

Abstract
We describe the development of a programmable microfluidic cell culture array that integrates on-chip generation of different drug concentrations with parallel culture of cells for drug candidate screening applications. The device has 64 individually addressable cell culture chambers in which, cells can be cultured and exposed to different drugs, either sequentially (eight concentrations of one drug followed by eight concentrations of a second drug) or simultaneously to 64 pair-wise concentration combinations of two drugs. For sequential exposure, a simple microfluidic diffusive mixer is used to generate different concentrations of drugs from two inputs. For generation of 64 pair-wise combinations from two drug inputs, a time dependent variable concentration scheme is used in conjunction with the simple diffusive mixer to generate the desired combinations. The generation of drug combinations and exposure to specific cell culture chambers is controlled using a LabView interface. The utility of this platform is demonstrated for inducing loss of viability of PC3 prostate cancer cells using combinations of either doxorubicin or mitoxantrone with TRAIL (TNF-alpha Relatd Apoptosis Inducing Ligand) either in a sequential or simultaneous format. Our results demonstrate that the device can capture the synergy between different sensitizer drugs and TRAIL and demonstrates the potential of the microfluidic cell array for screening and optimizing combinatorial drug treatments for cancer therapy.
INTRODUCTION
It is widely accepted that chemotherapeutic drugs are most effective when administered together, either sequentially or together in combination. Since different chemotherapeutic agents exert their effects through different mechanism(s), the use of combination therapies can potentially lead to increased efficacies at significantly lower doses and side-effects, compared to what would be observed with a high dose of a single chemotherapeutic agent. Combination therapies have been widely investigated and applied for curative and palliative care in prostate cancer, particularly in cases of hormone refractory and metastatic disease states1.
One recently investigated modality for the destruction of prostate cancer cells involves the induction of apoptosis through administration of the Tumor Necrosis Factor-related Apoptosis Inducing Ligand (TRAIL). TRAIL is a member of the Tumor Necrosis Factor (TNF) super-family of cytokines that induces apoptosis in cancer cells upon specific binding to death receptors (DR) 4 and 5 on the cell surface2. Recombinant TRAIL induces apoptosis in a variety of human cancer cell lines3–5, while causing minimal toxicity in non-malignant cells. TRAIL has also demonstrated potent anti-tumor activity in a number of xenograft models including those of colon and breast carcinomas6, 7. Despite the promise of TRAIL in cancer therapy, many tumor cells, including prostate cancer cells, are inherently resistant or acquire resistance to TRAIL-mediated apoptosis. As a result, therapeutic strategies involving DNA-damaging radiotherapy8, genotoxins9, and peptides10 have been investigated for enhancing cancer cell sensitivity to TRAIL9 and / or agonistic antibodies against DR4/DR511. Doxorubicin is one such agent that has been used to sensitize cells to subsequent TRAIL administration12, 13 so that efficacy can be improved at lower TRAIL concentrations. We have recently identified an FDA-approved drug, mitoxantrone, as having potent sensitization activity in prostate and pancreatic cancer cell lines14.
Identification of effective drug combinations in vitro (against cultured cells) requires the development of a large pool of potential drug combinations (“leads”) that can be pursued in animal studies or clinical trials. However, the generation of this pool of potential combinations is often a limiting step in the drug development process. This is because the efficacy of chemotherapeutic drugs, as well as interaction between different drugs, is dose-dependent6, 15. Therefore, the experimental space that needs to be covered for identification of potential combinations is quite large and requires expensive high-throughput screening equipment (e.g., automated liquid dispensing systems16) as well as large amounts of the different drugs.
A major focus of microfluidic chip-based drug screening has been the development of high-throughput assays for identifying potential combinations17, 18. Several groups19–22 have reported the development of different cell culture platforms for investigating cell responses and gene expression profiling. While these systems offer the ability to culture cells in a parallel manner, they are not suitable for screening and identifying combination therapy candidates as they do not facilitate the on-chip generation of concentration ranges needed for such experiments. Neils et al.23 demonstrated a 4 × 4 combinatorial mixing system that accepted two inputs and generated 16 output streams, but this does not have sufficient throughput and was not used for screening studies. More recently, Jang et al.24 developed a microfluidic active injection system for generating up to 100 combinatorial dilutions from two input streams. While this system achieves a sufficient level of throughput needed for microfluidic screening applications, their design does not allow for each chamber to be completely isolated as medium flows from one chamber to another in series. This leads to metabolites and other secreted by-products being transferred from one chamber to another, which in turn, can impact the response of the cells being studied. Importantly, while potential drug screening applications are discussed, this system has also not been applied for screening studies.
Here, we report the development of a programmable microfluidic cell array that integrates parallel culture of cells on-chip with generation of drug concentrations. Our microfluidic device has 64 individually addressable cell culture chambers in which, cells can be cultured and exposed to different drugs either sequentially or simultaneously to pair-wise concentration combinations. The utility of this platform is demonstrated by first sensitizing PC3 human bone-metastatic prostate cancer cells to TRAIL-induced death using two clinically-relevant sensitizer drugs (doxorubicin or mitoxantrone) or by simultaneously exposing cancer cells to pair-wise combinations of a sensitizer and TRAIL.
MATERIALS AND METHODS
Cells and Reagents
Human prostate cancer PC3 cells (ATCC, VA) were cultured and propagated in RPMI medium with 10% fetal bovine serum and 100 U/mL penicillin/100 μg/μL streptomycin at 37°C under 5% CO2 and 95% atmospheric air using standard ATCC protocols. Doxorubicin was purchased from MP Biomedicals and stored at a stock concentration of 3mM. TRAIL was purchased from R&D Systems (Minneapolis, MN). Mitoxantrone was purchased from Sigma.
Design and Fabrication of Microfluidic devices
Microfluidic devices were fabricated in the Materials Characterization Facility at Texas A&M University using routine soft lithography methods as previously described25, 26. The device consists of an upstream concentration generation module connected to an array of downstream cell culture chambers (Figure 1). The cell culture module consists of eight rows of chambers, each containing eight distinct culture chambers, for a total of 64 chambers. Access to each row of eight cell culture chambers was individually controlled using a valve array through a pneumatic channel, such that a specific combination of drug molecules can be delivered to each row.
Figure 1. Schematic of the microfluidic array.
