

Abstract
Background
Aging and tumorigenesis share intricate regulatory processes, that alter the genome, epigenome, transcriptome and immune landscape of tissues. Discovering the link between aging and cancer in terms of multiomics characteristics remains a challenge for biomedical researchers.
Methods
We collected high-throughput datasets for 57 human tumors and 20 normal tissues, including 23,125 samples with age information. On the basis of these sufficient omics data, we introduced six useful modules including genomic (somatic mutation and copy number variation), gene expression, DNA methylation, hallmarks (aging and cancer), immune landscape (immune infiltration, immune pathways, immune signatures, and antitumor immune activities) and survival analysis. Correlation and differential analyses were performed for the multiomic signatures associated with aging at the gene level.
Results
We developed Aging2Cancer (http://210.37.77.200:8080/Aging2Cancer/index.jsp), which is a comprehensive database and analysis platform for revealing the associations between aging and cancer. Users can search for and visualize the results of genes of interest to explore the relationships between aging and cancer at the gene level for different omics levels.
Conclusions
We believe that Aging2Cancer is a valuable resource for identifying novel biomarkers and will serve as a bridge for linking aging to cancer.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-024-11150-z.
Keywords: Aging, Cancer, Database, Multiomics
Background
Aging is the leading risk factor for cancer, as the incidence and mortality of cancers increase with age [1, 2]. Aging and tumorigenesis are both complex processes that are influenced by the genome, epigenome, transcriptome, and various regulatory elements [3, 4]. Recently, the availability of high-throughput technologies has led to the generation of large amounts of cancer-related omics data, which shed light on precision medicine for cancer treatment. However, cancer disparities involving age are often neglected in these studies. Therefore, an integrated database and analysis platform that focuses on the links between aging and cancer needs to be developed.
Several aging-related resources have been developed, including Aging Atlas [5], Human Ageing Genomic Resources (HAGR) [6], AgeAnno [7], AgingBank [8], Open Genes [9], and ClockBase [10]. Among them, AgingBank is a comprehensive and newly developed database that provides the first Cancer & Aging module. However, AgingBank supported only transcriptome-level (genes, lncRNAs, and miRNAs) analysis for The Cancer Genome Atlas (TCGA) project. A comprehensive resource that allows multiomics data analysis exploring the relationships between aging and cancer is urgently needed.
For this purpose, we established Aging2Cancer, an integrated resource for linking aging data to tumor multiomics data. Aging2Cancer includes multiomics data from 23,125 samples from the TCGA, International Cancer Genome Consortium (ICGC), and Genotype-Tissue Expression (GTEx) databases, covering 57 cancer types and 20 normal tissues. Aging2Cancer provides interactive modules to explore: (i) the correlation between transcriptome expression and aging in tumor and normal samples; (ii) the distribution of genomic alterations between younger and older patients; (iii) the correlation between DNA methylation at the gene-level and aging in tumor and normal samples; (iv) the activities of markers related to cancer- and aging-related hallmarks; (v) tumor immunity features including immune cell infiltration, immune pathways, immune signatures, and antitumor immune activity; and (vi) survival analysis based on multiomics features in different age groups. These modules allow users to explore the links between aging and multiomic signatures as well as clinical implementations. All the information about Aging2Cancer is freely accessible at http://210.37.77.200:8080/Aging2Cancer/.
Construction and content
Data collection and processing
The multiomics data in TCGA Pan-Cancer (PANCAN) and International Cancer Genome Consortium (ICGC) were downloaded from UCSC Xena (http://xena.ucsc.edu/) and the ICGC Data Portal (https://dcc.icgc.org/, release 7.0), respectively. The TCGA and pediatric tumor samples were filtered out from the ICGC cohort. The gene expression and DNA methylation data of normal tissues were downloaded from the Genotype-Tissue Expression (GTEx) portal. Only samples with age information were included in the analysis. We also retained cancer types with more than 15 cases for subsequent analyses. The information concerning the cancer types and normal tissues used in Aging2Cancer are listed in Supplementary Tables S1-S3. The RNA-seq profiles from TCGA and GTEx were recomputed via the TOIL pipeline, and the ICGC transcriptomes were processed via STAR, HISAT2, and Picard. All the gene expression levels were normalized to TPM values and log2 transformed (TPM + 0.001).
