

Abstract
Background
Teat number is one of the most important indicators to evaluate the lactation performance of sows, and increasing the teat number has become an important method to improve the economic efficiency of farms. Therefore, it is particularly important to deeply analyze the genetic mechanism of teat number traits in pigs. In this study, we detected Single Nucleotide Ploymorphism (SNP), Insertion-Deletion (InDel) and Structural variant (SV) by high-coverage whole-genome resequencing data, and selected teat number at birth and functional teat number as two types of teat number traits for genome-wide association study (GWAS) to reveal candidate genes associated with pig teat number traits.
Results
In this study, we used whole genome resequencing data from 560 Yorkshire sows to detect SNPs, InDels and SVs, and performed GWAS for the traits of born teat number and functional teat number, and detected a total of 85 significant variants and screened 214 candidate genes, including HEG1, XYLT1, SULF1, MUC13, VRTN, RAP1A and NPVF. Among them, HEG1 and XYLT1 were the new candidate genes in this study. The co-screening and population validation of multiple traits suggested that HEG1 may have a critical effect on the born teat number.
Conclusion
Our study shows that more candidate genes associated with pig teat number traits can be identified by GWAS with different variant types. Through large population validation, we found that HEG1 may be a new key candidate gene affecting pig teat number traits. In conclusion, the results of this study provide new information for exploring the genetic mechanisms affecting pig teat number traits and genetic improvement of pigs.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-024-11109-0.
Keywords: GWAS, Teat number, Whole genome sequencing, Structural variation
Introduction
As a reproductive trait, pig teat number trait determines the lactation performance of sows, and is an important target for genetic improvement of breeding pigs [1]. Higher born teat number help sows develop more functional teats as adult. Functional teat number is one of the limiting factors in improving sows’ lactation capacity [2]. The poverty teat number of sows, underdeveloped teats and uneven arrangement of teats in the herd will reduce the piglet survival rate the economic efficiency of the farm [3]. Therefore, it is necessary to increase the number of teats in sows through genetic improvement as a way to increase the lactation capacity of sows [4].
The pig teat number trait is regulated by multiple genes [5]. With the continuous maturation of sequencing technology, genome-wide association analysis has become an important tool for screening variants related to economic traits in livestock and poultry. In recent years, GWAS has been applied to a wide range of applications in pigs, and key genes related to important economic traits such as reproduction, growth and meat quality have been identified [6–8]. The genetic variants and genes associated with pig teat number have also been extensively studied [5, 9]. So far, a total of 977 QTLs associated with the pig teat number trait have been included in the PigQTL database (https://www.animalgenome.org/cgi-bin/QTLdb/SS/browse, 2024-2-5, interview). Genes such as VRTN [10], TOX3 [11], ABCD4 [12] have been identified as candidate genes for the teat number traits. Signaling pathways related to teat or mammary gland development have also been reported, such as the Wnt signaling pathway, which affects mammary gland differentiation and determines mammary gland primordia [13]. The TBX3 signaling pathway, which regulates mammary lineage [14], and the FGF pathway, which is closely related to mammary gland cells [15]. It is clear that identifying more genes will help us to deepen our understanding of the genetic mechanisms associated with the teat number trait. This could lead to a more appropriate breeding program to optimize the reproductive performance of the pig.
With the reduction of sequencing costs and the development of structural variant detection algorithms in recent years, GWAS based on structural variants have begun to be applied. Compared with SNPs, structural variants can carry richer genetic information due to longer sequences, and the explained heritability is higher than that of SNPs [16]. In a GWAS for porcine growth traits using SNP, InDel, SVs and tandem repeat (TR), Blaj et al. found that despite physical co-localization. SNPs or InDels did not always capture the effects of SVs and TRs on complex traits [17]. Zong et al. constructed a high-quality SV map using high-coverage sequencing data on a population of cross-bred Eurasian pigs, and performed GWAS for 25 carcass traits, 7 skeletal traits, and 4 meat quality traits, identifying that SV may affect pig body size between European and Chinese pig breeds [18]. In previous breeding work on pig teat number traits, most of the GWAS were performed using chip data, and resolution of teat number traits using resequencing data is rare.
In this context, to screen for candidate genes associated with teat number traits, this study utilized whole genome resequencing data with an average depth of more than 15× and obtained high-quality SNP, InDel and SV maps using different typing software. We then performed GWAS for born teat number and functional teat number, and finally validated the candidate genes affecting teat number traits. In conclusion, this study will provide a theoretical basis for the development of assisted breeding using SV as a molecular marker to further promote the genetic improvement of pig teat number traits in breeding.
Materials and methods
Animals and phenotypes
In this study, the selected Danish Yorkshire pigs were reared in the Shaanxi Shunxin Danish Breeding Pig Farm (Hanzhong City, Shaanxi Province, China). For 49,818 pigs born at the farm from 2016 to 2022, teat number related traits were collected. Including the born left teat number (LTN), the born right teat number (RTN), and the born total teat number (TTN). We also collected the date of birth, parity, sex, place of birth, and pedigree information. We traced back 3 generations of pedigree and kept 50,001 pigs in pedigree records for the next step. From July to November 2022, traits related to the number of functional teats in 430 pregnant sows on the farm were measured and recorded. We excluded undeveloped teats, damaged teats, and degraded teats, and retained the record of functional left teat number (FLTN), functional right teat number (FRTN) and functional total teat number (FTTN), as well as the information on the gestation parity, age and pedigree of the sows. Subsequently, the phenotypic data were cleaned and the abnormal values and phenotypically missing individuals were excluded for subsequent analysis. The maximum, minimum, mean, standard deviation and coefficient of variation of each trait were calculated.
Estimation of genetic parameters
To explore the level of heritability of teat number traits in this population, genetic parameters were estimated to calculate heritability of teat number traits in Yorkshire pigs by the maternal effect model using ASReml (version 4.1.0) software. The model was used for the born teat number traits as follows:
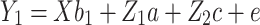 |
Where the 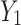 is the vector of born teat number phenotypes,
is the vector of born teat number phenotypes, 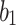 fixed effects vector includes pig farm-year-season, parity, and sex effects,
fixed effects vector includes pig farm-year-season, parity, and sex effects, 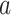 is the vector of individual additive effects,
is the vector of individual additive effects, 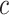 is the vector of maternal environmental effects,
is the vector of maternal environmental effects, 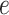 is the residual effects, and
is the residual effects, and 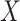 ,
, 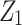 and
and 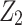 are the corresponding structure matrices.
are the corresponding structure matrices.
