

Abstract
Background
The innate immune system serves as the host’s first line of defense against invading pathogens. Stimulator of interferon genes (STING) is a key component of this system, yet its relationship with glucose metabolism, particularly in antiviral immunity, remains underexplored.
Methods
Metabolomics analysis was used for detecting metabolic alterations in spleens from STING knockout (KO) and wild-type (WT) mice. Co-immunoprecipitation was employed for determining ubiquitination of TKT. Mass spectrometry was used for detecting interaction proteins of STING. Enzyme activity kits were used for detecting the activities of TKT and G6PD.
Results
In this study, we demonstrate that herpes simplex virus (HSV) infection activates the pentose phosphate pathway (PPP) in host cells, thereby initiating an antiviral immune response. Using STING-manipulated cells and systemic knockout mice, we show that STING positively regulates PPP, which, in turn, limits HSV infection. Inhibition of the PPP significantly reduced the production of antiviral immune factors and dampened STING-induced innate immune responses. Mechanistically, we discovered that STING interacts with transketolase (TKT), a key enzyme in the non-oxidative branch of the PPP, and reduces its ubiquitination via the E3 ubiquitin ligase UBE3A, stabilizing TKT. Silencing TKT or inhibiting its activity with oxythiamine diminished antiviral immune factor production.
Conclusion
Our findings reveal that the PPP plays a synergistic role in generating antiviral immune factors during viral infection and suggest that PPP activation could serve as an adjunct strategy for antiviral therapy.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12964-024-01983-2.
Keywords: STING, PPP, Transketolase, Antiviral immune response, Inflammatory factors
Introduction
Viruses are acellular infectious pathogens that can trigger inflammatory reactions and cause damage to the host [1, 2]. They are broadly classified into DNA and RNA viruses, each employing distinct mechanisms to infect their hosts [3, 4]. Viral infection alters the host cell environment, creating conditions conducive to viral replication and persistence [5]. DNA virus infections, in particular, have been associated with the development of chronic lesions [6].
In response to viral invasion, the host mounts an immune response to combat infection [7, 8]. Innate immunity serves as the host’s first line of defense against viral pathogens. This process is initiated when pathogen-associated molecular patterns (PAMPs) interact with pattern recognition receptors (PRRs), triggering a cascade of signaling events that ultimately lead to the secretion of type I interferon (IFN-I), chemokines, and pro-inflammatory cytokines [9, 10]. The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling pathway is a crucial DNA-sensing mechanism in innate immunity and antiviral defense [11, 12]. Upon activation by DNA viruses, this pathway induces downstream signaling that promotes the expression of interferons, chemokines, and inflammatory mediators, effectively inhibiting viral replication [13, 14].
Interferon (IFN) is central to the host’s antiviral defense. Upon detecting viral invasion, IFN binds to its receptor and activates the transcription of numerous interferon-stimulated genes (ISGs) to counteract viral infection [15–17]. In addition, viral infection can activate the release of inflammatory cytokines, such as interleukin-6 (IL-6), via the cGAS-STING pathway, which drives immune cell differentiation and enhances antiviral immunity [18, 19]. Chemokines, secreted during viral infections, recruit immune cells to the site of infection, thereby limiting viral spread [20, 21].
Viral infection also induces significant metabolic changes within host cells [22]. DNA viruses have been shown to reprogram host cell metabolism, altering the efficiency of various metabolic pathways to facilitate viral replication [23, 24]. Immune cells can exploit these metabolic shifts, harnessing available energy and biosynthetic molecules to enhance their antiviral activity [25]. Studies have reported changes in glycolysis, the tricarboxylic acid cycle (TCA), the pentose phosphate pathway (PPP), fatty acid metabolism, and amino acid metabolism following viral infections [26–29]. PPP, a branch of glucose metabolism, plays a crucial role in generating intermediate metabolites that contribute to lipid synthesis and antioxidant defenses, while its end products are involved in nucleotide biosynthesis [30]. This pathway also links to glycolysis, highlighting its importance in the broader metabolic network. PPP consists of two phases: an oxidative phase, which generates NADPH for fatty acid synthesis and cellular redox maintenance, and a non-oxidative phase, which involves a series of reversible reactions catalyzed by the key enzyme transketolase (TKT) [31–33]. The non-oxidative PPP provides over 85% of the ribose required for de novo nucleotide biosynthesis [34, 35]. Researchers reported that DNA virus, such as HPV16, promoted cervical cancer cell proliferation by activating PPP [27]. Proteomics data analysis indicates that the level of TKT is elevated in cells infected with SARS-CoV-2, suggesting the activation of the non-oxidative PPP [36]. However, the role of PPP in antiviral immunity remains unclear.
In the present study, we investigated the relationship between the innate immune regulator STING and PPP, focusing on the role of PPP metabolism in antiviral responses. We found that herpes simplex virus (HSV) infection activates PPP in host cells, triggering an antiviral immune response. Using STING-modulated cells and systemic knockout mice, we demonstrated that STING positively regulates PPP, thereby restricting HSV infection. Inhibition of PPP significantly reduced the production of antiviral factors and attenuated STING-induced antiviral responses. Mechanistically, we identified that STING interacts with TKT in the non-oxidative PPP and reduces its ubiquitination through the E3 ubiquitin ligase UBE3A, stabilizing TKT. Collectively, our findings suggest that PPP plays a synergistic role in generating antiviral immune factors and that PPP activation may serve as a potential therapeutic strategy for viral infections.
Results
STING upregulates the pentose phosphate pathway (PPP)
To investigate whether STING expression influences cellular metabolism, we performed metabolomic profiling using spleens from STING knockout (KO) and wild-type (WT) mice. The analysis revealed substantial alterations in several metabolic pathways in STING KO mice. Notably, STING knockout led to an upregulation in amino acid and nucleotide metabolism, while pentose and coenzyme metabolism were markedly downregulated (Fig. 1A, B). Among the affected metabolites, key intermediates of the PPP, such as ribose-5-phosphate (R5P) and glucose-6-phosphate (G6P), were significantly downregulated in STING-deficient mice (Fig. 1A, B). The enrichment factor and raw data from metabolic assays further corroborated these findings, highlighting significant changes in PPP metabolites following STING knockout (Fig. 1A-C). These results prompted us to hypothesize that STING positively regulates the PPP.
Fig. 1.