Identification of age-related multi-omics signatures
Gene expression
To explore the associations between aging and gene expression, we performed a multivariate linear regression model as described by Lee et al.(https://zenodo.org/records/5576639) [11]. Specifically, we used the following formula:
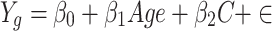 |
where the dependent variable (Y) represents the expression level of a given gene, the independent variable (age) represents the age at initial pathological diagnosis of the sample, and the covariates (C) include tumor pathologic stage, sex, and race. For normal tissues and tumor types lacking these covariates, simple linear regression was applied to estimate the associations.
DNA methylation
Only the Illumina HumanMethylation450 BeadChip (450 K) was used in this study, and missing values were imputed via the knnImputation function from the DMwR2 R package. The CpG probes were mapped to the promoter region of each gene (4-kb regions centered at the TSS) according to the annotations from GENCODE (V42, GRCh38). The DNA methylation levels for each gene were subsequently assigned on the basis of the average beta value of the CpG probes located in the promoter region. To estimate the associations between aging and gene methylation, multivariate and simple linear regression models similar to those used for gene expression were used for each cancer type and for normal tissue.
Genomic alteration
Single nucleotide variant (SNV) calls of TCGA patients were obtained from the Multi-Center Mutation Calling in Multiple Cancers (MC3) project [12]. The SNV calls of ICGC from multiple pipelines were integrated [13]. The gene-level copy number variation (CNV) was calculated via GISTIC 2.0 [14], where categories 1 and 2 considered copy number amplification, and categories − 2 and − 1 considered copy number deletion. Subjects who were under a given age (50 years by default) at the initial pathologic diagnosis were classified as younger, whereas others were classified as older. The chi-square test was used to compare the frequency of genomic alterations between the younger and older groups.
Cancer and aging hallmarks
The gene sets related to cancer hallmarks were derived from a previous study [15], and the gene sets related to aging hallmarks were collected from the Aging Atlas database (https://ngdc.cncb.ac.cn/aging/index). Single-sample GSEA (ssGSEA) was applied to estimate the activities related to each cancer and aging hallmark in tumors [16].
Tumor immunity
The immune module is divided into four tabs: immune infiltration, immune pathway, immune signature, and antitumor immune activity. The immune infiltration tab shows the components of the immune microenvironment, which are calculated by seven computational frameworks namely, CIBERSORT [17], CIBERSORT-ABS [17], QUANISEQ [18], TIMER [19], EPIC [20], MCPCOUNTER [21], and xCell [22]. The immune pathway tab provides the activities of 17 immune pathways collected from the ImmPort project [23] The immune signature tab presents the activities of immune checkpoints, human leukocyte antigen (HLA), tumor-infiltrating lymphocytes (TILs), macrophages/monocytes, lymphocyte infiltration, the TGF-β response, the IFN-γ response, and wound healing. Finally, the antitumor immune activity tab includes the ESTIMATE score, immune score, MHC score, and CYT score as previously described [24].
Database implementation
We chose Eclipse as the development environment. HTML, CSS, and JS were used to design and write the front-end interface, and the Bootstrap, Spring, and jQuery frameworks were further used to simplify the front-end development process. We used the SQL server to store and retrieve data. The data visualization was performed by Plotly.js. Java 11 served as the runtime environment to write business logic, and the website was deployed on a Tomcat 9 server.
