

Abstract
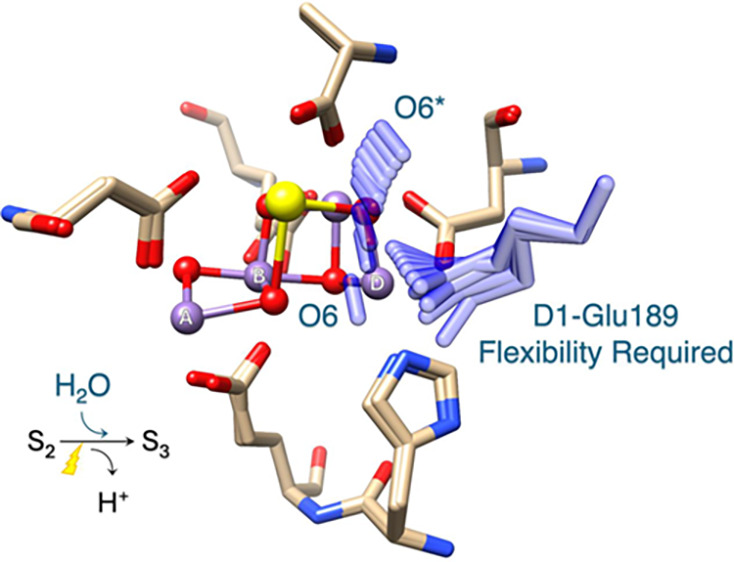
Photosynthetic water oxidation is a vital process responsible for producing dioxygen and supplying the energy necessary to sustain life on Earth. This fundamental reaction is catalyzed by the oxygen-evolving complex (OEC) of photosystem II, which houses the Mn4CaO5 cluster as its catalytic core. In this study, we specifically focus on the D1-Glu189 amino acid residue, which serves as a direct ligand to the Mn4CaO5 cluster. Our primary goal is to explore, using density functional theory (DFT), how the conformational flexibility of the D1-Glu189 side chain influences crucial catalytic processes, particularly the selection, positioning, and stabilization of a substrate water molecule within the OEC. Our investigation is based on a hypothesis put forth by Li et al. (Nature, 2024, 626, 670), which suggests that during the transition from the S2 to S3 state, a specific water molecule temporarily coordinating with the Ca ion, referred to as O6*, may exist as a hydroxide ion (OH–). Our results demonstrate a key mechanism by which the detachment of the D1-Glu189 carboxylate group from its coordination with the Ca ion allows the creation of a specialized microenvironment within the OEC that enables the selective attraction of O6* in its deprotonated form (OH–) and stabilizes it at the catalytic metal (MnD) site. Our findings indicate that D1-Glu189 is not only a structural ligand for the Ca ion but may also play an active and dynamic role in the catalytic process, positioning O6* optimally for its subsequent participation in the oxidation sequence during the water-splitting cycle.
1. Introduction
Most life activities in nature rely on photosynthesis, a fundamental biological process that harnesses solar energy to convert carbon dioxide into organic compounds essential for living organisms.1 Photosynthetic organisms such as cyanobacteria, algae, and higher plants have evolved remarkably sophisticated mechanisms to extract electrons from water, enabling their widespread distribution on Earth. Conversely, aerobic organisms have acquired the ability to use dioxygen, a byproduct of photosynthesis, through respiratory functions that resist the toxicity of oxygen and facilitate substantial energy transformations. This adaptation supports various metabolic processes that are essential for growth, development, and life maintenance, while also enabling the recycling of limited carbon between the inorganic environment and the biological realm using the boundless energy of sunlight, thereby sustaining the delicate balance of ecosystems. Given that photosynthesis is the only process in the biosphere capable of splitting water to produce dioxygen, deciphering its mechanism is of utmost importance, not only for its intrinsic scientific value but also for addressing environmental and energy challenges related to global warming and fossil fuel depletion.2,3
At the heart of the photosynthetic machinery is photosystem II (PSII), a multisubunit protein–cofactor complex located in the thylakoid membrane of chloroplasts and cyanobacterial cells. PSII plays a crucial role in supplying energized electrons necessary for the photosynthetic processes by splitting water molecules.4−7 The initiation of PSII function begins with a light-induced charge separation between a specialized chlorophyll dimer (P680) and a pheophytin (Pheo) electron acceptor, forming a P680+Pheo– radical pair. The electron that is transferred to Pheo– is then relayed through a series of redox reactions, moving first to a primary quinone electron acceptor and eventually to a secondary quinone electron acceptor on the stromal side of the thylakoid membrane. Simultaneously, the electron hole generated on P680+ is passed to the catalytic core of the oxygen-evolving complex (OEC), comprised of an oxo-bridged heteronuclear Mn4CaO5 cluster, on the luminal side of the membrane via a redox-active tyrosine residue, D1-Tyr161 (YZ). This cluster serves as the catalytic center for water oxidation, a complex four-electron oxidation process that is facilitated by YZ. The Mn4CaO5 cluster undergoes a cycle of oxidation states, known as the S-state cycle, which consists of five distinct intermediates denoted as Si states (i = 0–4). Each state represents a specific number of accumulated oxidizing equivalents or holes within the cluster, which are crucial for driving the water-splitting reaction.
Recent advancements made possible by serial femtosecond X-ray crystallography have provided unprecedented insights into the structural dynamics within PSII, particularly regarding the Mn cluster and its surrounding protein environment. These studies have offered high-resolution views of their structural evolution across key intermediates of the water-splitting cycle, namely the S1, S2, S3, and S0 states.8−15 With high spatial and temporal resolution, this technique has shed light not only on the static structural features but also on the intricate interplay of protein dynamics that govern critical functional events, such as water ingress and binding, proton transfer, and dioxygen formation. This is especially evident in the S2 to S3 transition; over a slower time scale, extensive shifts in amino acid side chains and backbone configurations take place, while faster, more localized displacements of the Mn and Ca ions within the Mn4CaO5 cluster occur simultaneously. One of the most significant events during this transition is the binding and activation of a new water molecule, referred to as O6 or OX, in the vicinity of the existing O5 oxygen atom within the Mn cluster. This event is a key step toward the formation of the O5–O6 bond, which is essential for the oxygen-evolving mechanism. The identity of O5 and O6 continues to be a subject of ongoing investigation. However, X-ray crystallography studies have measured O–O distances ranging from 1.9 to 2.2 Å,9−11,15 suggesting that O5 and O6 might exist predominantly in an oxyl–oxo form poised for bond formation or as a mixture of oxyl–oxo and hydroxo–oxo forms.
