

Abstract
Background:
Aripiprazole and risperidone, widely used atypical antipsychotics, are commonly adjunctively prescribed in clinical practice. When aripiprazole was combined with risperidone, the genotype of drug-metabolizing enzymes and liver impairment may lead to complex pharmacokinetic changes. The Physiologically Based Pharmacokinetic (PBPK) model can predict the influence of these factors on plasma concentration and optimize dosage regimens.
Objectives:
This study aims to investigate the pharmacokinetic changes of aripiprazole caused by various influencing factors when it was co-administered with risperidone through PBPK models.
Design:
The PBPK models of aripiprazole and risperidone were developed by gathering physicochemical parameters and drug-specific parameters. Then, by combining the inhibitory parameters, the enzymatic kinetic parameters of CYP2D6 genotypes, and the changes in anatomical and physiological parameters when liver function is damaged, the corresponding PBPK models were further established. Finally, this study put forward dosage optimization recommendations for situations where risks may exist.
Methods:
The comparison between predicted and observed plasma concentration data, along with the assessment of pharmacokinetic parameters, was utilized to evaluate the fit performance of the models.
Results:
The simulations of the PBPK model revealed that co-administration of risperidone did not result in significant changes in aripiprazole pharmacokinetics. However, in individuals with mild hepatic impairment and CYP2D6 normal metabolizer, a dose reduction of approximately 11% was advised when aripiprazole was combined with risperidone. When individuals with mild liver damage have CYP2D6 genotypes of intermediate metabolizer (IM) and poor metabolizer (PM), aripiprazole doses should be further reduced to 61% and 51%, respectively.
Conclusion:
The co-administration of aripiprazole and risperidone is generally considered safe from a pharmacokinetic perspective. However, if individuals have a CYP2D6 genotype of IM or PM and/or if they have mild hepatic impairment, adjusting the dose of aripiprazole is advisable to mitigate potential risks when combining it with risperidone.
Keywords: aripiprazole, CYP2D6 genotype, physiologically based pharmacokinetic model, risperidone
Plain language summary
Develop a physiologically based pharmacokinetic model of co-administration of aripiprazole and risperidone to predict the exposure of aripiprazole pharmacokinetics in subjects with different CYP2D6 genotype and individuals with hepatic impairment
Why was the study done? Aripiprazole and risperidone are both atypical antipsychotics, primarily used for the treatment of common psychiatric disorders such as schizophrenia and bipolar disorder. When used together, genetic enzyme variants and liver dysfunction can cause complex pharmacokinetics. The Physiologically Based Pharmacokinetic (PBPK) model helps predict these changes and guides dosage adjustments.
What did the researchers do? We developed PBPK models using the reported physicochemical properties of aripiprazole and clinical data. We validated the PBPK models.
What did the researchers find? We found that the individuals with mild hepatic impairment and (or) different CYP2D6 genotype need dosage adjustments: 1. In individuals with mild hepatic impairment, a dose reduction of approximately 11% was advised when aripiprazole is combined with risperidone. 2. When individuals with mild liver damage have CYP2D6 genotypes of intermediate metabolizer and poor metabolizer, aripiprazole doses should be further reduced to 61% and 51% when combined with risperidone, respectively.
What did the findings mean? If individuals have a CYP2D6 genotype of intermediate metabolizer or poor metabolizer and/or if they have mild hepatic impairment, adjusting the dose of aripiprazole is advisable to mitigate potential risks when combining it with risperidone.
Introduction
Schizophrenia is a common and severe mental disorder with high disability rates and a significant disease burden in clinical practice. Antipsychotic medications are the primary treatment for schizophrenia.1–3 Atypical antipsychotic drugs often cause adverse drug reactions such as metabolic syndrome, hyperprolactinemia, and QT prolongation. Risperidone, a widely prescribed atypical antipsychotic, is known to carry a significant risk of inducing hyperprolactinemia. Aripiprazole is another atypical antipsychotic medication commonly employed in clinical settings. Several studies have shown that risperidone and aripiprazole adjunctive treatment can resolve hyperprolactinemia induced by risperidone, so psychiatrists often add aripiprazole to risperidone therapy to enhance the therapeutic effect and reduce the incidence of hyperprolactinemia.4–6 A meta-analysis published in 2021 demonstrated that adjunctive use of aripiprazole, metformin, and paeoniae-glycyrrhiza decoction is beneficial in reducing hyperprolactinemia induced by atypical antipsychotic drugs, with aripiprazole showing the most significant effect. 7
Both risperidone and aripiprazole are mainly transformed through CYP2D6 and CYP3A4 into their metabolites,8,9 indicating that there may be competitive inhibition leading to potential drug–drug interaction (DDI). Currently, there is no literature evidence to support the safety of the concurrent use of the two medications. Previous studies have suggested that risperidone can increase the concentration of aripiprazole when used together, 10 and pharmacological databases such as DrugBank also suggested an increased risk of adverse reactions when the two drugs are combined. 11 However, some studies indicated that co-administration did not lead to significant pharmacokinetic changes.5,12
The genetic polymorphism of CYP2D6 is another crucial factor influencing the exposure levels of aripiprazole and risperidone. 13 Guidelines of the Dutch Pharmacogenetics Working Group (DPWG) and Clinical Pharmacogenetics Implementation Consortium (CPIC) have clearly specified that doses of antipsychotic medications like aripiprazole and risperidone should be adjusted according to the individual’s CYP2D6 genotype.14–16 In addition to the aforementioned factors, hepatic impairment is another main factor affecting drug metabolism, and this process is called drug–disease interaction (DDZI). 17 Mallikaarjun et al. 18 found that subjects with mild hepatic impairment exhibited a 33% increase in the area under the curve (AUC) of aripiprazole compared to healthy subjects after a single dose of 15 mg aripiprazole, whereas in cases of moderate or severe hepatic impairment, the AUC was minimal increase or decreased.
Previous studies have shown that hepatic function impairment or CYP2D6 genotype may further lead to changes in drug concentrations, increasing the risk of individual DDIs. Therefore, it is necessary to explore the changes in the exposure level of aripiprazole in combination with risperidone under the influence of multiple factors for dose adjustment.
The physiologically based pharmacokinetic model (PBPK) model can be used to study the influence of various factors on the process of drugs in vivo, including genetic polymorphisms of metabolic enzymes, hepatic dysfunction, and DDI.19–21 Therefore, using the PBPK model, our study aimed to predict the pharmacokinetic characteristics of aripiprazole and risperidone in combination under various influencing factors and to evaluate the efficacy and safety of antipsychotic treatment.
Materials and methods
Software
PK-Sim® software (version 10.0; Bayer Technology Services, Leverkusen, Germany) was used for PBPK model building and parameter optimization, and the software WebPlotDigitizer (version 4.5; Ankit Rohatgi, Austin, TX, USA) was used to extract data of drug concentration–time curves from published articles and/or relevant databases. Data analysis and graphic editing of simulation results are performed by OriginPro® (version 9.5.5; OriginLab, Northampton, Massachusetts, USA).
