/s41591-022-02061-1. [DOI] [PubMed] [Google Scholar]
- Hwang, H. J. , and Kim Y. K.. 2024. “Molecular Mechanisms of Circular RNA Translation.” Experimental & Molecular Medicine 56, no. 6: 1272–1280. 10.1038/s12276-024-01220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingels, J. , De Cock L., Mayer R. L., et al. 2022. “Small‐Scale Manufacturing of Neoantigen‐Encoding Messenger RNA for Early‐Phase Clinical Trials.” Cytotherapy 24, no. 2: 213–222. 10.1016/j.jcyt.2021.08.005. [DOI] [PubMed] [Google Scholar]
- Ishida, T. , and Kiwada H.. 2008. “Accelerated Blood Clearance (ABC) Phenomenon Upon Repeated Injection of PEGylated Liposomes.” International Journal of Pharmaceutics 354, no. 1: 56–62. 10.1016/j.ijpharm.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Islam, M. A. , Xu Y., Tao W., et al. 2018. “Restoration of Tumour‐Growth Suppression In Vivo via Systemic Nanoparticle‐Mediated Delivery of PTEN mRNA.” Nature Biomedical Engineering 2, no. 11: 850–864. 10.1038/s41551-018-0284-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanafrooz, Z. , Baradaran B., Mosafer J., et al. 2020. “Comparison of DNA and mRNA Vaccines Against Cancer.” Drug Discovery Today 25, no. 3: 552–560. 10.1016/j.drudis.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman, J. , Mellody M. P., Hou A. J., et al. 2020. “CAR‐T Design: Elements and Their Synergistic Function.” eBioMedicine 58: 102931. 10.1016/j.ebiom.2020.102931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. , Zhang Y., Liu C., et al. 2024. “Tumor‐Activated IL‐2 mRNA Delivered by Lipid Nanoparticles for Cancer Immunotherapy.” Journal of Controlled Release 368: 663–675. 10.1016/j.jconrel.2024.03.016. [DOI] [PubMed] [Google Scholar]
- Jokerst, J. V. , Lobovkina T., Zare R. N., and Gambhir S. S.. 2011. “Nanoparticle PEGylation for Imaging and Therapy.” Nanomedicine 6, no. 4: 715–728. 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff, P. W. , Higano C. S., Shore N. D., et al. 2010. “Sipuleucel‐T Immunotherapy for Castration‐Resistant Prostate Cancer.” New England Journal of Medicine 363, no. 5: 411–422. 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- Kastenmüller, W. , Kastenmüller K., Kurts C., and Seder R. A.. 2014. “Dendritic Cell‐Targeted Vaccines—Hope or Hype?” Nature Reviews Immunology 14, no. 10: 705–711. 10.1038/nri3727. [DOI] [PubMed] [Google Scholar]
- Kerr, M. D. , McBride D. A., Chumber A. K., and Shah N. J.. 2021. “Combining Therapeutic Vaccines With Chemo‐ and Immunotherapies in the Treatment of Cancer.” Expert Opinion on Drug Discovery 16, no. 1: 89–99. 10.1080/17460441.2020.1811673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirolomoom, A. , Kare A. J., Ingham E. S., et al. 2022. “In Situ T‐Cell Transfection by Anti‐CD3‐Conjugated Lipid Nanoparticles Leads to T‐Cell Activation, Migration, and Phenotypic Shift.” Biomaterials 281: 121339. 10.1016/j.biomaterials.2021.121339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. J. , Seo S. K., and Park H. Y.. 2022. “Physical and Chemical Advances of Synthetic Delivery Vehicles to Enhance mRNA Vaccine Efficacy.” Journal of Controlled Release 345: 405–416. 10.1016/j.jconrel.2022.03.029. [DOI] [PubMed] [Google Scholar]
- Kitte, R. , Rabel M., Geczy R., et al. 2023. “Lipid Nanoparticles Outperform Electroporation in mRNA‐Based CAR T Cell Engineering.” Molecular Therapy ‐ Methods & Clinical Development 31: 101139. 10.1016/j.omtm.2023.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klampatsa, A. , Leibowitz M. S., Sun J., Liousia M., Arguiri E., and Albelda S. M.. 2020. “Analysis and Augmentation of the Immunologic Bystander Effects of CAR T Cell Therapy in a Syngeneic Mouse Cancer Model.” Molecular Therapy ‐ Oncolytics 18: 360–371. 10.1016/j.omto.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klichinsky, M. , Ruella M., Shestova O., et al. 2020. “Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy.” Nature Biotechnology 38, no. 8: 947–953. 10.1038/s41587-020-0462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop, K. , Hoogenboom R., Fischer D., and Schubert U. S.. 2010. “Poly(Ethylene Glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives.” Angewandte Chemie International Edition 49, no. 36: 6288–6308. 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- Koch, S. D. , Hong H., Feyerabend S., et al. 2014. “A Randomized, Double‐Blind, Placebo‐Controlled, Phase I/II Trial of RNActive®‐Vaccine cv9104 in Patients With Metastatic Castrate‐Refractory Prostate Cancer (MCRPC): First Results of the Phase I Part.” Journal for Immunotherapy of Cancer 2, no. Suppl 3: P85. 10.1186/2051-1426-2-S3-P85. [DOI] [Google Scholar]