The OEC exhibits remarkable structural and electronic flexibility, enabling it to accommodate sequential oxidation state changes while efficiently binding and chemically transforming substrate water molecules throughout the catalytic cycle. This flexibility is largely due to the well-coordinated environment that supports the catalytic activity of the Mn4CaO5 cluster, comprising six carboxylate ligands (D1-Asp170, D1-Glu189, D1-Glu333, D1-Asp342, D1-Ala344, and CP43-Glu354), along with a histidine residue (D1-His332), as illustrated in Figure 1.16−18 Among these ligands, D1-Glu189 is particularly notable for its dynamic behavior during the S2 to S3 transition. In lower oxidation states, D1-Glu189 serves as a structural ligand by coordinating with both the Ca and MnD ions within the Mn4CaO5 cluster. However, upon the application of a second flash of light (2F) that advances the OEC to the S3 state, D1-Glu189 undergoes a displacement of approximately 0.2 Å away from the Ca ion, which appears to be a critical adjustment that facilitates the catalytic progression.8−12,14,15 A recent time-resolved crystallographic study conducted by Li et al. has further provided new insights into this transition. Their observations detected the emergence of new electron density, designated as O6*, within the coordination sphere of the Ca ion approximately 200 ns after 2F.15 Notably, this newly identified electron density gradually diminishes after around 200 μs, a time frame that coincides with the appearance and stabilization of increased O6 electron density at the MnD site within the Mn4CaO5 cluster. These findings suggest that the initially coordinated O6* is likely a water-derived species that is subsequently repositioned from its temporary location near the Ca ion to the MnD site, where it becomes incorporated as a substrate water molecule essential for catalytic activity.
Figure 1.
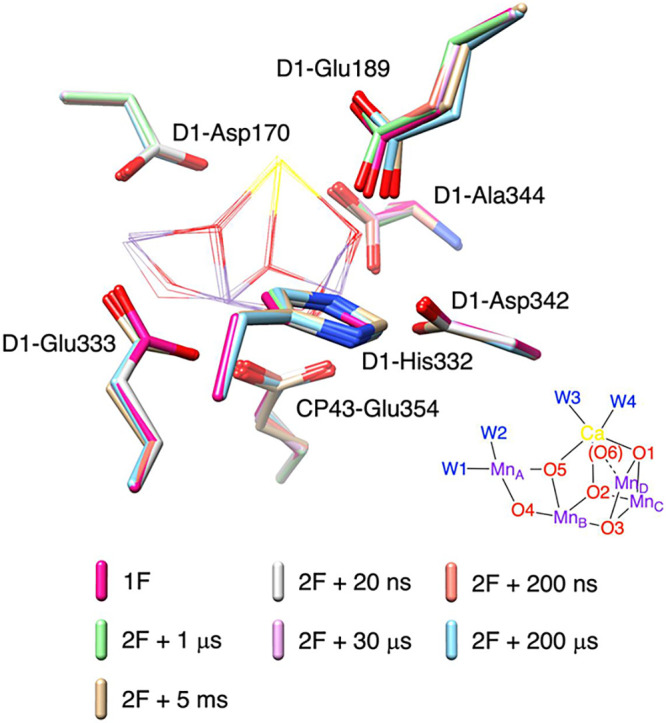
Conformational changes of amino acid residues within the primary coordination sphere of the Mn cluster observed experimentally during the S2 to S3 transition.15 Structural models captured at 1F (red), and time points at 20 ns (gray), 200 ns (orange), 1 μs (light green), 30 μs (pink), 200 μs (cyan), and 5 ms (gold) after 2F are superimposed. The inorganic Mn4CaO5 cluster is displayed in wireframe, while seven amino acid residues in the primary coordination sphere are shown in stick representation.
Based on these findings, the present study focuses on the specific role of D1-Glu189 within the OEC. Assuming, as proposed by Li et al., that the transient O6*species could exist in the form of a hydroxide ion (OH–),15 we use density functional theory (DFT) calculations to investigate how the conformational flexibility of the D1-Glu189 side chain contributes to the selection, guidance, and binding of the transient O6* within the OEC. Through this analysis, we aim to demonstrate that D1-Glu189 goes beyond serving merely as a coordinating ligand for the Ca and MnD ions and plays an active and dynamic role in catalytic processes that have been challenging to directly capture using current XFEL-based experimental techniques.
2. Computational Details
We constructed an OEC model using the XFEL crystallographic model of PSII (PDB ID 6JLL, monomer A), as illustrated in Figure S1. This model includes a total of 407 atoms comprising the inorganic Mn4CaO5 cluster, four terminal aqua/hydroxo ligands coordinated to Ca and MnA, an additional O6* ligand near the Ca site, 15 crystallographic water molecules, a chloride ion (Cl–), and the following amino acid residues: D1-Asp61, D1-Asn87, D1-Tyr161 (TyrZ), D1-Gln165, D1-Ser169, D1-Asp170, D1-Asn181, D1-Val185, D1-Phe182 (backbone only), D1-Glu189, D1-His190, D1-Asn298 (fragment), D2-Lys317 (fragment), D1-His332, D1-Glu333, D1-Ala336, D1-His337, D1-Asp342, D1-Ala344 (C-terminus), CP43-Glu354, CP43-Arg357, D1-Asn296, CP43-Leu401, CP43-Val410, and CP43-Ala411. Four Mn ions in the Mn4CaO5 cluster are labeled as MnA, MnB, MnC and MnD, as shown in Figures 1 and S1, which correspond to Mn4A, Mn3B, Mn2C, and Mn1D in the crystallographic structures. The oxidation state of MnD is assigned as MnIII, while MnA, MnB, and MnC are in the MnIV oxidation state. Geometry optimizations were performed with a spin multiplicity of 14 using the B3LYP functional19−21 enhanced by the D3 version of Grimme’s empirical dispersion correction and the Becke–Johnson (BJ) damping function,22,23 as implemented in Gaussian 16.24 The Los Alamos (LANL2DZ) pseudopotential basis set was used to represent Ca and Mn,25−27 while all other atoms were described using the 6-31G(d) basis set. These basis sets are labeled as BS1. The impact of steric effects resulting from the protein fold was taken into account by constraining the atomic positions of the backbone and the entire residues of D1-Gln165, D1-Asn298, D2-Lys317, D1-Asn296, CP43-Leu401, CP43-Val410, CP43-Ala411, and D1-Asn296, CP43-Leu401, CP43-Val410, and CP43-Ala411 located at the periphery of the model to their X-ray structure coordinates during the optimization of geometry. Single-point energies were computed employing the nonstandard B3LYP* functional, with a 5% reduction in the percentage of the Hartree–Fock exchange from 20 to 15%,28 in combination with the extended basis set comprising the Stuttgart/Dresden (SDD) pseudopotential basis set29,30 for Ca and Mn, and 6-311G(d,p) for all other atoms, designated as BS2. For these single-point calculations, the model system was immersed in a solvent using the IEFPCM model31 with a dielectric constant of 5.7 (corresponding to chlorobenzene) to approximate the low polarizability of the protein interior.
