

Abstract
Sphingoid bases are important bioactive lipids found in a variety of organisms, serving as the backbone of sphingolipids, which regulate essential physiological processes. Here we describe the total synthesis and structure revision of halisphingosine A, a sphingoid base initially isolated from marine sponges. To address inconsistencies in the NMR interpretation of this natural product, we developed a synthetic route involving a late‐stage enantioselective Henry reaction that allows access to multiple stereoisomers of the proposed halisphingosine A core structure. Our library of 32 fully characterized synthetic stereoisomers enabled us to rectify the structure of halisphingosine A as (2R,3R,8R,Z)‐2‐aminooctadec‐9‐ene‐1,3,8‐triol, and to pursue further structure–activity relation (SAR) studies regarding their antimicrobial and cytotoxic potential. In summary, our study offers a yet unreported compound library along with validated analytical datasets of marine sphingoid base derivatives, which significantly affects future ecometabolomic marine research and will facilitate the identification of inhibitors of sphingolipid metabolism or antagonists of sphingolipid base‐sensing receptors.
Keywords: Sphingolipid bases, Marine natural products, Structure Revision, Total Synthesis, Henry reaction
Synthesis of 32 stereoisomers of halisphingosine A in 11–13 steps with good overall yields. Comparative NMR studies resolved a longstanding structural misconception, leading to the revision of halisphingosine A. Our synthetic analytical study provides an unparalleled level of detail on the challenges associated with determining the structures of non‐model sphingoid bases, and will catalyse future research in marine metabolomics and sphingolipid metabolism.
Sphingoid bases represent an important class of bioactive lipids, which are found in bacteria, fungi, plants and animals (Figure 1). [1] They constitute the structural basis of sphingolipids, which influence a multitude of physiological relevant mechanisms ranging from membrane integrity to signal transduction. [2] Sphingoid bases also act as modulators of sphingolipid metabolism; as shown for the fungal derivatives fumonisins [3] and sphingofungins, [4] which inhibit important steps of mammalian sphingolipid biosynthesis and can deplete cells of sphingolipids making them essential tools for cell biology. Since an aberrant sphingolipid metabolism can cause fatal hereditary diseases [5] and is involved in cancer progression [6] as well as autoimmune diseases, [7] sphingoid bases represent interesting lead structures for drug development.
Figure 1.
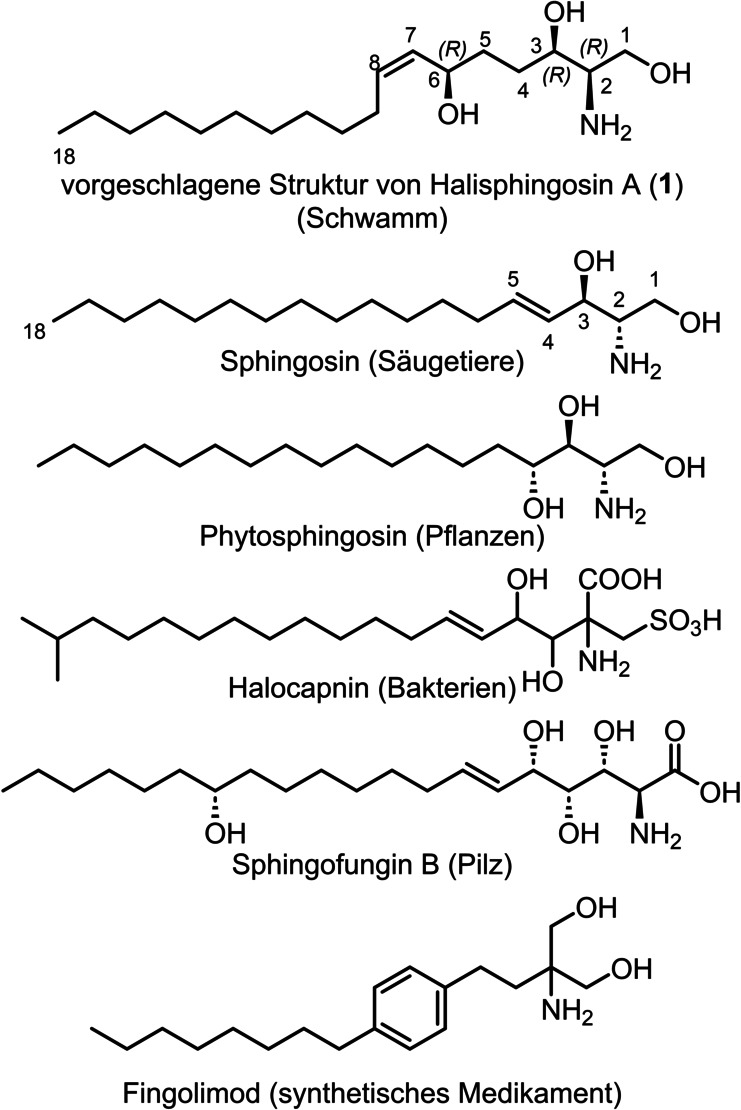
Different examples of sphingoid bases of mammalian, plant, bacterial and fungal origin, and fingolimod, which is a synthetic S1PR antagonist.