For the functional teat number traits, the model was used as follows:
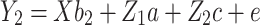 |
Where 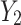 is the vector of functional teat number phenotypes,
is the vector of functional teat number phenotypes, 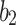 is a vector of fixed effects including sow parity of pregnancy and age effects,
is a vector of fixed effects including sow parity of pregnancy and age effects, 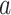 is an individual additive effects vector,
is an individual additive effects vector, 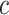 is the vector of maternal environmental effects,
is the vector of maternal environmental effects, 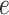 is the residual effects, and
is the residual effects, and 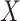 ,
, 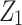 and
and 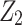 are the corresponding structure matrices.
are the corresponding structure matrices.
To explore potential relationships prior to the traits of born teat number and functional teat number, we calculated genetic correlations between traits using ASReml (version 4.1.0) and estimated the heritability for 430 sows with same traits.
Genotypic data and quality control
In this study, whole genome resequencing data were collected from 560 pig samples that had published in our previous studies (Sun et al. Unpublished). Briefly, after collecting ear tissue samples from 560 Yorkshire sows, DNA was extracted and whole-genome resequencing was performed using the DNBSEQ-T7 platform, yielding sequencing data with an average sequencing depth of 15.6×. We processed the raw data to remove low-quality reads using the default parameters of Fastp (version 0.20.1) [19]. Then, the clean reads were aligned to the Sscrofa11.1 (https://www.ncbi.nlm.nih.gov/) reference genome from NCBI using BWA (version 0.7.15) (BWA-MEM algorithm, default parameters) [20]. And we obtained the clean Binary Alignment Map format (bam) files by removing duplicate reads and sorting them. Subsequently, these files were used to generate the gvcf file using the HaplotypeCaller program in the GATK (version 4.1.1) pipeline [21]. Then the gvcf files were merged by the GenomicsDBImport module. Finally, GenotypeGVCFs was used to convert database into a vcf file. We filtered the variant data-sets of SNP and InDel (Insertion and deletion,2–50 bp), using the best practice parameters “QUAL < 30.0 | QD < 2.0 | MQ < 40.0 | FS > 60.0 | SOR > 3.0 | MQRankSum < -12.5 | ReadPosRankSum < -8.0” and “QD < 2.0 | QUAL < 30.0 | FS > 200.0 | ReadPosRankSum < -20.0”. We then retained biallelic SNPs and InDels.
In terms of SV detection, we used four software, including Manta (version 1.6.0) [22], Delly (version 1.16) [23], Tiddit (version 3.3.2) [24] and Breakdancer (version 3.0.1 ) [25], to detect the SVs from 560 pig samples, and then we used SURVIVOR (version 1.0.3) [26] to merge the SVs site that were called by at least two tools. Finally we used Smoove (version 0.2.8) [27] software to genotype SV type and used Bcftools (version 1.10.2) [28] to merge sample SV genotype files(Fig. 1).
Fig. 1.

SV discovery process overview
SNPs sets: PLINK (version 1.9.0) [29] was used for quality control, based on the following exclusion criteria: SNPs with miss genotype call rate > 0.1, samples with miss genotype call rate > 0.1, minor allele frequency(MAF) < 0.01, and Hardy-Weinberg equilibrium P < 1 × 10− 5, and using the “--indep-pairwise 50 5 0.5” parameter to prune SNPs by linkage-disequilibrium(LD). Finally, 18 autosomal 678,204 SNPs were retained for further analysis.
InDels sets: PLINK (version 1.9.0) was also utilized for quality control with the parameters of “--gene 0.01 –maf 0.05”, and the “--indep-pairwise 50 5 0.1” was used to prune InDels by LD. A total of 560 samples and 103,491 variants in 18 autosomes were retained for GWAS.
SVs sets: SV lengths were filtered to retain SVs within the range of 50 bp-100,000 bp, and only three types of SVs, INV, INS and DEL, were retained, and a total of 12,671 SVs from 18 autosomes were finally used for GWAS.
Genome-wide association analysis for each trait
To efficiently detect the variation associated with the low heritability teat number trait in this study, we used the Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway (BLINK) for GWAS by GAPIT (version 3.0) [30]. The BLINK model used the phenotype of each teat trait as the response variables and the numeric variants matrix as the genetic data. The significance threshold line 0.05/N (N is the number of variants) and the potential threshold line 1/N in the results of GWAS were determined by Bonferroni-corrected, and the variant data included SNP, InDel, and SV obtained after quality control. We used the R package CMplot (version 4.2.0) to gain the Manhattan plots and QQ-plots [31].
Candidate gene search and functional enrichment analysis
We searched the candidate genes which including or close to significant variants using BedTools (version 2.27.1) [32] based on Sscrofa11.1. The screening range was 0.5 Mb region upstream and downstream of the significant genetic variants. Subsequently, we used the gProfiler [33] website (https://biit.cs.ut.ee/gprofiler/gost) to enrich and analyze the candidate genes to reveal the functions of the candidate genes and the involvement of the candidate genes through Gene Ontology (GO)) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
In order to obtain genes screened together by different variant types, we selected the intersection of candidate genes screened by SNPs, InDels and SVs, and then used R package to draw the Venn map. LDBlockShow (version 2.6) [34] was used to generate and visualize the local linkage disequilibrium (LD) pattern of haplotype block between significant variants and genes.
Population validation of candidate genes
For the same candidate genes screened multiple times by GWAS, we selected 1,169 Yorkshire sows from another farm, collected their born total teat number, and genotyped them by target sequencing chip (GenoBaits Porcine 50 K Chip). We found the variant located in the candidate gene region in the validation population, extracted the variant and genotype of them for significance test with the phenotype of the validation population, and confirmed their significance by rank sum test.
Result
Descriptive statistics of phenotype and heritability
Descriptive statistics of the phenotypes of born teat number traits and functional teat number traits of Danish Yorkshire sows collected in this study are shown in Table 1. We found that the number of teats traits in pigs was a low heritability trait with heritabilities ranging from 0.085 to 0.173, while their coefficient of variation was less than 10%. The genetic correlation between traits were appearing positive correlation. Although the heritability of the born teat number traits and functional teat number traits were different in shared trait population (N = 430), both of them were at low heritability level (Table 2).
Table 1.
Descriptive statistics of traits
| Trait | N | Max | Min | Mean (SD) | CV (%) | h2 |
|---|---|---|---|---|---|---|
| LTN | 49,752 | 9 | 6 | 7.059 ± 0.283 | 4.007 | 0.104 |
| RTN | 49,714 | 9 | 6 | 7.175 ± 0.409 | 5.706 | 0.168 |
| TTN | 49,664 | 18 | 12 | 14.235 ± 0.581 | 4.083 | 0.173 |
| FLTN | 427 | 8 | 6 | 7.019 ± 0.464 | 6.615 | 0.085 |
| FRTN | 430 | 9 | 6 | 7.114 ± 0.532 | 7.478 | 0.112 |
| FTTN | 427 | 18 | 12 | 14.138 ± 0.800 | 5.661 | 0.151 |
N: number of samples; Max: maximum value; Min: minimum value; Mean: arithmetic mean; SD: standard deviation; CV: coefficient of variation; h2: heritability
Table 2.