3. Results and Discussion
Our study focuses on the role of the specific amino acid residue D1-Glu189 within the OEC. This residue is characterized by its relatively long side chain terminating in a carboxylate group, which allows it to influence the local protein environment by modulating the hydrogen-bond network and coordinating interactions. The main objectives of this research are to understand how the flexible nature of this long side chain manifests within the OEC and to evaluate its impact on the selective attraction, precise guidance, and effective stabilization of a substrate water molecule. A key hypothesis underlying this investigation is that during the S2 to S3 transition, a specific water molecule (O6*) may temporarily interact with a Ca ion in the form of a hydroxide ion (OH–).15 This transient association is assumed to be essential for its eventual incorporation into the Mn4CaO5 cluster, making it crucial to investigate its detailed mechanism.
The side chain of the glutamate residue is connected with the Cα atom of the backbone via two sp3 hybridized carbon atoms (Cβ and Cγ), enabling it to potentially orient in various directions, as depicted in Figure 2. The side chain orientation can be described with a set of three dihedral angles (D1, D2, D3), with D1 somewhat restricted due to steric interference between Cγ and the main chain. In the actual protein environment, the ranges of D2 and D3 are further limited by steric hindrance from neighboring residues, water molecules, and cofactors. To investigate the range of motion of the D1-Glu189 side chain within the OEC environment, we examined energy changes with respect to the dihedral angles D2 and D3 using the hydrogen-bonding network (arrangement I) shown in Figure S1B. The result, presented in Figure 2, indicates that the spatial configuration of D1-Glu189 exhibits a broader range of motion than anticipated. The energy difference (ΔE), compared to a metastable structure with O6* (OH–) bound to Ca, as depicted in Figure S1B, remains relatively low (<∼10 kcal mol–1) while D2 varies from 120° to 210° and D3 from −80° to 30°. Notably, regions A and B in the energy landscape, as indicated in Figure 2, draw our attention, as they provide the opportunity for O6* to engage in physical interactions with MnD. In the region A, the carboxylate group of D1-Glu189 adopts a twisted conformation oriented toward D1-Val185, and entry into this area is achievable with an energy of around 7 kcal/mol or less. On the other hand, reaching the region B involves a twist in the carboxylate group of D1-Glu189 toward O6*. Crossing a dashed line in the energy landscape results in a substantial shift of around 20° in the D1 dihedral angle, causing the carboxylate group to extend well beyond O6*, leaving a substantial space between O6* and MnD. However, this structural motion encounters an energy barrier of around 20 kcal mol–1 or higher, making it an impractical motion. Even with considerable space between O6* and MnD in the regions A and B, no spontaneous movement of O6* toward the MnD site was observed, as illustrated in Figure 2. This indicates that a small amount of energy is likely required to rearrange or disrupt the surrounding hydrogen bonding network formed by five water molecules W10, W20, W21, W22, and W23 (arrangement I in Figure S1B), which stabilizes O6* in its anionic form (OH–) and traps it at the Ca site.
Figure 2.
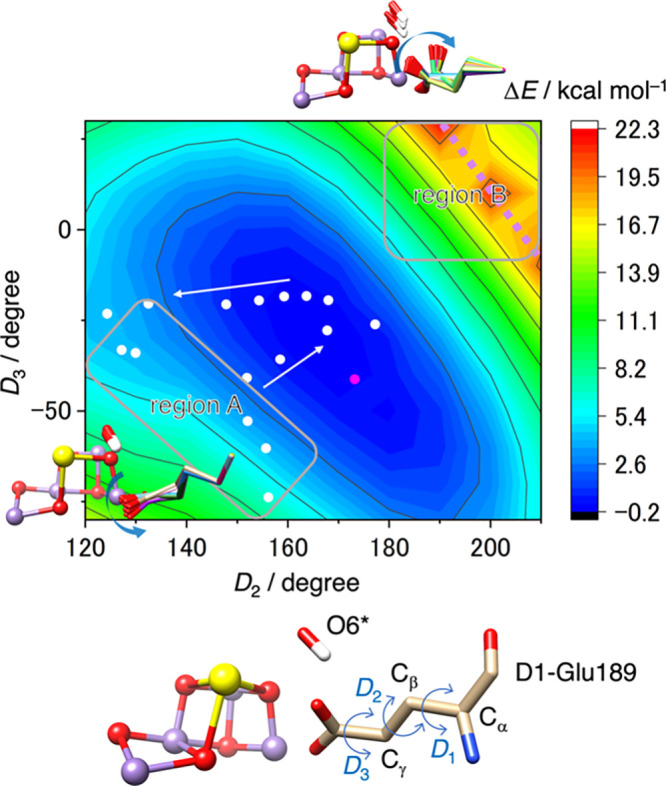
A potential energy surface representing the energy change (ΔE) as a function of the dihedral angles D2 and D3 of D1-Glu189. These calculations were based on the PDB coordinates (6JLL, monomer A),10 with an additional O6* remaining attached to the Ca ion of the Mn cluster. The relative energy ΔE is referenced to the metastable structure depicted in Figure S1B. White circles indicate the D2 and D3 values corresponding to the motion of the D1-Glu189 side chain when coupled with O6* binding to MnD (see Figure 3). A magenta circle represents the D2 and D3 values observed in the S3 state, in which O5 and O6 either form an oxyl-oxo bond or exist as bridging oxo and terminal hydroxo ligands.