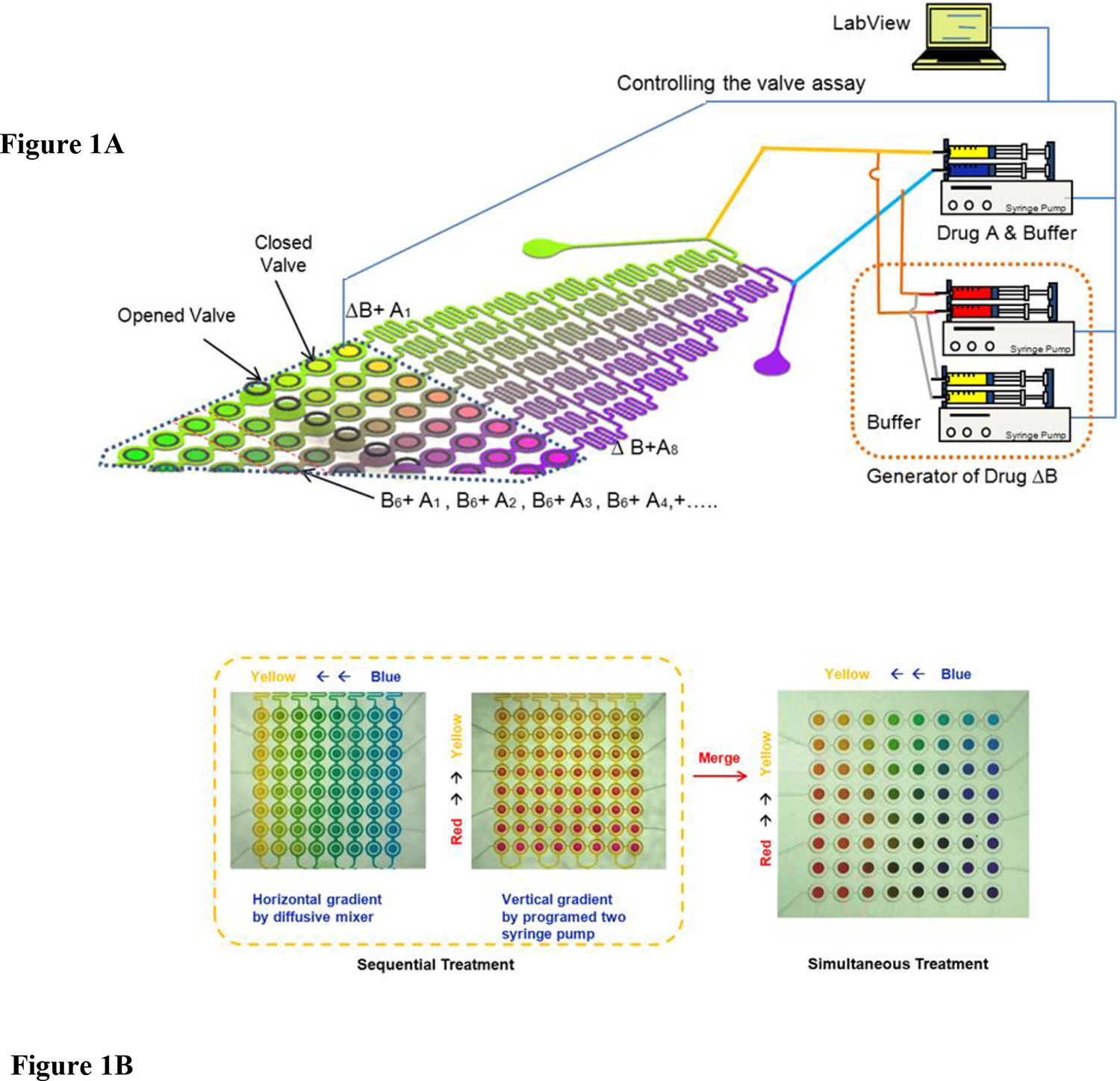
(A) Different concentrations of Drugs A and B are generated in the diffusive gradient mixer, and used, either sequentially or in combination, to perfuse cells cultured in downstream microchambers. The mixing operation for generating different drug combinations and the opening and closing of valves for perfusing cells in the microchambers are controlled though a LabView interface. (B) Depiction of the range of concentrations that can be generated for sequential and simultaneous treatment using color dyes. In the left panel, yellow and blue color dye solutions (representing the minimum and maximum concentrations of a drug) are mixed to generate eight outlet concentrations (“horizontal gradient” of colors between yellow and blue), and represent the gradient used in sequential exposure experiments. In the middle panel, yellow and red streams (representing the minimum and maximum concentrations of the second drug) are mixed together to generate a “vertical gradient” of colors between yellow and red. Merging the two color gradients (vertical direction concentration gradient: yellow to blue; horizontal direction concentration gradient: yellow to red) yields an array of pair-wise combinations, and represents the gradient used in simultaneous exposure experiments (right panel).
The microfluidic device consisted of two PDMS layers – a network layer containing the fluidic channels required for generating different concentrations of drug molecules and the cell culture chambers, and a pneumatic layer for controlling fluid access to cell culture chambers – that were assembled and bonded to a glass slide. A thin PDMS (Sylgard 184, Dow corning) membrane was fabricated by casting the pre-polymer on a master mold, spinning at 1200 rpm for 1 minute, followed by curing the pre-polymer. The PDMS layer for fluidic channels and cell culture micro-chambers was 150μm thick and the pneumatic layer for controlling the micro-chamber was 4 mm in thickness. The channel height for fluidic channels and cell culture micro-chambers was 100μm tall, and the height of pneumatic layer for controlling the micro-chamber was 200μm. Before replicating, the SU-8 mold was treated with tridecafluoro-1,2,2,2-tetrahydrooctyl-1-trichlorosilane so that the PDMS membrane could be peeled off from the SU-8 mold without any creating any defects.
Microfluidic devices were assembled by exposing the different PDMS layers and glass slides to oxygen plasma (150 mTorr, 100 W, 40 sec) and bonding them in a reactive ion etcher. The fluidic layer membrane was aligned and bonded to a PDMS bas-relief plate. In order to enable actuation of the PDMS micro-chamber, tubing was connected to the pneumatic layer and vacuum was applied when the PDMS structure was bonded to glass to prevent irreversible bonding between the PDMS micro-chamber and the glass. This protocol facilitates fabrication of the two layers without the need for glass etching27 or formation of complex multilayer structures28. Access ports were punched into the pneumatic layer prior to bonding to the fluidic layer. Holes for access to the fluidic layer were punched prior to bonding to the glass slide.
The valve array consisted of eight independently controllable valve groups, with each group containing eight valves that were operated simultaneously. The chamber regions can be either isolated (or exposed) to fluid by lowering (or raising) a ~ 25 nL volume microchamber to the channel bottom surface using a pneumatic source. The chamber valve system was designed such that the center of the valve is fixed and only the boundary wall moves during operation. This method is advantageous as it minimizes the volume (~ 8 nL) of liquid lost during opening and closing of the valve as well as the pressure on cells during valve operation.
Generation of drug concentrations in the microfluidic array
Different concentrations of drugs were generated in the array using a diffusive mixer in conjunction with a syringe pump and automatic valve synchronization system, depending on the specific experiment (Figure 1). For experiments involving sequential exposure of cells to two drugs, a diffusive mixer with two inputs was used to generate eight different concentrations of a drug20, 29. A stock solution of the drug and a buffer solution were introduced into the inlets of a diffusive mixer to generate eight output concentrations (“horizontal gradient”). After a stable concentration gradient was attained ~60 sec), a set of valves (that control eight cell culture chambers in a row were opened for 10 s and closed to capture the drug containing solution in the cell culture chambers. Valves that control different rows of cell culture chambers were sequentially operated from row #1 to #8. After 24 hours of exposure to the first drug, concentration gradients of the second drug were generated in the same manner. The concentrations of doxorubicin generated in the diffusive mixer for this study were 0.0, 0.9, 1.7, 2.6, 3.5, 4.2, 5.2, and 6.0 μM. The concentrations of mitoxantrone generated were 0.0, 1.4, 2.9, 4.3, 5.7, 7.1, 8.6, and 10 μM. The concentration range for TRAIL was 0.0, 2.9, 5.7, 8.6, 11.4, 14.3, 17.1, and 20.0 ng /mL for the sequential exposure experiments.
For experiments where cells were simultaneously exposed to pair-wise combinations of two drugs, the diffusive mixer was used along with a syringe pump and automatic valve synchronization system (Figure 1). In this case, the diffusive mixer was used as described above to first generate eight concentrations of the first drug (A). A single concentration of the second drug (B) was generated by mixing a stock solution of drug B with a buffer solution at a specific flow rate to generate a single concentration of drug B. This stream of drug B was introduced upstream into both streams entering of the diffusive mixer used for generating the concentration range of the first drug A (i.e., into streams containing the stock solution of drug A and its buffer). This combined solution (i.e., containing a stock concentration of A and a fixed concentration of B) was diluted in the diffusive mixer to generate 8 pair-wise concentrations. Since drug B is present at the same concentration in both inlet streams, its concentration is the same in all the streams exiting the diffusive mixer whereas drug A is diluted to eight concentrations. This arrangement generates eight pair-wise combinations (i.e., eight concentrations of drug A, each with a single concentration of drug B) and used to expose cells cultured in one row of eight cell culture chambers (Figure 1).