- Kong, B. , Kim Y., Kim E. H., Suk J. S., and Yang Y.. 2023. “mRNA: A Promising Platform for Cancer Immunotherapy.” Advanced Drug Delivery Reviews 199: 114993. 10.1016/j.addr.2023.114993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, F. , Zhou F., Ge L., Liu X., and Wang Y.. 2012. “Mannosylated Liposomes for Targeted Gene Delivery.” International Journal of Nanomedicine 7: 1079–1089. 10.2147/IJN.S29183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, N. , Tao W., Ling X., et al. 2019. “Synthetic mRNA Nanoparticle‐Mediated Restoration of p53 Tumor Suppressor Sensitizes p53‐Deficient Cancers to mTOR Inhibition.” Science Translational Medicine 11, no. 523: eaaw1565. 10.1126/scitranslmed.aaw1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski, P. S. , Rudra A., Miao L., and Anderson D. G.. 2019. “Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery.” Molecular Therapy 27, no. 4: 710–728. 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz, L. M. , Diken M., Haas H., et al. 2016. “Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy.” Nature 534, no. 7607: 396–401. 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- Kübler, H. , Scheel B., Gnad‐Vogt U., et al. 2015. “Self‐Adjuvanted mRNA Vaccination in Advanced Prostate Cancer Patients: A First‐In‐Man Phase I/IIa Study.” Journal for Immunotherapy of Cancer 3, no. 1: 26. 10.1186/s40425-015-0068-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landesman‐Milo, D. , and Peer D.. 2014. “Toxicity Profiling of Several Common RNAi‐Based Nanomedicines: A Comparative Study.” Drug Delivery and Translational Research 4, no. 1: 96–103. 10.1007/s13346-013-0158-7. [DOI] [PubMed] [Google Scholar]
- Lang, F. , Schrörs B., Löwer M., Türeci Ö., and Sahin U.. 2022. “Identification of Neoantigens for Individualized Therapeutic Cancer Vaccines.” Nature Reviews Drug Discovery 21, no. 4: 261–282. 10.1038/s41573-021-00387-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Moignic, A. , Malard V., Benvegnu T., et al. 2018. “Preclinical Evaluation of mRNA Trimannosylated Lipopolyplexes as Therapeutic Cancer Vaccines Targeting Dendritic Cells.” Journal of Controlled Release 278: 110–121. 10.1016/j.jconrel.2018.03.035. [DOI] [PubMed] [Google Scholar]
- Le, N. D. , Nguyen B. L., Patil B. R., et al. 2024. “Antiangiogenic Therapeutic mRNA Delivery Using Lung‐Selective Polymeric Nanomedicine for Lung Cancer Treatment.” ACS Nano 18, no. 11: 8392–8410. 10.1021/acsnano.3c13039. [DOI] [PubMed] [Google Scholar]
- Lee, K. , Kim T.‐S., Seo Y., Kim S. Y., and Lee H.. 2020. “Combined Hybrid Structure of siRNA Tailed IVT mRNA (ChriST mRNA) for Enhancing DC Maturation and Subsequent Anticancer T Cell Immunity.” Journal of Controlled Release 327: 225–234. 10.1016/j.jconrel.2020.08.009. [DOI] [PubMed] [Google Scholar]
- Lee, Y. , Jeong M., Park J., Jung H., and Lee H.. 2023. “Immunogenicity of Lipid Nanoparticles and Its Impact on the Efficacy of mRNA Vaccines and Therapeutics.” Experimental & Molecular Medicine 55, no. 10: 2085–2096. 10.1038/s12276-023-01086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, S. , Zhang X., Men K., et al. 2020. “Efficient Colorectal Cancer Gene Therapy With IL‐15 mRNA Nanoformulation.” Molecular Pharmaceutics 17, no. 9: 3378–3391. 10.1021/acs.molpharmaceut.0c00451. [DOI] [PubMed] [Google Scholar]
- Li, B. , Zhang X., and Dong Y.. 2019. “Nanoscale Platforms for Messenger RNA Delivery.” WIREs Nanomedicine and Nanobiotechnology 11, no. 2: e1530. 10.1002/wnan.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Zhang X.‐Q., Ho W., et al. 2023a. “mRNA Lipid Nanoparticle‐Mediated Pyroptosis Sensitizes Immunologically Cold Tumors to Checkpoint Immunotherapy.” Nature Communications 14, no. 1: 4223. 10.1038/s41467-023-39938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Ng D. S. W., Mah W.‐C., et al. 2015. “A Unique Role for p53 in the Regulation of M2 Macrophage Polarization.” Cell Death and Differentiation 22, no. 7: 1081–1093. 10.1038/cdd.2014.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , and Szoka F. C.. 2007. “Lipid‐Based Nanoparticles for Nucleic Acid Delivery.” Pharmaceutical Research 24, no. 3: 438–449. 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]