Standard input parameters for Aging2Cancer
There are several standard input parameters for Aging2Cancer, including ‘Gene Search’, ‘Age Selection’, ‘Data Source’, and ‘Cancer/Tissue Type’. The webserver collects the gene information that users enter in the gene search parameter with a gene symbol or Ensembl ID. The default value of the age selection parameter is set to 50 according to the Lee et al. study, which distinguished younger adult cancer patients from later-onset cancer patients [11]. In addition, users can choose any age cutoff at which they want to divide cancer patients into younger and older groups. The data source and cancer/tissue type parameters represent the data cohort and cancer/tissue type of interest to the user in the TCGA, ICGA, and GTEx databases.
Utility and discussion
Database overview
Aging2Cancer collected multiomics data on tumor and normal tissues as well as corresponding age information. On the basis of the genomic, transcriptomic, and epigenomic profiles of 23,125 samples which cover 57 cancer types and 20 normal tissues (Fig. 1), Aging2Cancer is designed to link age to tumor multiomics data. Users can explore the associations between age and multiomic signatures at the gene-level (including expression, genomic alteration, DNA methylation, cancer and aging hallmarks, and tumor immunology) for cancer types or tissues of interest (Fig. 1). In addition, Aging2Cancer can be used for survival analysis according to multiomic features in both the younger and older groups. We systematically compared the general features of Aging2Cancer with those of other databases in the aging field. As shown in Supplementary Table S4, Aging2Cancer focused on the multiomic data for Homo sapiens. Among the databases compared, AgingBank and our Aging2Cancer are the only two tools that provide a Cancer and Aging module allowing users to explore the associations between aging and cancer at the gene level. However, AgingBank only supports only the transcriptome-level analysis of the TCGA cohorts. Our tool incorporates other omics data, including genome, epigenome, and immunome data, from both the TCGA and ICGC datasets. In addition to the data, we provide several useful visualization tools, including scatter plots, waterfall plots, boxplots, and Kaplan-Meier curves, which can help users easily explore the links between aging and cancer in terms of multiomic characteristics.
Fig. 1.

Overview of Aging2Cancer. The upper panel contains the database content, which includes the collection and processing of tumor and normal multi-omics datasets and the construction of web tools. The lower panel contains the web tools of Aging2Cancer. The analysis of genome, transcriptome, epigenome, hallmarks, immune regulation, and survival about aging are provided
User interface
The interface of Aging2Cancer can be freely accessed at http://210.37.77.200:8080/Aging2Cancer/. The database provides several interactive modules including expression, DNA methylation, genomic alteration, cancer and aging hallmarks, tumor immunity, and survival analysis.
Expression
The expression module allows users to select a tumor type or normal tissue with specific genes and perform linear regression to identify associations between gene expression and the age of samples (Fig. 2, Expression). The user can visualize the relationship on the basis of the continuous age variable or under different age groups via scatter plots or violin plots. The corresponding image can be accessed by choosing the TCGA, ICGC, and GTEx cohorts and the gene symbol and color options. We also offer multiple input genes analyses in the expression module so that users can assess the difference in the linear regression coefficients of multiple genes between the younger and older groups.
Fig. 2.

Content and user interface of Aging2Cancer. (i) Search by a gene or gene list in a specific cancer type with an interested age threshold. (ii) Expression/Methylation module. The results page of this module includes a scatter plot that indicates the relationship between aging and gene expression/methylation and a violin plot exhibits the distribution of gene expression/methylation level in different aging groups. The heatmap plots shows the difference in the linear regression coefficients of multiple genes between the younger and older groups. (iii) Genomic module. The results page of this module included tables of the count and frequency of CNV/SNV in different aging groups. The differences in genomic alterations in different groups are estimated by the chi-square test. Waterfall plots shows the differences in the genomic patterns of multiple genes between the younger and older groups. (iv) Hallmarks/Immune module. The results page of this module included box plots that exhibit the scores of cancer- and aging-related hallmarks, immune-related scores, and user’s custom gene set scores in different aging groups, and scatter plots that indicate the relationship between hallmarks/immune scores and gene expression in different aging groups. (v) Survival module. The results page of this module included survival curves that consider both gene expression and aging information
DNA methylation
Users can search this module by gene name for specific tumor types or normal tissues. The output shows the associations between aging and DNA methylation levels in the gene promoter region via scatter plots or violin plots, similar to those in the expression module (Fig. 2, Methylation).