Clinical data
The data used for modeling are sourced from pharmacological databases such as DrugBank, PharmGKB, and FDA, as well as published literature. Plasma concentration–time profiles of aripiprazole and its main metabolite were digitized from published literature.12,18,22,23 All studies were conducted with participants covering a wide dosing range, single dose, and peroral administration. The datasets for validation preferably included profiles reporting many measurement points over a long period of time. A comprehensive list of all profiles utilized is shown in Supplemental Table S2.
Workflow
Figure 1 depicts the workflow for exploring the DDI, and the interactions between aripiprazole and risperidone under the influence of CYP2D6 genotype and/or mild hepatic impairment. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement. 24
Figure 1.

Schematic workflow for the development of a physiologically based pharmacokinetic model. Child-Pugh A is a classification used to assess the severity of chronic liver disease, which indicates the mildest level of liver dysfunction.
DDI, drug–drug interaction; DDGI, drug–drug–gene interaction; DDZI, drug–disease interaction; DGI, drug–gene interaction; PBPK, physiologically based pharmacokinetic.
PBPK initial model development
Our study focuses on predicting changes in serum concentrations of aripiprazole, which can fluctuate when it is used in combination with risperidone, with aripiprazole serving as the victim drug. 10 The absorption, distribution, metabolism, and excretion of aripiprazole were determined by inputting the physicochemical properties and dissolution curve parameters of aripiprazole and its main metabolite into PK-Sim. The physicochemical property parameters were gathered from literature and databases (Table 1). The dissolution curve parameters (b = 0.91, T1/2 = 5 min) were determined by integrating the dissolution curve of aripiprazole in gastrointestinal fluid, as reported in the literature, with the Weibull model.25,26
Table 1.
Physicochemical properties of aripiprazole and dehydro-aripiprazole for building PBPK model.
| Parameter | Aripiprazole | Dehydro-aripiprazole | Source/method |
|---|---|---|---|
| logP | 3.60 | 3.40 | Vieira et al.
27
Parameter identification |
| f u | 0.01 | 0.02 | DrugBank Parameter identification |
| MW (g/mol) | 448.4 | 446.4 | DrugBank |
| pKa | 7.46 | 7.46 | DrugBank |
| Solubility (µg/mL) | 4.00 (pH = 7) | 6.00 (pH = 7) | Mihajlovic et al.
28
DrugBank |
| CLH (mL/h/kg) | 47.96 | 40.00 | DrugBank Parameter identification |
| CLR (mL/h/kg) | 0.04 | 0.06 | Mallikaarjun et al.
18
Parameter identification |
Parameter identification refers to the process in which software calculates and determines the parameters that result in the best fit between the simulated values and the observed values.
CLH, specific hepatic clearance, CLR, specific renal clearance, fu, fraction unbound, logP, lipophilicity; MW, molecular weight; pKa, acid dissociation constant.
System-specific parameters obtained from the built-in PK-Sim database (i.e., physiological and anatomical parameters of the virtual population) were fixed to suit the corresponding clinical data.12,18,22,23 Details on the virtual populations are listed in Supplemental Table S1. The model was validated using clinical data for aripiprazole, as detailed in Supplemental Table S2.
| (1) |
“a” describes the scale parameter, “b” indicates the shape parameter, “m” denotes the fraction of the dissolved drug at time t, and Tlag is the lag time of drug dissolution.
DGI, DDI, and DDZI model development
Drug–gene interaction model development
The establishment of the drug–gene interaction (DGI) model requires enzymatic kinetic parameters of aripiprazole in different CYP2D6 genotypes (Table 2). Due to the enzyme activity of CYP2D6 poor metabolizer (PM) being 0, 29 the enzyme activity parameters for other genotypes can be calculated using the following formula based on the clearance of different genotypes from the literature (Supplemental Materials).11,27
Table 2.
Parameters of enzymatic kinetic parameters of aripiprazole in different CYP2D6 genotypes.
| Parameter | NM | IM | PM | UM | Source |
|---|---|---|---|---|---|
| Km,CYP2D6 (µM) | 26.20 | 26.20 | 26.20 | 26.20 | FDA |
| Kcat,CYP2D6,dehydrogenation (1/min) | 14.99 | 10.49 | 0.00 | 14.99 | Calculated |
| Kcat,CYP2D6,hydroxylation (1/min) | 57.17 | 40.02 | 0.00 | 57. 17 | Calculated |
| Km,CYP3A4 (µM) | 298.00 | 298.00 | 298.00 | 298.00 | FDA |
| Kcat,CYP3A4,dehydrogenation (1/min) | 54.85 | 54.85 | 73.79a | 54.85 | Calculated/parameter identification |
| Kcat,CYP3A4,other (1/min) | 44.24 | 44.24 | 59.38a | 44.24 | Calculated/parameter identification |
Parameter identification refers to the process in which software calculates and determines the parameters that result in the best fit between the simulated values and the observed values.
IM, intermediate metabolizer; Kcat, number describing maximum reaction rates per recombinant enzyme; Km, Michaelis constant; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer.
| (2) |
| (3) |
Kcat indicates the number describing maximum reaction rates per recombinant enzyme, Km indicates Michaelis constant, and [E] is the concentration of enzyme.
DDI model development
The DDI model was constructed by connecting the physicochemical parameters of aripiprazole collected earlier with the physicochemical parameters of risperidone provided by previous literature, 30 using inhibition constant (Ki).
The area under the curve ratio (AUCR) of aripiprazole in the absence and presence of risperidone was used to determine whether the change in aripiprazole exposure caused by risperidone was clinically significant. When the 90% confidence interval (CI) of the AUCR in the plasma completely falls within the equivalent range of 80%–125%, it can be considered that there will be no clinically significant DDI.27,31–33 Dehydro-aripiprazole is the main metabolite of aripiprazole and has pharmacological activity. Therefore, the active moiety (aripiprazole + dehydro-aripiprazole) can also serve as an indicator for evaluating DDI.
DDZI model development
To model the effect of DDZI, the difference in Child-Pugh A (CP-A) liver function was expressed by system-specific parameters (Table 3). In addition to the above parameters, the unbound fraction (fu) in the building blocks of aripiprazole and risperidone also changed. The change of unbound fraction can be calculated according to equation (4), and the fu of aripiprazole and risperidone in patients with CP-A liver function impairment was 1.23% and 2.10%, respectively. 34
Table 3.