- Li, Z. , Amaya L., Pi R., et al. 2023b. “Charge‐Altering Releasable Transporters Enhance mRNA Delivery In Vitro and Exhibit In Vivo Tropism.” Nature Communications 14, no. 1: 6983. 10.1038/s41467-023-42672-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenegger, F. S. , Schnorfeil F. M., Rothe M., et al. 2020. “Toll‐Like Receptor 7/8‐Matured RNA‐Transduced Dendritic Cells as Post‐Remission Therapy in Acute Myeloid Leukaemia: Results of a Phase I Trial.” Clinical & Translational Immunology 9, no. 3: e1117. 10.1002/cti2.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickefett, B. , Chu L., Ortiz‐Maldonado V., et al. 2023. “Lymphodepletion—An Essential but Undervalued Part of the Chimeric Antigen Receptor T‐Cell Therapy Cycle.” Frontiers in Immunology 14: 1303935. 10.3389/fimmu.2023.1303935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, S. , Khoo R., Peh K. M., et al. 2020. “bioPROTACs as Versatile Modulators of Intracellular Therapeutic Targets Including Proliferating Cell Nuclear Antigen (PCNA).” Proceedings of the National Academy of Sciences 117, no. 11: 5791–5800. 10.1073/pnas.1920251117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y.‐X. , Wang Y., Ding J., et al. 2021. “Reactivation of the Tumor Suppressor PTEN by mRNA Nanoparticles Enhances Antitumor Immunity in Preclinical Models.” Science Translational Medicine 13, no. 599: eaba9772. 10.1126/scitranslmed.aba9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares‐Fernández, S. , Lacroix C., Exposito J.‐Y., and Verrier B.. 2020. “Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response.” Trends in Molecular Medicine 26, no. 3: 311–323. 10.1016/j.molmed.2019.10.002. [DOI] [PubMed] [Google Scholar]
- Liu, G. W. , Guzman E. B., Menon N., and Langer R. S.. 2023. “Lipid Nanoparticles for Nucleic Acid Delivery to Endothelial Cells.” Pharmaceutical Research 40, no. 1: 3–25. 10.1007/s11095-023-03471-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.‐Q. , Zhang C., Zhang X., et al. 2022. “Intratumoral Delivery of IL‐12 and IL‐27 mRNA Using Lipid Nanoparticles for Cancer Immunotherapy.” Journal of Controlled Release 345: 306–313. 10.1016/j.jconrel.2022.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Wang Y., Miao L., et al. 2018. “Combination Immunotherapy of MUC1 mRNA Nano‐Vaccine and CTLA‐4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer.” Molecular Therapy 26, no. 1: 45–55. 10.1016/j.ymthe.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, M. , Hu S., Yan N., Popowski K. D., and Cheng K.. 2024. “Inhalable Extracellular Vesicle Delivery of IL‐12 mRNA to Treat Lung Cancer and Promote Systemic Immunity.” Nature Nanotechnology 19, no. 4: 565–575. 10.1038/s41565-023-01580-3. [DOI] [PubMed] [Google Scholar]
- Luke, J. J. , Donahue H., Nishino M., et al. 2015. “Single Institution Experience of Ipilimumab 3 mg/kg With Sargramostim (GM‐CSF) in Metastatic Melanoma.” Cancer Immunology Research 3, no. 9: 986–991. 10.1158/2326-6066.CIR-15-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, H. , Zhang S., Wang B., Cui S., and Yan J.. 2006. “Toxicity of Cationic Lipids and Cationic Polymers in Gene Delivery.” Journal of Controlled Release 114, no. 1: 100–109. 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Mackensen, A. , Haanen J. B. A. G., Koenecke C., et al. 2023. “CLDN6‐Specific CAR‐T Cells Plus Amplifying RNA Vaccine in Relapsed or Refractory Solid Tumors: The Phase 1 BNT211‐01 Trial.” Nature Medicine 29, no. 11: 2844–2853. 10.1038/s41591-023-02612-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee, M. S. , De Iaco A., Rodriguez J., et al. 2023. “Deploying an RNA‐Based Gene Writer System and Lipid Nanoparticle (LNP) Delivery to Generate Functional Chimeric Antigen Receptor (CAR) T Cells With In Vitro and In Vivo Anti‐Tumor Activity.” Blood 142, no. Supplement 1: 2073. 10.1182/blood-2023-184755. [DOI] [Google Scholar]
- Matos, A. I. , Carreira B., Peres C., et al. 2019. “Nanotechnology Is an Important Strategy for Combinational Innovative Chemo‐Immunotherapies Against Colorectal Cancer.” Journal of Controlled Release 307: 108–138. 10.1016/j.jconrel.2019.06.017. [DOI] [PubMed] [Google Scholar]
- Matos, A. I. , Peres C., Carreira B., et al. 2023. “Polyoxazoline‐Based Nanovaccine Synergizes With Tumor‐Associated Macrophage Targeting and Anti‐PD‐1 Immunotherapy Against Solid Tumors.” Advanced Science 10, no. 25: 2300299. 10.1002/advs.202300299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinlay, C. J. , Benner N. L., Haabeth O. A., Waymouth R. M., and Wender P. A.. 2018. “Enhanced mRNA Delivery Into Lymphocytes Enabled by Lipid‐Varied Libraries of Charge‐Altering Releasable Transporters.” Proceedings of the National Academy of Sciences 115, no. 26: E5859–E5866. 10.1073/pnas.1805358115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero, I. , Castanon E., Alvarez M., Champiat S., and Marabelle A.. 2021. “Intratumoural Administration and Tumour Tissue Targeting of Cancer Immunotherapies.” Nature Reviews Clinical Oncology 18, no. 9: 558–576. 10.1038/s41571-021-00507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, C. , Chen Z., Li G., Welte T., and Shen H.. 2021. “Nanoplatforms for mRNA Therapeutics.” Advanced Therapeutics 4, no. 1: 2000099. 10.1002/adtp.202000099. [DOI] [Google Scholar]
