

Abstract
Background
Glucocerebrosidase 1 (GBA1) mutations are associated with reduced survival in Parkinson's disease but their effect on survival in dementia with Lewy bodies (DLB) is unclear.
Objective
To assess the impact of GBA1 mutations on survival among Ashkenazi Jews with DLB, while controlling for APOE status.
Methods
One hundred and forty participants from Tel Aviv Medical Center, Israel were genotyped for GBA1 mutations and APOE polymorphisms. Survival rates and follow‐up cognitive screening scores were analyzed.
Results
GBA1 mutation carriers had a two‐fold increased risk of death (HR = 1.999), while APOE status did not independently affect survival. In a subset of patients with available clinical data (N = 63), carriers of the APOE ε4 allele showed faster cognitive deterioration, while GBA1 mutation carriers also declined more rapidly albeit not significantly.
Conclusion
Understanding the genetic effects on survival and progression is crucial for patient counseling and inclusion in clinical trials.
Dementia with Lewy bodies (DLB) is a clinically heterogeneous neurodegenerative disease characterized by the accumulation of Lewy bodies and neurites and often by concomitant Alzheimer's disease (AD) pathology. 1 Mutations in the GBA1 gene are the most common genetic factor associated with Parkinson's disease (PD) and are frequent among patients with DLB.2, 3, 4 Previous studies show that PD and DLB patients with GBA1 mutations have younger motor symptom onset and a more severe disease phenotype.5, 6 In PD, carriers of GBA1 mutations have a more aggressive disease course, with faster progression and reduced survival7, 8, 9, 10; however, the effect of GBA1 mutations on survival in DLB is still unclear, with conflicting results in the literature.11, 12 The APOE ε4 allele is a significant risk factor for developing AD, 13 and is the top hit in genome association studies in DLB. 14 A study examining the influence of these genetic factors in PD demonstrated that carriers of either GBA1 mutations or the APOE ε4 allele had faster cognitive decline and were at a higher risk of progressing to dementia than non‐carriers. Furthermore, carriers of both genetic factors had the most significant risk of progressing to dementia. 15 In a previous study in our cohort, which included follow‐up data until the start of 2020, we did not find a clear effect on cognitive deterioration of either factor. 5 We were also unable to examine survival due to insufficient years of follow‐up. The aim of the current study was to examine the effects of these common genetic factors on survival and progression in a cohort of Ashkenazi Jewish (AJ) patients with DLB, known to have high carrier rates of GBA1 mutations. 4
1. Methods
Study protocols were approved by the Institutional Review Board (IRB) Committees for Genetic Studies. Participants provided informed consent. Survival analysis included consecutively recruited patients with DLB of AJ descent, who attended the Tel Aviv Medical Center between July 2013 and November 2023. Baseline research evaluation, including comprehensive neuropsychological and motor testing, was conducted for all patients. The results of these evaluations for 100 of the patients included in this study appeared in our previous publication. 5 Follow‐up Mini‐Mental State Examination (MMSE) data were collected in routine clinic visits, which varied between 4 and 22 months and were not conducted at set intervals. Due to the tertiary nature of our center, patients were also followed up in the community, with less frequent visits to the hospital clinic. Death was confirmed via the Israeli National Registry in which date but not cause of death are available.