Different concentrations of drug B are generated by changing the flow rate of the buffer and thereby diluting drug B to different levels. This stream containing a single concentration of drug B is mixed with eight concentrations of drug A to generate a second set of eight pair-wise concentrations and used to expose a second row of cell culture chambers to a unique pair-wise drug combination. This process was repeated by continuously varying the flow rate of the buffer solution to generate eight different concentrations of drug B and mixed with eight concentrations of drug A, to generate sixty four pair-wise concentrations of the two drugs. The concentrations of doxorubicin, mitoxantrone, and TRAIL used for generating the pair-wise combinations are as given above.
Operation of the syringe pumps and the valve array was carried out using a programmable LabView (Austin, TX) interface (Figure 1A). Each group of valves (controlling a single row of cell culture chambers) in the valve array was individually opened or closed by applying vacuum and compressed air through a single port on the pneumatic controller. Since different valve groups were connected to different ports on the pneumatic controller, each valve group could be operated without affecting other valve groups.
Cell culture in microfluidic devices
The glass slide (bottom) in all devices were coated with 50 μg/mL collagen I (BD Biosciences, San Jose, CA) for 16 h at 4°C and excess collagen I was removed by washing with 500 μL of RPMI medium. PC3 cells were trapped in each chamber sequentially using the LabView controlled syringe pump and pneumatic controller. The pneumatically controlled chambers were formed by applying positive pressure to lower a PDMS barrier which resulted in a small area being isolated from the surrounding regions. When this operation as performed with a cell suspension (~ 5 × 106 cells/mL) flowing through the device, cells were trapped in specific locations (i.e., in a single column). Cells were captured in each chamber sequentially from bottom to top (column #8 to column #1) for ~ 2 sec each at a total flow rate of 2 mL/h (or a flow rate of 0.25 mL/h in each column) to ensure uniform cell seeding. Cells that were excluded from the culture chamber (i.e., around the PDMS wall) were washed out. Seeded cells were allowed to attach onto the collagen-coated glass surface and proliferate for 24 h. To support growth of PC3 cells, fresh RPMI medium was trapped in each chamber every 3 h using the procedure described above. After 24 h, cells were exposed to different drugs either sequentially or simultaneously. The growth medium in each chamber (containing a specific pair-wise combination of two drugs) was replenished every 3 h for the duration of the experiment. Prior to each round of media refreshment, the channels were washed with buffer for 30 seconds to remove any residual drug in the media present outside the cell culture chambers.
At the end of drug treatment, cells in each of the drug-treated cell culture chambers were stained with calcein AM (i.e., the Live dye component of the Live/Dead cell viability stain, Invitrogen, CA) and green-fluorescent cells enumerated by counting. The cell viability after treatment was determined as the ratio of number of live cells in each drug-treated chamber to the number of live cells in the untreated control. Since dead cells are likely to be washed away with the periodic media change, the number of dead cells was not enumerated. Thus, this approach provides an indicator of loss of viability of cancer cells in the device.
Cell experiments in 96-well plates
Well plates (96 wells/plate) were pretreated with 50 μg/mL of collagen I per well for 12 h at 4°C and excess collagen was removed. Cells were plated at a density of 8,400 cells/well and incubated at 37°C and 5% CO2 for approximately 24 h. For sequential exposure treatments, cells were first treated with a sensitizer drug for 24 h. The media was then removed, replaced with fresh serum-containing media, and the cells were treated with TRAIL. Cells were incubated for an additional 24 h after which cell viability was assessed using the calcein AM stain. Simultaneous exposure treatments were carried out by treating cells with the sensitizer drug and TRAIL at the same time for 24 h at which point, calcein AM was used to determine viability.
RESULTS AND DISCUSSION
3.1. Characterization of concentration gradients in the device
Figure 1B demonstrates using color dyes the two types of concentration gradients generated in the microfluidic device. The simplest gradient generated is a range of concentrations generated by mixing two streams in a standard diffusive mixer29. This is demonstrated in Figure 1B (left panel) where yellow and blue color dye solutions (representing the minimum and maximum concentrations of a drug) were injected through the two inlets of a diffusive mixer and resulted in eight outlet concentrations (“horizontal gradient” of colors between yellow and blue) were generated. In this arrangement, each cell in a row of chambers has a different drug concentration and this pattern is repeated in all the rows. This arrangement represents the gradient used in experiments where cells were sequentially exposed to gradients of two chemotherapeutic agents (i.e., exposure to eight concentrations of one drug, followed by exposure to eight concentrations of a second drug).
The second type of concentration gradient involved generating an array of pair-wise concentrations of two chemotherapeutic agents. This is shown in Figures 1B (middle panel) where yellow and red streams (representing the minimum and maximum concentrations of the second drug) were mixed together to generate a “vertical gradient” of colors between yellow and red. Merging the two color gradients (vertical direction concentration gradient: yellow to blue; horizontal direction concentration gradient: yellow to red) as described in Materials & Methods yields an array of pair-wise combinations, and represents the gradient used in experiments where cells were simultaneously exposed to two chemotherapeutic drugs at eight concentrations each, for a total of 64 pair-wise concentrations (Figure 1B right panel).
The sequence of steps involved in operation of the microfluidic device is shown in Figure 2. Violet dye was initially flowed through the channels with the cell chambers closed so that no violet dye was captured in the cell culture chambers and only present outside the cell chamber area (Figure 2A). Next, the cell culture chambers were opened to trap violet dye as described in Materials & Methods (Figure 2B) in a manner that mimics cell seeding in the device. A concentration gradient between yellow and blue colors was established in the diffusive mixer and the solution trapped in the cell culture chambers by opening the valve controlling a specific set of chambers for 10 sec. This is shown in Figure 2C where the first three columns on the left have a color solution trapped inside the chambers, a single column where the violet dye is being replaced with the color solution gradient, and four columns on the right still have violet dye in them (i.e., represents media not yet refreshed).
Figure 2. Sequence of steps in the operation of the combinatorial array.