STING upregulates the pentose phosphate pathway (PPP). A Analysis of metabolomics in spleens of wild-type and STING knockout mice. B Metabolic pathway enrichment analysis in spleens of wild-type and STING knockout mice. C Levels of R-5P, 6-PG, and F-6P in spleens of wild-type and STING knockout mice. D E Whole cell extracts (WCEs) from wild-type and STING-overexpressing KYSE-30 cells were analyzed for PPP metabolites (NADPH, GSH, R5P) and key enzymes (TKT, G6PD) using ELISA kits. F, G WCEs from wild-type and STING knockdown KYSE-30 cells were similarly analyzed. Data are expressed as mean ± SEM from three biological replicates. **p < 0.01, *** p < 0.001
To validate this hypothesis, we generated lentivirus-mediated STING overexpression and knockdown cell lines. We measured the levels of key PPP metabolites, including R5P, reduced glutathione (GSH), and nicotinamide adenine dinucleotide phosphate (NADPH), as well as the activities of glucose-6-phosphate dehydrogenase (G6PD) and TKT, the critical enzymes of the oxidative and non-oxidative branches of the PPP, respectively. In line with our metabolomic data, metabolite quantification confirmed significant changes in PPP activity (Fig. 1D-G, S1 and S2). In STING-overexpressing cell lines, G6PD and TKT activities, along with the levels of GSH, NADPH, and R5P, were markedly upregulated compared to WT cells (Fig. 1D, E and Fig. S2A, B). Conversely, STING knockdown resulted in a pronounced downregulation of these metabolites and enzymes (Fig. 1F, G and Fig. S2C, D). These results provide strong evidence that STING upregulates the pentose phosphate pathway, positioning STING as a critical regulator of this metabolic pathway during viral infection.
PPP participates in the antiviral immune response
As a key interferon-stimulating factor, STING is activated during DNA virus infections, triggering the production of antiviral immune factors to resist viral invasion. While the role of the PPP in tumorigenesis is well documented, its involvement in viral infection remains largely unexplored. Since STING upregulates PPP metabolites (Fig. 1), we hypothesized that the PPP might play a role in the antiviral immune response. To test this hypothesis, we injected herpes simplex virus (HSV), a DNA virus, into the tail veins of mice and performed metabolomic analyses on spleens from both HSV-infected and uninfected mice. Heatmap and enrichment factor analyses, along with raw data evaluations, revealed a significant increase in PPP metabolites in the HSV-infected group compared to the control group (Fig. 2A-C). To further validate these findings, we transfected KYSE-30 cells with poly(dA:dT), a viral DNA mimic, and measured the levels of key PPP metabolites in cell lysates using metabolite assay kits. The results showed that PPP metabolites, including ribose-5-phosphate (R5P) and reduced glutathione (GSH), as well as the activities of G6PD and TKT-key enzymes of the oxidative and non-oxidative branches of the PPP-were upregulated in poly(dA:dT)-treated cells (Fig. 2D). Subsequently, we analyzed the mRNA expression of antiviral immune factors, such as IFN-β, IL-6, and CCL5, as well as G6PD and TKT, in poly(dA:dT)-transfected cells. The mRNA levels of IFN-β, IL-6, CCL5, and TKT were significantly upregulated, while the mRNA level of G6PD remained unchanged (Fig. 2E).
Fig. 2.

PPP participates in the antiviral immune response. A, B Analysis of metabolomics and metabolic pathway enrichment in spleens of control (PBS) and HSV-treated mice (HSV-1, 5 × 10^6 PFU/mouse, tail vein injection). Spleens were collected six days post-injection for analysis. C ELISA detection of PPP pathway metabolites (TKT, R-5P, GSH) in KYSE-30 cells transfected with poly(dA:dT). D qPCR detection of inflammatory factors (IL-6, CCL5, IFN-β) and PPP enzymes (TKT, G6PD) mRNA in poly(dA:dT)-transfected KYSE-30 cells. E Levels of IL-6, CCL5, IFN-β, and TKT in KYSE-30 cells treated with poly(dA:dT) and/or the PPP inhibitor 6-AN (10 µM, 8 h). F Levels of IL-6, CCL5, IFN-β, and TKT in KYSE-30 cells treated with HSV (MOI = 1, 8 h) and/or 6-AN (10 µM, 8 h). Data are mean ± SEM from three biological replicates. * p < 0.05, ** p < 0.01, *** p < 0.001
To further explore the role of the PPP in the antiviral immune response, we treated KYSE-30 cells with 6-aminonicotinamide (6-AN) [37], a known PPP inhibitor, after transfecting them with poly(dA:dT). We then measured the mRNA levels of TKT and antiviral immune factors. 6-AN treatment reduced the poly(dA:dT)-induced upregulation of TKT, IFN-β, CCL5, and IL-6 (Fig. 2F). Additionally, in cells treated with HSV (MOI = 1) and 6-AN, we observed a marked increase in TKT and immune factor expression following HSV infection. However, administration of 6-AN diminished the increase in both TKT and antiviral immune factors (Fig. 2G). These findings suggest that the PPP regulates the expression of antiviral immune factors and plays a crucial role in the antiviral immune response.
PPP participates in STING-mediated antiviral immune response
To investigate the relationship between STING and PPP in antiviral immunity, we performed glycometabolomic analysis on spleen tissues from WT and STING knockout mice following HSV infection (5 × 10^6 PFU per mouse). The analysis revealed a significant reduction in PPP metabolites in STING knockout mice compared to WT mice post-HSV infection (Fig. 3A). This suggests that STING may positively regulate the PPP, indicating a synergistic role between the STING-mediated antiviral immune response and PPP activity.
Fig. 3.

PPP mediates STING-facilitated antiviral immune response. A Analysis of glucose metabolomics in spleens from wild-type and STING knockout mice injected with HSV via tail vein. B mRNA expression of inflammatory factors (IFN-β, IL-6, CCL5) in STING-overexpressed KYSE-30 cells transfected with poly(dA:dT). C Levels of TKT, G6PD, R-5P, and GSH in STING-overexpressed KYSE-30 cells treated with poly(dA:dT) and 6-AN or OT (10 mM, TKT inhibitor) for 10 h. D Levels of STING and inflammatory cytokines (IFN-β, CCL5, IL-6) in STING-overexpressed KYSE-30 cells treated with poly(dA:dT) and 6-AN. E mRNA expression of TKT and inflammatory cytokines (IFN-β, CCL5, IL-6) in STING-overexpressed KYSE-30 cells treated with ISD (DNA virus analogue) and OT. Data are mean ± SEM from three biological replicates. * p < 0.05, ** p < 0.01, *** p < 0.001
The role of STING in mediating antiviral immunity has been extensively documented. To validate STING's involvement, we transfected STING-overexpressing cells with poly(dA:dT), a viral DNA mimic, and assessed the production of antiviral immune factors (Fig. 3B). Subsequently, to investigate whether the PPP is directly involved in STING-mediated antiviral immunity, we transfected KYSE-30 cells overexpressing STING with poly(dA:dT), followed by treatment with the PPP inhibitor 6-AN and the non-oxidative PPP inhibitor oxythiamine (OT) for 10 h [30]. The metabolite content assay confirmed that these inhibitors effectively suppressed PPP activity (Fig. 3C). More importantly, qPCR analysis revealed that the addition of 6-AN diminished the upregulation of inflammatory factors induced by STING overexpression (Fig. 3D). To further corroborate this, we used interferon stimulatory DNA (ISD), another DNA viral mimic, to activate the viral response pathway in STING-overexpressing cells, followed by OT treatment for 10 h. As with poly(dA:dT), OT treatment reduced the expression of antiviral immune factors (Fig. 3E). These results collectively demonstrate that the PPP plays a crucial role in STING-mediated antiviral immune responses. The inhibition of key PPP enzymes significantly attenuates the immune response induced by STING, highlighting the importance of PPP in antiviral defense mechanisms.
