

Abstract
Denosumab is a human IgG2 monoclonal antibody against receptor activator of nuclear factor kappa‐B ligand (RANKL) for the treatment of osteoporosis and bone loss. HLX14 is a proposed biosimilar of denosumab. This randomized, parallel‐group, two‐part, phase I study aimed to compare the pharmacokinetics, pharmacodynamics, safety, and immunogenicity of HLX14 with reference denosumab in Chinese healthy adult male participants. In Part 1, participants were randomized 1:1 and given HLX14 or reference denosumab sourced from the European Union (EU). In double‐blind Part 2, participants were randomized 1:1:1:1 to receive HLX14 or denosumab sourced from the United States, EU, or China. All study drugs were administered via subcutaneous injection at a single dose of 60 mg. The primary endpoints were area under the serum drug concentration–time curve from time 0 to the last concentration‐quantifiable time t (AUC0–t), maximum serum drug concentration (C max), and area under the serum drug concentration–time curve from time 0 to infinity (AUC0–inf). Twenty‐four participants were randomized in Part 1 and 228 in Part 2. The 90% confidence intervals of geometric mean ratio of AUC0–t , C max, and AUC0–inf between HLX14 and denosumab from different sources fell within the pre‐specified similarity margins of 0.80–1.25 (AUC0–t , 0.91–1.13; C max, 0.91–1.13; AUC0–inf, 0.91–1.12), demonstrating pharmacokinetic similarity. No notable difference was observed among treatment groups in pharmacodynamics, safety, or immunogenicity. HLX14 demonstrated highly similar pharmacokinetic characteristics with comparable pharmacodynamics, safety, and immunogenicity to denosumab, supporting its further investigation as a potential denosumab biosimilar.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Denosumab is a human IgG2 monoclonal antibody against receptor activator of nuclear factor kappa‐B ligand for the treatment of osteoporosis and bone loss. HLX14 is a proposed biosimilar of denosumab.
WHAT QUESTION DID THIS STUDY ADDRESS?
This randomized, parallel‐group, two‐part, phase I study aimed to compare the pharmacokinetics, pharmacodynamics, safety, and immunogenicity of HLX14 with reference denosumab in Chinese healthy adult male participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
HLX14 demonstrated highly similar pharmacokinetic characteristics with comparable pharmacodynamics, safety, and immunogenicity to denosumab.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
HLX14 has shown promise as a potential denosumab biosimilar, warranting further investigation.
INTRODUCTION
Osteoporosis is a bone disease characterized by the decrease of bone mineral density and the loss of bone mass, as well as changes in the structure and strength of the bones. 1 It mainly affects postmenopausal women and older men, leading to fragility fractures most frequently in the thoracic and lumbar spine, wrist, and hip. 2 The global prevalence of osteoporosis in population aged >50 years was 26.0% in women (27.4% in postmenopausal) and 11.2% in men, representing a major health issue. 3
For those who achieve inadequate improvement with lifestyle and dietary measures for the management of osteoporosis, pharmacological treatment is highly recommended. Anti‐resorptive drugs are usually the first‐line treatment, among which bisphosphonates (e.g., oral alendronate and intravenous zoledronate) and monoclonal antibodies against receptor activator of nuclear factor kappa‐B ligand (RANKL, e.g., denosumab) are the most widely used. 4 , 5 , 6 , 7 Other treatment options include anabolic drugs (e.g., parathyroid hormone analogs), and sclerostin inhibitors (e.g., romosozumab); both classes of these drugs are required to be followed by anti‐resorptive treatment. 4 , 5 , 6 , 8
Oral bisphosphonates are known to cause upper gastrointestinal symptoms in 20%–30% of individuals and thus are contraindicated in those with active upper gastrointestinal disease. 8 Intravenous bisphosphonates avoid such adverse events, yet produce an acute‐phase flu‐like illness in around 1/3 of patients after the first dose, with symptoms that can be severe in 2%–3% of patients. 8 Additionally, use of bisphosphonates is restricted in individuals with severe renal impairment. 4 , 5 , 6 , 8 RANKL inhibitors, however, have a generally better safety profile. 4 , 5 , 6 , 8 RANKL is a transmembrane ligand belonging to the tumor necrosis factor superfamily; the inhibition of RANKL/RANK signaling downregulates osteoclast differentiation and activity, thus reducing bone resorption and increasing bone density. 9 The first RANKL inhibitor, denosumab (Prolia®), was approved by the European Medicines Agency in 2010, 10 by the United States (US) Food and Drug Administration (FDA) shortly thereafter, 11 and then by the China (CN) National Medical Products Administration (NMPA) in 2020. 12 A meta‐analysis has shown that denosumab could significantly improve bone mineral density more than bisphosphonate at the lumbar spine, total hip, and femoral neck. 7 Currently, denosumab is approved for the treatment of postmenopausal women and men with osteoporosis at high risk for fracture in the European Union (EU), the US, and CN. 13 , 14 , 15 Additional indications for the treatment of bone loss at increased risk of fractures associated with hormone ablation in men with prostate cancer, or associated with long‐term systemic glucocorticoid therapy in women and men are approved in the EU and the US. 13 , 14 The US FDA also approved denosumab in women at high risk for fracture receiving aromatase inhibitor for breast cancer. 14
Considering the increase in the prevalence of osteoporosis due to a global aging population, the development of new treatment options is important in managing osteoporosis and improving the quality of life in the elderly. Biosimilars, biological products that are highly similar to licensed reference biologics, can potentially increase accessibility to essential medications. HLX14 is a recombinant, fully human, anti‐RANKL monoclonal IgG2 antibody developed by Shanghai Henlius Biotech, Inc., and is a proposed biosimilar of denosumab. 16 Preclinical studies of HLX14 showed comparable pharmacokinetic (PK), pharmacodynamic (PD), and safety profiles with the reference denosumab. A two‐part phase I clinical study was conducted to compare the PK and PD characteristics, as well as safety and immunogenicity of HLX14 with those of reference denosumab sourced from the US, the EU, and CN in Chinese healthy adult male participants. Results from Part 1 were presented at the American Society for Clinical Pharmacology and Therapeutics 2022 annual meeting, 16 and the full results are herein reported.