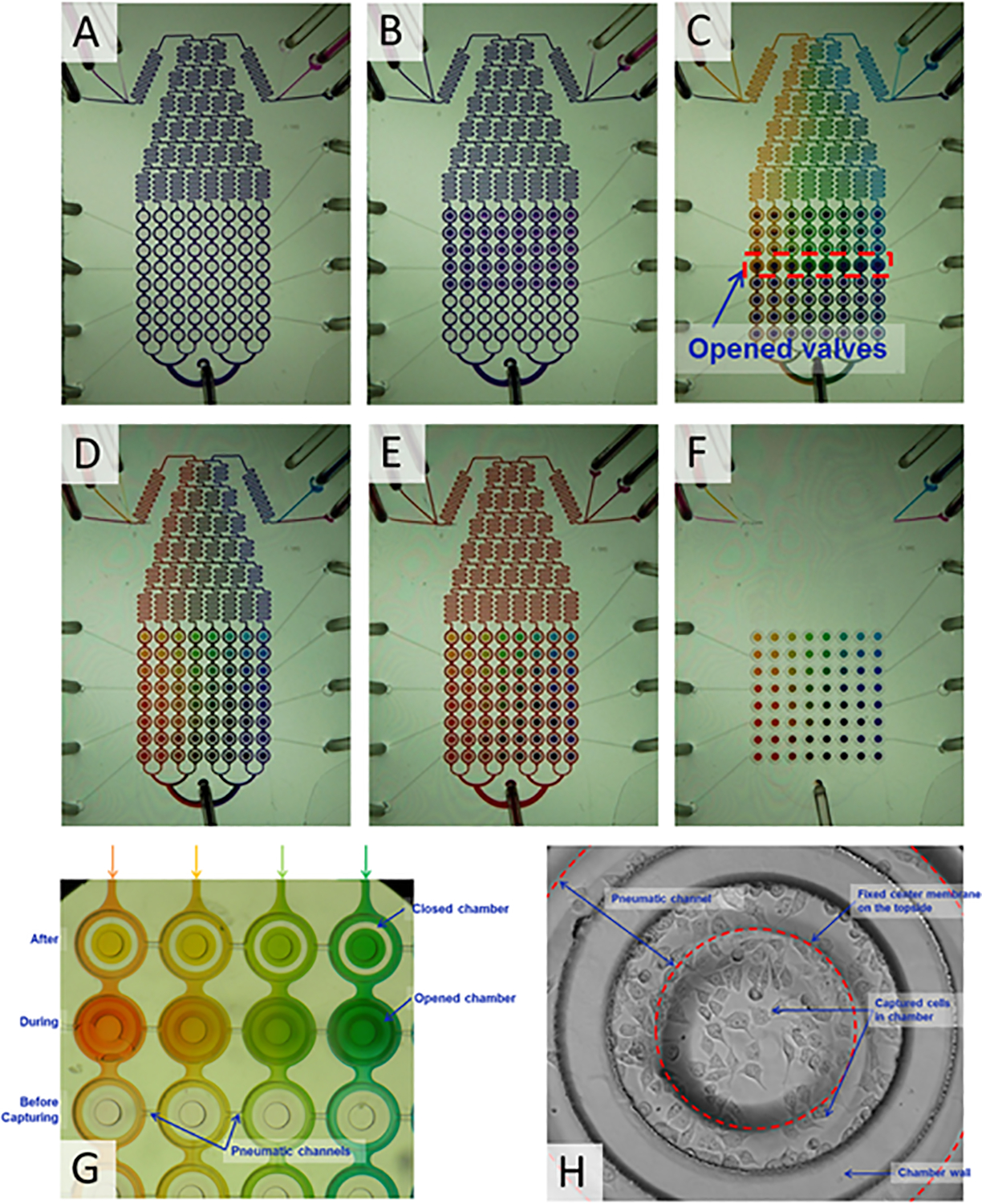
(A) Isolation of cell culture chambers shown by flowing violet dye around it. (B) Trapping of violet dye in the cell culture chambers. (C) Sequential trapping of different drug concentrations (represented by different colors) in columns of cell culture chambers. (D) Array of colors trapped in the chambers, with each color representing a pair-wise combination of two drugs. (E) No mixing occurs between the color dye present in the cell culture chamber and the color dye flowing outside. (F) Color dyes combinations trapped in the chambers without any liquid flowing around it. (G) Sequential operation of pneumatically controlled trapping system. Three sets of chambers are shown: empty chambers prior to trapping of color dye solution, chambers during trapping, and chambers after trapping of solution. (H) Representative cell culture chamber with PC3 cells trapped and grown for 24 h.
The “merging” of horizontal and vertical gradients to generate an array of pair-wise concentrations was carried out as described below. Red and yellow dyes were mixed at a ratio of 7:1 (i.e., mimics generation of a single concentration of drug B) (Figure 1A) and the resultant orange dye stream was added to the blue and yellow streams upstream of the gradient mixer. The color (ranging from violet to orange) was used to replace the blue-yellow color solution in the cell culture chambers as described in Materials & Methods. Figure 2D shows the resultant array of colors trapped in the device, with each color representing a single pair-wise concentration automatically generated using the LabView-controlled system. Next, the entire channel was filled with orange dye to mimic the media flowing around the cell culture chamber. No mixing between the outside orange solution and the solution trapped inside the chambers was observed after 24 h (Figures 2E). The color dyes trapped in the chamber without any liquid surrounding it is shown in Figure 2F. The operation of the pneumatically controlled trapping system is demonstrated in Figure 2G using color dyes. Three sets of chambers are shown: empty chambers prior to trapping of color dye solution, chambers during trapping, and chambers after trapping of solution. Before trapping, each chamber is empty and liquid flows around the closed chamber without entering. This demonstrates the fidelity of the valve system in trapping liquid and maintaining it separate from the surrounding liquid.
Most microfluidic systems19, 30 using a diffusive mixer need continuous perfusion of two components to generate and maintain concentration gradients. On the other hand, the pneumatically-controlled system in our device does not require continuous perfusion for keeping cells exposed to concentration gradients. This approach has several advantages. First, it minimizes the amount of drug used per experiment, which is significant considering that the compound libraries used for screening typically contain limited amounts of material and are expensive. Second, cells cultured in the chamber are not exposed to significant shear stress continuously as the time required for trapping solutions inside the cell culture chamber is only ~10 sec. Therefore, this system would be especially advantageous when working with cells (e.g. primary cells) that are sensitive to shear stress. Moreover, different concentration gradients (e.g., non-linear or exponential concentration gradient) can be easily generated by programming the Labview interface which gives the system flexibility in designing drug screening experiments.
3.2. Cell culture in the microfluidic device
The prostate cancer cell line PC3 was used for these studies. PC3 cells were captured in the array of cell culture chambers and maintained for 24 h (with media refreshed every 3 h) in a 5% CO2 incubator at 37°C for adaptation to the microfluidic environment prior to drug exposure. PC3 cells proliferated in the device and were not adversely affected by the microfluidic environment for 5 days (not shown). Figure 2H shows a representative cell culture chamber with PC3 cells grown for 24 h.
3.3. Sequential treatment with TRAIL sensitizer drugs and TRAIL
We employed the microfluidic cell array for investigating the effect of the combinatorial treatment of chemotherapeutic drugs and TRAIL on the viability of PC3 human prostate cancer cells. PC3 cells were cultured on-chip and exposed sequentially to different concentrations of doxorubicin and TRAIL or mitoxantrone and TRAIL. Combination treatments were carried out either in simultaneous or sequential formats. In case of the former, the components of the combination treatment (i.e., doxorubicin and TRAIL or mitoxantrone and TRAIL) were delivered together in a single formulation. In the case of sequential treatments, the second drug treatment (e.g. TRAIL) is administered after the completion of a first drug dose (e.g. doxorubicin or mitoxantrone). In order to carry out sequential treatments, PC3 cells were first exposed to different concentrations of doxorubicin or mitoxantrone as listed in Materials & Methods. These concentrations were chosen based on their effect on PC3 cells14 and were chosen such that they spanned both sides of the previously identified optimum concentration (3 μM for doxorubicin and 5 μM for mitoxantrone).
Different concentrations of drugs were generated by mixing two streams containing 0 and 6 μM doxorubicin or 0 and 10 μM mitoxantrone using the diffusive mixer described in Figure 1. After 24 h exposure to doxorubicin or mitoxantrone, a gradient of 0 – 20 ng/mL TRAIL was generated and sensitized cells were exposed for an additional 24 h. The cell viability after 24 h exposure to TRAIL was determined as described in Materials & Methods. Figure 3 shows representative fluorescent micrographs from the array of cells chambers in a single experiment. The data in the insets (Figure 3B) show that cells not exposed to either doxorubicin or TRAIL have proliferated and are confluent in the cell culture chamber (top left). Exposure to 6 μM of either doxorubicin (top right) or 20 ng/mL of TRAIL (bottom left) alone demonstrate a small loss of viability whereas cells that were exposed sequentially to 6 μM doxorubicin followed by exposure to 20 ng/mL TRAIL have very few live cells. Images for only the doxorubicin/TRAIL combination are shown, and similar results were obtained with the mitoxantrone/TRAIL combination as well.
Figure 3. Cell culture in the combinatorial array.