The genetic correlation between teat number traits
| Trait | LTN | RTN | TTN | FLTN | FRTN | FTTN |
|---|---|---|---|---|---|---|
| LTN | 0.005 | 0.779 | 0.905 | 0.937 | 0.738 | 0.916 |
| RTN | 0.253 | 0.972 | 0.971 | 0.912 | 0.983 | |
| TTN | 0.005 | 0.794 | 0.890 | 0.950 | ||
| FLTN | 0.085 | 0.721 | 0.858 | |||
| FRTN | 0.112 | 0.970 | ||||
| FTTN | 0.151 |
Genetic correlation above the diagonal. Heritability on the diagonal
Genome-wide association analysis for the born teat number traits
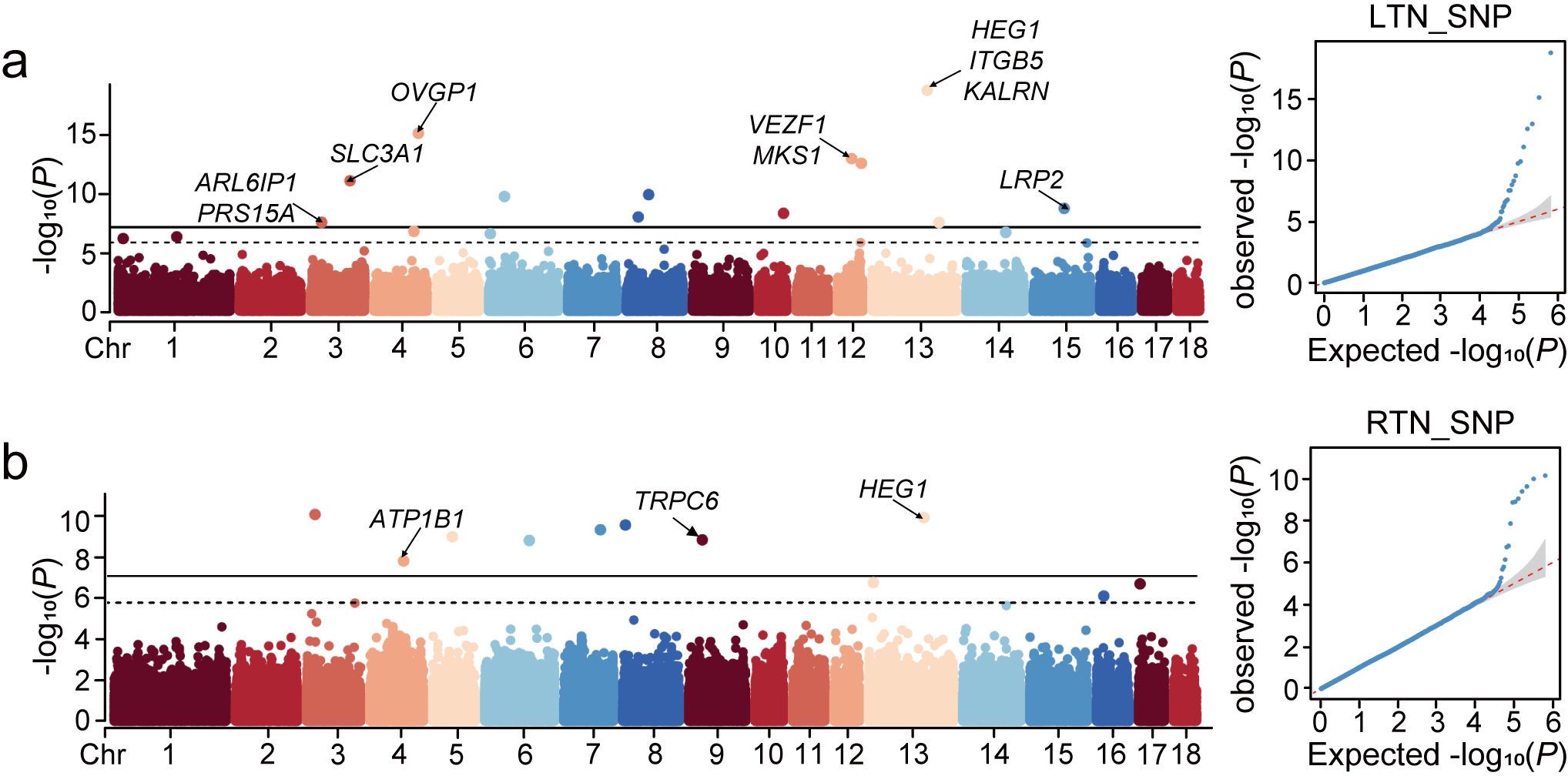
In this study, 560 Yorkshire sows were used for GWAS of born teat number traits and 27, 26, and 20 significant variants were detected in the GWAS results for SNP, InDel, and SV, respectively. This significant SNPs were distributed on several chromosomes, e.g., Chr3, Chr4, Chr12, Chr13(Fig. 2a). Within Chr13, 134.7 Mb-136.3 Mb, nine genes were identified to be associated with born teat number traits, including HEG1, MUC13, UMPS, OSBPL11, SNX4, ZNF148, SLC12A8, ITGB5, and KALRN. The significant SNP chr13:135307011 was screened for both LTN and TTN, and this significant SNP was located in an exon of the HEG1 gene (Fig. 2b). Interestingly, the HEG1 gene was also screened within 60Kb upstream of the significant SNP chr13:135418265, which affects the number of teats on the right side. After enrichment analysis, HEG1 was enriched for epithelium development, which is strongly associated with mammary gland development. To validate the effect of HEG1 on teat number in a sow population, we evaluated the effect of variation on this gene on total teat number in target sequencing from another Yorkshire sow population (n = 1169), and found that mutations in HEG1 gene region were significantly associated with born total teat number (P < 0.001) in this population (Fig. 2c). The candidate genes of TTN by annotation are listed in Table 3.
Fig. 2.

GWAS of born teat number based on SNPs and population validation. (a) GWAS of TTN based on SNPs. N = 560 Yorkshire pigs. The horizontal solid line represents Bonferroni threshold (-Log10(0.05/678,204) = 7.1324). The horizontal dashed line represents the suggestive significance threshold line (-Log10(1/678,204) = 5.8314). (b) Magnification of the gene region with significant variants in TTN, RTN and LTN GWAS result. Different color and shaped points distinguish three traits. (c) Population validation in 1169 Yorkshire pigs base on target sequencing with GWAS candidate significant SNP in TTN. The blue box represent sow population with reference variant; and the red box is sow population with mutation
Table 3.