Genotyping of founder GBA1 mutations was performed as previously described. 16 Briefly, patients were tested for the 84GG, IVS2 + IG > A, p.N370S, p.L444P, p.V394L, p.R496H, and 370Rec GBA1 mutations, using the “Gaucher Kit” (Catalog number 800672, Savyon Diagnostics, Ashdod, Israel, https://www.savyondiagnostics.com/product/nanochip‐gaucher). The E326K and T369M GBA1 variants were genotyped using gene‐specific TaqMan assay, followed by Sanger sequencing of polymerase chain reaction (PCR) products, to confirm carriers. GBA1 pathogenic variants were divided into three groups: (1) non‐Gaucher disease (GD) causing (E326K and T369M), (2) variants associated with mild Gaucher disease (N370S and R496H), and (3) variants associated with severe Gaucher disease (84GG, Ivs2, V349L). 17 Existence of the LRRK2 G2019S mutation was also examined. All samples were genotyped for rs429358 and rs7412, using TaqMan assays (C___3084793_20 and C____904973_10, respectively; Applied Biosystems) to establish their APOE haplotypes. Samples that carried E2 or E4 haplotypes were also Sanger sequenced as a second independent method to confirm ApoE haplotypes. Primers used to amplify the ApoE specific fragment for sequencing were: forward 5′ GGCACGGCTGTCCAAGGAGCT 3′ and reverse 5′ GCCCCGGCCTGGTACACTGC 3′.
1.1. Statistical Analysis
Descriptive statistics (means, standard deviations, Student's t‐test, and ANOVA for continuous variables; proportions, frequencies, and chi‐square test for categorical variables) were used to compare the demographics of GBA1 carriers and non‐carriers. In survival analyses, time to event was defined as survival in months. Participants were censored at the age of our last contact if they did not reach an event (death). We used log‐rank tests and Cox proportional hazards models, adjusted for sex and age at symptom onset, to compare survival between GBA1 carriers and non‐carriers and between APOE ε4 carriers and non‐carriers.
In order to assess deterioration in MMSE scores per year on an individual basis, we subtracted the last MMSE score recorded for each patient from the first MMSE score recorded and divided it by years of follow‐up. We used an analysis of covariance (ANCOVA) with GBA1 mutations and APOE polymorphisms as between‐subject factors and the deterioration per year as the dependent variable, with age at onset, disease duration until first MMSE, sex, and years of education as covariates. Eta squared served to estimate effect size. All analyses were conducted with Statistical Package for the Social Sciences (SPSS) software (Version 29; SPSS, Inc., Chicago, IL, USA), and the alpha level was set at 0.05.
2. Results
2.1. Participants’ Survival Analysis
One hundred and forty patients (n = 106, 75.7% men) were included in the survival analysis. Of these, 45 (32.1%) were carriers of mutations in the GBA1 gene, 38 carried mutations considered to be mild and 5 carried mutations considered severe, 1 was heterozygote for the T369M variant, and 1 was heterozygote for the E326K variant. Forty‐two (30%) patients were carriers of an APOE ε4 allele and, of these, 11 patients (7.8%) were carriers of both a GBA1 mutation and the APOE ε4 allele (Table 1). There were no LRRK2 mutation carriers in the cohort. One of the patients who was a GBA1 mutation carrier ended her life by assisted suicide overseas and was therefore excluded from further analyses. During the 2013–2023 period, 54 of the remaining 139 patients died, 20 of whom were GBA1 mutation carriers. Multivariable analysis, performed after adjusting for sex and age at onset, showed a significantly increased risk of death in GBA1 carriers compared with non‐carriers (hazard ratio [HR] = 1.999, P = 0.02; Fig. 1). APOE status had no independent effect on survival or age of onset (HR = 1.226, P = 0.482). The effect of GBA1 mutations on survival persisted, albeit with borderline significance, even when the severe mutation carriers were excluded (HR = 1.830, P = 0.05), demonstrating an effect of all GBA1 mutations on survival. Most patients died in the community, and therefore exact cause of death was unavailable in hospital records. As was found in previous studies, whole‐group analysis demonstrated that the GBA1 mutation carriers were younger at symptom onset (mean [SD] age, 66.93 [8.56]vs. 70.37 [6.20] years; t = 2.394, P = 0.020). Within the group of patients who died (N = 54), age of onset did not differ significantly between GBA1 and non‐GBA1 carriers (mean [SD] 68.4 [9.06] vs. 71.63 [6.131] years; t = 1.415, P = 0.168) nor did age at death (mean [SD] 76.2 [7.66] vs. 79.14 [6.2]; t = 2.780, P = 0.15).
TABLE 1.