System-specific parameters associated with Child-Pugh A liver function.
| Parameter | Normal liver function | Child-Pugh A | Proportional coefficient | Source |
|---|---|---|---|---|
| Hematocrit value | 0.46 | 0.39 | 0.85 | Wong et al. 35 |
| Plasma protein scale factor | 1.00 | 0.85 | 0.85 | Gerner and Scherf-Clavel 36 |
| Albumin | 1.00 | 0.81 | 0.81 | Woitas et al. 37 |
| α1-Acid glycoprotein | 1.00 | 0.60 | 0.60 | Barry et al. 38 |
| Fractional liver mass (L) | 2.13 | 1.47 | 0.69 | Edginton and Willmann 34 |
| Blood flow (L/min) | ||||
| Portal | 0.40 | Annet et al. 39 | ||
| Hepatic arterial | 1.30 | Annet et al. 39 | ||
| Renal | 1.38 | 1.21 | 0.88 | Dincer et al. 40 |
| Brain | 0.77 | 0.77 | 1.00 | Edginton and Willmann 34 |
| Other organs | 1.75 | Edginton and Willmann 34 | ||
| Enzyme abundance (μmol/L) | ||||
| CYP2D6 | 0.40 | 0.31 | 0.76 | Heimbach et al. 21 |
| CYP3A4 | 4.32 | 3.41 | 0.79 | Heimbach et al. 21 |
| Gastric residence time (min) | 24.00 | 28.80 | 1.20 | Heimbach et al. 21 |
Proportional coefficient means the ratio of individuals with mild liver dysfunction and individuals with normal liver function. Child-Pugh A refers to individuals with the mildest level of liver dysfunction.
| (4) |
Kprotein indicates the plasma binding proteins (albumin or α1-acid glycoprotein) partition coefficient of the compound, fprotein is the volume fraction of plasma binding proteins, and fu indicates the unbound fraction of the drug.
DDI model under the influence of multiple factors
Based on the model constructed above, our study predicted the exposure levels of aripiprazole when combined with risperidone, considering the CYP2D6 genotype and/or mild hepatic impairment: individuals with different CYP2D6 genotypes combined use of aripiprazole and risperidone (drug–drug–gene interaction, DDGI); patients with CP-A liver injury combined use of aripiprazole and risperidone (DDI and DDZI); combined use of aripiprazole and risperidone in patients with CP-A liver injury of different CYP2D6 genotypes (DDI, DGI, and DDZI).
Evaluation of the PBPK model
The quality of the PBPK model evaluation was analyzed using mean percentage error (MPE) and mean absolute percentage error (MAPE) of all predicted and observed plasma concentrations which were calculated according to equations (5) and (6). Predictions with MPE ⩽ 10% and MAPE < 25% were considered accurate predictions. 41 Aside from that, by comparing the predicted pharmacokinetic parameters with the observed pharmacokinetic parameters, the prediction accuracy of the model can also be evaluated. When the above ratios are within 0.8–1.25 folds, the predicted values are considered to be consistent with the observed values. 42 The clinical data needed for model validation are sourced from the corresponding clinical trial, as detailed in Supplemental Table S2.12,18,22,23
| (5) |
| (6) |
| (7) |
yi indicates the observed values, indicates the predicted values.
Dose optimization
When the trough concentration of aripiprazole exceeds the therapeutic reference range (100–350 ng/mL), it tends to increase risk, necessitating dose optimization. Populations that are not affected by factors such as concomitant medications, CYP2D6 genetic polymorphisms, and mild hepatic impairment are considered references. The AUC of the affected population differs from that of the reference population, expressed by a correction factor (CF). After correction, an appropriate dosage can be determined, ensuring that the AUC for individuals affected by the aforementioned factors remains consistent with that of the reference population (equation (9)). 43
| (8) |
| (9) |
CF indicates correction factors describing the effect of liver cirrhosis on hepatic metabolic activity, AUC′ and D′ represent the AUC of the plasma concentration–time curve and the dose under the influence of various factors, respectively.
Results
DGI models
PBPK mimics in vivo metabolic process of aripiprazole in individuals with different CYP2D6 genotypes. The simulation results showed that there were significant differences in the metabolic process of aripiprazole in four genotypes (Figure 2). Based on the reference of aripiprazole exposure levels in CYP2D6 normal metabolizer (NM), the fold change and 90% CI for AUC and peak concentration (Cmax) in CYP2D6 intermediate metabolizer (IM) were 1.22 (1.17–1.28) and 1.16 (1.12–1.20), respectively. For PM, the fold change and 90% CI for AUC and Cmax were 1.75 (1.68–1.82) and 1.14 (1.12–1.17), which were consistent with the information in the drug label provided by the FDA. 44
Figure 2.

Plasma concentration–time curves of aripiprazole and dehydro-aripiprazole after the administration of an oral single 10-mg dose of aripiprazole in healthy adults with CYP2D6 NM (a), IM (b), PM (c), and UM (d).
The curve above represents the predicted plasma concentration–time curves of aripiprazole and its 95% confidence interval, while the curve below represents the predicted plasma concentration–time curves of dehydro-aripiprazole and its 95% confidence interval. Simulations were compared with the corresponding observed clinical data. 22 Dots represent observed plasma concentration of aripiprazole, triangles represent dehydro-aripiprazole.
IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer.
The internal validation results showed that the MAPE values of the four single-dose models with different genotypes were all less than 25%, indicating that the prediction performance of the models was good, but the MPE values of the two models of CYP2D6 PM and CYP2D6 ultrarapid metabolizer (UM) were greater than 10% when predicting dehydro-aripiprazole, indicating that the prediction effect of these two models on dehydro-aripiprazole is poor (Figure 3; Table 4). To further verify the predictive performance of these models, external validation for the steady-state PBPK model was carried out, and the measured values were all within the predicted plasma concentration–time profiles 23 (Supplemental Figure S1).
Figure 3.
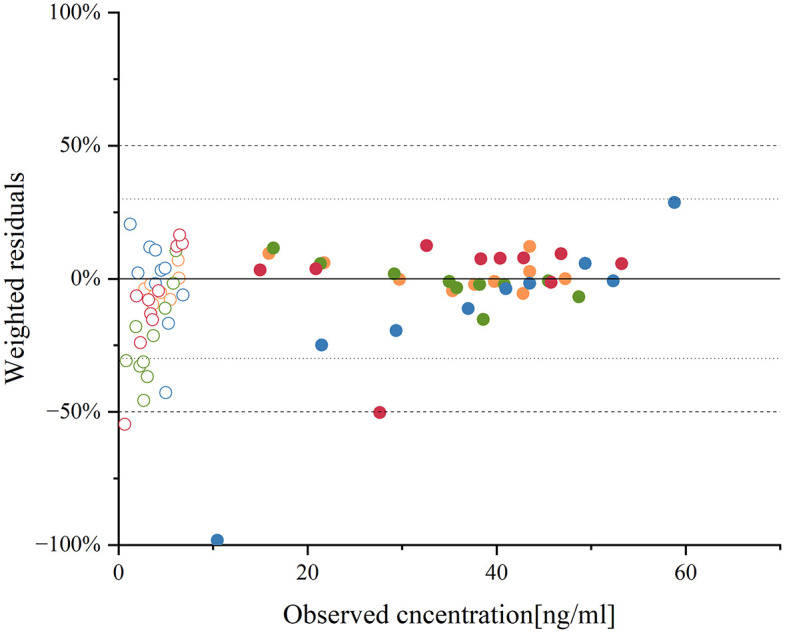
Residue plot of aripiprazole and dehydro-aripiprazole after the administration of an oral single 10-mg dose of aripiprazole in healthy adults according to CYP2D6 genotype with CYP2D6 NM, IM, PM, and UM.