Significant SNPs for TTN
| Traits | SNPs | Pos (bp) | P-value | Candidate Gene |
|---|---|---|---|---|
| TTN | chr4:65608738 | 65,608,738 | 3.56 × 10− 8 | PRDM14, SLCO5A1, SULF1, NCOA2 |
| chr4:69022640 | 69,022,640 | 6.31 × 10− 9 | TRIM55, ARMC1, PDE7A | |
| chr4:79455541 | 79,455,541 | 1.70 × 10− 14 | CEBPD, MCM4, SPIDR, SNTG1, PRKDC | |
| chr13:135307011 | 135,307,011 | 1.77 × 10− 8 | HEG1, MUC13, OSBPL11, SLC12A8, ITGB5, KALRN | |
| chr15:136826587 | 136,826,587 | 2.63 × 10− 11 | PRLH | |
| chr15:136853009 | 136,853,009 | 4.19 × 10− 8 | PRLH | |
| chr16:16346505 | 16,346,505 | 2.65 × 10− 11 | —— |
A total of 5 significant InDels and 10 significant SVs were detected in TTN (Fig. 3a and b). In addition, a total of 16 significant InDels and 14 significant SVs, and 5 significant InDels and 3 significant SVs were detected in RTN and LTN, respectively (Fig. S2). The information of significant InDels, SVs and the genes obtained after annotation are listed in Table S1 and Table S2.
Fig. 3.

GWAS of born teat number traits. (a) TTN GWAS base on InDels. (b) TTN GWAS base on SVs. (c) Candidate genes from GWAS screens for SNPs, Indels, and SVs, respectively, plotted on a Wayne diagram. (d) Magnified Manhattan plot of LTN GWAS result and LD heatmap of the magnified region. (e) Magnified Manhattan plot of GWAS result in TTN
For GWAS of born teat number traits, the genes obtained based on the annotation of different types of genetic variants were taken to the intersection (Fig. 3c). We found that SNP and InDel co-screened nine genes, namely HEG1, ITFGB5, KALRN, MUC13, OSBPL11, SLC12A8, SNX4, UMPS, and ZNF148. And 17 genes, including ARL6IP1, RPS15A, SMG1, XYLT1 and others, were screened together in GWAS based on SNP and SV. Within the 26.600 Mb-27.550 Mb region of chromosome 3, we found that the significant SV3:27195761 was located within the XYLT1 gene region, while SNP3:27053393, although already adjacent to XYLT1, still did not directly detect significant variants within the gene region base on SNP GWAS in LTN (Fig. 3d). Compared with SV3:27195761, SNP3:27053393 is more distant from XYLT1. In the analysis of TTN trait, we also screened the SV3:27195761 hotspot and annotated ARL6IP1, RPS15A, SMG1, XYLT1(Fig. 3e). However, the GWAS based on SNP of TTN did not find this hotspot. So, GWAS based on SV has more potential to capture the target genes.
Genome-wide association analysis for the functional teat number traits
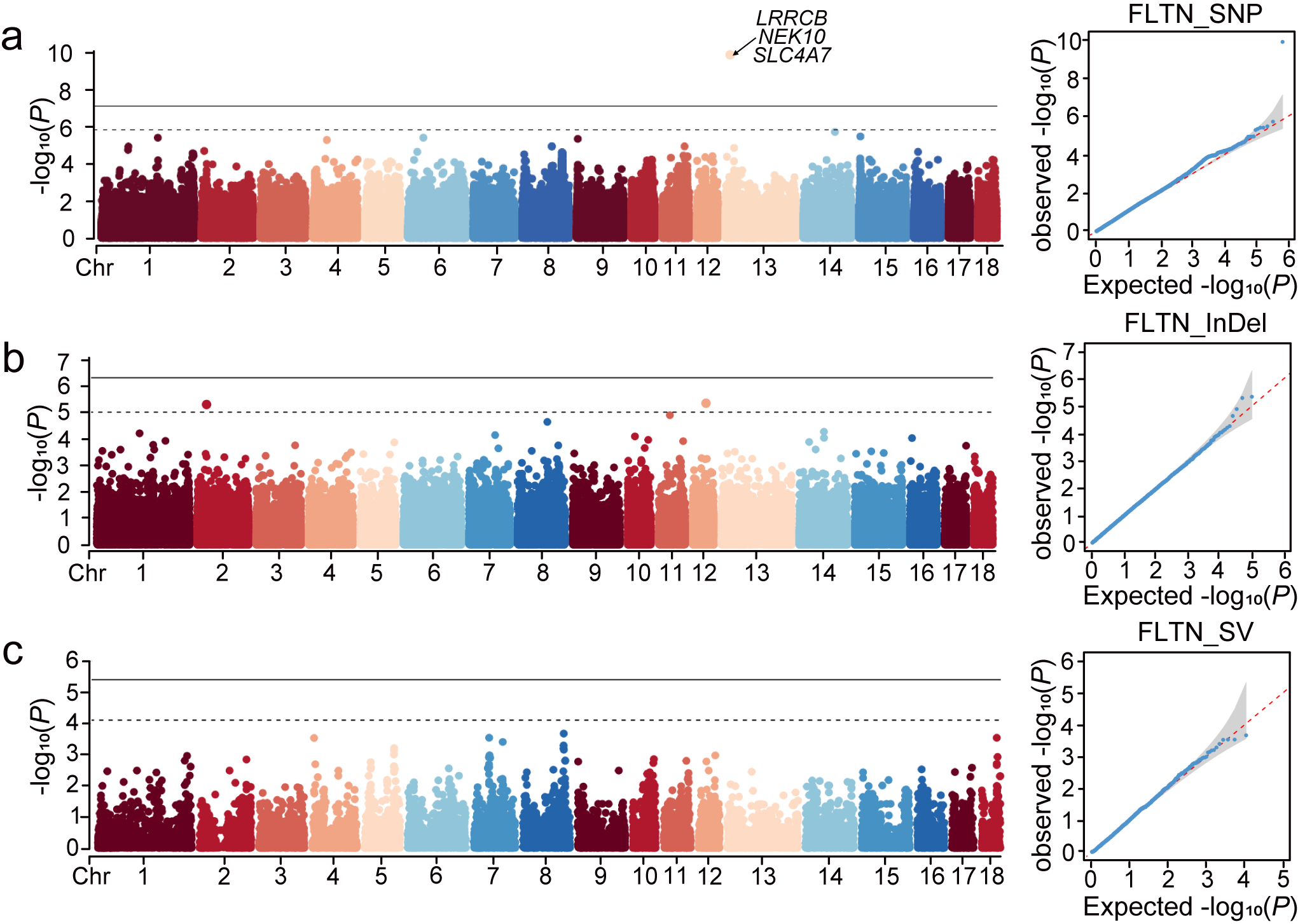
The functional teat number of sows determines the ability of lactation, and the number of functional teats is closely related to the survival rate of lactating piglets. Therefore, we collected functional teat number trait records from 430 sows and performed GWAS combining three types variant after removing outlier phenotypes. Overall, the number of significant variants detected by GWAS for functional teat number was 2, 3, and 7 for SNPs, InDels, and SVs, respectively. FTTN was not screened for genome-wide significant SNPs and InDels (Fig. 4a and b). But SV-based GWAS identified three significant regions, annotated to genes such as NPVF, SWI5, URM1(Fig. 4c; Table 4).