STING upregulates TKT
PPP consists of both oxidative and non-oxidative branches, regulated by the rate-limiting enzyme G6PD and TKT, respectively. Our previous results indicate that DNA mimics specifically upregulate the mRNA expression of TKT, the key enzyme of the non-oxidative PPP, without affecting G6PD mRNA levels in the oxidative branch (Fig. 2E). To further clarify how STING influences the PPP, we conducted transcriptome sequencing on intestinal tissues from WT and STING knockout mice. Interestingly, transcriptomic data revealed no significant changes in G6PD or TKT mRNA expression between WT and STING knockout mice, suggesting that STING may not directly regulate the transcription of these enzymes (Fig. 4A and table S1). This observation was further corroborated by qPCR analysis in KYSE-30 cells overexpressing STING, where no elevation in G6PD or TKT mRNA levels was detected compared to parental cells (Fig. 4B).
Fig. 4.

STING upregulates TKT. A Transcriptome sequencing analysis revealed mRNA expression of key PPP enzymes (TKT, G6PD) in wild-type and STING knockout mice. B mRNA expression of TKT and G6PD in wild-type and STING-overexpressed cells detected by qPCR. C Co-IP using an anti-STING antibody, followed by mass spectrometry to identify proteins interacting with STING. D Interaction between TKT and STING detected by anti-TKT and anti-STING antibodies. E Expression of G6PD and TKT in STING-overexpressing KYSE-30 cells and HSV-infected cells. F IHC staining of STING and TKT in liver tissues from wild-type and STING knockout mice. G TKT enzyme activity and mRNA expression of STING in KYSE-30 cells treated with poly(dA:dT). Data are mean ± SEM from three biological replicates. *** p < 0.001; ns, no significance
To investigate whether STING physically interacts with PPP enzymes, we performed immunoprecipitation using an anti-STING antibody, followed by mass spectrometry analysis of the interacting proteins. Notably, TKT was identified as a binding partner of STING in this analysis (Fig. 4C). Reciprocal co-immunoprecipitation (Co-IP) assays confirmed the interaction between STING and TKT in KYSE-30 cells (Fig. 4D). Moreover, STING overexpression enhanced the protein levels of TKT, while the expression of G6PD, despite being upregulated by HSV infection, remained unaffected by STING (Fig. 4E). Immunohistochemical analysis of liver tissues from WT and STING knockout mice further demonstrated a reduction in TKT expression in STING-deficient mice (Fig. 4F). Additionally, the enzymatic activity of TKT increased following STING overexpression (Fig. 4G), indicating that STING can upregulate the non-oxidative PPP by enhancing both the expression and activity of TKT. Collectively, these results suggest that STING directly promotes TKT function, thereby augmenting the non-oxidative branch of PPP during the antiviral immune response.
STING stabilizes TKT through inhibiting its ubiquitination
STING enhances both TKT expression and its enzymatic activity (Fig. 4E-G), but it does not appear to affect TKT transcription (Fig. 4A, B). This led us to investigate the role of post-translational modifications (PTMs) in regulating TKT stability via STING. We immunoprecipitated TKT and analyzed its complex via SDS-PAGE electrophoresis, but no significant phosphorylation was detected using a pan-phosphorylation antibody (Fig. 5A). Previous studies have reported that ubiquitination of TKT can occur [38]. To explore whether STING inhibits the ubiquitination of TKT, thereby stabilizing its expression, we transfected His-tagged ubiquitin (His-Ub) into 293 T cells for 24 h to enhance ubiquitination, followed by treatment with the proteasome inhibitor MG132 for 10 h to block proteasome-mediated degradation. Immunoprecipitation of TKT with anti-TKT antibodies followed by SDS-PAGE revealed that TKT is ubiquitinated, and its ubiquitination level was elevated by MG132 treatment (Fig. 5B). Ubiquitination typically occurs via K48- or K63-linked polyubiquitin chains. K48-linked ubiquitination often leads to proteasomal degradation, while K63-linked ubiquitination is associated with processes such as DNA damage response, protein sorting, autophagy, and neurodegeneration. We determined the specific type of ubiquitination in TKT and found that TKT primarily undergoes K48-linked ubiquitination, rather than K63-linked (Fig. 5C). Given that K48-linked ubiquitination typically promotes degradation, we hypothesized that STING may influence TKT protein levels by modulating this process. In STING-knockdown cells, K48-linked ubiquitination of TKT was increased, whereas in STING-overexpressing cells, it was reduced (Fig. 5D, E).
Fig. 5.

STING stabilizes TKT by inhibiting UBE3A-mediated TKT ubiquitination. A Pan-phosphorylation antibody of serine and threonine was used to detect TKT phosphorylation. B, C Identification of K48-linked ubiquitination of TKT in 293 T cells transfected with His-Ub and treated with MG132. D, E STING downregulates K48-linked ubiquitination of TKT. F, G Detection of interactions among STING, TKT, and UBE3A using specific antibodies. H Detection of the correlation between STING and UBE3A by western blots in KYSE-30 cells with STING knockdown and overexpression. I Effect of UBE3A on the ubiquitination of TKT was detected by immunoprecipitation in KYSE-30 cells transfected with His-Ub and shUBE3A. J K48-linked ubiquitination of TKT was detected by immunoprecipitation in STING-knocked down KYSE-30 cells transfected with His-Ub and shUBE3A. Data are mean ± SEM from three biological replicates
TKT has been identified as a target of the E3 ubiquitin ligase UBE3A [39]. Mass spectrometry analysis of proteins pulled down by STING identified UBE3A as an interacting partner (Fig. 4C). Since both TKT (Fig. 4C) and UBE3A were found to interact with STING, we hypothesized that STING, UBE3A, and TKT may form a complex. UBE3A likely mediates the ubiquitination and degradation of TKT under STING regulation. To test this, we used co-immunoprecipitation with antibodies against STING, TKT, and UBE3A to examine whether these three proteins interact. Our results confirmed the formation of a STING-TKT-UBE3A complex (Fig. 5F, G). Next, we assessed whether STING influences UBE3A expression. In STING-overexpressing cells, UBE3A protein levels were reduced, while STING knockout increased UBE3A expression (Fig. 5H). To further investigate UBE3A's role in TKT ubiquitination, we knocked down UBE3A in 293 T cells using shUBE3A and transfected His-Ub to detect TKT ubiquitination. UBE3A knockdown reduced TKT ubiquitination (Fig. 5I). Since STING knockdown increases TKT K48-linked ubiquitination (Fig. 5D), we next co-transfected shUBE3A and His-Ub into shSTING KYSE-30 cells and examined K48-linked ubiquitination of TKT. UBE3A knockdown attenuated the shSTING-induced increase in TKT ubiquitination (Fig. 5J). These results demonstrate that UBE3A mediates STING-regulated TKT ubiquitination. STING stabilizes TKT by inhibiting UBE3A-mediated K48-linked ubiquitination, thus preventing TKT degradation.