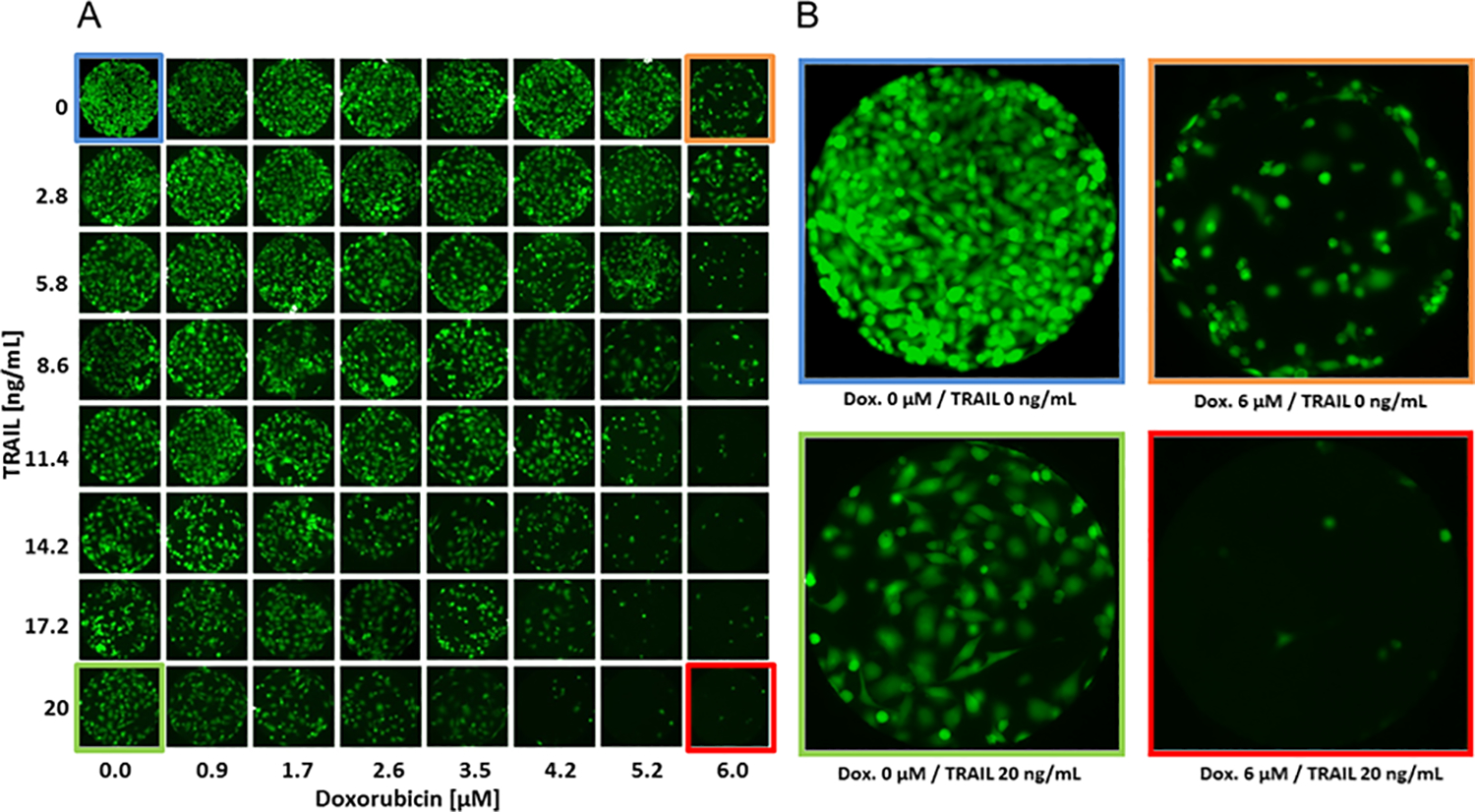
(A) Representative fluorescent micrographs of PC3 cells cultured in an array for 24 h. (B) Micrographs of cells in the four corners of the array after sequential exposure to Doxorubicin and TRAIL (top left: no Doxorubicin, no TRAIL; top right: Doxorubicin, no TRAIL; bottom left: No Doxorubicin, TRAIL; bottom right: Doxorubicin, TRAIL).
The fraction of live cells in the different cell chambers was determined as described in Materials & Methods. Figure 4A shows that exposure to TRAIL alone without doxorubicin sensitization led to a dose-dependent decrease in cell viability, with ~40% decrease in viability at the highest concentration tested (20 ng/mL). Doxorubicin by itself also induces a loss in PC3 cell viability as seen from a dose-dependent decrease in cell viability even in the absence of TRAIL (Figure 4A). The effect of sensitization is evident from the data on PC3 cell exposure to 2.7 μM of doxorubicin. The cell viability decreases from ~ 60% in the absence of TRAIL to ~5% with an increase in the concentration of TRAIL. This extent of decrease is smaller at lower and higher doxorubicin concentrations, as may be expected. In the former case, the concentration of doxorubicin used is not sufficient to fully sensitize cells to TRAIL-mediated cell death whereas in the latter case, doxorubicin by itself decreases cell viability (Figure 4A). Since the objective of combination therapy is to achieve maximum possible decrease in cell viability with the lowest concentration of sensitizer or chemotherapeutic molecule, sensitizing PC3 cells with ~3 μM doxorubicin followed by exposure to ~14 ng/mL of TRAIL is sufficient to reduce cell viability by ~95%. Sensitization with mitoxantrone prior to TRAIL exposure was somewhat less effective than doxorubicin, with a maximum decrease from ~65% to 20% cell viability when PC3 cells were sensitized with 4.3 μM of mitoxantrone (Figure 4B). This indicates that doxorubicin was a marginally more effective sensitizer than mitoxantrone in these in vitro investigations. These results are along the lines of what is anticipated, since different drugs will possess differential activities depending on the cancer cell lines investigated14.
Figure 4. PC3 cell viability after sequential exposure to Doxorubicin and TRAIL.