Fig. 4.

GWAS of FTTN base on SNPs, InDels, SVs, respectively. (a) GWAS result of FTTN base on SNPs. N = 427 pig samples. (b) GWAS result of FTTN base on InDels. (c) GWAS result of FTTN base on SVs
Table 4.
Significant SVs for functional teat number relevant traits
| Traits | Chr | Start | End | Type | P-value | Candidate Gene |
|---|---|---|---|---|---|---|
| FTTN | 1 | 268,509,186 | 268,509,485 | DEL | 1.11 × 10− 7 | LCN2, ODF2, SWI5, TRUB2, URM1, WDR34 |
| 13 | 127,891,501 | 127,891,579 | DEL | 6.32 × 10− 7 | CLDN1, CLDN16, P3H2, SDK1, GMNC | |
| 18 | 47,349,893 | 47,350,429 | DEL | 2.01 × 10− 6 | NPVF, OSBPL3 |
We identified VRTN in the SV-based GWAS for FRTN trait (Fig. 5c; Table 5), and VRTN has been reported in several studies related to pig teat number [35]. Although this gene was not identified in the SNP and InDel-based GWAS, we could see a clear peak trend on chromosome 7 (Fig. 5a and b, Table S4, Table S5). For new candidate genes, RAP1A was a potential gene of functional teat number, which was detected by InDel-based GWAS of FRTN. We screened only one significant SNP locus associated with FLTN trait, annotated to genes such as SLC4A7 and NEK10, and unfortunately, no relevant genetic markers were detected based on InDel and SV (Fig. S3, Table S4).
Fig. 5.

GWAS of FRTN base on SNPs, InDels, SVs, respectively. N = 430 pig samples. The dotted red box indicates that the same region of interest was detected by the three variant types GWAS results in Chr7. (a) GWAS of FRTN base on SNPs. There is no significant variation on Chr7, but trends can be detected. (b) GWAS of FRTN base on InDels. (c) GWAS of FRTN base on SVs
Table 5.
Significant SVs for functional right teat number trait
| Traits | Chr | Start | End | Type | P-value | Candidate Gene |
|---|---|---|---|---|---|---|
| FRTN | 7 | 97,757,361 | 97,757,668 | DEL | 7.76 × 10− 8 | ABCD4, ALDH6A1, AREL1, COQ6, EIF2B2, FAM161B, FCF1, LIN52, LTBP2, NPC2, PROX2, SYNDIG1L, VRTN, YLPM1, VSX2 |
| 12 | 51,093,256 | 51,093,314 | DEL | 3.44 × 10− 6 | MIS12, WSCD1, NUP88 | |
| 13 | 130,756,600 | 130,756,898 | DEL | 1.42 × 10− 8 | HES1, HRASLS | |
| 14 | 24,058,687 | 24,059,355 | DEL | 1.86 × 10− 6 | ADGRD1, SFSWAP, STX2 |
Enrichment analysis of candidate genes
Table S6 shows the results of the GO term enrichment and KEGG pathway of candidate genes. The GO term enrichment was involved in terms such as Wnt signaling pathway (GO:0016055), animal organ formation (GO:0048645), epithelium development (GO:0060429), cellular process involved in reproduction in multicellular organism (GO:0022412), regulation of developmental process (GO:0050793). Furthermore, these candidate genes are involved in the cG/MP-PKG signaling pathway (KEGG:04022), JAK-STAT signaling pathway (KEGG:04630), PI3K-Akt signaling pathway (KEGG:04151) and other pathways.
Discussion
Sow teat number affects the survival rate of lactating piglets and is an important indicator of lactation ability, and increasing the teat number can improve sow reproductive performance [36, 37]. Related studies have shown that there is genetic variation in the teat number and some of the reproductive traits of sows, which has a profound effect on the weight of piglets 10 days after birth [38, 39]. When sows do not have enough teats to satisfy piglets’ nursing needs, producers opt for fostering methods or even euthanize piglets that are not viable [40]. There are many factors that influence teat traits and there are numerous research reports on them. There are inter- and intra-breed differences in pig teat numbers, and generally breeds with high reproductive performance have relatively high teat numbers [41]. The hormone levels of the animal’s organism will determine the development of mammary glands and the formation of teats, such as growth hormone, prolactin, estrogen, etc [42–44].
In this study, we collected far more the born teat number traits records than the functional teat number traits due to the varying ease of phenotype collection. Although the genetic correlation between the functional teat number traits and the born teat number traits suggests a similar mechanism of inheritance for both traits, the heritability of the traits in the shared traits population also implies that it may have genetic differences between the born teat number traits and the functional teat number traits.
For the TNN trait, we identified SULF1, SLCO5A1 and NCOA2 candidate genes on chromosome 4. It has been shown that these genes are the candidate genes of rib number trait in pigs, and the number of ribs determines the body length of pigs [45]. Therefore, SULF1, SLCO5A1, and NCOA2 genes may be involved in the growth and development process of pigs, which affects the number of ribs and increases the body length, thereby favoring the increase in teat number. After conducting GWAS based on SNP for TTN, RTN and LTN traits, we found that these results co-localized with the HEG1 gene. Interestingly, we also identified this gene in an InDel-based GWAS for the RTN trait. HEG1 has previously been reported to be involved in the regulation of the Wnt signaling pathway [46], which is closely associated with mammary gland development. Moreover, HEG1 is enriched in epithelium development, and teat development is also similarly regulated by this type of pathway [47]. And we found variant typing within the HEG1 region in chip data from another Yorkshire sow population and when the locus in this region was mutated, the number of teats in the population was significantly higher than in sows that were not mutated. Therefore, the HEG1 gene may be a key candidate gene affecting the pig teat number trait, and its specific molecular mechanism needs to be further explored.
It has been shown that combining SNP, InDel and SV explains higher heritability of traits and improves the power to identify genetic factors underlying complex traits [16]. In this study, we co-located a candidate gene XYLT1 by GWAS based on SNP and SV in the LTN trait. This gene has been associated with systemic wrinkle in the aromatic pig in previous studies [48], and we hypothesized that it is likely that this gene also affects embryonic teat formation; it has also been suggested that XYLT1 is a key regulator of bone length [49], and considering that many proteins that regulate mammary gland development have dual roles in regulating both mammary gland development and bone development [50], we suggest that XYLT1 is a key candidate gene associated with the pig teat number trait. We also identified the ACP1 gene, which has been reported to be involved in growth factor signaling in mammary gland development and is associated with inverted teats in pig [51]. The AHCTF1 gene determines live piglet production traits [52], and a high number of teats prevents piglets from competing for teats due to insufficient teats. CTBP1 has previously been reported to be involved in mammary gland development in mice [53]. TRIML1, TRIML2 and ZFP42 genes were identified in a study of teat number in local pigs [54]. RPS15A was screened by two variants together, and it was reported that this gene is closely related to mammary gland development and is also expressed in porcine mammary tissue [55].