When the Ca-bound O6* (OH–) attempts to establish a bonding interaction with MnD, it faces significant hindrance due to the presence of D1-Glu189, located 3.09 Å from Ca (200 ms after 2F).10 To gain insight into how D1-Glu189 navigates the transfer of O6* toward MnD, we deliberately inserted O6* into the tight space between Ca and D1-Glu189 by systematically adjusting the angle of Ca···O6*···O–-Glu189 from 60° to 160° in 10° increments. Figure 3A depicts the coordinated movements of D1-Glu189 and O6*, with their color-coding representing ΔE at each angle. The result clearly indicates that alterations in the side-chain conformation of D1-Glu189 play a guiding role, allowing O6* to enter the confined region between Ca and D1-Glu189. The dihedral angles D2 and D3 of the side chain shift, as illustrated by white circles in Figure 2, moving toward the region A along the most gradual slope of the potential surface when the motion of O6* is not coordinated. The energy reaches its maximum (ΔE ∼ 13 kcal mol–1) at around an angle of 140° and a Ca···O–-Glu189 distance of 4.81 Å, beyond which O6* spontaneously binds to MnD. The substantial conformational change of D1-Glu189 (approaching the region A in Figure 2) is essential, though not exaggerated, for enabling O6* to move. This is more clearly understood when Ca and O–-Glu189 oxygen atoms are represented as spheres, as in Figure 3C,D. Without a conformational change in D1-Glu189, there would be significant interference between D1-Glu189 and the moving O6*, as seen in Figure 3D. However, the extensive motion of D1-Glu189 creates a narrow passage that minimally allows O6* to pass through, as depicted in Figure 3C. As O6* moves, the position of Ca fluctuates, exhibiting a maximum displacement of 0.7 Å, consistent with the experimental observation of a single negative difference density attributed to Ca disorder at time points 30 μs to 5 ms after 2F.15 The MnD-bound O6*, now referred to as O6, serves not only as a hydrogen bond donor to D1-Glu189, but also continues to act as a hydrogen bond acceptor for W21 (following the water numbering in ref (15)), which has been pulled into a close proximity to O6 by the movement of O6*, as indicated in Figure 3E. The presence of W21 in this position hinders D1-Glu189 from returning to its original configuration. By manually disconnecting the hydrogen bond between W21 and O6 and restoring W21 to its original position, along with altering the hydrogen-bonding pattern from arrangement I (Figure S1B) to II (Figure S1C), we confirmed that D1-Glu189 can revert to its original conformation and energetically cluster around the observed configuration (in magenta), as demonstrated in Figure 3B. We believe that such a hydrogen-bonding reorganization is likely to occur easily at room temperature. The return motion of D1-Glu189 (escaping from the region A) appears to descend a steep slope on the potential energy surface when O6* is uncoordinated (Figure 2).
Figure 3.
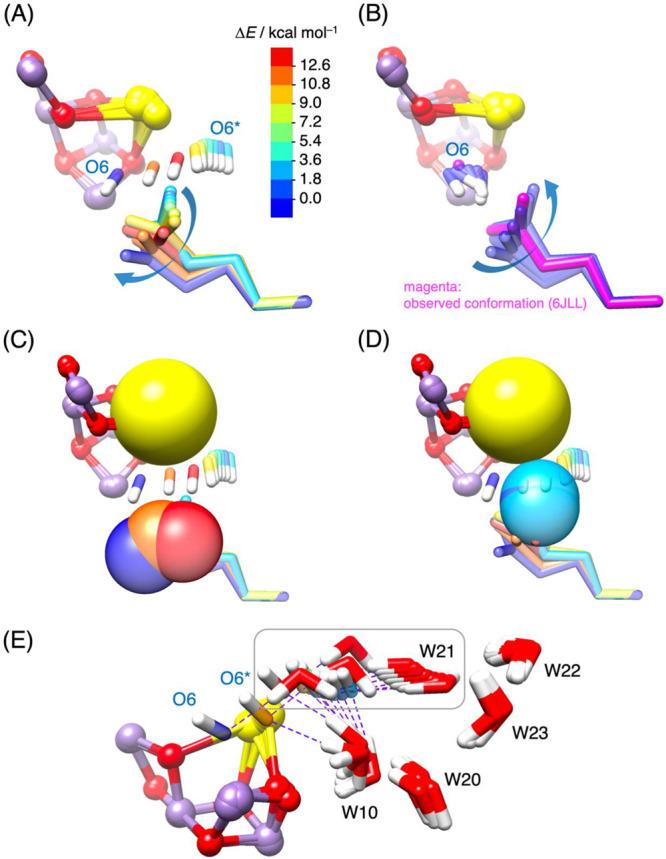
(A) Conformational changes in the side chain of D1-Glu189 as O6* moves toward MnD. The colors of O6* and D1-Glu189 correspond to the relative energy (ΔE) compared to the metastable structure shown in Figure S1B. (B) Conformational changes in the side chain of D1-Glu189 as O6* (renamed O6) binds to MnD, followed by the return of D1-Glu189 to its original orientation. The experimentally observed conformation in the S3 state (6JLL, monomer A)10 is highlighted in magenta. (C, D) A sphere representation illustrates Ca and the coordinating oxygen atoms of D1-Glu189 during O6* binding to MnD, aiding in the comprehension of the steric hindrance resulting from repulsive forces between neighboring Ca, D1-Glu189, and moving O6*. (E) Fluctuations of five water molecules (W10, W20, W21, W22, and W23) accompanying the movement of O6* toward MnD. Purple dashed lines represent hydrogen bonds between O6* and W21, as well as between O6* and W10, contributing to the stabilization of the anionic form of O6* (OH–). As O6* moves closer to MnD, W21 is also pulled toward the space between Ca and D1-Glu189 due to its interaction with O6*, while interference from D1-Glu189 (not shown) causes the hydrogen bond between O6* and W10 to break just before O6* binds to MnD (O6* → O6).