TKT participates in STING-mediated antiviral immune response
We have demonstrated that STING upregulates TKT and activates the non-oxidative PPP (Fig. 4). Next, we investigated whether this upregulation of TKT and activation of the non-oxidative PPP mediates the antiviral immune response initiated by STING. TKT was downregulated using siTKT (Fig. 6A, B), and cells were activated with the STING activator poly(dA:dT). The cytokine expression levels in the supernatant of poly(dA:dT)-transfected cells were then analyzed. After TKT knockdown, we observed a decrease in the levels of IL-6 and IFN-β (Fig. 6C). To further confirm the role of TKT in the antiviral immune response, we transiently transfected KYSE-30 cells with TKT siRNA and measured the mRNA levels of antiviral immune factors after poly(dA:dT) co-treatment. TKT knockdown significantly reduced the production of these antiviral immune factors (Fig. 6D), while TKT overexpression increased the levels of antiviral immune factors in poly(dA:dT)-treated cells (Fig. 6E). Additionally, overexpressing TKT in STING-knockdown KYSE-30 cells restored the antiviral immune response, reversing the reduction of antiviral immune factors caused by STING knockdown (Fig. 6F, G). These findings suggest that TKT plays a key role in mediating STING-driven antiviral immune responses (Fig. 7).
Fig. 6.

TKT mediates the antiviral immune response of STING. A Expression and enzyme activity of TKT in control and TKT siRNA KYSE-30 cells. B ELISA detection of antiviral immune factors (IL-6, IFN-β) in control and TKT siRNA KYSE-30 cells with or without poly(dA:dT). C qPCR detection of inflammatory factors (CCL-5, IL-6, IFN-β) in control and TKT siRNA KYSE-30 cells with or without poly(dA:dT). D qPCR detection of inflammatory factors in control and TKT overexpressing KYSE-30 cells with or without poly(dA:dT). E qPCR detection of inflammatory factors in control and STING-knocked down KYSE-30 cells transfected with TKT expressing vector and/or poly(dA:dT). Data are mean ± SEM from three replicates in each group. ** p < 0.01; *** p < 0.001
Fig. 7.
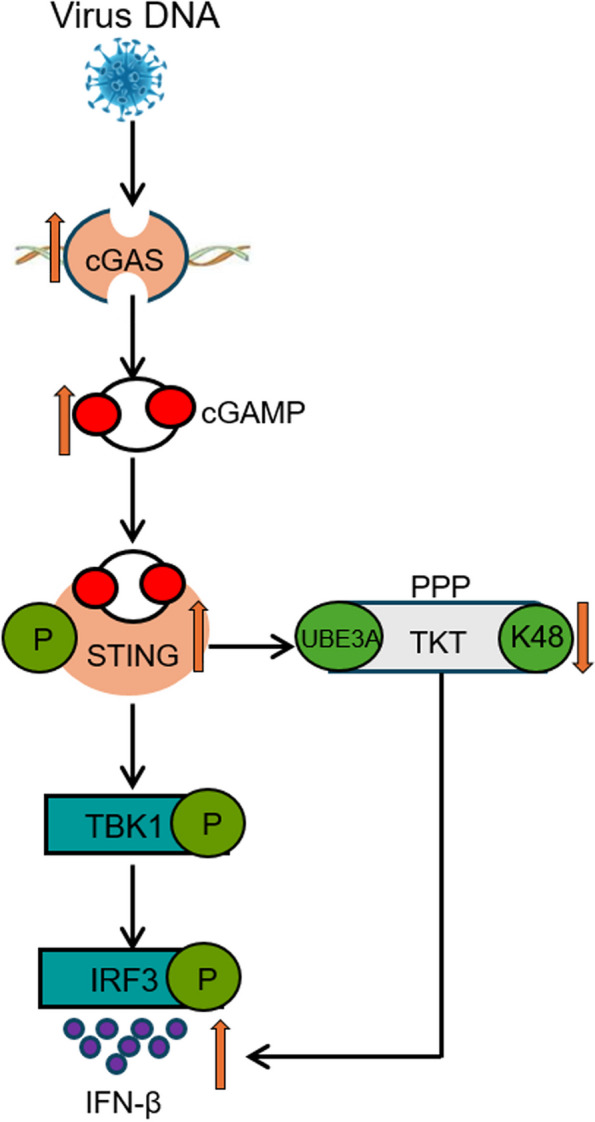
Schematic diagram depicting that STING exerts antiviral innate immune response by activating pentose phosphate pathway. DNA virus infection activates the PPP in host cells, thereby initiating STING-mediated antiviral immune response. STING interacts with TKT, a key enzyme in the non-oxidative branch of the PPP, and reduces its ubiquitination via the E3 ubiquitin ligase UBE3A, stabilizing TKT and upregulating PPP
Discussion
In this study, we confirmed that HSV infection activates STING, leading to upregulation of the expression and activity of TKT, a key enzyme in the non-oxidative PPP. Silencing TKT during DNA virus infection reduced the production of antiviral immune factors triggered by STING activation, suggesting that TKT and the PPP are at least partially involved in the antiviral immune response induced by STING. Mechanistically, STING stabilizes TKT by downregulating K48-linked ubiquitination, which typically marks proteins for degradation (Fig. 7). Contrary to the traditional view that increased host cell metabolism due to viral invasion primarily provides energy for viral replication, our findings demonstrate that virus-infected host cells activate the PPP to promote innate immune responses against viral infections.
To establish successful infection, viruses like HSV-1 and human cytomegalovirus (HCMV) have evolved various strategies to evade host antiviral immunity, promoting viral infection and replication [40]. Studies have indicated that viral infections lead to metabolic abnormalities in host cells, favoring conditions for viral replication by boosting cellular energy production. However, the potential for metabolic changes to aid in resisting viral infections remains unclear. Our findings support the notion that host cell metabolic reprogramming can positively influence the restriction of viral infection and replication.
The innate immune system serves as the first line of defense against pathogens, with type I interferons, inflammatory cytokines, and chemokines playing essential roles in early viral resistance [41]. The cGAS pathway has garnered significant attention as a crucial signaling route in resisting DNA virus infections. After the cytosolic DNA sensor cGAS detects dsDNA, it releases a second messenger, initiating the translocation of STING from the endoplasmic reticulum (ER) to the ER-Golgi intermediate compartment (ERGIC), where STING is phosphorylated by TBK1. This phosphorylation activates IRF3, leading to the production of type I interferons, cytokines, and chemokines [42, 43]. In this study, we found viral infection also activates the PPP, a metabolic pathway essential for the antiviral immune response. Metabolomic analysis of HSV-infected mice revealed that PPP is upregulated during infection, with STING acting as a positive regulator of this pathway. Interestingly, STING does not affect the transcription of key PPP enzymes like G6PD and TKT. Instead, it interacts with TKT, influencing its expression and enzymatic activity. Further experiments demonstrated that STING stabilizes TKT by inhibiting K48-linked ubiquitination, mediated by the E3 ubiquitin ligase UBE3A.