Functional teat number is a key factor in determining the lactation power of sows. In the GWAS for FTTN, we did not screen for significant genetic markers. There are two possible reasons for this: on the one hand, the phenotype may have a skewed in distribution due to the small sample size (N = 427), and on the other hand, it may be that the trait of functional teat number itself is more sensitive to the environment, leading to the mixing of noise in the process of performing the GWAS. However, we found a clear peak trend on chromosome 7 in the FRTN analysis. We also identified the VRTN gene, which has been repeatedly reported to be associated with pig teat number [11, 56]. On effective teat number, we also identified the HEG1 gene, which is not only involved in the Wnt signaling pathway, but also associated with cardiovascular development [46, 57]. Therefore, we speculate that the HEG1 gene may play a role in the vascularization of the mammary gland, which affects the vascularization, and when the mammary gland is underdeveloped, the phenomenon of blind teat and inverted teat occurs in production, which indirectly contributes to the number of effective teats.
Among other genes of our interest, RAP1A is essential for epithelial acinar structure and lumen formation in the human breast [58], and it is required for angiogenesis [59]. NPVF is a candidate gene capable of regulating reproduction, which could regulate breeding season of mare [60], and it could also regulate hypothalamic hormone expression. So, we considered that NPVF is a new candidate gene of functional teat number traits. The SLC4A7 gene has been previously reported to be a good candidate for the intramuscular fat content of the Beijing black pig [61], which determines fat deposition in cattle and is associated with backfat thickness [62]. Milk is rich in nutrients, and regulation of SLC4A7 gene would improve milk composition and contribute to the health of lactating piglets. The CHIA gene is highly expressed in udder tissues, which is a favorable condition for it to be a key candidate gene. FLVCR2 has not only been reported to be associated with porcine teat number, but also as a candidate gene for porcine rib number [63, 64]. TGFB3 expression plays a role in the growth and development of follicular membrane cell and granulosa cell growth and development, regulates the proliferative state and cholesterol homeostasis of the developing mammary gland in infancy, and also determines embryonic gonadal development, and has also been reported in breast cancer studies [65–67]. Nonetheless, we did not explore the effect of sex chromosomes on functional teat number traits, and we will delve into the trait of sex chromosomes on teat number.
Conclusion
We performed a GWAS based on the teat number trait using three genetic variants, SNP, InDel and SV, and screened candidate genes such as HEG1 and XYLT1. For the number of functional teats, we proposed two new genes related to the number of sows’ teats, RAP1A and NPVF. These genes provide theoretical references and new targets for molecular marker breeding of high-breeding sows.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Not applicable.
Author contributions
TY, GY and JS conceived and designed the experiments. JW, JS, YW, MC and RD performed the data analysis. YP, JW and RD edited and revised the manuscript. YP, MC and RH collected the phenotypes. XD provided validation population. YP, AG, TY and RD prepared the figures, and tables. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (32202629), the National Key Research and Development Plan of China (2021YFD1301200), the Shaanxi Province Key R&D Plan (S2024-YF-YBNY-0752) and the Shaanxi Livestock and Poultry Breeding Double-chain Fusion Key Project (2022GD-TSLD-46).
Data availability
The variation data analysed in this paper have been deposited in the European Variation Archive (EVA) at EMBL-EBI under accession number PRJEB82721.
Declarations
Ethics approval and consent to participate
The study was approved by the Institutional Animal Care and Use Committee of the Northwest A & F University (approval number: NWAFU-314021167). All experiments strictly followed the guidelines of this committee. The experimental animals were not anesthetized or euthanized in this study. All experimental protocols were approved by the aforementioned committee and in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jialin Wei and Jingchun Sun authors who contributed equally to this study.
References
- 1.Hirooka H, de Koning DJ, Harlizius B, van Arendonk JA, Rattink AP, Groenen MA, Brascamp EW, Bovenhuis H. A whole-genome scan for quantitative trait loci affecting teat number in pigs. J Anim Sci. 2001;79(9):2320–6. [DOI] [PubMed] [Google Scholar]
- 2.Trost LS, Zeidler S, Ammer S, Rosengart S, Wendt M, Visscher C, Tetens J, Traulsen I. Development of a new grading system to assess the foster performance of lactating sows. Animal. 2022;16(11):100655. [DOI] [PubMed] [Google Scholar]
- 3.Andersen IL, Naevdal E, Boe KE. Maternal investment, sibling competition, and offspring survival with increasing litter size and parity in pigs (Sus scrofa). Behav Ecol Sociobiol. 2011;65(6):1159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rohrer GA, Nonneman DJ. Genetic analysis of teat number in pigs reveals some developmental pathways independent of vertebra number and several loci which only affect a specific side. Genet Sel Evol. 2017;49(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li T, Wan P, Lin Q, Wei C, Guo K, Li X, Lu Y, Zhang Z, Li J. Genome-Wide Association Study Meta-Analysis Elucidates Genetic Structure and identifies candidate genes of Teat Number traits in pigs. Int J Mol Sci 2023, 25(1). [DOI] [PMC free article] [PubMed]
- 6.Pan Z, Yao Y, Yin H, Cai Z, Wang Y, Bai L, Kern C, Halstead M, Chanthavixay G, Trakooljul N, et al. Pig genome functional annotation enhances the biological interpretation of complex traits and human disease. Nat Commun. 2021;12(1):5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan G, Liu X, Xiao S, Xin W, Xu W, Li Y, Huang T, Qin J, Xie L, Ma J, et al. An imputed whole-genome sequence-based GWAS approach pinpoints causal mutations for complex traits in a specific swine population. Sci China Life Sci. 2022;65(4):781–94. [DOI] [PubMed] [Google Scholar]