- Metzloff, A. E. , Padilla M. S., Gong N., et al. 2024. “Antigen Presenting Cell Mimetic Lipid Nanoparticles for Rapid mRNA CAR T Cell Cancer Immunotherapy.” Advanced Materials 36: 2313226. 10.1002/adma.202313226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, L. , Li L., Huang Y., et al. 2019. “Delivery of mRNA Vaccines With Heterocyclic Lipids Increases Anti‐Tumor Efficacy by STING‐Mediated Immune Cell Activation.” Nature Biotechnology 37, no. 10: 1174–1185. 10.1038/s41587-019-0247-3. [DOI] [PubMed] [Google Scholar]
- Miao, L. , Zhang Y., and Huang L.. 2021. “mRNA Vaccine for Cancer Immunotherapy.” Molecular Cancer 20, no. 1: 41. 10.1186/s12943-021-01335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, V. K. , and Kopetz S.. 2022. “Don't Blame the Messenger: Lessons Learned for Cancer mRNA Vaccines During the COVID‐19 Pandemic.” Nature Reviews Cancer 22, no. 6: 317–318. 10.1038/s41568-022-00463-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortezaee, K. 2020. “Immune Escape: A Critical Hallmark in Solid Tumors.” Life Sciences 258: 118110. 10.1016/j.lfs.2020.118110. [DOI] [PubMed] [Google Scholar]
- Mukalel, A. J. , Hamilton A. G., Billingsley M. M., et al. 2024. “Oxidized mRNA Lipid Nanoparticles for In Situ Chimeric Antigen Receptor Monocyte Engineering.” Advanced Functional Materials 34: 2312038. 10.1002/adfm.202312038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima, I. , Saito S., Akahoshi E., Yagyu S., Sugano‐Ishihara M., and Nakazawa Y.. 2022. “Non‐Viral Inducible Caspase 9 mRNA Delivery Using Lipid Nanoparticles Against Breast Cancer: An In Vitro Study.” Biochemical and Biophysical Research Communications 635: 144–153. 10.1016/j.bbrc.2022.09.105. [DOI] [PubMed] [Google Scholar]
- Neo, S. Y. , Xu S., Chong J., Lam K.‐P., and Wu J.. 2023. “Harnessing Novel Strategies and Cell Types to Overcome Immune Tolerance During Adoptive Cell Therapy in Cancer.” Journal for Immunotherapy of Cancer 11, no. 4: e006434. 10.1136/jitc-2022-006434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neshat, S. Y. , Chan C. H. R., Harris J., et al. 2023. “Polymeric Nanoparticle Gel for Intracellular mRNA Delivery and Immunological Reprogramming of Tumors.” Biomaterials 300: 122185. 10.1016/j.biomaterials.2023.122185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, C. K. Y. , Dazert E., Boldanova T., et al. 2022. “Integrative Proteogenomic Characterization of Hepatocellular Carcinoma Across Etiologies and Stages.” Nature Communications 13, no. 1: 2436. 10.1038/s41467-022-29960-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix, M. A. , and Wiita A. P.. 2024. “Alternative Target Recognition Elements for Chimeric Antigen Receptor (CAR) T Cells: Beyond Standard Antibody Fragments.” Cytotherapy 26: 729–738. 10.1016/j.jcyt.2024.02.024. [DOI] [PubMed] [Google Scholar]
- Nogueira, S. S. , Schlegel A., Maxeiner K., et al. 2020. “Polysarcosine‐Functionalized Lipid Nanoparticles for Therapeutic mRNA Delivery.” ACS Applied Nano Materials 3, no. 11: 10634–10645. 10.1021/acsanm.0c01834. [DOI] [Google Scholar]
- Palucka, K. , and Banchereau J.. 2013. “Dendritic‐Cell‐Based Therapeutic Cancer Vaccines.” Immunity 39, no. 1: 38–48. 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papukashvili, D. , Rcheulishvili N., Liu C., Ji Y., He Y., and Wang P. G.. 2022. “Self‐Amplifying RNA Approach for Protein Replacement Therapy.” International Journal of Molecular Sciences 23, no. 21: 21. 10.3390/ijms232112884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parayath, N. N. , Stephan S. B., Koehne A. L., Nelson P. S., and Stephan M. T.. 2020. “In Vitro‐Transcribed Antigen Receptor mRNA Nanocarriers for Transient Expression in Circulating T Cells In Vivo.” Nature Communications 11, no. 1: 6080. 10.1038/s41467-020-19486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passariello, M. , Yoshioka A., Takahashi K., et al. 2022. “Novel Tri‐Specific Tribodies Induce Strong T Cell Activation and Anti‐Tumor Effects In Vitro and In Vivo.” Journal of Experimental & Clinical Cancer Research 41, no. 1: 269. 10.1186/s13046-022-02474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor, F. , Berraondo P., Etxeberria I., et al. 2018. “An RNA Toolbox for Cancer Immunotherapy.” Nature Reviews Drug Discovery 17, no. 10: 751–767. 10.1038/nrd.2018.132. [DOI] [PubMed] [Google Scholar]