Patient demographics.
| Parameter | GBA1 mutation carriers (n = 45) | GBA1 non‐carriers (n = 95) | P‐value |
|---|---|---|---|
| Sex (male) | 32 (71.11%) | 73 (76.84%) | 0.53* |
| APOE ε4 carriers | 11 (24.44%) | 31 (32.63%) | 0.43* |
| ε3/ε4 carriers | 9 (20.00%) | 29 (30.52%) | |
| ε4/ε4 carriers | 2 (4.44%) | 2 (2.10%) | |
| Age at onset a (years) (n = 139) | 66.93 [8.56] | 70.37 [6.20] | 0.02** |
| Died (n = 140) | 21 (46.66%) | 34 (35.78%) | |
| Patients who died (n = 54) | |||
| Survival after diagnosis until death (months) | 93.60 [30.80] | 90.17 [33.43] | 0.71** |
| Age at death (years) | 76.20 [7.66] | 79.14 [6.20] | 0.15** |
| Age at onset a (years) | 68.40 [9.06] | 71.63 [6.13] | 0.16** |
| Sex (male) | 12 (57.14%) | 28 (82.35%) | 0.18* |
| APOE ε4 carriers | 6 (28.57%) all of them ε3/ε4 carriers | 13 (38.23%) all of them ε3/ε4 carriers | 0.57* |
Chi‐square test.
Independent sample two‐sided t‐test.
Age at onset refers to age at symptom onset.
FIG. 1.
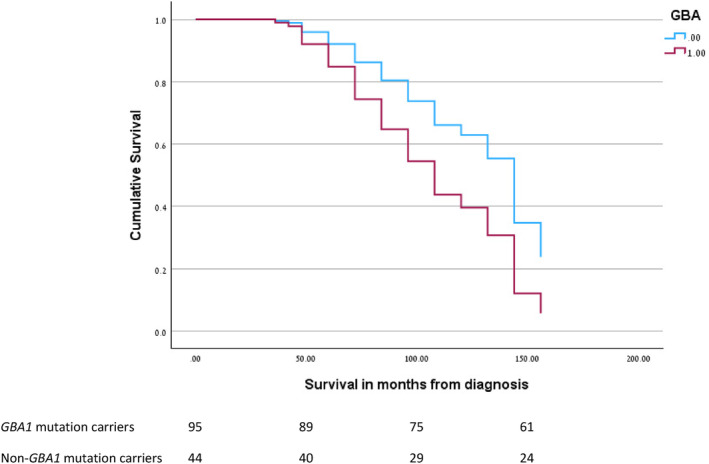
Cox proportional hazard regression for survival in months from diagnosis in GBA1 mutation carriers and non‐carriers adjusted for sex and age at onset. [Color figure can be viewed at wileyonlinelibrary.com]
2.2. Participants’ Follow‐Up Analysis
Follow‐up MMSE data were available for 63 of the 140 participants. Of these subjects, 19 were carriers of GBA1 mutations, 15 were carriers of APOE ε4 polymorphisms, and 8 patients were carriers of both genetic factors. Average disease duration at baseline was 37.57 months (SD 25.75). Average number of clinic visits was 3.78 (SD 1.90), and average time between visits was 13.7 months (SD 8.5). Average follow‐up time was 36.7 months (SD 22.62). Average deterioration on the MMSE per year was 0.22 (SD 0.31) points. ANCOVA revealed that carriers of the APOE ε4 allele had a faster cognitive deterioration than non‐carriers: 3.90 vs. 2.04 per year, F(1,62) = 5.32, P = 0.025, ŋ2 = 0.88. Carriers of the GBA1 mutation deteriorated more rapidly compared with non‐carriers with a trend towards significance: 3.44 vs. 2.17 per year, F(1,62) = 3.85, P = 0.055, ŋ2 = 0.066. The interaction between the two mutations was not significant: F(1,62) = 2.03, P = 0.159, ŋ2 = 0.036.