The intricate dynamics highlighted here emphasize the pivotal role of the conformational flexibility of D1-Glu189 in facilitating the movement of O6* and its functional importance in accessing to the S3 state with the hydroxo–oxo form (Stotal = 3) at room temperature.32 However, this mechanism alone does not account for the formation of the oxyl–oxo form (EPR silent, Stotal = 6),33−36 which is expected to emerge around 200 μs after 2F and become predominant in the low-temperature S3 state.10 One explanation for the discrepancy with the EPR results is the hypothesis that a reversible proton transfer may occur between O6 (OH–) bound to MnD and the carboxylate oxygen of D1-Glu189, which is positioned within hydrogen bonding distance. However, previous computational studies found no evidence to support such a proton transfer.37−39 To further investigate, we compared the relative stabilities of three chemical species in the S3 state: hydroxo–oxo, oxyl–oxo, and deprotonated hydroxo–oxo (i.e., oxo–oxo with protonated D1-Glu189), as illustrated in Figure S2. Our analysis revealed that the hydroxo–oxo and oxyl–oxo species are nearly equally stable, which is consistent with the EPR observation that two species (Stotal = 3 and EPR silent) could coexist or interconvert under physiological conditions.33 In contrast, the deprotonation of the hydroxo ligand to form the deprotonated hydroxo–oxo (oxo–oxo) with protonated D1-Glu189 was found to be energetically unfavorable, with an uphill energy cost exceeding 14 kcal mol–1 (Figure S2A), in agreement with the previous studies.38,39 This remains true even when considering potential changes in the dihedral angles (D1, D2, and D3) of the D1-Glu189 side chain (Figure S2B,C). Therefore, it is highly unlikely that D1-Glu189 is involved in storing the proton from O6 (OH–) or mediating its release into the bulk.
Many research groups have undertaken theoretical investigations to elucidate the sequence of water molecule binding and Mn oxidation during the S2 to S3 transition.40−48 The proposed sequences derived from the high-valent scheme,49,50 in which the Mn oxidation state in the S2 state is MnAIVMnBIVMnCIVMnDIII, intricately intertwine with the structural rearrangements of the Mn cluster. The basis for this deduction lies in the fact that a five-coordinated MnDIII cannot undergo oxidation unless one of the three amino acid ligands (D1-Glu189, D1-Asp342, and D1-His332) is replaced by a water-derived ligand. This conclusion remains robust, with no successful instances of realizing a five-coordinated MnDIV in any theoretical investigations. Building upon this computational evidence grounded in the high-valent scheme, we recently proposed a reaction scheme outlined in Figure 4.44,45 If there are no significant changes in the framework of the Mn cluster, known as the open cubane structure, the sequence comprises water binding as the initial step, followed by Mn oxidation, as demonstrated by the order indicated above the dashed line in Figure 4. While it has been conventionally assumed that the bound water originates from W3 or W2, the latest XFEL study suggests a novel possibility that O6* could serve as the direct source of the bound water.15 This proposition holds significant implications if O6* indeed represents an OH– ion, as illustrated in Figure 3. However, current knowledge does not entirely rule out the possibility that O6* might actually be H2O. The uncertainty surrounding this issue stems from the inherent characteristics of Ca2+, which typically exhibit relatively low Lewis acidity compared to Mn. In this scenario, the specificity of O6* may be diminished. As W2, W3, and O6* are all water molecules (H2O), it becomes unclear which one is responsible for the MnD-bound O6. The ambiguity arises because, in all cases, they produce the same final products, making it difficult to distinguish their individual contributions to the reaction. The propensity of Ca2+ and O6* (H2O) to dissociate at the current level of theory (B3LYP/BS1) further complicates the assessment of whether O6* (H2O) serves as the substrate for the ongoing cycle or the subsequent one. Therefore, the ambiguous nature of O6* (whether it is OH– or H2O) adds a layer of complexity to the mechanistic understanding of the S2–S3 transition, intertwined with the sequence of water binding and Mn oxidation, as well as the structural rearrangement of the Mn cluster. Considering the ligand-exchange activity of MnIII and the ligand-exchange inertness of MnIV, it is reasonable to suggest that the attachment of O6* to MnDIII may be reversible. This opens up the possibility for an alternative mechanistic pathway where Mn oxidation precedes water binding to the MnIV site. Such a pathway would require the transient formation of a closed cubane structure,51−53 as exemplified by the sequence indicated below the dashed line in Figure 4. This dual-pathway model suggests a level of mechanistic flexibility that enables the OEC to adapt to varying physiological conditions. The reversibility for O6* motion is in line with the persistent presence of residual density for O6*, which remains detectable even in the S3 state (at a time point of 5 ms after 2F),15 as well as membrane-inlet mass spectrometry (MIMS) measurements, which detected substrate water 16O/18O exchange after the incubation of the PSII in the S3 state in the presence of H218O.54−56
Figure 4.

A proposed reaction mechanism for the S2 to S3 transition, based on the high-valent scheme.
In these contexts, future research should adopt a broader perspective that explores the distinct structural characteristics extending beyond the immediate coordination spheres, while also assessing how these features interact with physiological conditions such as pH, temperature, and the presence of cryoprotectants or other environmental additives. The catalytic environment of the OEC differs across organisms like cyanobacteria and higher plants due to differences in protein composition, secondary ligand environments, and cofactors. These differences, combined with environmental factors, may influence the efficiency and mechanistic pathways involved in the uptake and binding of water molecules to the OEC.33,57−65 A comprehensive approach that considers these aspects may provide valuable insights into how nature optimizes water uptake and coordination, as well as associated processes such as electron transfer and proton transport, across different species and environmental contexts, ultimately advancing our understanding of biological water oxidation.
4. Conclusions
In this study, we focused on elucidating the specific role of the D1-Glu189 amino acid residue within the OEC to better understand how the conformational flexibility of its side chain influences the selective attraction, guidance, and stabilization of a substrate water molecule. This study is based on the currently tested hypothesis that during the S2 to S3 transition, a specific water molecule (O6*) may transiently associate with a Ca ion, stabilizing as a hydroxide ion (OH–).15 Our detailed analysis demonstrates a critical mechanism in the catalytic function of the OEC, specifically highlighting the detachment of the D1-Glu189 carboxylate group from its coordination with the Ca ion. This detachment triggers a significant reorganization of the local protein environment, resulting in the formation of a transient but well-defined binding passage within the OEC. This passage serves as a selective gateway, effectively attracting O6* in its deprotonated form from the surrounding pool of water molecules and directing its transition from a loosely coordinated state at the Ca ion to a more stable, catalytically active position at the MnD site. Once at this site, O6* undergoes further structural adjustments, acquiring the necessary chemical properties to act as a substrate water molecule, now referred to as O6. This optimal positioning of O6 at the catalytic center is crucial for enabling the series of subsequent oxidation steps required for the progression of the water-splitting catalytic cycle. These findings suggest that the conformational flexibility of D1-Glu189 is not merely a passive characteristic but could actively contribute to shaping the specialized microenvironment within the OEC and supporting key catalytic processes.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number JP22K05317. The computation was performed using Research Center for Computational Science, Okazaki, Japan.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c09981.