An example for the development of a clinical drug that originated from sphingoid research and mimics a central metabolite of the sphingolipid degradation pathway is the orally available immunomodulator fingolimod that is used today to treat multiple sclerosis. After phosphorylation in vivo fingolimod becomes a sphingosine‐1‐phosphate receptor (S1PR) agonist and limits inflammatory cell migration into the central nervous system. Currently, multiple drugs are in clinical development that target a diverse set of S1PR subtypes to treat a variety of severe conditions. [8]
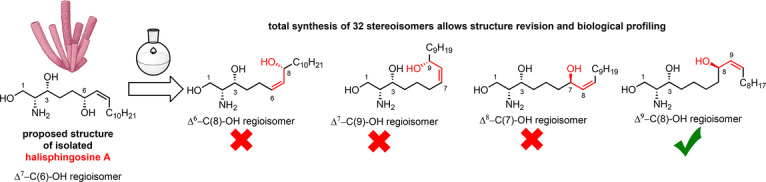
Our group has long been focused on the isolation, total synthesis, and physiological evaluation of complex lipids.[ 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 ] This ongoing interest led us to take particular note of the reported total synthesis of the cytotoxic sphingoid base halisphingosine A 1 by the group of Schobert, [18] which was isolated in small quantities from marine sponges of the genus Haliclona.[ 19 , 20 ] The synthetic proposed structure of halisphingosine A 1 showed inconsistencies in the NMR data when compared to the isolated natural product. Following up on these studies and based on our recent endeavors in sphingolipid total synthesis, we concluded that the configuration of the three stereocenters (2R, 3R, 6R) of halisphingosine A 1 have likely been erroneously assigned. Thus, we set out to develop a synthetic sequence that enables access to different stereoisomers of this sphingoid base to reveal its absolute configuration and provide ample quantities for biological and SAR studies. [21]
Given the importance of these bioactive lipids, extensive efforts have been devoted to synthesizing sphingosines, utilizing chiral pool approaches, chiral auxiliaries, and asymmetric reactions to build their adjacent stereogenic centers.[ 22 , 23 ] In our retrosynthetic analysis of the proposed structure of halisphingosine A 1 and its stereoisomers we envisioned a late stage enantioselective Henry reaction of a functionalized aldehyde 7 and 2‐nitroethanol 2 using chiral copper complexes to introduce the 1,2‐amino alcohol moiety in a syn‐ or anti‐ configuration (Scheme 1A). The necessary aldehyde 7 should be accessible via a reaction sequence starting from literature known hemiacetals R‐ and S‐3, which are accessible via the chiral pool (L‐ and D‐malic acid), the installation of the Z‐allylic alcohol moiety via a Wittig reaction and a one carbon chain elongation. Overall the reaction sequence would allow the synthesis of all possible stereoisomers 12–19 of the proposed halisphingosine A 1. The synthetic sequence commenced as planned with the opening of the hemiacetals R‐ and S‐3 [24] with triphenyl(undecyl)phosphonium bromide and sodium bis(trimethylsilyl)amide (NaHMDS) to yield Z‐allylic alcohols R‐ and S‐4 in 87 % and 96 % yield, respectively (Scheme 1B). The resulting primary alcohol was converted into the corresponding mesylate, which underwent nucleophilic substitution into the corresponding iodide 5 using Finkelstein reaction conditions. Finally, chain elongation with lithiated 1,3‐dithiane generated dithioacetal 6, which was hydrolyzed using an excess of MeI to afford the desired aldehydes R‐ and S‐7 in 82 % and 84 % yield over 4 steps, respectively. With aldehydes R‐ and S‐7 in hand, we used the stereoselective Henry reaction developed by the Chen laboratory, in which two enantiomeric sets of copper complexes introduce the remaining two stereocenters at C‐2 and C‐3.[ 25 , 26 ] Following the reported procedure all eight possible stereoisomers of nitroalcohol 8–11 (SI‐34, 35, 48, 49) were received after purification by column chromatography in satisfactory to good yields (56–78 %).