Genomic alteration
This function allows users to explore the differences in genomic alterations between aging groups. Human age in this study is divided into younger and older groups with a cutoff of a given age. The number and frequency of SNVs, copy number amplifications, and depletions at the gene level are subsequently calculated and compared between age groups via the chi-square test. This module provides a table form to present the distribution of genomic alterations and corresponding P-values (Fig. 2, Genomic). In addition, a waterfall plot is generated to visualize the differences in the genomic patterns of multiple genes between the younger and older groups.
Cancer and aging hallmarks
The hallmarks of cancer and aging have been comprehensively summarized, and a series of features are shared between carcinogenesis and the aging process [25]. We collected comprehensive gene sets related to cancer and aging hallmarks from the literature and estimated their activities in each tumor sample via the ssGSEA algorithm. Under this tab, users can select different tumors and compare the differences in cancer and aging hallmarks between age groups (with a user-defined age cutoff) (Fig. 2, Hallmarks). Moreover, the correlation between gene expression and hallmarks in different groups can be explored via this module (Fig. 2, Hallmarks). Users can also add and analyze their custom gene sets to explore their associations with aging in tumor patients.
Tumor immunity
The immune dysfunction that accompanies aging is linked to the onset of age-related diseases, including cancer [26]. To explore the link between tumor immunity and aging, we developed an immune module that include immune infiltration, the immune pathway, the immune signature, and antitumor immune activity. Users can select immune modules, genes and tumor types of interest for the purpose of their own research. The differences in immune infiltration and scores and their correlation with gene expression in both the younger and older groups (with a user-defined age cutoff) are exhibited in this module (Fig. 2, Immune).
Survival analysis
With this module, users can perform survival analysis according to gene expression levels in younger, older, and all patients (Fig. 2, Survival). This function gives users an opportunity to develop precision treatment strategies that consider age information. The survival curve and the outcome of the Kaplan-Meier analysis can be generated by selecting different cohorts, gene symbols and color options. In addition, we offer survival analysis based on the SNV, CNV, and methylation status in different groups.
Case study
To illustrate the various functions of Aging2Cancer, we first identified the 15 genes most strongly associated with aging in tumors [27] and explored age-related features in low-grade glioma (LGG) patients since its multiomic landscape is strongly affected by aging [28]. We observed high genomic alteration rates for these genes in both younger and older LGG patients. Among them, the mutation rates of IDH1, TP53, and ATRX decreased and the copy number variations of ARID1A and BRAF increased in older LGG patients (Fig. 3A). The expression of these genes also exhibited strong associations with aging (Fig. 3B). For example, the expression of ATRX increases with age in LGG patients (Fig. 3C). A previous study revealed that ATRX is an important regulator of human cell senescence [29]. Consistent with these results, we found that the cellular senescence score was increased in the older LGG group, and the correlations between ATRX and cellular senescence were significantly greater in the older patients than in the younger patients (Fig. 3D). Moreover, an opposite role of ATRX in LGG patient survival was observed, where the mutation of ATRX served as a risk factor in younger patients, whereas ATRX mutations confer better overall survival in older patients (Fig. 3E). These results suggest that Aging2Cancer not only accurately presents the multiomics characteristics of aging tumors but also provides powerful tools for identifying novel and valuable biomarkers for tumor therapy.
Fig. 3.