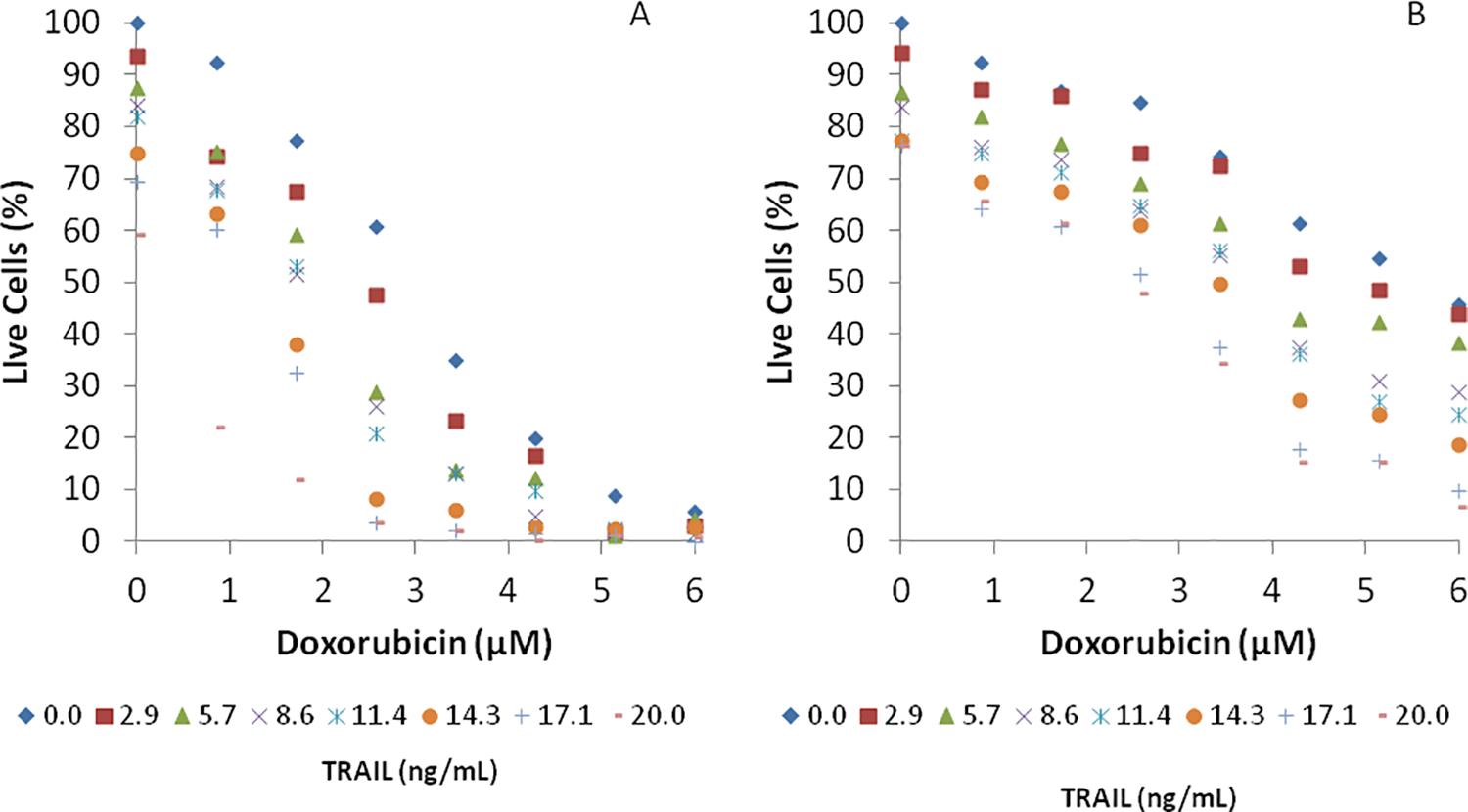
PC3 cells were initially exposed to (A) eight concentrations (0.0, 0.9, 1.7, 2.6, 3.5, 4.2, 5.2, and 6.0 μM) of Doxorubicin or (B) eight concentrations (0.0, 1.4, 2.9, 4.3, 5.7, 7.1, 8.6, and 10 μM) of Mitoxantrone for 24 h, followed by exposure to eight concentrations (0.0, 2.9, 5.7, 8.6, 11.4, 14.3, 17.1, and 20.0 ng /mL) of TRAIL for 24 h. Data shown are average of three independent experiments.
3.4. Simultaneous treatment with TRAIL sensitizer drugs and TRAIL
PC3 cells were also simultaneously exposed to 64 pair-wise concentrations of doxorubicin (0 – 6 μM) and TRAIL (0 – 20 ng/mL) or mitoxantrone (0 – 10 μM) and TRAIL (0 – 20 ng/mL) in the microfluidic device. Pair-wise concentrations were generated as described in Materials & Methods and as demonstrated in Figure 3 with color dyes. PC3 cells were exposed to different concentration pairs for 24 h. The media in the cell chambers was replenished with fresh media with a specific pair-wise combination of the two drugs (doxorubicin or mitoxantrone, along with TRAIL). Figure 5A shows that PC3 cells exposed simultaneously to pair-wise combinations of doxorubicin and TRAIL for 24 h demonstrate a dose-dependent decrease in cell viability. However, the decrease in cell viability observed with this treatment was less than that observed with sequential exposure to doxorubicin and TRAIL. The maximum decrease in cell viability was upon simultaneous exposure to doxorubicin and TRAIL was ~2-fold (Figure 5A), compared to the ~10-fold decrease observed with sequential exposure to the two drugs (Figure 4A). Similar results were also observed with simultaneous exposure of PC3 cells to mitoxantrone and TRAIL (Figure 5B); for example, the decrease in cell viability with 4.29 μM of mitoxantrone and 20 ng/mL of TRAIL was only ~ 40%. However, a higher decrease in PC3 cell viability was observed at the higher concentrations of mitoxantrone. These results are consistent with our previous findings in well-plate screens, which indicated that sequential treatments of mitoxantrone resulted in higher efficacies of TRAIL sensitization compared to those observed with simultaneous treatments14. It is possible that mitoxantrone needs a long time for sensitization activity and that it is necessary to treat cells with the drug for 24 hours, and also with TRAIL for an additional 24 h. We are currently following up on these observations in our laboratories in order to elucidate mechanisms underlying this phenomenon. Nevertheless, it is important to note that the microfludic device can be operated in a sequential as well as simultaneous drug treatment format, which makes this a powerful platform for screening as well as end-point and transient dosing studies.
Figure 5. PC3 cell viability after simultaneous exposure to pairwise combinations.
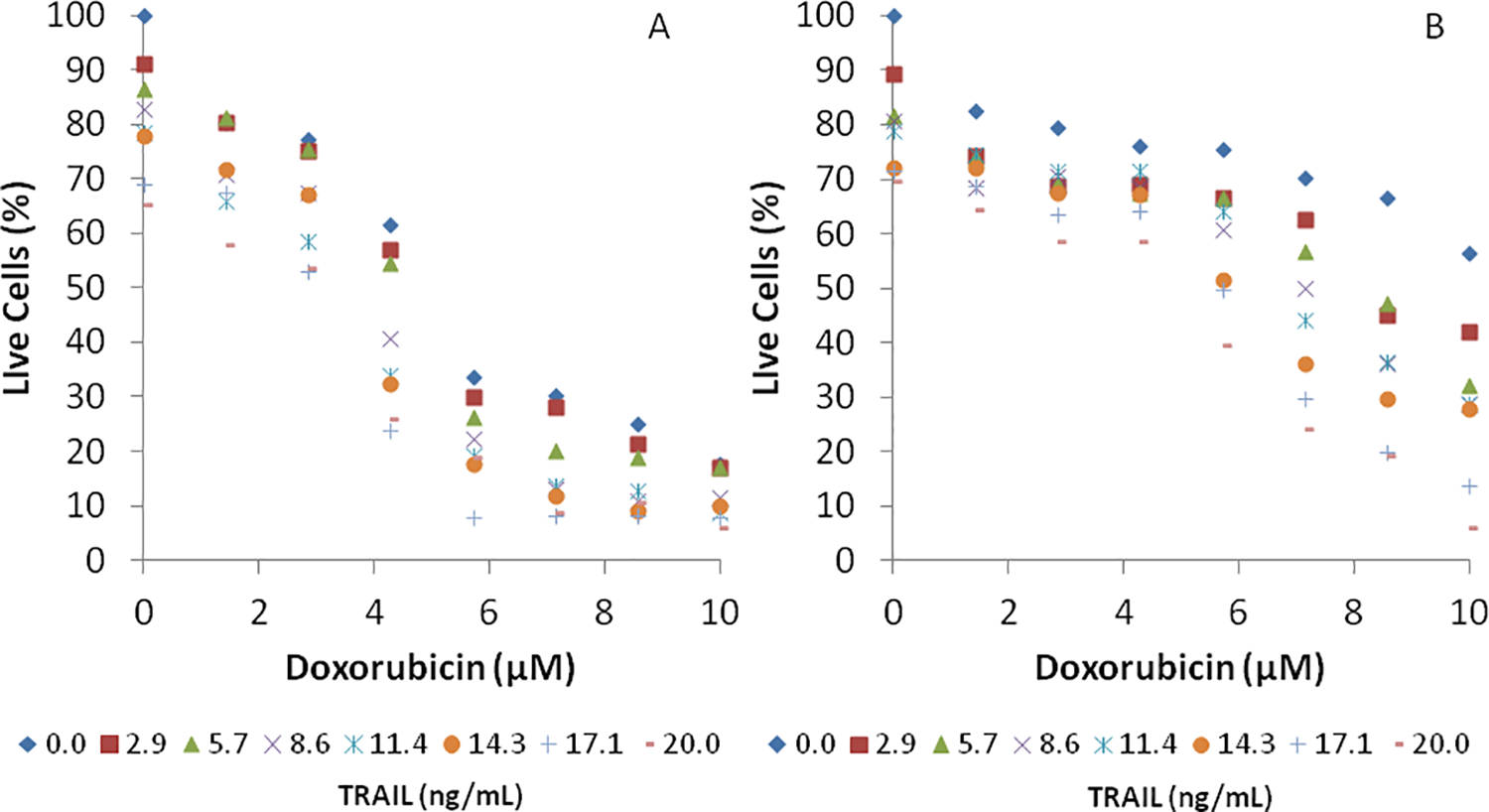
PC3 cells were exposed to 64 pair-wise combinations of (A) eight concentrations (0.0, 0.9, 1.7, 2.6, 3.5, 4.2, 5.2, and 6.0 μM) Doxorubicin and eight concentrations (0.0, 2.9, 5.7, 8.6, 11.4, 14.3, 17.1, and 20.0 ng /mL) of TRAIL or (B) eight concentrations (0.0, 1.4, 2.9, 4.3, 5.7, 7.1, 8.6, and 10 μM) of Mitoxantrone and eight concentrations (0.0, 2.9, 5.7, 8.6, 11.4, 14.3, 17.1, and 20.0 ng /mL) of TRAIL for 24 h. Data shown are average of three independent experiments.