- Pelechano, V. , Wei W., and Steinmetz L. M.. 2015. “Widespread Co‐Translational RNA Decay Reveals Ribosome Dynamics.” Cell 161, no. 6: 1400–1412. 10.1016/j.cell.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perche, F. , Benvegnu T., Berchel M., et al. 2011. “Enhancement of Dendritic Cells Transfection In Vivo and of Vaccination Against B16F10 Melanoma With Mannosylated Histidylated Lipopolyplexes Loaded With Tumor Antigen Messenger RNA.” Nanomedicine: Nanotechnology, Biology and Medicine 7, no. 4: 445–453. 10.1016/j.nano.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Peres, C. , Matos A. I., Carreira B., et al. 2024. “Multifunctional Nanovaccine Sensitizes Breast Cancer to Immune Checkpoint Therapy.” Advanced Functional Materials 34, no. 33: 2401749. 10.1002/adfm.202401749. [DOI] [Google Scholar]
- Peres, C. , Matos A. I., Moura L. I. F., et al. 2021. “Preclinical Models and Technologies to Advance Nanovaccine Development.” Advanced Drug Delivery Reviews 172: 148–182. 10.1016/j.addr.2021.03.001. [DOI] [PubMed] [Google Scholar]
- Perez, C. R. , and De Palma M.. 2019. “Engineering Dendritic Cell Vaccines to Improve Cancer Immunotherapy.” Nature Communications 10, no. 1: 5408. 10.1038/s41467-019-13368-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesch, T. , Bonati L., Kelton W., et al. 2019. “Molecular Design, Optimization, and Genomic Integration of Chimeric B Cell Receptors in Murine B Cells.” Frontiers in Immunology 10: 2630. 10.3389/fimmu.2019.02630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phua, K. K. L. , Staats H. F., Leong K. W., and Nair S. K.. 2014. “Intranasal mRNA Nanoparticle Vaccination Induces Prophylactic and Therapeutic Anti‐Tumor Immunity.” Scientific Reports 4, no. 1: 5128. 10.1038/srep05128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack, F. P. , Thomas S. J., Kitchin N., et al. 2020. “Safety and Efficacy of the BNT162b2 mRNA Covid‐19 Vaccine.” New England Journal of Medicine 383, no. 27: 2603–2615. 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovitz, J. , Sharma R., Hoshyar R., Soo Kim B., Murthy N., and Lee K.. 2023. “Gene Editing Therapeutics Based on mRNA Delivery.” Advanced Drug Delivery Reviews 200: 115026. 10.1016/j.addr.2023.115026. [DOI] [PubMed] [Google Scholar]
- Presnyak, V. , Alhusaini N., Chen Y.‐H., et al. 2015. “Codon Optimality Is a Major Determinant of mRNA Stability.” Cell 160, no. 6: 1111–1124. 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery, M. J. , Franzén A. S., and Pecher G.. 2023. “CAR NK Cells: The Future Is Now.” Annual Review of Cancer Biology 7: 229–246. 10.1146/annurev-cancerbio-061521-082320. [DOI] [Google Scholar]
- Ramishetti, S. , Kedmi R., Goldsmith M., et al. 2015. “Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles.” ACS Nano 9, no. 7: 6706–6716. 10.1021/acsnano.5b02796. [DOI] [PubMed] [Google Scholar]
- Riley, R. S. , June C. H., Langer R., and Mitchell M. J.. 2019. “Delivery Technologies for Cancer Immunotherapy.” Nature Reviews Drug Discovery 18, no. 3: 175–196. 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohner, E. , Yang R., Foo K. S., Goedel A., and Chien K. R.. 2022. “Unlocking the Promise of mRNA Therapeutics.” Nature Biotechnology 40, no. 11: 1586–1600. 10.1038/s41587-022-01491-z. [DOI] [PubMed] [Google Scholar]
- Rojas, L. A. , Sethna Z., Soares K. C., et al. 2023. “Personalized RNA Neoantigen Vaccines Stimulate T Cells in Pancreatic Cancer.” Nature 618, no. 7963: 144–150. 10.1038/s41586-023-06063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybakova, Y. , Kowalski P. S., Huang Y., et al. 2019. “mRNA Delivery for Therapeutic Anti‐HER2 Antibody Expression In Vivo.” Molecular Therapy 27, no. 8: 1415–1423. 10.1016/j.ymthe.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin, U. , Derhovanessian E., Miller M., et al. 2017. “Personalized RNA Mutanome Vaccines Mobilize Poly‐Specific Therapeutic Immunity Against Cancer.” Nature 547, no. 7662: 222–226. 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- Sahin, U. , Oehm P., Derhovanessian E., et al. 2020. “An RNA Vaccine Drives Immunity in Checkpoint‐Inhibitor‐Treated Melanoma.” Nature 585, no. 7823: 107–112. 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- Sanz, L. , and Álvarez‐Vallina L.. 2021. “Engineered mRNA and the Rise of Next‐Generation Antibodies.” Antibodies 10, no. 4: 4. 10.3390/antib10040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayour, E. J. , Boczkowski D., Mitchell D. A., and Nair S. K.. 2024. “Cancer mRNA Vaccines: Clinical Advances and Future Opportunities.” Nature Reviews Clinical Oncology 21: 1–12. 10.1038/s41571-024-00902-1. [DOI] [PubMed] [Google Scholar]