TKT is crucial in the non-oxidative arm of the PPP, where it produces phosphorylated ribose, a key component for DNA repair. The role of TKT in cancer progression is well-established, as TKT-driven metabolic reprogramming promotes malignancy and metastasis in various cancers [44, 45]. Our previous research revealed that TKT promotes the progression of esophageal cancer. However, the specific role of TKT in viral infections remains unclear. Although we demonstrated that TKT inhibition reduces the production of antiviral immune factors, future research should explore whether this occurs through STING’s classical interferon activation pathway or via alternative signaling mechanisms.
While previous studies on TKT have primarily focused on its enzymatic regulation, recent structural biology research suggests that TKT is a docking target for UBE3A, an E3 ubiquitin ligase [39]. Our mass spectrometry and immunoprecipitation assays confirmed that STING, TKT, and UBE3A form a ternary complex, with UBE3A mediating K48-linked ubiquitination and degradation of TKT. This provides insight into how STING negatively regulates UBE3A, and hence enhances TKT by reducing K48-linked ubiquitination. Disruptions in immune and metabolic homeostasis underlie various inflammation-related pathologies [46]. For instance, a high-fat diet can induce STING-related cytokine production and alter anabolic balance [47]. As downstream effectors of STING, TBK1 and IRF3 are also known to regulate metabolic pathways [48], including fatty acid metabolism [49]. Additionally, fatty acid biosynthesis has been shown to modulate the expression of interferon by regulating the expression of interferon stimulated genes (ISGs) [50]. Our research suggests that classical immune signaling may be interconnected with metabolic pathways, providing new insights in the antiviral responses.
The classical STING pathway activates TBK1 and IRF3, inducing the production of type I interferons and other antiviral immune factors. Our investigation into STING glycosylation revealed that HSV-1 infection enhances glucose metabolism in host cells, significantly activating the hexosamine biosynthetic pathway (HBP) and supplying donor glucosamine for O-GlcNAc glycosylation. Glycosylation of STING at serine 229, followed by K63-linked ubiquitination, promotes its antiviral activity [51]. However, mutations in STING’s S365 phosphorylation site have been shown to impair downstream signaling, reducing its antiviral potency [52]. STING has been reported to exhibit interferon-independent antiviral functions in immune cells, indicating that its role extends beyond the pathway [53, 54]. While we did not perform in vivo validation of PPP agonists or inhibitors to assess survival in virus-infected mice, our cell-based studies confirm that STING enhances TKT expression, thereby contributing to antiviral immunity. Future research should explore the specific mechanisms through which the PPP influences the production of antiviral immune factors and validate these findings in animal models.
Methods
Experimental animals
All animal experiments were conducted in accordance with the National Science and Technology Commission of China’s Guidelines for the Care, Use, and Welfare of Experimental Animals. C57BL/6 J mice and STING−/− mice were obtained from Cyagen Biological Technology Co. (Suzhou, China) and acclimatized under controlled conditions (22—24 °C, 50—60% humidity, 12-h light/dark cycle) with ad libitum access to food and water. Mice were randomly assigned to experimental groups for drug treatment. Validation of STING knockout status was confirmed by qPCR and Western blotting before starting the experiments [55].
HSV infection of mice
Seven-to-ten-week-old WT and STING−/− mice were infected intravenously with 5 × 106 plaque-forming units (PFU) of HSV-1 (Cat# H129-G4, genechem, Shanghai, China). Six days post-injection, serum and tissue samples (spleen, liver, lungs) were harvested for immune profiling and histological analysis, including immune assays and tissue-specific changes in response to HSV-1 infection [56].
Metabolomics sequencing
Weight- and age-matched WT and STING knockout (STING−/−) mice were divided into six experimental groups (five mice per group) along with gender-matched controls. Mice were injected intravenously with 5 × 106 PFU of HSV-1. Growth and health conditions of mice were monitored daily. After approximately one week, mice were euthanized, and the spleens were collected. Metabolomic analyses were conducted on three independent replicates from each group to compare the metabolic profiles of WT and STING−/− mice, with and without HSV-1 infection [30].
Cell culture
KYSE-30 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and streptomycin (Life Technologies, Carlsbad, CA), and incubated at 37 °C with 5% CO2. 293 T cells were maintained in DMEM supplemented with 10% FBS under the same conditions.
Cell line construction
The full-length STING gene was cloned into the V3 vector, tagged with Flag and HA tags. Agarose gel electrophoresis and vector primer sequencing were used to confirm the successful construction of the plasmid. The V3 plasmid, along with the lentiviral packaging plasmid psPAX2 and envelope plasmid pMD2.G, was co-transfected into 293 T cells using Lipofectamine 2000 in 100-mm cell culture dishes. Supernatants of the cell culture were centrifuged and filtered and applied for the infection. Infected cells were selected with puromycin. Stable colonies were expanded, and the STING-expressing cell lines were verified by qPCR and Western blotting. STING knockdown vectors were obtained from GenePharma Company (Shanghai, China), and lentiviral transduction followed standard protocols [57].
siRNA transfection and PPP inhibition
KYSE-30 cells were transfected with small interfering RNAs (siRNAs) targeting TKT and control siRNAs using standard transfection protocols. The sequences of the siRNAs are listed in the Table 1. TKT silencing efficiency was verified by quantitative PCR (qPCR) and Western blotting 48 h post-transfection. After silencing TKT and infecting cells with poly(dA:dT), inflammatory factor expression was assessed using immunoblotting and qPCR. Additionally, PPP metabolites were quantified using enzyme-linked immunosorbent assays (ELISAs). PPP pathway inhibitors 6-AN and OT, purchased from Jiangsu Taozhu Biotechnology, were prepared according to the manufacturer’s instructions and used in subsequent experiments [30].
Table 1.
Primer sequences of gene/siRNA
| Gene/siRNA | Primers | Primer Sequences |
|---|---|---|
| TKT-human | forward primer | CGCCAATACAAAGGGTATCTG |
| reverse primer | TTTCTTTCTTCAGCAGTTCGG | |
| GAPDH-human | forward primer | AACGGATTTGGTCGTATTGG |
| reverse primer | TTGATTTTGGAGGGATCTCG | |
| STING-human | forward primer | AGCATTACAACAACCTGCTACG |
| reverse primer | GTTGGGGTCAGCCATACTCAG | |
| CCL-5-human | forward primer | CCAGCAGTCGTCTTTGTCAC |
| reverse primer | CTCTGGGTTGGCACACACTT | |
| IL-6-human | forward primer | TGGCTGCAGGACATGACAACT |
| reverse primer | ATCTGAGGTGCCCATGCTACA | |
| IFN-β-human | forward primer | CAACAAGTGTCTGCTCGAAAT |
| reverse primer | TCTCCTCAGGGATGTCAAAG | |
| G6PD-human | forward primer | AACATCGCCTGCGTTATCCTC |
| reverse primer | ACGTCCCGGATGATCCCAA | |
| siRNA-TKT | forward primer | CCGGCAAAUACUUCGACAATTA |
| reverse primer | UUGUCGAAGUAUUGCCGGTT |
Western blotting
Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% skim milk (Santa Cruz Biotechnology) and incubated overnight with primary antibodies (Table 2). After washing with tris-buffered saline with Tween 20 (TBST), HRP-conjugated secondary antibodies were added, and protein bands were visualized using chemiluminescence [58].