Scheme 1.

A) Retrosynthetic analysis of the proposed Δ7‐C(6)‐OH halisphingosine A 1. B) Synthetic Scheme showing the preparation of aldehyde R‐ and S‐7. C) Final steps in the total synthesis of diastereomers 12–15 of the proposed Δ7‐C(6)‐OH halisphingosine A 1 and D) chemical structures of the corresponding synthesized enantiomers 16–19 following the same reaction procedure.
Different protocols for the reduction of the nitro group were studied (NaBH4/NiCl2, [27] LiAlH4, SnCl2, Zn/NH4Cl, Fe/NH4Cl) before we discovered that Zn [28] and acetic acid in ethyl acetate yielded the corresponding amines in nearly quantitative yields (Scheme 1C). Final deprotection of the crude amines using tetra‐n‐butylammonium fluoride (TBAF) afforded all eight possible stereoisomers 12–19 of the proposed structure of halisphingosine A 1 in satisfactory to excellent yields (57–85 %) over 2 steps. Comparative nuclear magnetic resonance (NMR) analysis of 12–19 showed significant differences of multiple 13C‐spectrum signals (▵δ >4 ppm, see Table S1 and S2) compared to those described by Jung and co‐workers. [20] The nature of the strong chemical shift differences and a re‐analysis of the originally reported dataset let us to the conclusion that the naturally occurring halisphingosine A should be a regioisomer of the proposed structure (Figure 1). Here, we first proposed that either a C(7)‐OH, a C(8)‐OH regioisomer or a C(9)‐OH homolog with or without an inverted allylic system could be likely structural candidates (Scheme 2A).
Scheme 2.

A) Retrosynthesis of different regioisomers of halisphingosine A. B) Synthesis of precursor aldehydes R/S‐23 ([Ru]: RuCl[(R,R)‐TsDPEN](mesitylene) or RuCl[(S,S)‐TsDPEN](mesitylene)), C) Synthesis of all eight Δ6‐C(8)‐OH stereoisomers 24–31 based on the previously established three‐step reaction sequence i. 2‐nitroethanol 2, chiral copper complex; ii. Zn, HOAc; iii. TBAF.
In order to test our hypothesis, we decided to use the stereoselective Henry reaction in combination with different chiral aldehydes 23, 32, 37, and 42 followed by our reduction and deprotection protocol, to obtain the different isomers of halisphingosine A. To streamline the access to the differently substitute aldehydes, we designed a synthetic route consisting of an alkynylation reaction to generate the carbon backbone, a stereoselective reduction to install the Z‐configured double bond and an asymmetric reduction to prepare the chiral alcohol moiety.