- 8.Zheng X, Zhao P, Yang K, Ning C, Wang H, Zhou L, Liu J. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J Anim Sci Biotechnol. 2020;11:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang F, Li J, Guo M, Mei Q, Yu M, Liu H, Legarra A, Xiang T. Genomic evaluation and genome-wide association studies for total number of teats in a combined American and Danish Yorkshire pig populations selected in China. J Anim Sci 2022, 100(7). [DOI] [PMC free article] [PubMed]
- 10.Yang J, Huang L, Yang M, Fan Y, Li L, Fang S, Deng W, Cui L, Zhang Z, Ai H, et al. Possible introgression of the VRTN mutation increasing vertebral number, carcass length and teat number from Chinese pigs into European pigs. Sci Rep. 2016;6:19240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moscatelli G, Dall’Olio S, Bovo S, Schiavo G, Kazemi H, Ribani A, Zambonelli P, Tinarelli S, Gallo M, Bertolini F, et al. Genome-wide association studies for the number of teats and teat asymmetry patterns in large White pigs. Anim Genet. 2020;51(4):595–600. [DOI] [PubMed] [Google Scholar]
- 12.Yang L, Li X, Zhuang Z, Zhou S, Wu J, Xu C, Ruan D, Qiu Y, Zhao H, Zheng E et al. Genome-Wide Association Study Identifies the Crucial Candidate Genes for Teat Number in Crossbred Commercial Pigs. Animals (Basel) 2023, 13(11). [DOI] [PMC free article] [PubMed]
- 13.Roarty K, Serra R. Wnt5a is required for proper mammary gland development and TGF-beta-mediated inhibition of ductal growth. Development. 2007;134(21):3929–39. [DOI] [PubMed] [Google Scholar]
- 14.Tumpel S, Sanz-Ezquerro JJ, Isaac A, Eblaghie MC, Dobson J, Tickle C. Regulation of Tbx3 expression by anteroposterior signalling in vertebrate limb development. Dev Biol. 2002;250(2):251–62. [PubMed] [Google Scholar]
- 15.Mailleux AA, Spencer-Dene B, Dillon C, Ndiaye D, Savona-Baron C, Itoh N, Kato S, Dickson C, Thiery JP, Bellusci S. Role of FGF10/FGFR2b signaling during mammary gland development in the mouse embryo. Development. 2002;129(1):53–60. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Y, Zhang Z, Bao Z, Li H, Lyu Y, Zan Y, Wu Y, Cheng L, Fang Y, Wu K, et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature. 2022;606(7914):527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaj I, Tetens J, Bennewitz J, Thaller G, Falker-Gieske C. Structural variants and tandem repeats in the founder individuals of four F(2) pig crosses and implications to F(2) GWAS results. BMC Genomics. 2022;23(1):631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zong W, Wang J, Zhao R, Niu N, Su Y, Hu Z, Liu X, Hou X, Wang L, Wang L, et al. Associations of genome-wide structural variations with phenotypic differences in cross-bred eurasian pigs. J Anim Sci Biotechnol. 2023;14(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao C, Su KJ, Wu C, Cao X, Sha Q, Li W, Luo Z, Qin T, Qiu C, Zhao LJ et al. Multi-View Variational Autoencoder for Missing Value Imputation in Untargeted Metabolomics. ArXiv 2024. [DOI] [PMC free article] [PubMed]
- 22.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, Saunders CT. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–2. [DOI] [PubMed] [Google Scholar]
- 23.Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisfeldt J, Vezzi F, Olason P, Nilsson D, Lindstrand A. TIDDIT, an efficient and comprehensive structural variant caller for massive parallel sequencing data. F1000Res. 2017;6:664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6(9):677–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, Balloux F, Dessimoz C, Bahler J, Sedlazeck FJ. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun. 2017;8:14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen B. Smoove: structural variant calling and genotyping with existing tools, but, smoothly. In.; 2018.
- 28.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Zhang Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genomics Proteom Bioinf. 2021;19(4):629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin L, Zhang H, Tang Z, Xu J, Yin D, Zhang Z, Yuan X, Zhu M, Zhao S, Li X, et al. rMVP: a Memory-efficient, Visualization-enhanced, and parallel-accelerated Tool for Genome-wide Association study. Genomics Proteom Bioinf. 2021;19(4):619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolberg L, Raudvere U, Kuzmin I, Adler P, Vilo J, Peterson H. G:profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023;51(W1):W207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong SS, He WM, Ji JJ, Zhang C, Guo Y, Yang TL. LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief Bioinform 2021, 22(4). [DOI] [PubMed]
- 35.Duijvesteijn N, Veltmaat JM, Knol EF, Harlizius B. High-resolution association mapping of number of teats in pigs reveals regions controlling vertebral development. BMC Genomics. 2014;15(1):542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pumfrey RA, Johnson RK, Cunningham PJ, Zimmerman DR. Inheritance of teat number and its relationship to maternal traits in swine. J Anim Sci. 1980;50(6):1057–60. [DOI] [PubMed] [Google Scholar]
- 37.Kim JS, Jin DI, Lee JH, Son DS, Lee SH, Yi YJ, Park CS. Effects of teat number on litter size in gilts. Anim Reprod Sci. 2005;90(1–2):111–6. [DOI] [PubMed] [Google Scholar]
- 38.Bidanel JP, Rosendo A, Iannuccelli N, Riquet J, Gilbert H, Caritez JC, Billon Y, Amigues Y, Prunier A, Milan D. Detection of quantitative trait loci for teat number and female reproductive traits in Meishan x large White F2 pigs. Animal. 2008;2(6):813–20. [DOI] [PubMed] [Google Scholar]
- 39.Zaalberg RM, Chu TT, Bovbjerg H, Jensen J, Villumsen TM. Genetic parameters for early piglet weight, litter traits and number of functional teats in organic pigs. Animal. 2023;17(3):100717. [DOI] [PubMed] [Google Scholar]
- 40.Rangstrup-Christensen L, Schild SA, Pedersen LJ, Sorensen JT. Causes of preweaning mortality in organic outdoor sow herds. Res Vet Sci. 2018;118:171–80. [DOI] [PubMed] [Google Scholar]
- 41.Martins TF, Braga Magalhaes AF, Verardo LL, Santos GC, Silva Fernandes AA, Gomes Vieira JI, Irano N, Dos Santos DB. Functional analysis of litter size and number of teats in pigs: from GWAS to post-GWAS. Theriogenology. 2022;193:157–66. [DOI] [PubMed] [Google Scholar]
- 42.Chomwisarutkun K, Murani E, Ponsuksili S, Wimmers K. Gene expression analysis of mammary tissue during fetal bud formation and growth in two pig breeds–indications of prenatal initiation of postnatal phenotypic differences. BMC Dev Biol. 2012;12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H, Fan M, Chen X, Jiang X, Loor JJ, Aboragah A, Zhang C, Bai H, Fang Z, Shen T, et al. Activated autophagy-lysosomal pathway in dairy cows with hyperketonemia is associated with lipolysis of adipose tissues. J Dairy Sci. 2022;105(8):6997–7010. [DOI] [PubMed] [Google Scholar]