Table 2.
Source and dilution of antibody
| Antibody | Company (item number) | Dilution |
|---|---|---|
| STING | Abcam (ab129153) | IHC: 1:200 WB: 1:10,000 |
| STING | Proteintech (19,851–1-AP) | WB:1:1000 IHC:1:2000 |
| TKT | Santa Cruz (sc-390179) | IHC: 1:100 Co-IP: 1:50 |
| TKT | Abcam (ab56788) | WB: 1:1000 |
| UBE3A | ZEN-BIOSCIENCE (R27404) | WB: 1:1000 |
| β-actin | Proteintech (60,008–1-lg) | WB: 1:10,000 |
| IgG(M) | Santa Cruz (sc-2025) | IHC: 1:200 |
| K48-ub | Abcam (ab140601) | WB:1:2000 |
| His-ub | Proteintech (66,005–1-lg) | WB:1:1000 |
| K63-ub | Abcam (ab179434) | WB:1:1000 |
| IgG(R) | Proteintech (30,000–0-AP) | IP: 0.5 ug-1 ug |
RNA extraction and qPCR
Total RNA was extracted from cells using the Trizol method, following the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA). The extracted RNA was reverse transcribed into complementary DNA (cDNA) using a reverse transcription kit (Vazyme, Nanjing, China). qPCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) with GAPDH as the internal control. Data were analyzed using the 2^-ΔΔCT method. All experiments were repeated at least three times for consistency [58].
Co-Immunoprecipitation (Co-IP)
Cell lysates were prepared and incubated with primary antibodies overnight at 4 °C. Protein A/G agarose beads (Cat# 37478, proteintech, China) were added, and the mixtures were incubated for 2 h at 4 °C. After washing, protein-bound beads were subjected to SDS-PAGE followed by immunoblotting to detect protein interactions [59].
ELISA determination of cytokines
Cells were treated with poly(dA:dT) or PPP inhibitors and harvested via trypsin digestion. Cell pellets were washed twice with PBS and lysed using RIPA buffer. Cytokines was quantified using ELISA kits (Solarbio, China) according to the manufacturer’s protocols [51].
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were deparaffinized and rehydrated, followed by antigen retrieval in sodium citrate buffer. After blocking endogenous peroxidase with hydrogen peroxide and washing with PBS, sections were blocked with 3% bovine serum albumin. Primary antibodies (Table 2) conjugated with HRP were applied overnight at 4 °C. The next day, sections were incubated with secondary antibodies, and signal development was carried out using 3,3'-diaminobenzidine (DAB) (Dako, Heverlee, Belgium). Counterstaining was performed with hematoxylin [60].
His-Ub transfection and ubiquitination detection
STING-overexpressing or knockdown cells were co-transfected with the His-Ub plasmid and shUBE3A plasmid. Cells were treated with MG132 (10 μM) for 12 h to block proteasomal degradation. Ubiquitinated TKT was detected by immunoprecipitation using anti-His antibodies, followed by Western blotting. The shUBE3A plasmid was obtained from the Wang Lab at Jilin University, and transfections were carried out following the manufacturer's instructions [27].
RNA-mediated interference
siRNAs targeting human TKT and UBE3A, along with non-targeting control siRNAs, were synthesized by GenePharma (Shanghai, China) and transfected into 293 T cells using Lipofectamine 2000 (Invitrogen, Carlsad, CA, USA). The final siRNA concentration was 30 nM. Gene silencing efficiency was assessed using qPCR and western blotting [27].
Enzyme activity detection
The stably transfected KYSE-30 cells were counted and seeded into a six-well plate at a density of 1 × 105 cells per milliliter. The cells were then treated according to the experimental design. After completion of the treatment, the cells were digested with trypsin to obtain cell suspension, which was subsequently washed twice with PBS. RIPA lysis buffer was added to lyse the cells for 30 min. Then the lysate was centrifuged at 3,000 rpm for 20 min at 4 °C. The supernatant was collected, and the protein concentration was quantified. Enzyme activity kits (Solarbio, China) were used for detecting the activities of TKT and G6PD [27].
Identification of STING interacting proteins
STING antibodies were used to pull down STING and its interaction proteins. SDS-PAGE was performed to isolate the proteins. The gel was stained with Coomassie bright blue overnight. After washing, differential bands in the gel were cut and analyzed by the mass spectrometry platform of Henan University for identifing the interacting proteins of STING.
Statistical analysis
Statistical analyses were conducted using SPSS 19.0. One-way ANOVA and t-test were used for the significant test. All data were obtained from three independent biological experiments. Statistical significance was defined as p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001), whereas statistical insignificance was defined as p > 0.05.
Supplementary Information
Supplementary Material 1: Fig. S1 Knockdown and overexpression of STING in KYSE-30 cells. A, B. mRNA and protein expression of STING in STING-overexpression KYSE-30 cells. C, D. mRNA and protein expression of STING in STING-knocked down KYSE-30 cells. Data are expressed as mean ± SEM from three biological replicates. *** p < 0.001.
Supplementary Material 2: Fig. S2 STING upregulates PPP in HEK 293 cells. A, B. Whole cell extracts (WCEs) from wild-type and STING-overexpressing HEK 293 cells were analyzed for PPP metabolites (NADPH, GSH, R5P) and key enzymes (TKT, G6PD) using ELISA kits. C, D. WCEs from wild-type and STING-knocked down HEK 293 cells were similarly analyzed. Data are expressed as mean ± SEM from three biological replicates. ** p< 0.01, *** p< 0.001.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFE0109800), the National Natural Science Foundation of China (Nos. 82020108024, 82400658 and 82200596), and the China Postdoctoral Science Foundation (No. 2022M721014). We thank scientists in Xu lab for technical supports and effective discussions.
Authors’ contributions
D.W. wrote the manuscript. Z.Z. and W.Y. designed the study. H.L., X.X., L.Z., J.L., Z.Y. and Y.L. performed the experiments. Y.J. participated in organizing figures. Z.X. and Y.J. contributed to the conception and writing. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2023YFE0109800), the National Natural Science Foundation of China (Nos. 82020108024, 82400658 and 82200596), and the China Postdoctoral Science Foundation (No. 2022M721014).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Protocols for animal usage were approved by the institutional animal care and use committee (IACUC) at Henan University, China. All animal experiments were conducted on the basis of the institutional guidelines, and were approved by the Laboratory Animal Center of Henan University.
Consent for publication
All authors consent to submit and publish this article.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dan-Hui Wu, Zi-Long Zhao and Wei-Tao Yin contributed equally to this work.
Contributor Information
Yong-Ping Jian, Email: yongpingjian123@163.com.
Zhi-Xiang Xu, Email: zhixiangxu08@gmail.com.