The circles represent the ratio of the difference between the predicted and observed values 22 to the observed values, with solid circles representing aripiprazole and circles representing dehydro-aripiprazole. Different colors represent different CYP2D6 genotypes, with orange representing normal metabolizer, green representing IM, blue representing PM, and red representing ultrarapid metabolizer.
IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer.
Table 4.
Summary of quantitative measures of model performance for aripiprazole and its metabolite.
| Performance measures | CYP2D6 genotype | Aripiprazole | Dehydro-aripiprazole |
|---|---|---|---|
| MPE (%) | NM | 1.69 | −3.06 |
| IM | −1.26 | −21.92 | |
| PM | −13.95 | −1.51 | |
| UM | 0.58 | −8.41 | |
| MAPE (%) | NM | 4.39 | 5.11 |
| IM | 5.06 | 24.02 | |
| PM | 21.62 | 11.99 | |
| UM | 10.93 | 16.81 |
IM, intermediate metabolizer; MAPE, mean absolute percentage error; MPE, mean percentage error; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer.
DDZI models
The model simulated the pharmacokinetic profiles of CP-A liver injury patients after a single dose of 15 mg of aripiprazole. The AUC of aripiprazole and dehydro-aripiprazole were 5485.65 and 1797.11 ng h/mL, respectively (Figure 4). The internal validation of the DDZI model showed an MPE of −10% and a MAPE of 18% (Figure 5), indicating a high level of prediction accuracy. The steady-state blood concentrations of aripiprazole and its active moiety in the group with mild hepatic impairment after a single dose of the same amount were 1.65 ± 0.12 times and 1.50 ± 0.09 times higher than population with normal liver function, indicating that there is no difference in the drug exposure between the two populations. The result of the model evaluation is that the MAPE is within 25% (Figure 5).
Figure 4.
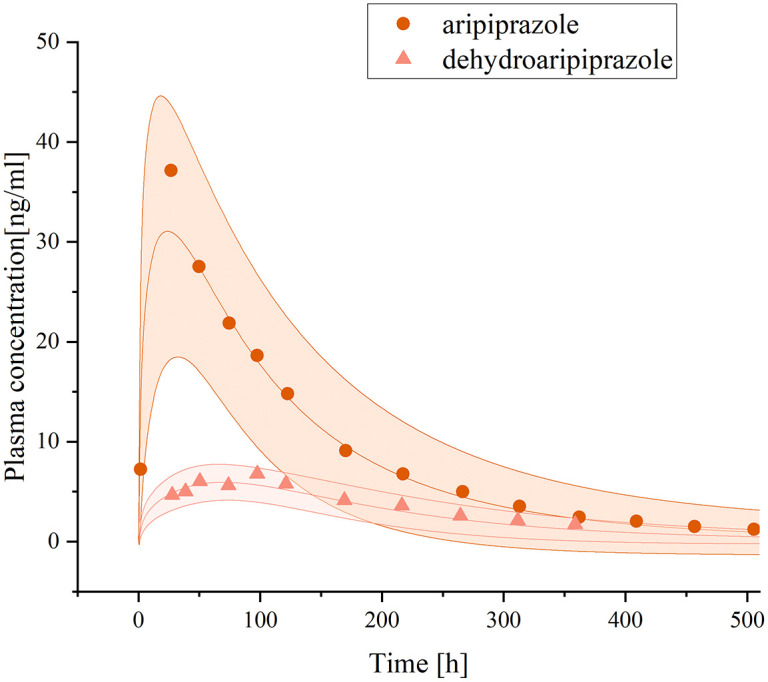
Plasma concentration–time curves of aripiprazole and dehydro-aripiprazole after the administration of an oral single 15-mg dose of aripiprazole in adults with mild hepatic impairment.
The curve above represents the predicted plasma concentration–time curves of aripiprazole and its 95% confidence interval, while the curve below represents the predicted plasma concentration–time curves of dehydro-aripiprazole and its 95% confidence interval. Simulations were compared with the corresponding observed clinical data. 18 Dots represent observed plasma concentration of aripiprazole, triangles represent dehydro-aripiprazole.
Figure 5.
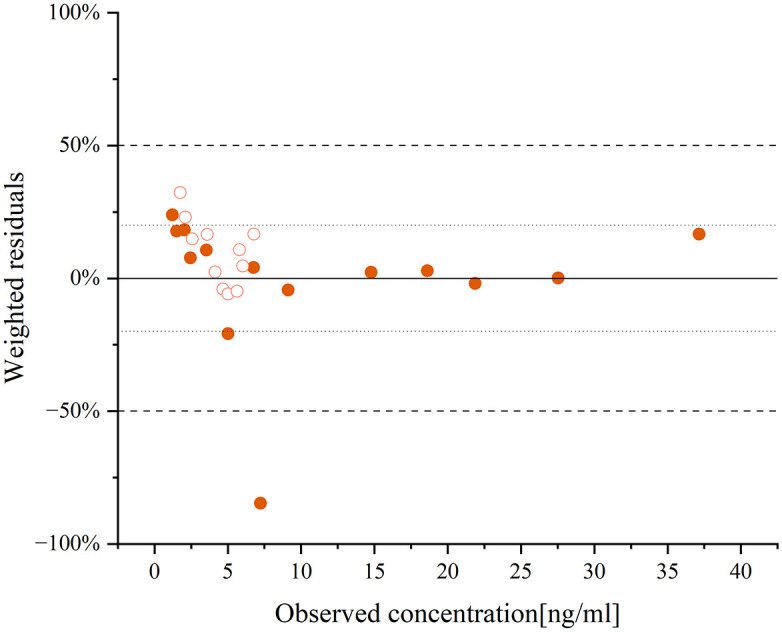
The residue plot of aripiprazole and dehydro-aripiprazole after the administration of an oral single 15-mg dose of aripiprazole in adults with mild hepatic impairment.
The circles represent the ratio of the difference between the predicted and observed values 18 to the observed values, with solid circles representing aripiprazole and circles representing dehydro-aripiprazole.