The utility of the microfluidic array is clearly evident from the PC3 viability data. While maximum decrease in cell viability (i.e., complete or near-complete cell death) can be achieved by simply using high concentrations of doxorubicin or TRAIL, this is counter-productive as the side-effects observed with use of high drug concentration often outweigh the benefits. Since combination therapy seeks to identify the lowest concentrations where maximum possible cancer cell death is achieved, the ability to generate different dose-response curves in a high-throughput manner is of significant interest. In our studies, sensitizing PC3 cells with ~3 μM of doxorubicin resulted in a nearly 10-fold increase in cell death with subsequent TRAIL exposure (Figure 4A). Since only a ~30% decrease in viability was observed with single-agent doxorubicin at this concentration, this could possibly represent the optimal concentration of doxorubicin to be used in sensitization studies with PC3 cells. A similar analysis suggests that the optimal concentration of mitoxantrone for sensitizing PC3 cells is ~3 μM.
Comparison of microfluidic device and well plate cultures
The results obtained with the microfluidic device were compared against those obtained from exposing PC3 cells to the different drugs under the two treatment regimes (sequential and simultaneous) in a 96-well plate. Figure 6 shows the results of these comparisons with doxorubicin and mitoxantrone. In general, the combination treatments in the device lead to a smaller decrease in PC3 cell viability than those in well plates. While sequential treatment efficacies were more comparable between the two formats, the differences were more pronounced for simultaneous exposure to a sensitizer and TRAIL.
Figure 6. Comparison of PC3 viability in microfluidic array and tissue culture plates.
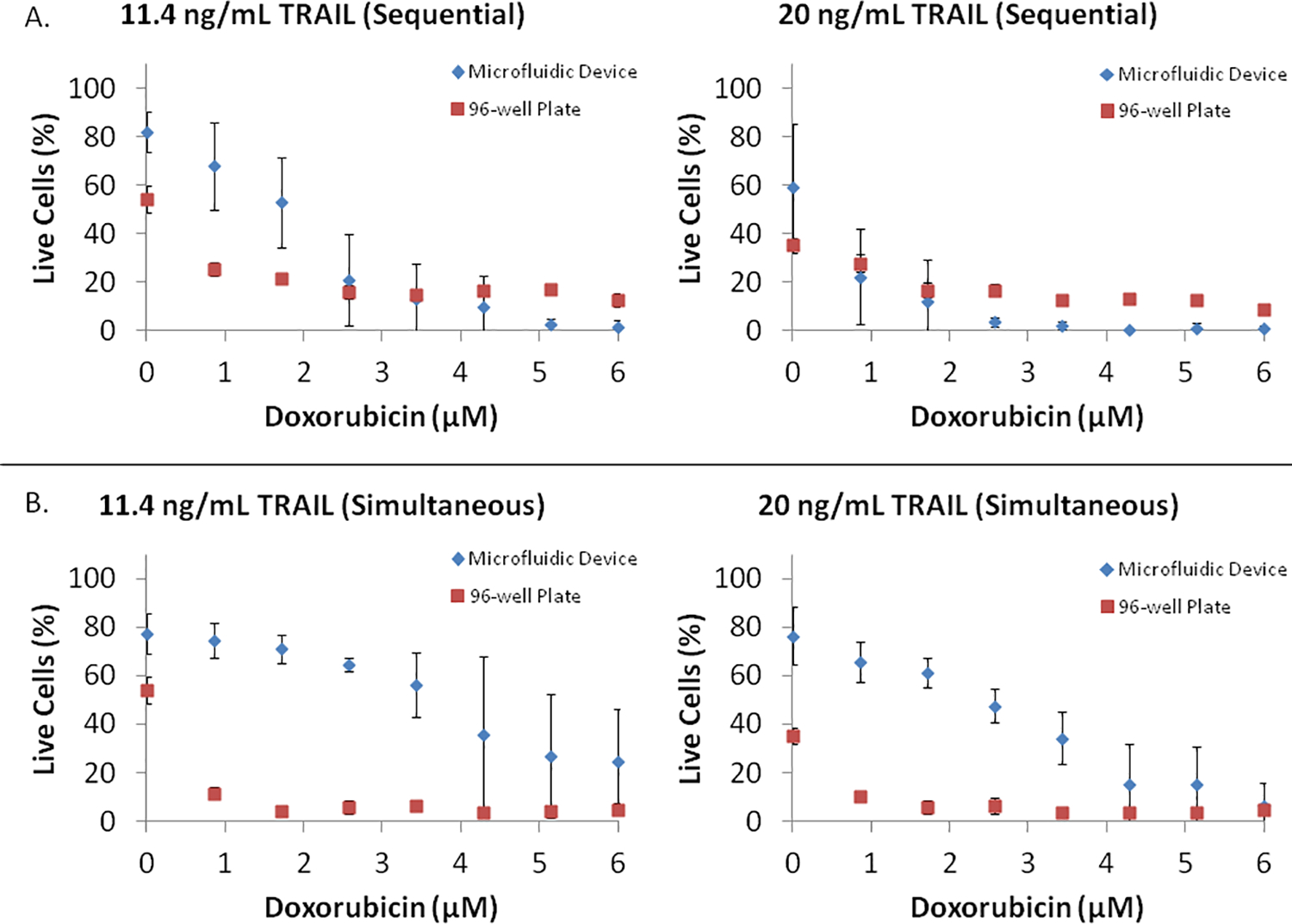
PC3 cell viability in response to (A) sequential exposure to eight concentrations (0.0, 0.9, 1.7, 2.6, 3.5, 4.2, 5.2, and 6.0 μM) of Doxorubicin for 24 h, followed by exposure to 11.4 ng/mL or 20 ng/mL TRAIL for 24 h, and (B) simultaneous exposure to eight concentrations (0.0, 0.9, 1.7, 2.6, 3.5, 4.2, 5.2, and 6.0 μM) of Doxorubicin and 11.4 ng/mL or 20 ng/mL TRAIL for 24 h, was compared between the microfluidic array and tissue culture plates. Data shown are average of three independent experiments and one standard deviation.