References
- 1.Girdhar K, Powis A, Raisingani A, Chrudinová M, Huang R, Tran T, et al. Viruses and metabolism: the effects of viral infections and viral insulins on host metabolism. Annu Rev Virol. 2021;8:373–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, et al. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature. 2019;569:663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao J, Wan W, Yu K, Lemey P, Pettersson JH, Bi Y, et al. Farmed fur animals harbour viruses with zoonotic spillover potential. Nature. 2024;634:228–33. [DOI] [PubMed] [Google Scholar]
- 4.Kupferschmidt K. Stem cells hint at how bats live with viruses. Science. 2023;379:746. [DOI] [PubMed] [Google Scholar]
- 5.Yan M, Yu Z. Viruses contribute to microbial diversification in the rumen ecosystem and are associated with certain animal production traits. Microbiome. 2024;12:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beaumont É, Larochette V, Preisser L, Miot C, Pignon P, Blanchard S, et al. IL-26 inhibits hepatitis C virus replication in hepatocytes. J Hepatol. 2022;76:822–31. [DOI] [PubMed] [Google Scholar]
- 7.Guo Y, Jiang F, Kong L, Wu H, Zhang H, Chen X, et al. OTUD5 promotes innate antiviral and antitumor immunity through deubiquitinating and stabilizing STING. Cell Mol Immunol. 2021;18:1945–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B, Xu S, Liu M, Wei Y, Wang Q, Shen W, et al. The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy. 2023;19:1916–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Šestan M, Mikašinović S, Benić A, Wueest S, Dimitropoulos C, Mladenić K, et al. An IFNγ-dependent immune-endocrine circuit lowers blood glucose to potentiate the innate antiviral immune response. Nat Immunol. 2024;25:981–93. [DOI] [PubMed] [Google Scholar]
- 10.Brown Harding H, Kwaku GN, Reardon CM, Khan NS, Zamith-Miranda D, Zarnowski R, et al. Candida albicans extracellular vesicles trigger type I IFN signalling via cGAS and STING. Nat Microbiol. 2024;9:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan M, Hu T, Lyu J, Yin Y, Sun J, Wang Q, et al. CSNK1A1/CK1α suppresses autoimmunity by restraining the CGAS-STING1 signaling. Autophagy. 2024;20:311–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulen MF, Samson N, Keller A, Schwabenland M, Liu C, Glück S, et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature. 2023;620:374–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang Y, Hepojoki J, Maldonado RS, Mito T, Terzioglu M, Manninen T, et al. Ancestral allele of DNA polymerase gamma modifies antiviral tolerance. Nature. 2024;628:844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh M, Saha S, Li J, Montrose DC, Martinez LA. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol Cell. 2023;83:266–80.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erttmann SF, Swacha P, Aung KM, Brindefalk B, Jiang H, Härtlova A, et al. The gut microbiota prime systemic antiviral immunity via the cGAS-STING-IFN-I axis. Immunity. 2022;55:847–61.e10. [DOI] [PubMed] [Google Scholar]
- 16.Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, et al. Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity. 2022;55:308–23.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa B, Becker J, Krammer T, Mulenge F, Durán V, Pavlou A, et al. Human cytomegalovirus exploits STING signaling and counteracts IFN/ISG induction to facilitate infection of dendritic cells. Nat Commun. 2024;15:1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong C, Schubert M, Tijhuis AE, Requesens M, Roorda M, van den Brink A, et al. cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature. 2022;607:366–73. [DOI] [PubMed] [Google Scholar]
- 19.Xue Q, Lai H, Zhang H, Li G, Pi F, Wu Q, et al. Selenium attenuates radiation colitis by regulating cGAS-STING signaling. Adv Sci (Weinh). 2024;11:e2403918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talbot EJ, Joshi L, Thornton P, Dezfouli M, Tsafou K, Perkinton M, et al. cGAS-STING signalling regulates microglial chemotaxis in genome instability. Nucleic Acids Res. 2024;52:1188–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Z, Wang X, Fu Y, Wu W, Hu Z, Lin Q, et al. YTHDF2 in peritumoral hepatocytes mediates chemotherapy-induced antitumor immune responses through CX3CL1-mediated CD8(+) T cell recruitment. Mol Cancer. 2024;23:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bappy SS, Haque Asim MM, Ahasan MM, Ahsan A, Sultana S, Khanam R, et al. Virus-induced host cell metabolic alteration. Rev Med Virol. 2024;34: e2505. [DOI] [PubMed] [Google Scholar]
- 23.Meade N, Toreev HK, Chakrabarty RP, Hesser CR, Park C, Chandel NS, et al. The poxvirus F17 protein counteracts mitochondrially orchestrated antiviral responses. Nat Commun. 2023;14:7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Liang X, Jiang J, Yang L, Xin J, Shi D, et al. PBMC transcriptomics identifies immune-metabolism disorder during the development of HBV-ACLF. Gut. 2022;71:163–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia W, Jiang P. p53 promotes antiviral innate immunity by driving hexosamine metabolism. Cell Rep. 2024;43: 113724. [DOI] [PubMed] [Google Scholar]
- 26.Guarnieri JW, Dybas JM, Fazelinia H, Kim MS, Frere J, Zhang Y, et al. Core mitochondrial genes are down-regulated during SARS-CoV-2 infection of rodent and human hosts. Sci Transl Med. 2023;15: eabq1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng Q, Zhang Y, Sun H, Yang X, Hao S, Liu B, et al. Human papillomavirus-16 E6 activates the pentose phosphate pathway to promote cervical cancer cell proliferation by inhibiting G6PD lactylation. Redox Biol. 2024;71: 103108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palanki R, Yamagata H, Mitchell MJ. OLAH connects fatty acid metabolism to the severity of respiratory viral disease. Cell. 2024;187:4549–51. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Hook JS, Ding Q, Xiao X, Chung SS, Mettlen M, et al. Neutrophil metabolomics in severe COVID-19 reveal GAPDH as a suppressor of neutrophil extracellular trap formation. Nat Commun. 2023;14:2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu MJ, Zhao Y, Li QT, Lei XY, He KY, Guo JR, et al. HMGA1 promotes the progression of esophageal squamous cell carcinoma by elevating TKT-mediated upregulation of pentose phosphate pathway. Cell Death Dis. 2024;15:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu H, Chen C, Hou ZS, He XD, Xie S, Ni J, et al. PI3Kγ maintains the self-renewal of acute myeloid leukemia stem cells by regulating the pentose phosphate pathway. Blood. 2024;143:1965–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang YT, Trzeciak AJ, Rojas WS, Saavedra P, Chen YT, Chirayil R, et al. Metabolic adaptation supports enhanced macrophage efferocytosis in limited-oxygen environments. Cell Metab. 2023;35:316–31.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lao Y, Cui X, Xu Z, Yan H, Zhang Z, Zhang Z, et al. Glutaryl-CoA dehydrogenase suppresses tumor progression and shapes an anti-tumor microenvironment in hepatocellular carcinoma. J Hepatol. 2024;81:847–61. [DOI] [PubMed] [Google Scholar]