The authors declare no competing financial interest.
Supplementary Material
References
- Photosystem II: The Light Driven Water: Plastoquinone Oxidoreductase; Wydrzynski T. J.; Satoh K., Eds.; Springer: Dordrecht, The Netherlands, 2005. [Google Scholar]
- Barber J.; Tran P. D. From natural to artificial photosynthesis. J. R. Soc., Interface 2013, 10, 20120984. 10.1098/rsif.2012.0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore J. D.; Crabtree R. H.; Brudvig G. W. Molecular catalysts for water oxidation. Chem. Rev. 2015, 115, 12974–13005. 10.1021/acs.chemrev.5b00122. [DOI] [PubMed] [Google Scholar]
- McEvoy J. P.; Brudvig G. W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006, 106, 4455–4483. 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- Cox N.; Pantazis D. A.; Neese F.; Lubitz W. Biological water oxidation. Acc. Chem. Res. 2013, 46, 1588–1596. 10.1021/ar3003249. [DOI] [PubMed] [Google Scholar]
- Yano J.; Yachandra V. K. Mn4Ca cluster in photosynthesis: where and how water is oxidized to dioxygen. Chem. Rev. 2014, 114, 4175–4205. 10.1021/cr4004874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J.-R. The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu. Rev. Plant Biol. 2015, 66, 23–48. 10.1146/annurev-arplant-050312-120129. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Sugahara M.; Kubo M.; Nakajima Y.; Nakane T.; Yamashita K.; Umena Y.; Nakabayashi M.; Yamane T.; Nakano T.; Suzuki M.; Masuda T.; Inoue S.; Kimura T.; Nomura T.; Yonekura S.; Yu L.-J.; Sakamoto T.; Motomura T.; Chen J.-H.; Kato Y.; Noguchi T.; Tono K.; Joti Y.; Kameshima T.; Hatsui T.; Nango E.; Tanaka R.; Naitow H.; Matsuura Y.; Yamashita A.; Yamamoto M.; Nureki O.; Yabashi M.; Ishikawa T.; Iwata S.; Shen J.-R. Light-induced structural changes and the site of O = O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. 10.1038/nature21400. [DOI] [PubMed] [Google Scholar]
- Kern J.; Chatterjee R.; Young I. D.; Fuller F. D.; Lassalle L.; Ibrahim M.; Gul S.; Fransson T.; Brewster A. S.; Alonso-Mori R.; Hussein R.; Zhang M.; Douthit L.; de Lichtenberg C.; Cheah M. H.; Shevela D.; Wersig J.; Seuffert I.; Sokaras D.; Pastor E.; Weninger C.; Kroll T.; Sierra R. G.; Aller P.; Butryn A.; Orville A. M.; Liang M.; Batyuk A.; Koglin J. E.; Carbajo S.; Boutet S.; Moriarty N. W.; Holton J. M.; Dobbek H.; Adams P. D.; Bergmann U.; Sauter N. K.; Zouni A.; Messinger J.; Yano J.; Yachandra V. K. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. 10.1038/s41586-018-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Yamashita K.; Nakajima Y.; Ueno G.; Li H.; Yamane T.; Hirata K.; Umena Y.; Yonekura S.; Yu L.-J.; Murakami H.; Nomura T.; Kimura T.; Kubo M.; Baba S.; Kumasaka T.; Tono K.; Yabashi M.; Isobe H.; Yamaguchi K.; Yamamoto M.; Ago H.; Shen J.-R. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser. Science 2019, 366, 334–338. 10.1126/science.aax6998. [DOI] [PubMed] [Google Scholar]
- Ibrahim M.; Fransson T.; Chatterjee R.; Cheah M. H.; Hussein R.; Lassalle L.; Sutherlin K. D.; Young I. D.; Fuller F. D.; Gul S.; Kim I.-S.; Simon P. S.; de Lichtenberg C.; Chernev P.; Bogacz I.; Pham C. C.; Orville A. M.; Saichek N.; Northen T.; Batyuk A.; Carbajo S.; Alonso-Mori R.; Tono K.; Owada S.; Bhowmick A.; Bolotovsky R.; Mendez D.; Moriarty N. W.; Holton J. M.; Dobbek H.; Brewster A. S.; Adams P. D.; Sauter N. K.; Bergmann U.; Zouni A.; Messinger J.; Kern J.; Yachandra V. K.; Yano J. Untangling the sequence of events during the S2 → S3 transition in photosystem II and implications for the water oxidation mechanism. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 12624–12635. 10.1073/pnas.2000529117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein R.; Ibrahim M.; Bhowmick A.; Simon P. S.; Chatterjee R.; Lassalle L.; Doyle M.; Bogacz I.; Kim I.-S.; Cheah M. H.; Gul S.; de Lichtenberg C.; Chernev P.; Pham C. C.; Young I. D.; Carbajo S.; Fuller F. D.; Alonso-Mori R.; Batyuk A.; Sutherlin K. D.; Brewster A. S.; Bolotovsky R.; Mendez D.; Holton J. M.; Moriarty N. W.; Adams P. D.; Bergmann U.; Sauter N. K.; Dobbek H.; Messinger J.; Zouni A.; Kern J.; Yachandra V. K.; Yano J. Structural dynamics in the water and proton channels of photosystem II during the S2 to S3 transition. Nat. Commun. 2021, 12, 6531. 10.1038/s41467-021-26781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Nakajima Y.; Nomura T.; Sugahara M.; Yonekura S.; Chan S. K.; Nakane T.; Yamane T.; Umena Y.; Suzuki M.; Matsuda T.; Motomura T.; Naitow H.; Matsuura Y.; Kimura T.; Tono K.; Owada S.; Joti Y.; Tanaka R.; Nango E.; Akita F.; Kubo M.; Iwata S.; Shen J.-R.; Suga M. Capturing structural changes of the S1 to S2 transition of photosystem II using time-resolved serial femtosecond crystallography. IUCrJ. 2021, 8, 431–443. 10.1107/S2052252521002177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick A.; Hussein R.; Bogacz I.; Simon P. S.; Ibrahim M.; Chatterjee R.; Doyle M. D.; Cheah M. H.; Fransson T.; Chernev P.; Kim I.-S.; Makita H.; Dasgupta M.; Kaminsky C. J.; Zhang M.; Gätcke J.; Haupt S.; Nangca I. I.; Keable S. M.; Aydin A. O.; Tono K.; Owada S.; Gee L. B.; Fuller F. D.; Batyuk A.; Alonso-Mori R.; Holton J. M.; Paley D. W.; Moriarty N. W.; Mamedov F.; Adams P. D.; Brewster A. S.; Dobbek H.; Sauter N. K.; Bergmann U.; Zouni A.; Messinger J.; Kern J.; Yano J.; Yachandra V. K. Structural evidence for intermediates during O2 formation in photosystem II. Nature 2023, 617, 629–636. 