- 44.Robinson GW. Cooperation of signalling pathways in embryonic mammary gland development. Nat Rev Genet. 2007;8(12):963–72. [DOI] [PubMed] [Google Scholar]
- 45.Liu K, Hou L, Yin Y, Wang B, Liu C, Zhou W, Niu P, Li Q, Huang R, Li P. Genome-wide association study reveals new QTL and functional candidate genes for the number of ribs and carcass length in pigs. Anim Genet. 2023;54(4):435–45. [DOI] [PubMed] [Google Scholar]
- 46.Zhao YR, Wang JL, Xu C, Li YM, Sun B, Yang LY. HEG1 indicates poor prognosis and promotes hepatocellular carcinoma invasion, metastasis, and EMT by activating Wnt/beta-catenin signaling. Clin Sci (Lond). 2019;133(14):1645–62. [DOI] [PubMed] [Google Scholar]
- 47.Spina E, Cowin P. Embryonic mammary gland development. Semin Cell Dev Biol. 2021;114:83–92. [DOI] [PubMed] [Google Scholar]
- 48.Xiaoli L, Fengbin H, Shihui H, Xi N, Sheng L, Zhou W, Xueqin R, Jiafu W. Detection of genomic structure variants associated with wrinkled skin in Xiang pig by next generation sequencing. Aging. 2021;13(22):24710–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mis EK, Liem KF Jr., Kong Y, Schwartz NB, Domowicz M, Weatherbee SD. Forward genetics defines Xylt1 as a key, conserved regulator of early chondrocyte maturation and skeletal length. Dev Biol. 2014;385(1):67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wysolmerski JJ. The evolutionary origins of maternal calcium and bone metabolism during lactation. J Mammary Gland Biol Neoplasia. 2002;7(3):267–76. [DOI] [PubMed] [Google Scholar]
- 51.Chomwisarutkun K, Murani E, Ponsuksili S, Wimmers K. Microarray analysis reveals genes and functional networks relevant to the predisposition to inverted teats in pigs. J Anim Sci. 2012;90(1):1–15. [DOI] [PubMed] [Google Scholar]
- 52.Bergfelder-Druing S, Grosse-Brinkhaus C, Lind B, Erbe M, Schellander K, Simianer H, Tholen E. A genome-wide association study in large white and landrace pig populations for number piglets born alive. PLoS ONE. 2015;10(3):e0117468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van de Moosdijk AA, van Amerongen R. Identification of reliable reference genes for qRT-PCR studies of the developing mouse mammary gland. Sci Rep. 2016;6:35595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z, Li H, Zhong Z, Jiang S. A whole genome sequencing-based genome-wide Association Study reveals the potential associations of Teat Number in Qingping pigs. Anim (Basel) 2022, 12(9). [DOI] [PMC free article] [PubMed]
- 55.Piantoni P, Bionaz M, Graugnard DE, Daniels KM, Akers RM, Loor JJ. Gene expression ratio stability evaluation in prepubertal bovine mammary tissue from calves fed different milk replacers reveals novel internal controls for quantitative polymerase chain reaction. J Nutr. 2008;138(6):1158–64. [DOI] [PubMed] [Google Scholar]
- 56.Duan Y, Zhang H, Zhang Z, Gao J, Yang J, Wu Z, Fan Y, Xing Y, Li L, Xiao S, et al. VRTN is required for the development of thoracic vertebrae in mammals. Int J Biol Sci. 2018;14(6):667–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Kreuk BJ, Gingras AR, Knight JD, Liu JJ, Gingras AC, Ginsberg MH. Heart of glass anchors Rasip1 at endothelial cell-cell junctions to support vascular integrity. Elife. 2016;5:e11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Itoh M, Nelson CM, Myers CA, Bissell MJ. Rap1 integrates tissue polarity, lumen formation, and tumorigenic potential in human breast epithelial cells. Cancer Res. 2007;67(10):4759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan J, Li F, Ingram DA, Quilliam LA. Rap1a is a key regulator of fibroblast growth factor 2-induced angiogenesis and together with Rap1b controls human endothelial cell functions. Mol Cell Biol. 2008;28(18):5803–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thorson JF, Prezotto LD, Cardoso RC, Sharpton SM, Edwards JF, Welsh TH Jr., Riggs PK, Caraty A, Amstalden M, Williams GL. Hypothalamic distribution, adenohypophyseal receptor expression, and ligand functionality of RFamide-related peptide 3 in the mare during the breeding and nonbreeding seasons. Biol Reprod. 2014;90(2):28. [DOI] [PubMed] [Google Scholar]
- 61.Yang M, Zhang R, Liu X, Shi G, Liu H, Wang L, Hou X, Shi L, Wang L, Zhang L. Integrating genome-wide association study with RNA-seq revealed DBI as a good candidate gene for intramuscular fat content in Beijing black pigs. Anim Genet. 2023;54(1):24–34. [DOI] [PubMed] [Google Scholar]
- 62.Martins R, Machado PC, Pinto LFB, Silva MR, Schenkel FS, Brito LF, Pedrosa VB. Genome-wide association study and pathway analysis for fat deposition traits in nellore cattle raised in pasture-based systems. J Anim Breed Genet. 2021;138(3):360–78. [DOI] [PubMed] [Google Scholar]
- 63.Verardo LL, Lopes MS, Wijga S, Madsen O, Silva FF, Groenen MA, Knol EF, Lopes PS, Guimaraes SE. After genome-wide association studies: gene networks elucidating candidate genes divergences for number of teats across two pig populations. J Anim Sci. 2016;94(4):1446–58. [DOI] [PubMed] [Google Scholar]
- 64.Zhang LC, Yue JW, Pu L, Wang LG, Liu X, Liang J, Yan H, Zhao KB, Li N, Shi HB, et al. Genome-wide study refines the quantitative trait locus for number of ribs in a large White x Minzhu intercross pig population and reveals a new candidate gene. Mol Genet Genomics. 2016;291(5):1885–90. [DOI] [PubMed] [Google Scholar]
- 65.Lin H, Chaudhury M, Sharma N, Bhattacharyya S, Elolimy AA, Yeruva L, Ronis MJJ, Mercer KE. MicroRNA profiles were altered in neonatal piglet mammary glands following postnatal infant formula feeding. J Nutr Biochem. 2020;83:108397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Memon MA, Anway MD, Covert TR, Uzumcu M, Skinner MK. Transforming growth factor beta (TGFbeta1, TGFbeta2 and TGFbeta3) null-mutant phenotypes in embryonic gonadal development. Mol Cell Endocrinol. 2008;294(1–2):70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen C, Zhao KN, Masci PP, Lakhani SR, Antonsson A, Simpson PT, Vitetta L. TGFbeta isoforms and receptors mRNA expression in breast tumours: prognostic value and clinical implications. BMC Cancer. 2015;15:1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The variation data analysed in this paper have been deposited in the European Variation Archive (EVA) at EMBL-EBI under accession number PRJEB82721.