- Sayour, E. J. , De Leon G., Pham C., et al. 2017. “Systemic Activation of Antigen‐Presenting Cells via RNA‐Loaded Nanoparticles.” Oncoimmunology 6, no. 1: e1256527. 10.1080/2162402X.2016.1256527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, M. D. , Rodriguez P. C., Koehn B. H., et al. 2018. “Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation Into Ly6c+CD103+ Monocytic Antigen‐Presenting Cells in Tumors.” Immunity 48, no. 1: 91–106.e6. 10.1016/j.immuni.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, D. , Beasock D., Fessler A., et al. 2022a. “To PEGylate or Not to PEGylate: Immunological Properties of Nanomedicine's Most Popular Component, Polyethylene Glycol and Its Alternatives.” Advanced Drug Delivery Reviews 180: 114079. 10.1016/j.addr.2021.114079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J. , Huang M.‐W., Lu Z.‐D., et al. 2022b. “Delivery of mRNA for Regulating Functions of Immune Cells.” Journal of Controlled Release 345: 494–511. 10.1016/j.jconrel.2022.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimosakai, R. , Khalil I. A., Kimura S., and Harashima H.. 2022. “mRNA‐Loaded Lipid Nanoparticles Targeting Immune Cells in the Spleen for Use as Cancer Vaccines.” Pharmaceuticals 15, no. 8: 8. 10.3390/ph15081017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, H. , Kang S., Won C., and Min D.‐H.. 2023. “Enhanced Local Delivery of Engineered IL‐2 mRNA by Porous Silica Nanoparticles to Promote Effective Antitumor Immunity.” ACS Nano 17, no. 17: 17554–17567. 10.1021/acsnano.3c06733. [DOI] [PubMed] [Google Scholar]
- Shin, H. E. , Han J.‐H., Park J. D., et al. 2024. “Enhancing CAR‐NK Cells Against Solid Tumors Through Chemical and Genetic Fortification With DOTAP‐Functionalized Lipid Nanoparticles.” Advanced Functional Materials 34: 2315721. 10.1002/adfm.202315721. [DOI] [Google Scholar]
- Short, L. , Holt R. A., Cullis P. R., and Evgin L.. 2024. “Direct In Vivo CAR T Cell Engineering.” Trends in Pharmacological Sciences 45, no. 5: 406–418. 10.1016/j.tips.2024.03.004. [DOI] [PubMed] [Google Scholar]
- Stadler, C. R. , Bähr‐Mahmud H., Celik L., et al. 2017. “Elimination of Large Tumors in Mice by mRNA‐Encoded Bispecific Antibodies.” Nature Medicine 23, no. 7: 815–817. 10.1038/nm.4356. [DOI] [PubMed] [Google Scholar]
- Stasi, A. D. , Tey S.‐K., Dotti G., et al. 2011. “Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy.” New England Journal of Medicine 365, no. 18: 1673–1683. 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock, S. , Klüver A.‐K., Fertig L., et al. 2023. “Mechanisms and Strategies for Safe Chimeric Antigen Receptor T‐Cell Activity Control.” International Journal of Cancer 153, no. 10: 1706–1725. 10.1002/ijc.34635. [DOI] [PubMed] [Google Scholar]
- Suk, J. S. , Xu Q., Kim N., Hanes J., and Ensign L. M.. 2016. “PEGylation as a Strategy for Improving Nanoparticle‐Based Drug and Gene Delivery.” Advanced Drug Delivery Reviews 99: 28–51. 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurs, F. V. , Lub‐de Hooge M. N., de Vries E. G. E., and de Groot D. J. A.. 2019. “A Review of Bispecific Antibodies and Antibody Constructs in Oncology and Clinical Challenges.” Pharmacology & Therapeutics 201: 103–119. 10.1016/j.pharmthera.2019.04.006. [DOI] [PubMed] [Google Scholar]
- Tahtinen, S. , Tong A.‐J., Himmels P., et al. 2022. “IL‐1 and IL‐1ra Are Key Regulators of the Inflammatory Response to RNA Vaccines.” Nature Immunology 23, no. 4: 532–542. 10.1038/s41590-022-01160-y. [DOI] [PubMed] [Google Scholar]
- Tang, Z. , You X., Xiao Y., et al. 2023. “Inhaled mRNA Nanoparticles Dual‐Targeting Cancer Cells and Macrophages in the Lung for Effective Transfection.” Proceedings of the National Academy of Sciences 120, no. 44: e2304966120. 10.1073/pnas.2304966120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thran, M. , Mukherjee J., Pönisch M., et al. 2017. “mRNA Mediates Passive Vaccination Against Infectious Agents, Toxins, and Tumors.” EMBO Molecular Medicine 9, no. 10: 1434–1447. 10.15252/emmm.201707678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin, M. N. , and Roni M. A.. 2021. “Challenges of Storage and Stability of mRNA‐Based COVID‐19 Vaccines.” Vaccine 9, no. 9: 9. 10.3390/vaccines9091033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoecke, L. , and Roose K.. 2019. “How mRNA Therapeutics Are Entering the Monoclonal Antibody Field.” Journal of Translational Medicine 17, no. 1: 54. 10.1186/s12967-019-1804-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint, S. , Goyvaerts C., Maenhout S., et al. 2012. “Preclinical Evaluation of TriMix and Antigen mRNA‐Based Antitumor Therapy.” Cancer Research 72, no. 7: 1661–1671. 10.1158/0008-5472.CAN-11-2957. [DOI] [PubMed] [Google Scholar]