As an example, the synthesis of aldehydes R‐ and S‐23, which are employed in the synthesis of the Δ6‐C(8) regioisomers of halisphingosine A, will be discussed in detail while the synthesis of the remaining building blocks 32, 37, and 42 can be found in the Supporting Information (Scheme 2B). The synthesis of aldehydes R‐ and S‐23 started from undecanoyl chloride, which was reacted with bis(trimethylsilyl)acetylene to yield ynone 20. Reduction of the ketone 20 using Noyori asymmetric hydrogenation catalysis [29] led to R‐ and S‐alcohols in excellent yields, which were protected with a tert‐butyldimethylsilyl (TBS) group. Afterwards the trimethylsilyl (TMS) group was cleaved under basic conditions, before the resulting alkynes were converted into alkynyl lithium reagents and coupled with (3‐bromopropoxy)(tert‐butyl)dimethylsilane. [30] The resulting alkynes 22 were stereoselectively reduced with in situ prepared nickel boride and ethylenediamine to exclusively obtain the R‐ and S‐Z‐alkenes in outstanding yields of 95 % and 96 %, respectively. [31] Removal of the primary TBS group using TBAF yielded alcohols, which, upon treatment with Dess–Martin periodinane (DMP), gave access to the desired aldehydes R‐ and S‐23. Finally, our previously established reaction sequence allowed the installation of the two remaining stereocenters via the diastereoselective Henry reaction, which was followed by reduction of the nitro group and cleavage of the TBS ether yielding again the eight possible stereoisomers of the Δ6‐C(8)‐OH regioisomers 24–31 in 44 %–66 % yield over three steps (Scheme 2C).
Unfortunately, none of the obtained analytical datasets for isomers 24–31 matched the previously published NMR data of natural halisphingosine A (▵δ >4 ppm for at least one 13C signal, see Table S3 and S4). Since the chemical shifts of compound 24 (all R‐configured stereocenters) exhibited the highest similarity to halisphingosine A, we speculated that the remaining chemical shift discrepancies were due to an incorrect assignment of the chain length between the 1,2‐amino alcohol motif and the allyl (or inverted allyl) group. Therefore, we continued synthesizing the remaining hypothesized regioisomers Δ7‐C(9)‐OH 33–36 and Δ8‐C(7)‐OH 38–41 of halisphingosine A following our reliable synthetic route (Scheme 3A and B). Again careful NMR‐analysis of the eight synthesized stereoisomers (33–36 and 38–41) revealed still notable, but minor 13C and 1H‐chemical shifts differences in the aliphatic region (Scheme 3A−C, and for details see Tables S5 and S6).
Scheme 3.

A) Synthesis of Δ7‐C(9)‐OH stereoisomers 33–36 of halisphingosine A based on the previously established three‐step reaction sequence i. 2‐nitroethanol, chiral copper complex; ii. Zn, HOAc; iii. TBAF. B) Synthesis of Δ8‐C(7)‐OH stereoisomers 38–41 of halisphingosine A based on the previously established three‐step reaction sequence i. 2‐nitroethanol, chiral copper complex; ii. Zn, HOAc; iii. TBAF. C) Stacked view of representative 1H and 13C NMR spectra of regioisomers 12, 24, 33, 38 and 43 showing chemical shift differences. D) Synthesis of all eight Δ9‐C(8)‐OH stereoisomers 43–50 of halisphingosine A using the previously established three‐step reaction sequence i. 2‐nitroethanol, chiral copper complex; ii. Zn, HOAc; iii. TBAF. E) Comparison of the optical rotation and 13C NMR values of (2R,3R,8R,Z)‐43 and (2S,3S,8S,Z)‐44 with literature data with Δδ 13C values next to the corresponding carbons in ppm.
Finally, we synthesized all of the eight Δ9‐C(8)‐OH stereoisomers (Scheme 3D), and were pleased to find that the NMR data for both, the Δ9‐C(8)‐OH (2R,3R,8R,Z) 43 as well as the enantiomer (2S,3S,8S,Z) 44 showed an almost perfect match with the reported signals for halisphingosine A (Tables S7 and S8). In addition, both compounds gave similar absolute optical rotation values as reported for the isolated sphingosine; however only Δ9‐C(8)‐OH (2R,3R,8R,Z) 43 exhibited the positive value as reported for the natural product (Scheme 3E). Thus, we concluded that (2R,3R,8R,Z)‐2‐aminooctadec‐9‐ene‐1,3,8‐triol 43 is the most likely structure of the natural product halisphingosine A.