DDI models
The PBPK model of aripiprazole combined with different doses of risperidone simulated single and multiple doses (Supplemental Figure S2). After a single dose of 20 mg aripiprazole, the AUC was 773.85 μmol min/L. When aripiprazole (20 mg) was co-administered with different doses of risperidone (2, 4, 6 mg), the AUC of aripiprazole was 776.38, 779.04, and 781.31 μmol min/L, respectively. The above DDI models were verified, and the model prediction performance was good (Supplemental Figure S3). 12 The simulation showed that the AUC ratio of aripiprazole before and after combined with different doses of risperidone (2, 4, 6 mg) was 1.00, 1.01, and 1.01, which was within the invalidation boundary. It can be observed that co-administration of 2, 4, or 6 mg of risperidone did not significantly impact the exposure level of aripiprazole compared to monotherapy.
DDI model under the influence of multiple factors and dose optimization
When co-administered with risperidone in the presence of other influencing factors, the exposure level of aripiprazole may vary, and the required dosage adjustments may also differ (Figures 6–8; Table 5). First, when co-administering the two drugs in different CYP2D6 genotype populations, using aripiprazole monotherapy in the CYP2D6 NM population as the reference (AUCcon = 748.25 μmol min/L), the exposure level of aripiprazole increased by 16% in the CYP2D6 IM population and by 39% in the CYP2D6 PM population, necessitating a dosage reduction of 14% and 28%, respectively (Figure 6; Table 5). Furthermore, when combining the two drugs in the mild hepatic impairment population, the exposure level of aripiprazole increased by 13% in the CYP2D6 NM genotype population, requiring a dosage reduction of 11% (Figure 7; Table 5); in the CYP2D6 IM and PM genotype populations, the exposure level increased by 65% and 96% respectively, necessitating dosage reductions of 39% and 49%, respectively (Figure 8; Table 5).
Figure 6.

Plasma concentration–time curves of aripiprazole and dehydro-aripiprazole after the administration of multiple oral 20 mg tablets of aripiprazole co-administration with risperidone in healthy adults with CYP2D6 NM (a), IM (b), PM (c), and UM (d).
The curve above represents the predicted plasma concentration–time curves of aripiprazole and its 95% confidence interval, while the curve below represents the predicted plasma concentration–time curves of dehydro-aripiprazole and its 95% confidence interval. Simulations were compared with the corresponding observed clinical data. Dots represent observed plasma concentration of aripiprazole, triangles represent dehydro-aripiprazole.
IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer
Figure 7.
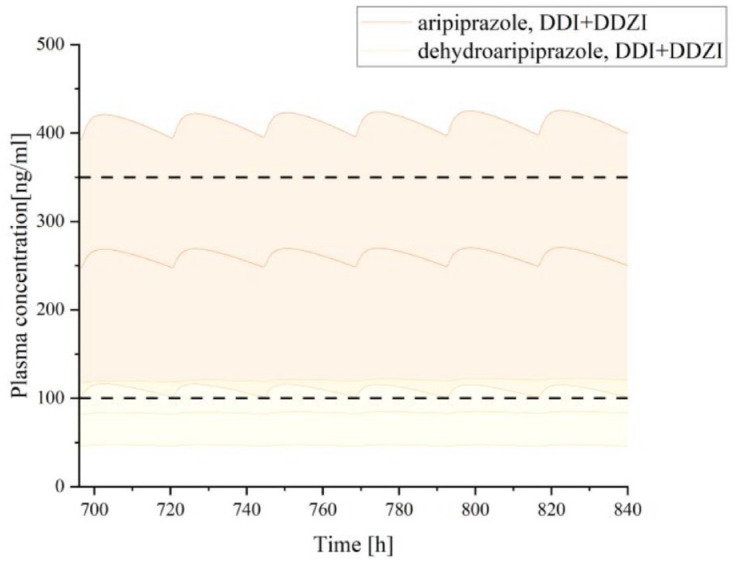
Plasma concentration–time curves of aripiprazole and dehydro-aripiprazole after the administration of multiple oral 20 mg tablets of aripiprazole co-administration with risperidone in adults with mild hepatic impairment.
CP-A, Child-Pugh A; DDI, drug–drug interaction; DDZI, drug–disease interaction.
Figure 8.

Plasma concentration–time curves of aripiprazole and dehydro-aripiprazole after the administration of multiple oral 20 mg tablets of aripiprazole co-administration with risperidone in adults with mild hepatic impairment according to CYP2D6 phenotype, including CYP2D6 NM (a), IM (b), PM (c), and UM (d).
CP-A, Child-Pugh A; IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer.
Table 5.
Summary of dose optimization for DDI under the influence of other factors.
| Factors | Genotype | AUC (μmol min/L) | CF | ΔD |
|---|---|---|---|---|
| Genotype | NM | 755.26 | 0.99 | 1% |
| IM | 865.09 | 0.86 | 14%※ | |
| PM | 1036.57 | 0.72 | 28%※ | |
| UM | 758.19 | 0.99 | 1% | |
| Mild hepatic impairment | NM | 842.20 | 0.89 | 11%※ |
| Genotype and mild hepatic impairment | NM | 842.20 | 0.89 | 11%※ |
| IM | 1231.96 | 0.61 | 39%※ | |
| PM | 1466.17 | 0.51 | 49%※ | |
| UM | 840.33 | 0.89 | 11%※ |
represents requiring a dosage reduction.
AUC, area under the cure; CF, correction factor indicates correction factors describing the effect of liver cirrhosis on hepatic metabolic activity; ΔD, changes of dose; DGI, drug–gene interaction; DDZI, drug–disease interaction; IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer.
Discussion
Currently, there is limited research on the pharmacokinetics of the combined use of aripiprazole and risperidone. Although a few studies have suggested that the combination did not increase the risk of adverse drug reaction, the reliability of the evidence is limited, and there is a lack of research on the risks under the influence of multiple factors.12,45 Our study mainly focused on two factors: DGI46,47 and hepatic dysfunction, 48 as they are important factors that affect drug metabolism and are commonly encountered in clinical practice in psychiatry. In this study, successful development of PBPK models was constructed for aripiprazole and dehydro-aripiprazole in scenarios where aripiprazole was co-administered with risperidone. Validated with all predictions fell within the expected range of error. These models were tailored for combination aripiprazole with risperidone for populations with varying CYP2D6 genotypes and individuals with mild hepatic impairment. Following the validation of model accuracy, variations in the exposure levels of aripiprazole were predicted, and optimization of dosage was performed.