- Van Lint, S. , Renmans D., Broos K., et al. 2016. “Intratumoral Delivery of TriMix mRNA Results in T‐Cell Activation by Cross‐Presenting Dendritic Cells.” Cancer Immunology Research 4, no. 2: 146–156. 10.1158/2326-6066.CIR-15-0163. [DOI] [PubMed] [Google Scholar]
- Veiga, N. , Goldsmith M., Granot Y., et al. 2018. “Cell Specific Delivery of Modified mRNA Expressing Therapeutic Proteins to Leukocytes.” Nature Communications 9, no. 1: 4493. 10.1038/s41467-018-06936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vom Berg, J. , Vrohlings M., Haller S., et al. 2013. “Intratumoral IL‐12 Combined With CTLA‐4 Blockade Elicits T Cell–Mediated Glioma Rejection.” Journal of Experimental Medicine 210, no. 13: 2803–2811. 10.1084/jem.20130678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa, A. , Aljabbari A., Lokras A., Foged C., and Thakur A.. 2020. “Opportunities and Challenges in the Delivery of mRNA‐Based Vaccines.” Pharmaceutics 12, no. 2: 2. 10.3390/pharmaceutics12020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, E. E. , Frenck R. W., Falsey A. R., et al. 2020. “Safety and Immunogenicity of Two RNA‐Based Covid‐19 Vaccine Candidates.” New England Journal of Medicine 383, no. 25: 2439–2450. 10.1056/NEJMoa2027906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Wang Y., Ding J., et al. 2020. “A Bioorthogonal System Reveals Antitumour Immune Function of Pyroptosis.” Nature 579, no. 7799: 421–426. 10.1038/s41586-020-2079-1. [DOI] [PubMed] [Google Scholar]
- Wang, W. , Liu Y., He Z., et al. 2024. “Breakthrough of Solid Tumor Treatment: CAR‐NK Immunotherapy.” Cell Death Discovery 10, no. 1: 1–16. 10.1038/s41420-024-01815-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tiruthani K., Li S., et al. 2021. “mRNA Delivery of a Bispecific Single‐Domain Antibody to Polarize Tumor‐Associated Macrophages and Synergize Immunotherapy Against Liver Malignancies.” Advanced Materials 33, no. 23: 2007603. 10.1002/adma.202007603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Zhang L., Xu Z., Miao L., and Huang L.. 2018. “mRNA Vaccine With Antigen‐Specific Checkpoint Blockade Induces an Enhanced Immune Response Against Established Melanoma.” Molecular Therapy 26, no. 2: 420–434. 10.1016/j.ymthe.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, E. W. , Parker K. R., Sotillo E., et al. 2021. “Transient Rest Restores Functionality in Exhausted CAR‐T Cells Through Epigenetic Remodeling.” Science 372, no. 6537: eaba1786. 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, J. S. , Carlino M. S., Khattak A., et al. 2024. “Individualised Neoantigen Therapy mRNA‐4157 (V940) Plus Pembrolizumab Versus Pembrolizumab Monotherapy in Resected Melanoma (KEYNOTE‐942): A Randomised, Phase 2b Study.” Lancet 403, no. 10427: 632–644. 10.1016/S0140-6736(23)02268-7. [DOI] [PubMed] [Google Scholar]
- Wei, J. , Yang Y., Wang G., and Liu M.. 2022. “Current Landscape and Future Directions of Bispecific Antibodies in Cancer Immunotherapy.” Frontiers in Immunology 13: 1035276. 10.3389/fimmu.2022.1035276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, J. , Yang M., Wang W., Xu X., and Tian Z.. 2020. “Revealing Thermodynamics and Kinetics of Lipid Self‐Assembly by Markov State Model Analysis.” Journal of the American Chemical Society 142, no. 51: 21344–21352. 10.1021/jacs.0c09343. [DOI] [PubMed] [Google Scholar]
- Wilk, A. J. , Weidenbacher N. L.‐B., Vergara R., et al. 2020. “Charge‐Altering Releasable Transporters Enable Phenotypic Manipulation of Natural Killer Cells for Cancer Immunotherapy.” Blood Advances 4, no. 17: 4244–4255. 10.1182/bloodadvances.2020002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Wu W., Zhou B., and Li B.. 2024. “Chimeric Antigen Receptor Therapy Meets mRNA Technology.” Trends in Biotechnology 42, no. 2: 228–240. 10.1016/j.tibtech.2023.08.005. [DOI] [PubMed] [Google Scholar]