10.1038/s41586-023-06038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Nakajima Y.; Nango E.; Owada S.; Yamada D.; Hashimoto K.; Luo F.; Tanaka R.; Akita F.; Kato K.; Kang J.; Saitoh Y.; Kishi S.; Yu H.; Matsubara N.; Fujii H.; Sugahara M.; Suzuki M.; Masuda T.; Kimura T.; Thao T. N.; Yonekura S.; Yu L.-J.; Tosha T.; Tono K.; Joti Y.; Hatsui T.; Yabashi M.; Kubo M.; Iwata S.; Isobe H.; Yamaguchi K.; Suga M.; Shen J.-R. Oxygen-evolving photosystem II structures during S1–S2–S3 transitions. Nature 2024, 626, 670–677. 10.1038/s41586-023-06987-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umena Y.; Kawakami K.; Shen J.-R.; Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- Tanaka A.; Fukushima Y.; Kamiya N. Two different structures oft he oxygen-evolving complex in the same polypeptide frameworks of photosystem II. J. Am. Chem. Soc. 2017, 139, 1718–1721. 10.1021/jacs.6b09666. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Hirata K.; Ueno G.; Murakami H.; Nakajima Y.; Shimizu T.; Yamashita K.; Yamamoto M.; Ago H.; Shen J.-R. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correction-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and accurate Ab Initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effects of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]
- Wadt W. R.; Hay P. J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. 10.1063/1.448800. [DOI] [Google Scholar]
- Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. 10.1063/1.448975. [DOI] [Google Scholar]
- Reiher M.; Salomon O.; Hess B. A. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. 10.1007/s00214-001-0300-3. [DOI] [Google Scholar]
- Dolg M.; Wedig U.; Stoll H.; Preuss H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. 10.1063/1.452288. [DOI] [Google Scholar]
- Kaupp M.; Schleyer P. v. R.; Stoll H.; Preuss H. Pseudopotential approaches to Ca, Sr, and Ba hydrides. Why are some alkaline earth MX2 compounds bent?. J. Chem. Phys. 1991, 94, 1360–1366. 10.1063/1.459993. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Cox N.; Retegan M.; Neese F.; Pantazis D. A.; Boussac A.; Lubitz W. Electronic structure of the oxygen-evolving complex in photosystem II prior to O–O bond formation. Science 2014, 345, 804–808. 10.1126/science.1254910. [DOI] [PubMed] [Google Scholar]
- Boussac A.; Rutherford A. W.; Sugiura M. Electron transfer pathways from the S2-state to the S3-state either after a Ca2+/Sr2+ or Cl–/I– exchange in photosystem II from Thermosynechococcus elongatus. Biochim. Biophys. Acta 2015, 1847, 576–586. 10.1016/j.bbabio.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Isobe H.; Shoji M.; Shen J.-R.; Yamaguchi K. Chemical equilibrium models for the S3 state of the oxygen-evolving complex of photosystem II. Inorg. Chem. 2016, 55, 502–511. 10.1021/acs.inorgchem.5b02471. [DOI] [PubMed] [Google Scholar]
- Isobe H.; Shoji M.; Suzuki T.; Shen J.-R.; Yamaguchi K. Spin, valence, and structural isomerism in the S3 state of the oxygen-evolving complex of photosystem II as a manifestation of multimetallic cooperativity. J. Chem. Theory Comput. 2019, 15, 2375–2391. 10.1021/acs.jctc.8b01055. [DOI] [PubMed] [Google Scholar]
- Rummel F.; O’Malley P. J. How nature makes O2: an electronic level mechanism for water oxidation in photosynthesis. J. Phys. Chem. B 2022, 126, 8214–8221. 10.1021/acs.jpcb.2c06374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narzi D.; Capone M.; Bovi D.; Guidoni L. Evolution from S3 to S4 states of the oxygen-evolving complex in photosystem II monitored by quantum mechanics/molecular mechanics (QM/MM) dynamics. Chem. – Eur. J. 2018, 24, 10820–10828. 10.1002/chem.201801709. [DOI] [PubMed] [Google Scholar]
- Mandal M.; Saito K.; Ishikita H. The nature of the short oxygen–oxygen distance in the Mn4CaO6 complex of photosystem II crystals. J. Phys. Chem. Lett. 2020, 11, 10262–10268. 10.1021/acs.jpclett.0c02868. [DOI] [PubMed] [Google Scholar]
- Malcomson T.; Rummel F.; O’Malley P. Hey ho, where’d the proton go? Final deprotonation of O6 within the S3 state of photosystem II. J. Photochem. Photobiol., B 2024, 257, 112946 10.1016/j.jphotobiol.2024.112946. [DOI] [PubMed] [Google Scholar]
- Narzi D.; Bovi D.; Guidoni L. Pathway for Mn-cluster oxidation by tyrosine-Z in the S2 state of photosystem II. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 8723–8728. 10.1073/pnas.1401719111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retegan M.; Krewald V.; Mamedov F.; Neese F.; Lubitz W.; Cox N.; Pantazis D. A. A five-coordinate Mn(IV) intermediate in biological water oxidation: spectroscopic signature and a pivot mechanism for water binding. Chem. Sci. 2016, 7, 72–84. 10.1039/C5SC03124A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E. M. The S2 to S3 transition for water oxidation in PSII (photosystem II), revisited. Phys. Chem. Chem. Phys. 2018, 20, 22926–22931. 10.1039/C8CP03720E. [DOI] [PubMed] [Google Scholar]
- Pushkar Y.; Ravari A. K.; Jensen S. C.; Palenik M. Early binding of substrate oxygen is responsible for a spectroscopically distinct S2 state in photosystem II. J. Phys. Chem. Lett. 2019, 10, 5284–5291. 10.1021/acs.jpclett.9b01255. [DOI] [PubMed] [Google Scholar]
- Isobe H.; Shoji M.; Suzuki T.; Shen J.-R.; Yamaguchi K. Exploring reaction pathways for the structural rearrangements of the Mn cluster induced by water binding in the S3 state of the oxygen evolving complex of photosystem II. J. Photochem. Photobiol., A 2021, 405, 112905 10.1016/j.jphotochem.2020.112905. [DOI] [Google Scholar]