METHODS
Study design and treatment
This phase I clinical trial was conducted from November 3, 2020 to September 12, 2023 and comprised two parts. Part 1 was a single‐center, open‐label, randomized, pilot study aiming to compare the PK, PD, safety, and immunogenicity of HLX14 with EU‐denosumab. Part 2 of the study was conducted at three hospitals and was a double‐blind, randomized, four‐arm study comparing the PK, PD, safety, and immunogenicity among HLX14, US‐denosumab, EU‐denosumab, and CN‐denosumab.
In Part 2 of this study, a single dose of 60 mg of HLX14, US‐denosumab, EU‐denosumab, or CN‐denosumab was administered via subcutaneous injection on day 1. Participants were required to take 600 mg of calcium and 400 IU of vitamin D daily after meals during the study (day 1–134), the doses of which could be adjusted by the investigators based on serum calcium levels. The study was initiated on the day of signing the informed consent form and was completed on day 183 (for Part 1) or day 274 (for Part 2) after dosing, or by the time of subject's withdrawal from the study, whichever occurred first.
The study was conducted in accordance with the International Council for Harmonization guidelines on Good Clinical Practice and all applicable regulatory requirements, including the Declaration of Helsinki, and was approved by the ethics committees at each participating center. Each subject provided written, informed consent prior to enrollment in the study.
Subjects
Eligible participants were Chinese males aged ≥18 and ≤65 years (for Part 1) or aged >28 and ≤65 years (for Part 2), and weighed ≥50 kg with a body mass index (BMI) of 19–26 kg/m2. Participants with no disease histories, or with a disease history that does not affect the study, and no abnormality in physical examinations, vital signs, electrocardiogram, chest imaging, or laboratory tests were enrolled. Key exclusion criteria included a history of allergy to the study drugs, calcium, and/or vitamin D; previously or currently suffering from jaw osteomyelitis or osteonecrosis of the jaw; fracture, or bone‐related surgery within 6 months prior to screening; abnormal serum calcium levels during screening; and being positive for tobacco test. Full eligibility criteria can be found in the Appendix S1.
Randomization and masking
In Part 1, participants were randomized at a 1:1 ratio. In Part 2, participants were randomized at a 1:1:1:1 ratio; randomization was stratified by weight (≤65 kg and >65 kg), and was performed using an interactive web response system with unique random numbers.
Part 1 employed an open‐label design. In Part 2, the investigators, relevant study personnel, and participants were masked to study allocation. The whole dosing procedure was performed by an unblinded nurse to ensure that all other relevant personnel remained blinded.
Study endpoints
The primary endpoints (i.e., the primary PK parameters) were the area under the serum drug concentration–time curve from time 0 to the last concentration‐quantifiable time t (AUC0–t ), maximum serum drug concentration (C max), and area under the serum drug concentration–time curve from time 0 to infinity (AUC0–inf). Secondary endpoints were secondary PK parameters, PD parameters of serum C‐terminal telopeptide of type I collagen (s‐CTX, a bone resorption marker 17 ), safety profile, and immunogenicity.