Case study of Aging2Cancer. (A) Genomic alterations of 15 essential genes in LGG younger and older patients. Left panel showed the genomic alteration distributions via oncoprint plot. Right panel showed the difference of genomic alterations between LGG groups via Chi-squared test. (B) The distribution of mutation counts for ATRX gene for LGG younger and older patients. (C) The difference in linear combinations of 10 essential genes expressions between younger and older LGG patients. (D) The relationship between aging and ATRX expression in LGG patients. (E) Violin plot showed the difference of cellular senescence scores between LGG younger and older groups. (F) The correlation coefficients between ATRX and cellular senescence between LGG younger and older groups. (G) Kaplan–Meier curves for overall survival in LGG younger and older patients according to the ATRX mutation status
Discussion and future directions
In summary, Aging2Cancer is a comprehensive resource for investigating the links between aging and the multiomics signatures across normal tissues and multiple cancer types. Aging2Cancer provides a user-friendly interface and serviceable modules to explore the associations between expression, genomic alterations, DNA methylation, cancer and aging hallmarks, tumor immunity, and clinical prognosis data and aging at the gene level. With these tools, users can easily screen potential aging-related biomarkers in cancer patients, to assist in the development of precise cancer treatment strategies. For example, the SNV of ATRX is reported to be more common in younger individuals [27], which can be easily explored by users in the genomic module of Aging2Cancer. In addition, users can identify age-related biomarkers for prognosis, such as CCNB2, BUB1, and CDK1 in breast cancer, which is consistent with the findings of a previous study [30]. In the future, Aging2Cancer will be continually updated to include more tumor-related multiomics samples with age information. Moreover, increasing numbers of aging- and cancer-related single-cell sequencing datasets have illuminated the development of relevant research fields. For example, a brain cell atlas across human brain regions was recently created via the integration of single-cell transcriptomes [31]. This atlas contains more than 26.3 million adult, fetal, organoid, and tumor cells. There are 234,295 brain tumor cells with aging information. Similarly, we will make efforts to integrate these valuable data to provide more valuable information at single-cell resolution.
Conclusions
In this study, we introduce Aging2Cancer, a comprehensive resource for linking aging to cancer. The database was constructed on the basis of large-scale multiomics data from 23,125 samples with age information, which included 57 cancer types and 20 normal tissues. We offer several functional tools including gene expression, DNA methylation, genomic alterations, cancer- and aging-related hallmarks, tumor immune regulation, and survival analysis, which consider the age of samples to provide in-depth biological insights. We believe that Aging2Cancer is a valuable and time-saving resource for biologists that provides novel insights into the link between aging and cancer.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
Not applicable.
Abbreviations
- TCGA
The Cancer Genome Atlas
- ICGC
International Cancer Genome Consortium
- GTEx
Genotype-Tissue Expression
- SNV
Single nucleotide variant
- CNV
Copy number variation
- ssGSEA
Single-sample GSEA
- HLA
Human leukocyte antigen
- TILs
Tumor-infiltrating lymphocytes
- LGG
Low-grade glioma
Author contributions
DX, YS, NZ, and GD contributed equally to this work. KL, BW and JX conceived and designed the study; KL, BW and JX supervised the study and data analysis; DX, YS, NZ, GD, DZ, PL, JC, GT, QW, and HJ performed the data analysis. DX, YS, NZ, and GD constructed the data portal, DX and KL wrote and revised the manuscript from input from all authors. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32160152, 32460161), Major Science and Technology Program of Hainan Province (ZDKJ2021040), Innovative research project for Graduate students in Hainan Province (Qhys2023-489, Qhys2023-447, HYYB2023A36, HYYB2023A06).
Data availability
All data in Aging2Cancer is available to the users without registration or login (http://210.37.77.200:8080/Aging2Cancer/index.jsp). Users can directly download the results of visualization in the corresponding module.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dahua Xu, Yutong Shen, Nihui Zhang and Guoqing Deng contributed equally to this work.