3. Discussion
We found that AJ patients with DLB who are carriers of GBA1 mutations had a two‐fold increased mortality risk. While some prior studies have not shown reduced survival in DLB patients with GBA1 mutations, our findings align with numerous studies in PD patients that indicated reduced survival among GBA1 mutation carriers.7, 8 In the present cohort, we found no main effect of the APOE ε4 allele on survival, in contrast to another study that found both a lower MMSE at baseline and a shorter disease duration among APOE ε4 carriers, 11 with no effect of GBA1 mutations. Differences in the type of GBA1 mutation across cohorts may explain the variations. The Dutch cohort had more E326K carriers, a milder variant unrelated to Gaucher disease in homozygous form. In contrast, our AJ cohort were mainly N370S carriers which is linked to type 1 Gaucher disease. Furthermore, the Dutch study did not find a more severe cognitive syndrome in GBA1 mutation carriers, unlike other cohorts,3, 5, 18 and thus patients might have been less severely affected at baseline. Another study on survival in patients with DLB has shown that the APOE ε4 allele was associated with shorter survival, but only in individuals with lower hippocampal volumes at baseline. 12
An analysis of a subgroup of patients for which clinical follow‐up data were available demonstrated that the presence of the APOE ε4 allele was associated with faster cognitive deterioration. Pathological studies indicate that the APOE ε4 allele is independently associated with more severe Lewy body pathology, regardless of AD pathology levels. This suggests that APOE ε4 may modify processes favoring Lewy body pathology spread. 19 A recent study that used a real‐time quaking‐induced conversion (RT‐QuIC) assay found that alpha‐synuclein seeding was exacerbated by APOE ε4 in a small cohort of LBD brains with minimal AD pathology. 20 The impact of the APOE ε4 allele on alpha‐synuclein spread might explain our observation of accelerated progression in carriers. Nevertheless, the APOE ε4 carriers did not show reduced survival. A possible explanation for this finding is that the main cause of death in DLB may not be directly related to the level of cognitive decline but to the severity of the motor dysfunction. Studies have shown that DLB patients with a higher burden of AD pathology tend to have a higher prevalence of the APOE ε4 allele. This association was linked to poorer cognitive performance compared with patients with a lower burden of AD pathology.21, 22, 23
GBA1 mutations are associated with more diffuse neocortical Lewy bodies in PD 24 and DLB, 25 which could explain the increased mortality. However, in DLB, the GBA1 mutation carriers have also been shown to have less coexistent amyloid pathology, 26 implying that survival in this disease may be mediated primarily by alpha‐synuclein pathology, a crucial consideration for designing trials of disease‐modifying therapies. Our previous findings revealed that DLB patients with GBA1 mutations have a more severe disease phenotype4, 5 and that the APOE ε4 allele acted in a complex interaction so that patients who carried both genetic risk factors had a more severe cognitive and motor involvement at presentation. However, the impact of these two common genetic factors on survival and clinical progression in patients with DLB was unclear. Here we demonstrate a clear impact on survival of GBA1 mutations and an impact of APOE ε4 polymorphisms on cognitive progression but not on survival. Further studies will need to examine whether different types of GBA1 mutations have a differential impact. The main limitation to our study is the limited cognitive follow‐up data that were available, potentially resulting in underpowered analysis. The frequency of follow‐up may have been influenced by disease severity; however, all patients diagnosed with DLB were offered follow‐up at the hospital, which aligns with their typical preference and therefore we believe it is unlikely that this introduced bias into our study. Another limitation is the absence of confirmatory post‐mortem pathology, with diagnoses relying on clinical criteria and supporting features. This is likely to have led to an underdiagnosis of cases as previously described. 27 A further limitation is that the cause of death was not available for most patients and that due to the wide variation in the number of clinic follow‐up visits and time points only the overall deterioration per year could be calculated. One of the main strengths of the study is the clinical follow‐up available for these patients that allowed for high clinical diagnostic accuracy.