- 34.Tong L, Chen Z, Li Y, Wang X, Yang C, Li Y, et al. Transketolase promotes MAFLD by limiting inosine-induced mitochondrial activity. Cell Metab. 2024;36:1013–29.e5. [DOI] [PubMed] [Google Scholar]
- 35.Guo X, Ji N, Guo Q, Wang M, Du H, Pan J, et al. Metabolic plasticity, essentiality and therapeutic potential of ribose-5-phosphate synthesis in Toxoplasma gondii. Nat Commun. 2024;15:2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bojkova D, Costa R, Reus P, Bechtel M, Jaboreck MC, Olmer R, et al. Targeting the pentose phosphate pathway for SARS-CoV-2 therapy. Metabolites. 2021;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang K, Nan F, Wang J, Zhang Y, Li J, Xue X, et al. A versatile nanozyme-based NADH circulating oxidation reactor for tumor therapy through triple cellular metabolism disruption. Small. 2024;20: e2311027. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Lin XT, Yu HQ, Fang L, Wu D, Luo YD, et al. Elevated FBXL6 expression in hepatocytes activates VRK2-transketolase-ROS-mTOR-mediated immune evasion and liver cancer metastasis in mice. Exp Mol Med. 2023;55:2162–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pandya NJ, Meier S, Tyanova S, Terrigno M, Wang C, Punt AM, et al. A cross-species spatiotemporal proteomic analysis identifies UBE3A-dependent signaling pathways and targets. Mol Psychiatry. 2022;27:2590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasegawa T, Oka T, Son HG, Oliver-García VS, Azin M, Eisenhaure TM, et al. Cytotoxic CD4(+) T cells eliminate senescent cells by targeting cytomegalovirus antigen. Cell. 2023;186:1417–31.e20. [DOI] [PubMed] [Google Scholar]
- 41.Fisch D, Pfleiderer MM, Anastasakou E, Mackie GM, Wendt F, Liu X, et al. PIM1 controls GBP1 activity to limit self-damage and to guard against pathogen infection. Science. 2023;382: eadg2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiménez-Loygorri JI, Villarejo-Zori B, Viedma-Poyatos Á, Zapata-Muñoz J, Benítez-Fernández R, Frutos-Lisón MD, et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat Commun. 2024;15:830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W, Li G, Zhou X, Liang H, Tong B, Wu D, et al. Disassembly of the TRIM56-ATR complex promotes cytoDNA/cGAS/STING axis-dependent intervertebral disc inflammatory degeneration. J Clin Invest. 2024;134:e165140. [DOI] [PMC free article] [PubMed]
- 44.Kirbas Cilingir E, Besbinar O, Giro L, Bartoli M, Hueso JL, Mintz KJ, et al. Small warriors of nature: novel red emissive chlorophyllin carbon dots harnessing fenton-fueled ferroptosis for in vitro and in vivo cancer treatment. Small. 2024;20:e2309283. [DOI] [PubMed] [Google Scholar]
- 45.Liu Q, Zhu F, Liu X, Lu Y, Yao K, Tian N, et al. Non-oxidative pentose phosphate pathway controls regulatory T cell function by integrating metabolism and epigenetics. Nat Metab. 2022;4:559–74. [DOI] [PubMed] [Google Scholar]
- 46.Vila IK, Chamma H, Steer A, Saccas M, Taffoni C, Turtoi E, et al. STING orchestrates the crosstalk between polyunsaturated fatty acid metabolism and inflammatory responses. Cell Metab. 2022;34:125–39.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang T, Qu X, Wang X, Xu D, Sheng M, Lin Y, et al. The macrophage STING-YAP axis controls hepatic steatosis by promoting the autophagic degradation of lipid droplets. Hepatology. 2024;80:1169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eren RO, Kaya GG, Schwarzer R, Pasparakis M. IKKε and TBK1 prevent RIPK1 dependent and independent inflammation. Nat Commun. 2024;15:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qin Y, Qiu D, Zhang Q. HNF1A regulates the crosstalk between innate immune responses and MAFLD by mediating autophagic degradation of TBK1. Autophagy. 2023;19:1026–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanno T, Nakajima T, Yokoyama S, Asou HK, Sasamoto S, Kamii Y, et al. SCD2-mediated monounsaturated fatty acid metabolism regulates cGAS-STING-dependent type I IFN responses in CD4(+) T cells. Commun Biol. 2021;4:820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, An W, Lu L, Yuan J, Wu D, Yang Q, et al. O-GlcNAc of STING mediates antiviral innate immunity. Cell Commun Signal. 2024;22:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JJ, Chen KJ, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun. 2020;11:3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lv B, Dion WA, Yang H, Xun J, Kim DH, Zhu B, et al. A TBK1-independent primordial function of STING in lysosomal biogenesis. Mol Cell. 2024;84:3979–96.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu J, Dobbs N, Yang K, Yan N. Interferon-independent activities of mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity. 2020;53:115–26.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He KY, Lei XY, Wu DH, Zhang L, Li JQ, Li QT, et al. Akkermansia muciniphila protects the intestine from irradiation-induced injury by secretion of propionic acid. Gut Microbes. 2023;15: 2293312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yin X, Zhang S, Lee JH, Dong H, Mourgkos G, Terwilliger G, et al. Compartmentalized ocular lymphatic system mediates eye-brain immunity. Nature. 2024;628:204–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang JY, Lei XY, He KY, Guo JR, Liu MJ, Li JQ, et al. HMGA1 drives chemoresistance in esophageal squamous cell carcinoma by suppressing ferroptosis. Cell Death Dis. 2024;15:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li T, Xu L, Wei Z, Zhang S, Liu X, Yang Y, et al. ELF5 drives angiogenesis suppression though stabilizing WDTC1 in renal cell carcinoma. Mol Cancer. 2023;22:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo JR, He KY, Yuan JL, An W, Yin WT, Li QT, et al. HMGA1 sensitizes esophageal squamous cell carcinoma to mTOR inhibitors through the ETS1-FKBP12 axis. Int J Biol Sci. 2024;20:2640–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lv H, Zong Q, Chen C, Lv G, Xiang W, Xing F, et al. TET2-mediated tumor cGAS triggers endothelial STING activation to regulate vasculature remodeling and anti-tumor immunity in liver cancer. Nat Commun. 2024;15:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1: Fig. S1 Knockdown and overexpression of STING in KYSE-30 cells. A, B. mRNA and protein expression of STING in STING-overexpression KYSE-30 cells. C, D. mRNA and protein expression of STING in STING-knocked down KYSE-30 cells. Data are expressed as mean ± SEM from three biological replicates. *** p < 0.001.
Supplementary Material 2: Fig. S2 STING upregulates PPP in HEK 293 cells. A, B. Whole cell extracts (WCEs) from wild-type and STING-overexpressing HEK 293 cells were analyzed for PPP metabolites (NADPH, GSH, R5P) and key enzymes (TKT, G6PD) using ELISA kits. C, D. WCEs from wild-type and STING-knocked down HEK 293 cells were similarly analyzed. Data are expressed as mean ± SEM from three biological replicates. ** p< 0.01, *** p< 0.001.
Data Availability Statement
No datasets were generated or analysed during the current study.