Contributor Information
Jiankai Xu, Email: jxu@ems.hrbmu.edu.cn.
Bo Wang, Email: wangqugans@163.com.
Kongning Li, Email: likongning@muhn.edu.cn.
References
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. [DOI] [PubMed] [Google Scholar]
- 2.White MC, Holman DM, Boehm JE, Peipins LA, Grossman M, Henley SJ. Age and cancer risk: a potentially modifiable relationship. Am J Prev Med. 2014;46(3 Suppl 1):S7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186:243–78. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31–46. [DOI] [PubMed] [Google Scholar]
- 5.Aging Atlas Consortium. Aging Atlas: a multi-omics database for aging biology. Nucleic Acids Res. 2021;49:D825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tacutu R, Thornton D, Johnson E, Budovsky A, Barardo D, Craig T, et al. Human Ageing Genomic Resources: new and updated databases. Nucleic Acids Res. 2018;46:D1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang K, Gong H, Guan J, Zhang L, Hu C, Zhao W, et al. AgeAnno: a knowledgebase of single-cell annotation of aging in human. Nucleic Acids Res. 2023;51:D805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y, Shang S, Guo S, Wang X, Zhou H, Sun Y et al. AgingBank: a manually curated knowledgebase and high-throughput analysis platform that provides experimentally supported multi-omics data relevant to aging in multiple species. Brief Bioinform. 2022;23. [DOI] [PubMed]
- 9.Rafikova E, Nemirovich-Danchenko N, Ogmen A, Parfenenkova A, Velikanova A, Tikhonov S, et al. Open Genes-a new comprehensive database of human genes associated with aging and longevity. Nucleic Acids Res. 2024;52:D950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ying K, Tyshkovskiy A, Trapp A, Liu H, Moqri M, Kerepesi C et al. ClockBase: a comprehensive platform for biological age profiling in human and mouse. 2023.
- 11.Lee W, Wang Z, Saffern M, Jun T, Huang K-L. Genomic and molecular features distinguish young adult cancer from later-onset cancer. Cell Rep. 2021;37:110005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018;6:271–e2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plaisier CL, Pan M, Baliga NS. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res. 2012;22:2302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017;77:e108–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6. [DOI] [PMC free article] [PubMed]
- 21.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J, et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data. 2018;5:180015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu D, Cao M, Wang B, Bi X, Zhang H, Wu D, et al. Epigenetically regulated lncRNAs dissect the intratumoural heterogeneity and facilitate immune evasion of glioblastomas. Theranostics. 2023;13:1490–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.López-Otín C, Pietrocola F, Roiz-Valle D, Galluzzi L, Kroemer G. Meta-hallmarks of aging and cancer. Cell Metab. 2023;35:12–35. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Liang Q, Ren Y, Guo C, Ge X, Wang L, et al. Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther. 2023;8:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li CH, Haider S, Boutros PC. Age influences on the molecular presentation of tumours. Nat Commun. 2022;13:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chatsirisupachai K, Lesluyes T, Paraoan L, Van Loo P, de Magalhães JP. An integrative analysis of the age-associated multi-omic landscape across cancers. Nat Commun. 2021;12:2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovatcheva M, Liao W, Klein ME, Robine N, Geiger H, Crago AM, et al. ATRX is a regulator of therapy induced senescence in human cells. Nat Commun. 2017;8:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ingebriktsen LM, Finne K, Akslen LA, Wik E. A novel age-related gene expression signature associates with proliferation and disease progression in breast cancer. Br J Cancer. 2022;127:1865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, Huang Y, Huang L, Huang Z, Hao Z-Z, Xu L, et al. A brain cell atlas integrating single-cell transcriptomes across human brain regions. Nat Med. 2024;30:2679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data in Aging2Cancer is available to the users without registration or login (http://210.37.77.200:8080/Aging2Cancer/index.jsp). Users can directly download the results of visualization in the corresponding module.