4. Conclusions
In this study we aimed to examine the effect of the two most common genetic factors associated with DLB, GBA1 mutations and APOE polymorphisms, on survival and clinical progression. The effect of various genetic factors on survival is key for assessing prognosis, planning clinical trials, developing disease‐modifying treatments, and advancing personalized clinical approaches. The precise effect of these genetic factors and possible interactions with other genetic aspects should be further explored with the use of advanced biomarkers for assessment of different pathologies in vivo.
Author Roles
(1) Research Project: A. Conception, B. Design, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
T.S.: 1A, 1B, 1C, 2A, 2B, 3A, 3B.
G.K.: 2B, 2C, 3B.
A.M.: 1B, 1C, 3B.
K.R.: 1C, 2B, 3B.
Y.P.: 1C, 2B, 3B.
O.G.: 1B, 1C, 3B.
M.G.W.: 1B, 1C, 3B.
A.B.‐S.: 1B, 1C, 3B.
T.G.: 1B, 1C, 3B.
A.O.‐U.: 1B, 1C, 3B.
R.N.A.: 2B, 2C, 3B.
N.G.: 1A, 1B, 2C, 3B.
N.B.: 1B, 1C, 2C, 3A, 3B.
Financial Disclosures of All Authors (For the Preceding 12 Months)
T.S. reports support from The Michael J. Fox Foundation and has received speaker fees from Eisai, Biogen, and Eli Lilly. G.K., A.M., K.R., Y.P., O.G., M.G.W., and A.B.‐S. report no disclosures. T.G. has served as advisor to AbbVie and Truemed and has received travel support for herself and her team from Alphamedix, AbbVie, Medison, and Medoc. She received research and educational support from the Movement Disorders Society and a grant from the Parkinson's Foundation Center of Excellence. A.O.‐U. reports support from The Michael J. Fox Foundation and Chaya Charitable Fund and has received honoraria from Sanofi Genzyme. R.N.A. reports research funding from The Michael J. Fox Foundation, the Silverstein Foundation, and the Parkinson's Foundation. He received consultation fees from Biogen, Biohaven, Capsida, Gain Therapeutics, Genzyme/Sanofi, Servier, Takeda, and Vanqua Bio. N.G. reports stock ownership in medically related fields: Lysosomal Therapeutic Ltd, Vibrant, BOL. Intellectual property rights: GaitBetter. Consultancies: Intec Pharma, NeuroDerm, Denali, AbbVie, Sanofi‐Genzyme, Biogen, Vibrant, BOL, LTI, Idorsia, and Pharma2B. Advisory boards: LTI, NeuroDerm, Sionara, Sanofi‐Genzyme, Biogen, Denali, Intec, Pharma, Idorsia. Honoraria: AbbVie, Sanofi‐Genzyme, Movement Disorder Society, Bial, and UCB. N.B. reports support from Ionis Pharmaceuticals and has received speaker fees from Eisai, Eli Lilly, and Biogen.
Acknowledgments
We would like to thank all the patients and their families for taking part in this study.
Relevant conflicts of interest/financial disclosures: All authors report no conflict of interests pertaining to the article.