To further validate our findings, we converted (2R,3R,8R,Z) 43 into the literature reported N‐tert‐butyloxycarbonyl (Boc) protected derivative 51 for which different NMR datasets are available (Scheme 3E Table S9–S11). [19] We were delighted to find that compound 51 displayed the same set of NMR shifts as reported for the Boc‐derivatized halisphingosine A, which serves as additional proof of the revised structure and indirectly of the reduced derivative halisphingosine B, which shows nearly identical spectroscopic characteristics as its congener. [19]
Due to the intrinsic bioactivities of sphingosine derivatives, we investigated the antibiotic and cytotoxic activity of all R‐configured regioisomers and halisphingosine A itself. Indeed, we observed moderate structure‐dependent antimicrobial activity against Gram‐negative Escherichia coli (16–64 μg mL−1) and Gram‐positive Staphylococcus aureus (32–128 μg mL−1, see Table S12), [32] as well antifungal activity against Candida albicans (32–64 μg mL−1). Consistent with the original studies on isolated halisphingosine A, which demonstrated activity against various cancer cell lines, we also observed cytotoxicity in the low micromolar range against lung and liver cell lines but with only moderate differences between the derivatives (see Table S12).
In summary, we successfully synthesized in total 32 stereoisomers of halisphingosine A within 11–13 steps in the longest linear sequence in good overall yields. Comparative NMR studies allowed us to answer a long standing structural question and finally revise the structure of the marine natural product halisphingosine A, and indirectly it's reduced congener halisphingosine B.
Our studies provide an unprecedented level of detail on the analytical challenges encountered when determining the absolute structures of non‐model sphingoid bases with unverified origin and limited sample availability. This is exemplified by the observation that e.g. all four regiosiomers 12, 24, 33, and 38 (all R‐configuration) of halisphingosine A (43) exhibit strikingly similar tandem mass spectra and retention times (Figure S10 and S11) and thus are not distinguishable by mass spectrometry methods alone. We therefore would like to emphasize that dereplication efforts concerning non‐model sphingoid bases should ideally be supported by spiking experiments or NMR analysis.
In this way, our compound library and analytical understanding serve as a valuable asset for the de‐replication of marine metabolomics and the identification of modulators of the sphingolipid metabolism.
Conflict of Interests
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
We would like to thank Heike Heinecke (Leibniz‐HKI) for measurement of NMR spectra, Antje Meyer and Nina Messerschmidt (Leibniz‐HKI) for preliminary cytotoxic and antiproliferative assays and Jeannine Jung and Jannine Seelbach (HIPS) for performing the cytotoxicity assays. CB greatly acknowledges funding from the European Union's Horizon 2020 research and innovation program (ERC Grant number: 802736, MORPHEUS). Open Access funding enabled and organized by Projekt DEAL.
Dedicated to Hans-Ulrich Reissig on the occasion of his 75th birthday
Sauer M., Kany A. M., Götze S., Müller R., Beemelmanns C., Angew. Chem. Int. Ed. 2024, 63, e202416036. 10.1002/anie.202416036
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Pruett S. T., Bushnev A., Hagedorn K., Adiga M., Haynes C. A., Sullards M. C., Liotta D. C., Merrill A. H., J. Lipid Res. 2008, 49, 1621–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Merrill A. H., Chem. Rev. 2011, 111, 6387–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gelderblom W. C., Jaskiewicz K., Marasas W. F., Thiel P. G., Horak R. M., Vleggaar R., Kriek N. P., Appl. Env. Microbiol 1988, 54, 1806–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. VanMiddlesworth F., Giacobbe R. A., Lopez M., Garrity G., Bland J. A., Bartizal K., Fromtling R. A., Polishook J., Zweerink M., Edison A. M., J. Antibiot. 1992, 45, 861–867. [DOI] [PubMed] [Google Scholar]