When the three aforementioned factors existed independently, different factors had different effects on the exposure levels of aripiprazole and dehydro-aripiprazole. The PBPK model suggested that DGI could significantly alter the exposure levels of aripiprazole and its active metabolite, which was consistent with the existing research conclusions on the impact of CYP2D6-based DGI on aripiprazole exposure levels.13,49,50 Our study further evaluated dose optimization in the presence of DGI. The prediction results suggested that when the patient’s CYP2D6 is IM, the dose of aripiprazole should be reduced by about 14%; while in patients with CYP2D6 PM, the dose of aripiprazole should be reduced by 28%, which is consistent with the recommended dose of DPWG. 51 The DDI (when co-administered with risperidone) or DDZI (in the presence of mild hepatic impairment) did not have a significant effect, which is consistent with existing research findings.10,17,18 Although there is limited and small-scale clinical trial data available, the literature search did not reveal the need for dose optimization of aripiprazole in cases of mild hepatic impairment or co-administration with risperidone. This is consistent with the information provided in the drug label of aripiprazole, 47 which indicated that these two factors did not significantly increase the occurrence rate of adverse drug reactions. Furthermore, previous theories suggested that the simultaneous action of multiple factors on an individual may result in synergistic effects19,52 or antagonistic effects. 53 Our research findings aligned with the former, indicating a synergistic effect. The exposure level of aripiprazole will change when co-administration is affected by DGI and/or DDZI. When co-administered to patients with mild hepatic impairment and CYP2D6 genotype NM, the change in aripiprazole exposure levels (ΔAUC) was 13%, necessitating a reduction in aripiprazole dosage by approximately 11%. This suggested that the combined effect of DDI (ΔAUC = 1%) and DDZI (ΔAUC = 8%) on aripiprazole exposure levels is greater than the sum of their individual effects. When DGI (ΔAUCIM = 14%, ΔAUCPM = 37%), DDZI (ΔAUC = 8%), and DDI (ΔAUC = 1%) act simultaneously, this synergistic effect became more pronounced. In patients co-administered with mild hepatic impairment and CYP2D6 genotypes IM and PM, the ΔAUC of aripiprazole was 65% and 96%, respectively, indicating a need to reduce the dose to 61% and 51%, respectively.
Traditional pharmacokinetic studies are mostly conducted through clinical trials or post-marketing surveillance reports. 54 There is a lack of research on the multi-factorial modulation of antipsychotic drug exposure levels, and currently, there is only limited research on DDGI in other antipsychotic drugs.55,56 However, in clinical practice, the exposure level of aripiprazole is often the result of the combined influence of multiple factors, which hinders the clinical application of previous research findings. Our study investigated the safety of co-administering aripiprazole and risperidone under various conditions using PBPK modeling. It proposed relative dose recommendations for aripiprazole based on different influencing factors, thereby providing a theoretical foundation for the clinical utilization of this drug combination from a pharmacokinetic standpoint. With further clinical validation, these results are more likely to be embraced by clinicians and more readily adopted in clinical practice. In terms of methodology, PBPK modeling offers a superior benefit–risk ratio compared to traditional pharmacokinetic research methods. It can serve as a foundation for designing and implementing clinical trial protocols while also reducing the required sample size in clinical trials.
Limitations
As a model prediction study, some potential limitations should be considered. First, although the basic models of aripiprazole and risperidone, DGI, DDI, and DDZI models have been verified by corresponding clinical trial data, the prediction model under the influence of multiple factors is limited by the lack of relevant clinic trials and has not been verified. It is necessary to further mine the corresponding data and verify the model through clinical trials. Meanwhile, our study used 80%–125% as the no-effect boundary to determine whether the exposure levels changed, which is one of the most conservative standards for measuring drug equivalence. This means that our standards may deviate from the actual concentration–effect relationship. A fuller understanding of the dose–concentration and/or concentration–effect relationships of drug effects may be beneficial in interpreting predicted results and further optimizing criteria for dose adjustment.
Conclusion
Through PBPK models, our study accurately predicted the plasma concentration profiles and pharmacokinetic parameters of aripiprazole and its active metabolite after co-administration with risperidone in various populations, thereby offering valuable insights for personalized treatment approaches. Our study demonstrated that the co-administration of aripiprazole and risperidone is generally considered safe. However, in individuals with a CYP2D6 genotype of IM or PM and/or those with mild hepatic impairment, co-administration of the two drugs could result in increased exposure levels of aripiprazole. Therefore, it is crucial to appropriately reduce the dose of aripiprazole to mitigate potential risks, particularly in individuals with a CYP2D6 IM or PM genotype or mild hepatic impairment.
Supplemental Material
Supplemental material, sj-docx-1-taw-10.1177_20420986241303432 for Physiologically based pharmacokinetic modeling to predict the effect of risperidone on aripiprazole pharmacokinetics in subjects with different CYP2D6 genotypes and individuals with hepatic impairment by Fan Mou, Zhiwei Huang, Yu Cheng, Xue Zhao, Xiujia Sun, Huafang Li and Shunying Yu in Therapeutic Advances in Drug Safety
Supplemental material, sj-docx-2-taw-10.1177_20420986241303432 for Physiologically based pharmacokinetic modeling to predict the effect of risperidone on aripiprazole pharmacokinetics in subjects with different CYP2D6 genotypes and individuals with hepatic impairment by Fan Mou, Zhiwei Huang, Yu Cheng, Xue Zhao, Xiujia Sun, Huafang Li and Shunying Yu in Therapeutic Advances in Drug Safety
Acknowledgments
None.
Footnotes
ORCID iD: Fan Mou  https://orcid.org/0009-0009-8485-9202
https://orcid.org/0009-0009-8485-9202
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Fan Mou, Genetics and Biochemistry Laboratory, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Zhiwei Huang, Drug Clinical Trial Institution, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Yu Cheng, Genetics and Biochemistry Laboratory, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Xue Zhao, Clinical Research Center, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Xiujia Sun, Genetics and Biochemistry Laboratory, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Huafang Li, Drug Clinical Trial Institution, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Shunying Yu, Genetics and Biochemistry Laboratory, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, 600 Wan Ping Nan Road, Shanghai 200030, China.
Declarations
Ethics approval and consent to participate: The data involved in this article are available from the public databases.
Consent for publication: All the authors consent for publication.
Author contributions: Fan Mou: Data curation; Investigation; Methodology; Validation; Writing – original draft.
Zhiwei Huang: Supervision; Validation; Writing – review & editing.
Yu Cheng: Data curation; Investigation; Validation; Writing – review & editing.
Xue Zhao: Investigation; Validation; Writing – review & editing.
Xiujia Sun: Data curation; Investigation; Validation.
Huafang Li: Funding acquisition; Supervision; Validation; Writing – review & editing.
Shunying Yu: Conceptualization; Funding acquisition; Methodology; Project administration; Supervision; Validation; Writing – review & editing.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the grant from key program of the Clinical Research Center at Shanghai Mental Health Center, China (No. CRC2021ZD02), the grant from interdisciplinary medical-engineering research under Shanghai Jiao Tong University’s “Star of Jiao Tong” Program, China (No. YG2023LC14), the General Program of Shanghai Mental Health Center “Academic Project” (2021-YJ04) and the Shanghai Jiao Tong University Medical-Industrial Interdisciplinary Fund (YG2021QN131).
Competing interests: The authors declare that there is no conflict of interest.
Availability of data and materials: All data and materials included in this study are available upon request by contact with the corresponding author.
References
- 1. Institute for Health Metrics and Evaluation. Global Health Data Exchange (GHDx), https://vizhub.healthdata.org/gbd-results/ (2022, accessed 14 May 2022).