Funding agency: The research was funded by internal hospital grants.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J‐P, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB cCnsortium. Neurology 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sidransky E, Nalls MA, Aasly JO, Aharon‐Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nalls MA, Duran R, Lopez G, Kurzawa‐Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 2013;70(6):727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shiner T, Mirelman A, Gana Weisz M, Bar‐Shira A, Ash E, Cialic R, et al. High frequency of GBA gene mutations in dementia with Lewy bodies among Ashkenazi Jews. JAMA Neurol 2016;73(12):1448–1453. [DOI] [PubMed] [Google Scholar]
- 5. Shiner T, Mirelman A, Rosenblum Y, Kavé G, Weisz MG, Bar‐Shira A, et al. The effect of GBA mutations and APOE polymorphisms on dementia with Lewy bodies in Ashkenazi Jews. J Alzheimers Dis 2021;80(3):1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alcalay RN, Caccappolo E, Mejia‐Santana H, Tang MX, Rosado L, Orbe Reilly M, et al. Cognitive performance of GBA mutation carriers with early‐onset PD: the CORE‐PD study. Neurology 2012;78(18):1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brockmann K, Srulijes K, Pflederer S, Hauser A‐K, Schulte C, Maetzler W, et al. GBA‐associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30(3):407–411. [DOI] [PubMed] [Google Scholar]
- 8. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA‐associated Parkinson's disease: the mutation matters. Ann Neurol 2016;80(5):662–673. [DOI] [PubMed] [Google Scholar]
- 9. Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen‐Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in parkinson disease. JAMA Neurol 2016;73(10):1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, et al. Features of GBA‐associated Parkinson's disease at presentation in the UK tracking Parkinson's study. J Neurol Neurosurg Psychiatry 2018;89(7):702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van der Lee SJ, van Steenoven I, van de Beek M, Tesi N, Jansen IE, van Schoor NM, et al. Genetics contributes to concomitant pathology and clinical presentation in dementia with Lewy bodies. J Alzheimers Dis 2021;83(1):269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graff‐Radford J, Lesnick TG, Boeve BF, Przybelski SA, Jones DT, Senjem ML, et al. Predicting survival in dementia with Lewy bodies with hippocampal volumetry. Mov Disord 2016;31(7):989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer's disease: a systematic review and meta‐analysis. Neuroepidemiology 2012;38(1):1–17. [DOI] [PubMed] [Google Scholar]
- 14. Rongve A, Witoelar A, Ruiz A, Athanasiu L, Abdelnour C, Clarimon J, et al. GBA and APOE ε4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Sci Rep 2019;9(1):7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szwedo AA, Dalen I, Pedersen KF, Camacho M, Bäckström D, Forsgren L, et al. GBA and APOE impact cognitive decline in Parkinson's disease: a 10‐year population‐based study. Mov Disord 2022;37(5):1016–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gan‐Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype‐phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008;70(24):2277–2283. [DOI] [PubMed] [Google Scholar]
- 17. Gan‐Or Z, Amshalom I, Kilarski LL, Bar‐Shira A, Gana‐Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015;84(9):880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lerche S, Machetanz G, Wurster I, Roeben B, Zimmermann M, Pilotto A, et al. Dementia with Lewy bodies: GBA1 mutations are associated with cerebrospinal fluid alpha‐synuclein profile. Mov Disord 2019;34(7):1069–1073. [DOI] [PubMed] [Google Scholar]
- 19. Dickson DW, Heckman MG, Murray ME, Soto AI, Walton RL, Diehl NN, et al. APOE ε4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 2018;91(12):e1182–e1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jin Y, Li F, Sonoustoun B, Kondru NC, Martens YA, Qiao W, et al. APOE4 exacerbates α‐synuclein seeding activity and contributes to neurotoxicity in Alzheimer's disease with Lewy body pathology. Acta Neuropathol 2022;143(6):641–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lemstra AW, de Beer MH, Teunissen CE, Schreuder C, Scheltens P, van der Flier WM, et al. Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2017;88(2):113–118. [DOI] [PubMed] [Google Scholar]
- 22. Ryman SG, Yutsis M, Tian L, Henderson VW, Montine TJ, Salmon DP, et al. Cognition at each stage of Lewy body disease with co‐occurring Alzheimer's disease pathology. J Alzheimers Dis 2021;80(3):1243–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Howard E, Irwin DJ, Rascovsky K, Nevler N, Shellikeri S, Tropea TF, et al. Cognitive profile and markers of Alzheimer disease‐type pathology in patients with Lewy body dementias. Neurology 2021;96(14):e1855–e1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 2009;132(7):1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clark LN, Kartsaklis LA, Wolf Gilbert R, Dorado B, Ross BM, Kisselev S, et al. Association of glucocerebrosidase mutations with dementia with Lewy bodies. Arch Neurol 2009;66(5):578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 2012;79(19):1944–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet 2015;386(10004):1683–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.