- Isobe H.; Shoji M.; Suzuki T.; Shen J.-R.; Yamaguchi K. Roles of the flexible primary coordination sphere of the Mn4CaOX cluster: what are the immediate decay products of the S3 state?. J. Phys. Chem. B 2022, 126, 7212–7228. 10.1021/acs.jpcb.2c02596. [DOI] [PubMed] [Google Scholar]
- Allgöwer F.; Gamiz-Hernandez A. P.; Rutherford A. W.; Kaila V. R. Molecular principles of redox-coupled protonation dynamics in photosystem II. J. Am. Chem. Soc. 2022, 144, 7171–7180. 10.1021/jacs.1c13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Messinger J.; Kloo L.; Sun L. Reversible structural isomerization of nature’s water oxidation catalyst prior to O–O bond formation. J. Am. Chem. Soc. 2022, 144, 11736–11747. 10.1021/jacs.2c03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Yang K. R.; Long Z.; Armstrong W. H.; Brudvig G. W.; Batista V. S. Water ligands regulate the redox leveling mechanism of the oxygen-evolving complex of the photosystem II. J. Am. Chem. Soc. 2024, 146, 15986–15999. 10.1021/jacs.4c02926. [DOI] [PubMed] [Google Scholar]
- Krewald V.; Retegan M.; Cox N.; Messinger J.; Lubitz W.; DeBeer S.; Neese F.; Pantazis D. A. Metal oxidation states in biological water splitting. Chem. Sci. 2015, 6, 1676–1695. 10.1039/C4SC03720K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah M. H.; Zhang M.; Shevela D.; Mamedov F.; Zouni A.; Messinger J. Assessment of the manganese cluster’s oxidation state via photoactivation of photosystem II microcrystals. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 141–145. 10.1073/pnas.1915879117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe H.; Shoji M.; Yamanaka S.; Umena Y.; Kawakami K.; Kamiya K.; Shen J.-R.; Yamaguchi K. Theoretical illumination of water-inserted structures of the CaMn4O5 cluster in the S2 and S3 states of the oxygen-evolving complex of photosystem II: full geometry optimizations by B3LYP hybrid density functional. Dalton Trans. 2012, 41, 13727–13740. 10.1039/c2dt31420g. [DOI] [PubMed] [Google Scholar]
- Pantazis D. A.; Ames W.; Cox N.; Lubitz W.; Neese F. Two interconvertible structures that explain the spectroscopic properties of the oxygen-evolving complex of photosystem II in the S2 state. Angew. Chem., Int. Ed. 2012, 51, 9935–9940. 10.1002/anie.201204705. [DOI] [PubMed] [Google Scholar]
- Bovi D.; Narzi D.; Guidoni L. The S2 state of the oxygen-evolving complex of photosystem II explored by QM/MM dynamics: spin surfaces and metastable states suggest a reaction path towards the S3 state. Angew. Chem., Int. Ed. 2013, 52, 11744–11749. 10.1002/anie.201306667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier W.; Wydrzynski T. Oxygen ligand exchange at metal sites–implications for the O2 evolving mechanism of photosystem II. Biochim. Biophys. Acta 2001, 1503, 197–209. 10.1016/S0005-2728(00)00225-5. [DOI] [PubMed] [Google Scholar]
- Hillier W.; Wydrzynski T. 18O-water exchange in photosystem II: substrate binding and intermediates of the water splitting cycle. Coord. Chem. Rev. 2008, 252, 306–317. 10.1016/j.ccr.2007.09.004. [DOI] [Google Scholar]
- Cox N.; Messinger J. Reflections on substrate water and dioxygen formation. Biochim. Biophys. Acta 2013, 1827, 1020–1030. 10.1016/j.bbabio.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Haddy A. EPR spectroscopy of the manganese cluster of photosystem II. Photosynth. Res. 2007, 92, 357–368. 10.1007/s11120-007-9194-9. [DOI] [PubMed] [Google Scholar]
- Pokhrel R.; Brudvig G. W. Oxygen-evolving complex of photosystem II: correlating structure with spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 11812–11821. 10.1039/c4cp00493k. [DOI] [PubMed] [Google Scholar]
- Boussac A.; Ugur I.; Marion A.; Sugiura M.; Kaila V. R. I.; Rutherford A. W. The low spin–high spin equilibrium in the S2-state of the water oxidizing enzyme. Biochim. Biophys. Acta 2018, 1859, 342–356. 10.1016/j.bbabio.2018.02.010. [DOI] [PubMed] [Google Scholar]
- Boussac A. Temperature dependence of the high-spin S2 to S3 transition in photosystem II. Biochim. Biophys. Acta 2019, 1860, 508–518. 10.1016/j.bbabio.2019.05.001. [DOI] [PubMed] [Google Scholar]
- Zimmermann J. L.; Rutherford A. W. Electron paramagnetic resonance properties of the S2 state of the oxygen-evolving complex of photosystem II. Biochemistry 1986, 25, 4609–4615. 10.1021/bi00364a023. [DOI] [Google Scholar]
- Beck W. F.; Brudvig G. W. Binding of Amines to the O2-evolving center of photosystem II. Biochemistry 1986, 25, 6479–6486. 10.1021/bi00369a021. [DOI] [PubMed] [Google Scholar]
- Boussac A.; Rutherford A. W.; Styring S. Interaction of ammonia with the water splitting enzyme of photosystem II. Biochemistry 1990, 29, 24–32. 10.1021/bi00453a003. [DOI] [PubMed] [Google Scholar]
- Retegan M.; Pantazis D. A. Interaction of methanol with the oxygen-evolving complex: atomistic models, channel identification, species dependence, and mechanistic implication. Chem. Sci. 2016, 7, 6463–6476. 10.1039/C6SC02340A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosou M.; Pantazis D. A. Comprehensive evaluation of models for ammonia binding to the oxygen evolving complex of photosystem II. J. Phys. Chem. B 2024, 128, 1333–1349. 10.1021/acs.jpcb.3c06304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.