- Wu, L. , Wang W., Tian J., et al. 2022. “Intravenous Delivery of RNA Encoding Anti‐PD‐1 Human Monoclonal Antibody for Treating Intestinal Cancer.” Journal of Cancer 13, no. 2: 579–588. 10.7150/jca.63991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Y. , Chen J., Zhou H., et al. 2022. “Combining p53 mRNA Nanotherapy With Immune Checkpoint Blockade Reprograms the Immune Microenvironment for Effective Cancer Therapy.” Nature Communications 13, no. 1: 758. 10.1038/s41467-022-28279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, W. , Chen B., and Wong J.. 2021. “Evolution of the Market for mRNA Technology.” Nature Reviews Drug Discovery 20, no. 10: 735–736. 10.1038/d41573-021-00147-y. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Zhu J., Sun J., et al. 2022. “Intratumoral Delivered Novel Circular mRNA Encoding Cytokines for Immune Modulation and Cancer Therapy.” Molecular Therapy ‐ Nucleic Acids 30: 184–197. 10.1016/j.omtn.2022.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. , and Lai S. K.. 2015. “Anti‐PEG Immunity: Emergence, Characteristics, and Unaddressed Questions.” WIREs Nanomedicine and Nanobiotechnology 7, no. 5: 655–677. 10.1002/wnan.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T. , Li C., Wang X., et al. 2020. “Efficient Hepatic Delivery and Protein Expression Enabled by Optimized mRNA and Ionizable Lipid Nanoparticle.” Bioactive Materials 5, no. 4: 1053–1061. 10.1016/j.bioactmat.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Nishimiya D., Löchte S., et al. 2023a. “Facile Repurposing of Peptide–MHC‐Restricted Antibodies for Cancer Immunotherapy.” Nature Biotechnology 41, no. 7: 932–943. 10.1038/s41587-022-01567-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Liu Y., Zhao K., et al. 2023b. “Dual mRNA Co‐Delivery for In Situ Generation of Phagocytosis‐Enhanced CAR Macrophages Augments Hepatocellular Carcinoma Immunotherapy.” Journal of Controlled Release 360: 718–733. 10.1016/j.jconrel.2023.07.021. [DOI] [PubMed] [Google Scholar]
- Ye, Z. , Chen J., Zhao X., et al. 2022. “In Vitro Engineering Chimeric Antigen Receptor Macrophages and T Cells by Lipid Nanoparticle‐Mediated mRNA Delivery.” ACS Biomaterials Science & Engineering 8, no. 2: 722–733. 10.1021/acsbiomaterials.1c01532. [DOI] [PubMed] [Google Scholar]
- Yin, D. , Zhong Y., Ling S., et al. 2024. “Dendritic‐Cell‐Targeting Virus‐Like Particles as Potent mRNA Vaccine Carriers.” Nature Biomedical Engineering 1–16. 10.1038/s41551-024-01208-4. [DOI] [PubMed] [Google Scholar]
- Yuan, M. , Han Z., Liang Y., et al. 2023. “mRNA Nanodelivery Systems: Targeting Strategies and Administration Routes.” Biomaterials Research 27, no. 1: 90. 10.1186/s40824-023-00425-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, C. , Zhang C., Walker P. G., and Dong Y.. 2022. “Formulation and Delivery Technologies for mRNA Vaccines.” In mRNA Vaccines, edited by Yu D. and Petsch B., 71–110. Cham: Springer International Publishing. 10.1007/82_2020_217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Zhang X., Zhao W., et al. 2019a. “Chemotherapy Drugs Derived Nanoparticles Encapsulating mRNA Encoding Tumor Suppressor Proteins to Treat Triple‐Negative Breast Cancer.” Nano Research 12, no. 4: 855–861. 10.1007/s12274-019-2308-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Parayath N. N., Ene C. I., et al. 2019b. “Genetic Programming of Macrophages to Perform Anti‐Tumor Functions Using Targeted mRNA Nanocarriers.” Nature Communications 10, no. 1: 3974. 10.1038/s41467-019-11911-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Wu L., Zhang Y., et al. 2024. “Aptamer‐Based Nongenetic Reprogramming of CARs Enables Flexible Modulation of T Cell‐Mediated Tumor Immunotherapy.” ACS Central Science 10, no. 4: 813–822. 10.1021/acscentsci.3c01511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Sun C., Wang C., Jankovic K. E., and Dong Y.. 2021. “Lipids and Lipid Derivatives for RNA Delivery.” Chemical Reviews 121, no. 20: 12181–12277. 10.1021/acs.chemrev.1c00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Zhang Y., Xia S., et al. 2020. “Gasdermin E Suppresses Tumour Growth by Activating Anti‐Tumour Immunity.” Nature 579, no. 7799: 415–420. 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Gan L., Ke D., Chen Q., and Fu Y.. 2023. “Mechanisms and Research Advances in mRNA Antibody Drug‐Mediated Passive Immunotherapy.” Journal of Translational Medicine 21, no. 1: 693. 10.1186/s12967-023-04553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , He H., Wang K., et al. 2020. “Granzyme A From Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells.” Science 368, no. 6494: eaaz7548. 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
- Zimmermannova, O. , Ferreira A. G., Ascic E., et al. 2023. “Restoring Tumor Immunogenicity With Dendritic Cell Reprogramming.” Science Immunology 8, no. 85: eadd4817. 10.1126/sciimmunol.add4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermannova, O. , Ferreira A. G., and Pereira C.‐F.. 2024. “Orchestrating an Immune Response to Cancer With Cellular Reprogramming.” Genes & Immunity 25, no. 1: 95–97. 10.1038/s41435-023-00237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.