Several factors could possibly contribute to the difference in drug efficacy between the two formats. First, the microfluidic device was operated with periodic media replenishment every 3 h (i.e. in a fed-batch mode) and removal of waste products whereas nutrients were not replenished and waste product levels build up for the duration of the experiment in the well plate cultures. Given that these treatments typically occur over a period of 24–48 hours, it is possible that the addition of fresh nutrients and/or removal of waste products lead to the lower drug efficacy (or higher cancer cell viability) seen in microfluidic devices. Second, the high local concentrations of growth factors present in the microchannel environment could also lead to an increase in cell viability as compared to the macro-scale well plate cultures31. Third, the cell density (number of cells per unit area) is different between the two systems (higher in the device compared to the well plate system), which can potentially contribute to the higher growth rate and cell viability observed in the device.
It is also possible that the absorption of drug molecules into PDMS32 leads to the difference between the two systems. However, this effect is likely to be minimal as we perfused collagen through the channels prior to seeding, which would reduce absorption of drugs into the PDMS (as has been demonstrated with bovine serum albumin by Whitesides and co-workers33). Secondly, fresh medium containing the drug molecule was perfused through the channels every 3 h over the duration of the experiment, which would be expected to saturate the PDMS and minimize absorption at later time points. Nevertheless, it is not possible to completely eliminate the possibility that absorption of drugs to the PDMS leads to decreased drug efficacy in the microfluidic device. It is important to note that despite the differences between the microfluidic device and the well plate culture, the microfluidic device does capture the synergy between doxorubicin and TRAIL and mioxantrone and TRAIL combination treatments, indicating the utility of this approach, for both screening and dosing studies. As with any screening methodology, additional lead validation methods will be necessary when selecting leads from microfluidic devices.
Summary
In summary, we have developed a microfluidic cell array capable of screening and optimizing combinatorial drug treatments. While the device has been described and characterized using chemotherapeutic drugs as sensitizer for TRAIL-induced apoptosis, this approach can be extended to identifying combinatorial drug treatments for a variety of diseases. Finally, the ability to carry out sequential and simultaneous treatments also facilitates exploration of diverse dosing studies in toxicology and biology.
Acknowledgements
The authors would like to acknowledge financial support from the National Cancer Institute (5R21CA131891-02). DT acknowledges a fellowship from the Achievement Rewards for College Scientists (ARCS) Foundation.
References
- 1.Beltran H, Beer TM, Carducci MA, de Bono J, Gleave M, Hussain M, Kelly WK, Saad F, Sternberg C, Tagawa ST and Tannock IF, Eur Urol, 2011, 60, 279–290. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L and Fang B, Cancer Gene Ther, 2005, 12, 228–237. [DOI] [PubMed] [Google Scholar]
- 3.Sheikh MS and Fornace AJ Jr, J Cell Physiol, 2000, 182, 171–181. [DOI] [PubMed] [Google Scholar]
- 4.Bouralexis S, Findlay DM and Evdokiou A, Apoptosis, 2005, 10, 35–51. [DOI] [PubMed] [Google Scholar]
- 5.Kelley SK and Ashkenazi A, Curr Opin Pharmacol, 2004, 4, 333–339. [DOI] [PubMed] [Google Scholar]
- 6.Naka T, Sugamura K, Hylander BL, Widmer MB, Rustum YM and Repasky EA, Cancer Res, 2002, 62, 5800–5806. [PubMed] [Google Scholar]
- 7.Singh AV, Xiao D, Lew KL, Dhir R and Singh SV, Cancer Res, 2003, 63, 5390–5400.14500373 [Google Scholar]
- 8.Shankar S, Singh TR and Srivastava RK, Prostate, 2004, 61, 35–49. [DOI] [PubMed] [Google Scholar]
- 9.Ohtsuka T, Ryu H, Minamishima YA, Ryo A and Lee SW, Oncogene, 2003, 22, 1678–1687. [DOI] [PubMed] [Google Scholar]
- 10.Barua S, Linton RS, Gamboa J, Banerjee I, Yarmush ML and Rege K, Cancer Lett, 2010, 293, 240–253. [DOI] [PubMed] [Google Scholar]
- 11.Sun SY, Yue P, Hong WK and Lotan R, Cancer Res, 2000, 60, 7149–7155. [PubMed] [Google Scholar]
- 12.El-Zawahry A, McKillop J and Voelkel-Johnson C, BMC Cancer, 2005, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly MM, Hoel BD and Voelkel-Johnson C, Cancer biology & therapy, 2002, 1, 520–527. [DOI] [PubMed] [Google Scholar]
- 14.Taylor DJ, Parsons CE, Han H, Jayaraman A and Rege K, BMC Cancer, 2011, Epub ahead of print Nov 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohgami T, Kato K, Kobayashi H, Sonoda K, Inoue T, Yamaguchi S, Yoneda T and Wake N, Cancer Sci, 2010, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Booth NL, Sayers TJ, Brooks AD, Thomas CL, Jacobsen K, Goncharova EI, McMahon JB and Henrich CJ, Cancer Immunol Immunother, 2009, 58, 1229–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugiura S, Edahiro J, Kikuchi K, Sumaru K and Kanamori T, Biotechnol Bioeng, 2008, 100, 1156–1165. [DOI] [PubMed] [Google Scholar]
- 18.Wu MH, Huang SB and Lee GB, Lab Chip, 2010, 10, 939–956. [DOI] [PubMed] [Google Scholar]
- 19.Lee PJ, Hung PJ, Rao VM and Lee LP, Biotech Bioeng, 2006, 94, 5–14. [DOI] [PubMed] [Google Scholar]
- 20.Thompson DM, King KR, Wieder KJ, Toner M, Yarmush L and Jayaraman A, Anal Chem, 2004, 76, 4098–4103. [DOI] [PubMed] [Google Scholar]
- 21.King KR, Wang S, Irimia D, Jayaraman A, Toner M and Yarmush ML, Lab Chip, 2007, 7, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wieder KJ, King KR, Thompson DM, Zia C, L YM and Jayaraman A, Biomed Microdevices, 2005, 7, 213–222. [DOI] [PubMed] [Google Scholar]
- 23.Neils C, Tyree Z, Finlayson B and Folch A, Lab Chip, 2004, 4, 342–350. [DOI] [PubMed] [Google Scholar]
- 24.Jang YH, Hancock MJ, Kim SB, Selimović Š, Sim WY, Bae H and Khademhosseini A, Lab Chip, 2011, 11, 3277–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitesides GM, Ostuni E, Takayama S, Jiang X and Ingber DE, Annu Rev Biomed Eng, 2001, 3, 335–373. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Hegde M and Jayaraman A, Lab Chip, 10, 43–50. [DOI] [PubMed] [Google Scholar]
- 27.Grover WH, Skelley AM, Liu CN, Lagally ET and Mathies RA, Sensors and Actuator B, 2003, 89, 315–323. [Google Scholar]
- 28.Jeon NL, Chiu DT, Wargo CJ, Wu H, Choi IS, Anderson JR and Whitesides GM, Biomed Microdevices, 2002, 4, 117–121. [Google Scholar]
- 29.Jeon NL, Baskaran H, Dertinger SKW, Whitesides GM, Water LVD and Toner M, Nature Biotechnology, 2002, 20, 826–830. [DOI] [PubMed] [Google Scholar]
- 30.Neils C, Tyree A, Finlayson B and Folch A, Lab Chip, 2004, 4, 342–350. [DOI] [PubMed] [Google Scholar]
- 31.Walkers GM, Ozers MS and Beebe DJ, Biomed Microdevices, 2002, 4, 161–166. [Google Scholar]
- 32.Toepke MW and Beebe DJ, Lab Chip, 2006, 6, 1484–1486. [DOI] [PubMed] [Google Scholar]
- 33.Ostuni E, Chen CS, Ingber DE and Whitesides GM, Langmuir, 2001, 17, 2828–2834. [Google Scholar]