The following secondary PK parameters were calculated in this study: time to reach maximum serum drug concentration (T max), total clearance (CL/F), apparent terminal elimination rate constant (λ Z), elimination half‐life (t 1/2), apparent volume of distribution (V d/F), area extrapolated from time t to infinity as a percentage of total AUC0–inf (%AUCex), mean residence time (MRT), area under the serum drug concentration–time curve from day 0 to day 28 (AUC0–28d), and area under the serum drug concentration–time curve from day 0 to day 112 (AUC0–112d). PD parameters included in the study were area under the effect–time curve from time 0 to the last concentration‐quantifiable time t of s‐CTX (AUEC0–t ), minimum observed concentration of s‐CTX (I min), maximum percent inhibition of s‐CTX (I max), and time to reach I min of s‐CTX (T min).
Assessments
Sampling timepoints for PK, PD, and immunogenicity assessments are provided in the Appendix S1.
The serum concentrations of HLX14 and denosumab were determined by a validated enzyme‐linked immunosorbent assay at WuXi AppTec (Shanghai) Co., Ltd. (Shanghai, China) for samples collected in Part 1, and at Shanghai Jollin Lab Co., Ltd. (Shanghai, China) for Part 2 samples.
The serum concentrations of s‐CTX were analyzed with β‐CrossLaps/serum Elecsys cobas e 100 test kit (cat# 11972308122) on Roche cobas® 6000 e601 immunoassay analyzer (Roche Diagnostics, IN, USA) at WuXi AppTec (Shanghai) Co., Ltd.
Only anti‐drug antibody (ADA)‐positive samples were further tested for neutralizing antibodies (NAbs). In Part 1, ADAs were assessed by a validated electrochemiluminescence immunoassay, and NAbs were detected by a validated functional cell‐based assay, both at WuXi AppTec (Shanghai) Co., Ltd. In Part 2, both ADAs and NAbs were assessed by validated electrochemiluminescence immunoassays at Shanghai Jollin Lab Co., Ltd.
Details of the analytical methods for PK, PD, and immunogenicity are provided in the Appendix S1.
Adverse events (AEs) were monitored during the study. After the last follow‐up visit, only serious AEs related to the study drugs were recorded. AEs were coded by Medical Dictionary for Regulatory Activities version 26.1, and graded per National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0. AEs of special interest (AESIs) in this study included hypersensitivity reactions, hypocalcemia, serious infections (including skin infections), osteonecrosis of the jaw, and atypical femur fracture.
Statistical analysis
A total of 24 participants were planned for Part 1, with 12 participants in each group. The sample size of Part 2 was recalculated based on the results from Part 1. In Part 1, the maximum coefficient of variation (CV) of the three primary endpoints (AUC0–t , C max, and AUC0–inf) was approximately 28%. Assuming the actual geometric mean ratio (GMR) of HLX14 to denosumab for the primary endpoints being 0.98, the significance level being α = 0.05 (two one‐tailed tests), the power for each primary endpoint of a single between‐group comparison being 97%, the power of a single between‐group comparison being above 91%, and the similarity margins for the primary endpoints being 80%–125%, 48 evaluable participants were needed in each treatment group. Considering that PK might not be evaluable in about 15% of participants, a total of 228 participants (57 per group) were planned to be enrolled in Part 2.
The definition of population sets used in this study is provided in the Appendix S1. For Part 2 of the study, PK similarity analysis was performed. The primary endpoints (AUC0–t , C max, and AUC0–inf) were log‐transformed and analyzed by analysis of variance (ANOVA). The factor in the ANOVA analysis was the treatment group. Least squares mean (LSM) and their 90% confidence intervals (CIs) were calculated for each treatment group. These point estimates and CIs were then exponentially back‐transformed to obtain GMR and the associated 90% CIs. If the 90% CIs of GMR of AUC0–t , C max, and AUC0–inf fell within the range of 0.80–1.25, PK similarity was demonstrated between the two treatment groups.
All statistical analyses were performed using SAS® software, version 9.4 or later (SAS Institute, NC, USA). The PK and PD parameters were calculated using Phoenix WinNonlin, version 8.2 (Certara, NJ, USA) with a non‐compartmental analysis.
Ethics statement
The study was conducted in accordance with the International Council for Harmonization guidelines on Good Clinical Practice and all applicable regulatory requirements, including the Declaration of Helsinki, and was approved by the ethics committees at each participating center.
RESULTS
Subjects and exposure
In Part 1, 155 healthy adult male participants were screened, of which 24 were enrolled and randomized to the HLX14 group (n = 12) and the EU‐denosumab group (n = 12) (Figure S1). All participants had completed the study and were included in all analysis sets (Table S1). In Part 2, a total of 1030 participants were screened, among whom 228 were enrolled and randomized to the HLX14 group (n = 58), the US‐denosumab group (n = 57), the EU‐denosumab group (n = 56), and the CN‐denosumab group (n = 57), comprising the full analysis set (FAS) (Figure 1). Details of reasons for screen failure were provided in Table S2. All participants received study treatment and were included in the safety set (SS) (Table S1). Three (1.3%) participants were excluded from the PK parameter set (PKPS), one (0.4%) due to pre‐dose serum drug concentration being >5% of C max, and the other two (0.9%) due to premature drop‐out before reaching C max. Two (0.9%) participants were excluded from the PD parameter set (PDPS) due to premature drop‐out before reaching I min. All participants were included in the PK concentration set (PKCS) and the PD concentration set (PDCS). 213 (93.4%) participants had completed Part 2 of the study (Figure 1); the reasons for discontinuation from the study were participant decision (seven [3.1%] participants), poor compliance (six [2.6%]), and loss to follow‐up (two [0.9%]).