- 5. Abed Rabbo M., Khodour Y., Kaguni L. S., Stiban J., Lipids Health Dis. 2021, 20, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ogretmen B., Nat. Rev. Cancer 2018, 18, 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pyne N. J., Adams D. R., Pyne S., Trends Pharmacol. Sci. 2017, 38, 581–591. [DOI] [PubMed] [Google Scholar]
- 8. McGinley M. P., Cohen J. A., Lancet 2021, 398, 1184–1194. [DOI] [PubMed] [Google Scholar]
- 9. Beemelmanns C., Woznica A., Alegado R. A., Cantley A. M., King N., Clardy J., J. Am. Chem. Soc. 2014, 136, 10210–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woznica A., Cantley A. M., Beemelmanns C., Freinkman E., Clardy J., King N., Proc. Natl. Acad. Sci. USA 2016, 113, 7894–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leichnitz D., Pflanze S., Beemelmanns C., Org. Biomol. Chem. 2019, 17, 6964–6969. [DOI] [PubMed] [Google Scholar]
- 12. Bissell A. U., Rautschek J., Hoefgen S., Raguž L., Mattern D. J., Saeed N., Janevska S., Jojić K., Huang Y., Kufs J. E., Herboeck B., Guo H., Hillmann F., Beemelmanns C., Valiante V., ACS Chem. Biol. 2022, 17, 386–394. [DOI] [PubMed] [Google Scholar]
- 13. Raguž L., Peng C.-C., Kaiser M., Görls H., Beemelmanns C., Angew. Chem. Int. Ed. 2022, 61, e202112616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leichnitz D., Peng C.-C., Raguž L., Rutaganira F. U. N., Jautzus T., Regestein L., King N., Beemelmanns C., Chemistry 2022, 28, e202103883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raguž L., Peng C.-C., Rutaganira F. U. N., Krüger T., Stanišić A., Jautzus T., Kries H., Kniemeyer O., Brakhage A. A., King N., Beemelmanns C., Angew. Chem. Int. Ed. 2022, 61, e202209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peng C.-C., Dormanns N., Regestein L., Beemelmanns C., RSC Adv. 2023, 13, 27520–27524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roman D., Meisinger P., Guillonneau R., Peng C.-C., Peltner L. K., Jordan P. M., Haensch V., Götze S., Werz O., Hertweck C., Chen Y., Beemelmanns C., Angew. Chem. Int. Ed. Engl. 2024, 63, e202401195. [DOI] [PubMed] [Google Scholar]
- 18. Bär A., Bär S. I., Schobert R., Org. Biomol. Chem. 2020, 18, 7565–7570. [DOI] [PubMed] [Google Scholar]
- 19. Molinski T. F., Biegelmeyer R., Stout E. P., Wang X., Frota M. L. C., Henriques A. T., J. Nat. Prod. 2013, 76, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mansoor T. A., Park T., Luo X., Hong J., Lee C. O., Jung J. H., Nat. Prod. Sci. 2007, 13, 247–250. [Google Scholar]
- 21. Sauer M., Beemelmanns C., Chem. Commun. 2022, 58 (64), 8990–8993. [DOI] [PubMed] [Google Scholar]
- 22. Koskinen P. M., Synthesis 1998, 8, 1075–1091. [Google Scholar]
- 23. Gao Y., He X., Ding F., Zhang Y., Synthesis 2016, 48,4017–4037. [Google Scholar]
- 24. Prévost M., St-Jean O., Guindon Y., J. Am. Chem. Soc. 2010, 132, 12433–12439. [DOI] [PubMed] [Google Scholar]
- 25. Xu K., Lai G., Zha Z., Pan S., Chen H., Wang Z., Chemistry 2012, 18, 12357–12362. [DOI] [PubMed] [Google Scholar]
- 26. Qin D.-D., Lai W.-H., Di Hu., Chen Z., Wu A.-A., Ruan Y.-P., Zhou Z.-H., Chen H.-B., Chemistry 2012, 18, 10515–10518. [DOI] [PubMed] [Google Scholar]
- 27. Loh C. C. J., Atodiresei I., Enders D., Chemistry 2013, 19, 10822–10826. [DOI] [PubMed] [Google Scholar]
- 28. Hauduc C., Bélanger G., J. Org. Chem. 2017, 82, 4703–4712. [DOI] [PubMed] [Google Scholar]
- 29. Fujii A., Hashiguchi S., Uematsu N., Ikariya T., Noyori R., J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar]
- 30. Moritz B. J., Mack D. J., Tong L., Thomson R. J., Angew. Chem. Int. Ed. 2014, 53, 2988–2991. [DOI] [PubMed] [Google Scholar]
- 31. Brown C. A., Ahuja V. K., J. Chem. Soc. Chem. Commun. 1973, 15, 553–554. [Google Scholar]
- 32. Rice L. B., J. Infect. Dis. 2008, 197, 1079–1081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.