- 2. Huang CY, Fang SC, Shao YJ. Comparison of long-acting injectable antipsychotics with oral antipsychotics and suicide and all-cause mortality in patients with newly diagnosed schizophrenia. JAMA Netw Open 2021; 4: e218810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taipale H, Tanskanen A, Mehtala J, et al. 20-Year follow-up study of physical morbidity and mortality in relationship to antipsychotic treatment in a nationwide cohort of 62,250 patients with schizophrenia (FIN20). World Psychiatry 2020; 19: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qiao Y, Yang F, Li C, et al. Add-on effects of a low-dose aripiprazole in resolving hyperprolactinemia induced by risperidone or paliperidone. Psychiatry Res 2016; 237: 83–89. [DOI] [PubMed] [Google Scholar]
- 5. Chen JX, Su YA, Bian QT, et al. Adjunctive aripiprazole in the treatment of risperidone-induced hyperprolactinemia: a randomized, double-blind, placebo-controlled, dose-response study. Psychoneuroendocrinology 2015; 58: 130–140. [DOI] [PubMed] [Google Scholar]
- 6. Kane JM, Correll CU, Goff DC, et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. J Clin Psychiat 2009; 70: 1348–1357. [DOI] [PubMed] [Google Scholar]
- 7. Zhang L, Qi H, Xie YY, et al. Efficacy and Safety of Adjunctive Aripiprazole, Metformin, and Paeoniae-Glycyrrhiza Decoction for Antipsychotic-Induced Hyperprolactinemia: A Network Meta-Analysis of Randomized Controlled Trials. Front Psychiatry 2021; 12: 728204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mannens G, Meuldermans W, Snoeck E, et al. Plasma protein binding of risperidone and its distribution in blood. Psychopharmacology (Berl) 1994; 114: 566–572. [DOI] [PubMed] [Google Scholar]
- 9. Bauman JN, Frederick KS, Sawant A, et al. Comparison of the bioactivation potential of the antidepressant and hepatotoxin nefazodone with aripiprazole, a structural analog and marketed drug. Drug Metab Dispos 2008; 36: 1016–1029. [DOI] [PubMed] [Google Scholar]
- 10. Kiss A, Menus A, Toth K, et al. Phenoconversion of CYP2D6 by inhibitors modifies aripiprazole exposure. Eur Arch Psychiatry Clin Neurosci 2020; 270: 71–82. [DOI] [PubMed] [Google Scholar]
- 11. DrugBank. Aripiprazole, https://go.drugbank.com/drugs/DB01238 (2005, accessed 14 May 2024).
- 12. Waade RB, Christensen H, Rudberg I, et al. Influence of comedication on serum concentrations of aripiprazole and dehydroaripiprazole. Ther Drug Monit 2009; 31: 233–238. [DOI] [PubMed] [Google Scholar]
- 13. Jukic MM, Smith RL, Haslemo T, et al. Effect of CYP2D6 genotype on exposure and efficacy of risperidone and aripiprazole: a retrospective, cohort study. Lancet Psychiatry 2019; 6: 418–426. [DOI] [PubMed] [Google Scholar]
- 14. Blazy C, Ellingrod V, Ward K. Variability between Clinical Pharmacogenetics Implementation Consortium (CPIC®) guidelines and a commercial pharmacogenetics laboratory in genotype to phenotype interpretations for patients utilizing psychotropics. Front Pharmacol 2022; 13: 939313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beunk L, Nijenhuis M, Soree B, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2D6, CYP3A4 and CYP1A2 and antipsychotics. Eur J Hum Genet 2024; 32: 278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bank PCD, Caudle KE, Swen JJ, et al. Comparison of the guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin Pharmacol Ther 2018; 103: 599–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simon N, Torrents R, Azorin JM. Comorbidities and the right dose: antipsychotics. Expert Opin Drug Metab Toxicol 2022; 18: 507–518. [DOI] [PubMed] [Google Scholar]
- 18. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet 2008; 47: 533–542. [DOI] [PubMed] [Google Scholar]
- 19. Marok FZ, Fuhr LM, Hanke N, et al. Physiologically based pharmacokinetic modeling of bupropion and its metabolites in a CYP2B6 drug-drug-gene interaction network. Pharmaceutics 2021; 13: 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Groen BD, Nicolai J, Kuik AC, et al. Ontogeny of hepatic transporters and drug-metabolizing enzymes in humans and in nonclinical species. Pharmacol Rev 2021; 73: 597–678. [DOI] [PubMed] [Google Scholar]
- 21. Heimbach T, Chen Y, Chen J, et al. Physiologically-based pharmacokinetic modeling in renal and hepatic impairment populations: a pharmaceutical industry perspective. Clin Pharmacol Ther 2021; 110: 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kneller LA, Zubiaur P, Koller D, et al. Influence of CYP2D6 phenotypes on the pharmacokinetics of aripiprazole and dehydro-aripiprazole using a physiologically based pharmacokinetic approach. Clin Pharmacokinet 2021; 60: 1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mallikaarjun S, Salazar DE, Bramer SL. Pharmacokinetics, tolerability, and safety of aripiprazole following multiple oral dosing in normal healthy volunteers. J Clin Pharmacol 2004; 44: 179–187. [DOI] [PubMed] [Google Scholar]
- 24. von Elm E, Altman DG, Egger M, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med 2007; 147: 573–577. [DOI] [PubMed] [Google Scholar]
- 25. Kambayashi A, Dressman JB. Towards virtual bioequivalence studies for oral dosage forms containing poorly water-soluble drugs: a physiologically based biopharmaceutics modeling (PBBM) approach. J Pharm Sci 2022; 111: 135–145. [DOI] [PubMed] [Google Scholar]
- 26. Langenbucher F. Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol 1972; 24: 979–981. [DOI] [PubMed] [Google Scholar]
- 27. Vieira MD, Kim MJ, Apparaju S, et al. PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin Pharmacol Ther 2014; 95: 550–557. [DOI] [PubMed] [Google Scholar]
- 28. Mihajlovic T, Kachrimanis K, Graovac A, et al. Improvement of aripiprazole solubility by complexation with (2-hydroxy)propyl-beta-cyclodextrin using spray drying technique. AAPS PharmSciTech 2012; 13: 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taylor C, Crosby I, Yip V, et al. A review of the important role of CYP2D6 in pharmacogenomics. Genes (Basel) 2020; 11: 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kneller LA, Abad-Santos F, Hempel G. Physiologically based pharmacokinetic modelling to describe the pharmacokinetics of risperidone and 9-hydroxyrisperidone according to cytochrome P450 2D6 phenotypes. Clin Pharmacokinet 2020; 59: 51–65. [DOI] [PubMed] [Google Scholar]
- 31. US Food and Drug Administration. In vitro metabolism- and transporter-mediated drug–drug interaction studies guidance for industry, https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm581965.pdf (2017, accessed 14 May 2022).
- 32. Einolfl HJ, Chen L, Fahmi OA, et al. Evaluation of various static and dynamic modeling methods to predict clinical CYP3A induction using in vitro CYP3A4 mRNA induction data. Clin Pharmacol Ther 2014; 95: 179–188. [DOI] [PubMed] [Google Scholar]
- 33. Administration UFaD. Clinical drug interaction studies with combined oral contraceptives guidance for industry, https://www.fda.gov/media/143849/download (2023, accessed 9 May 2024).
- 34. Edginton AN, Willmann S. Physiology-based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin Pharmacokinet 2008; 47: 743–752. [DOI] [PubMed] [Google Scholar]
- 35. Wong F, Girgrah N, Graba J, et al. The cardiac response to exercise in cirrhosis. Gut 2001; 49: 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gerner B, Scherf-Clavel O. Physiologically based pharmacokinetic modelling of cabozantinib to simulate enterohepatic recirculation, drug-drug interaction with rifampin and liver impairment. Pharmaceutics 2021; 13: 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woitas RP, Stoffel-Wagner B, Flommersfeld S, et al. Correlation of serum concentrations of cystatin C and creatinine to inulin clearance in liver cirrhosis. Clin Chem 2000; 46: 712–715. [PubMed] [Google Scholar]
- 38. Barry M, Keeling PW, Weir D, et al. Severity of cirrhosis and the relationship of alpha 1-acid glycoprotein concentration to plasma protein binding of lidocaine. Clin Pharmacol Ther 1990; 47: 366–370. [DOI] [PubMed] [Google Scholar]
- 39. Annet L, Materne R, Danse E, et al. Hepatic flow parameters measured with MR imaging and Doppler US: correlations with degree of cirrhosis and portal hypertension. Radiology 2003; 229: 409–414. [DOI] [PubMed] [Google Scholar]
- 40. Dincer D, Besisk F, Demirkol O, et al. Relationships between hemodynamic alterations and Child-Pugh Score in patients with cirrhosis. Hepatogastroenterology 2005; 52: 1521–1525. [PubMed] [Google Scholar]
- 41. Owen JS, Fiedler-Kelly J. Introduction to population pharmacokinetic/pharmacodynamic analysis with nonlinear mixed effects models. Hoboken: Wiley, 2014. [Google Scholar]
- 42. Hsueh CH, Hsu V, Pan Y, et al. Predictive performance of physiologically-based pharmacokinetic models in predicting drug-drug interactions involving enzyme modulation. Clin Pharmacokinet 2018; 57: 1337–1346. [DOI] [PubMed] [Google Scholar]
- 43. Ozbey AC, Bachmann F, Duthaler U, et al. Dose adjustment in patients with liver cirrhosis—comparison of two different modeling approaches. Clin Pharmacol Ther 2023; 113: 1346–1358. [DOI] [PubMed] [Google Scholar]
- 44. Food and Drug Administration. Abilify® (aripiprazole): U.S. full prescribing information, https://www.otsuka-us.com/media/static/Abilify-PI.pdf (2020, accessed 9 May 2024).
- 45. Jiang P, Sun X, Ren J, et al. Effects of the combination of second-generation antipsychotics on serum concentrations of aripiprazole and dehydroaripiprazole in Chinese patients with schizophrenia. Gen Psychiatr 2021; 34: e100423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 2013; 138: 103–141. [DOI] [PubMed] [Google Scholar]
- 47. Soria-Chacartegui P, Villapalos-Garcia G, Zubiaur P, et al. Genetic polymorphisms associated with the pharmacokinetics, pharmacodynamics and adverse effects of olanzapine, aripiprazole and risperidone. Front Pharmacol 2021; 12: 711940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Frye RF, Zgheib NK, Matzke GR, et al. Liver disease selectively modulates cytochrome P450-mediated metabolism. Clin Pharmacol Ther 2006; 80: 235–245. [DOI] [PubMed] [Google Scholar]
- 49. Belmonte C, Ochoa D, Román M, et al. Influence of CYP2D6, CYP3A4, CYP3A5 and ABCB1 polymorphisms on pharmacokinetics and safety of aripiprazole in healthy volunteers. Basic Clin Pharmacol 2018; 122: 596–605. [DOI] [PubMed] [Google Scholar]
- 50. Milosavljevic F, Bukvic N, Pavlovic Z, et al. Association of CYP2C19 and CYP2D6 poor and intermediate metabolizer status with antidepressant and antipsychotic exposure: a systematic review and meta-analysis. JAMA Psychiatry 2021; 78: 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Swen JJ, Nijenhuis M, de Boer A, et al. Pharmacogenetics: from bench to byte-an update of guidelines. Clin Pharmacol Ther 2011; 89: 662–673. [DOI] [PubMed] [Google Scholar]
- 52. Depta JP, Lenzini PA, Lanfear DE, et al. Clinical outcomes associated with proton pump inhibitor use among clopidogrel-treated patients within genotype groups following acute myocardial infarction. Pharmacogenomics J 2015; 15: 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vormfelde SV, Brockmöller J, Bauer S, et al. Relative impact of genotype and enzyme induction on the metabolic capacity of CYP2C9 in healthy volunteers. Clin Pharmacol Ther 2009; 86: 54–61. [DOI] [PubMed] [Google Scholar]
- 54. Hoots BE, Xu L, Kariisa M. 2018 annual surveillance report of drug-related risks and outcomes—United States. CDC National Center for Injury Prevention and Control, 2018. [Google Scholar]
- 55. Wojtyniak JG, Selzer D, Schwab M, et al. Physiologically based precision dosing approach for drug-drug-gene interactions: a simvastatin network analysis. New Jersey: Clin Pharmacol Ther 2021; 109: 201–211. [DOI] [PubMed] [Google Scholar]
- 56. Loer HLH, Feick D, Rüdesheim S, et al. Physiologically based pharmacokinetic modeling of tacrolimus for food-drug and CYP3A drug-drug-gene interaction predictions. CPT Pharmacometrics Syst Pharmacol 2023; 12: 724–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-taw-10.1177_20420986241303432 for Physiologically based pharmacokinetic modeling to predict the effect of risperidone on aripiprazole pharmacokinetics in subjects with different CYP2D6 genotypes and individuals with hepatic impairment by Fan Mou, Zhiwei Huang, Yu Cheng, Xue Zhao, Xiujia Sun, Huafang Li and Shunying Yu in Therapeutic Advances in Drug Safety
Supplemental material, sj-docx-2-taw-10.1177_20420986241303432 for Physiologically based pharmacokinetic modeling to predict the effect of risperidone on aripiprazole pharmacokinetics in subjects with different CYP2D6 genotypes and individuals with hepatic impairment by Fan Mou, Zhiwei Huang, Yu Cheng, Xue Zhao, Xiujia Sun, Huafang Li and Shunying Yu in Therapeutic Advances in Drug Safety