FIGURE 1.
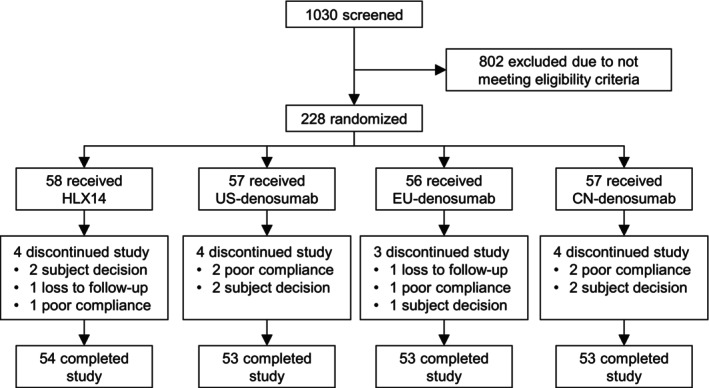
Subject disposition in Part 2 of the study. CN, China; EU, European Union; US, United States.
Demographic and baseline characteristics were well balanced between the two treatment groups in Part 1 (Table S3), as well as among the four treatment groups in Part 2 (Table 1). The actual exposures to HLX14 or denosumab in all treatment groups in Part 1 and Part 2 were 60 mg. Exposures to calcium and vitamin D during the study were comparable among groups (Table S4).
TABLE 1.
Demographics and baseline characteristics in Part 2 (FAS).
| HLX14 (n = 58) | US‐denosumab (n = 57) | EU‐denosumab (n = 56) | CN‐denosumab (n = 57) | |
|---|---|---|---|---|
| Median age, years (range) | 34.0 (28–48) | 34.0 (28–53) | 33.0 (28–50) | 34.0 (28–47) |
| Asian, n (%) | 58 (100) | 57 (100) | 56 (100) | 57 (100) |
| Median height, cm (range) | 169.1 (156.7–184.2) | 168.3 (150.6–182.7) | 168.6 (154.7–188) | 168.2 (151.5–178.3) |
| Median weight, kg (range) | 65.2 (53–77.8) | 65.1 (50.8–81.9) | 65.2 (56.9–83.8) | 65.4 (50.3–81.7) |
| Median BMI, kg/m2 (range) | 22.6 (19.3–26) | 23.2 (19.3–25.7) | 23.1 (19.3–26) | 23.7 (19.6–26) |
| ADA‐positive, n (%) | 1 (1.7) | 1 (1.8) | 3 (5.4) | 2 (3.5) |
Abbreviations: ADA, anti‐drug antibody; BMI, body mass index; EU, European Union; FAS, full analysis set.
Pharmacokinetics
In the PKCS, serum concentrations of HLX14 and denosumab were comparable at all planned time points of PK sample collection both in Part 1 and Part 2 of the study, as demonstrated in the mean serum drug concentration–time curves (Figure 2a,b and Figure S2A,B). Descriptive statistics of the primary PK parameters in the PKPS showed similar AUC0–t , C max, and AUC0–inf among treatment groups (Table 2 and Table S5). In Part 1, the CV of the primary endpoint AUC0–t was 25.6% in the HLX14 group and 26.7% in the EU‐denosumab group. The CV of the other two primary endpoints C max and AUC0–inf was 16.1% versus 18.7% and 25.8% versus 28.4%, respectively. The maximum CV of the three primary endpoints in Part 1 (approximately 28%) was used for recalculating the sample size of Part 2. In Part 2, the geometric mean (geometric CV [CVb]) of AUC0–t in the HLX14, US‐, EU‐, and CN‐denosumab groups was 324.4 (20.8%), 332.6 (25.5%), 309.0 (25.2%), and 323.7 (21.9%) day·μg/mL, respectively. For C max and AUC0–inf, the geometric mean (CVb) was 6.0 (17.4%) versus 6.0 (24.1%) versus 5.6 (24.4%) versus 6.1 (23.7%) μg/mL, and 335.0 (20.5%) versus 344.9 (25.1%) versus 321.3 (24.4%) versus 336.2 (21.5%) day μg/mL. Descriptive statistics of the secondary PK parameters in the PKPS were also comparable among groups (Table 2 and Table S5).
FIGURE 2.
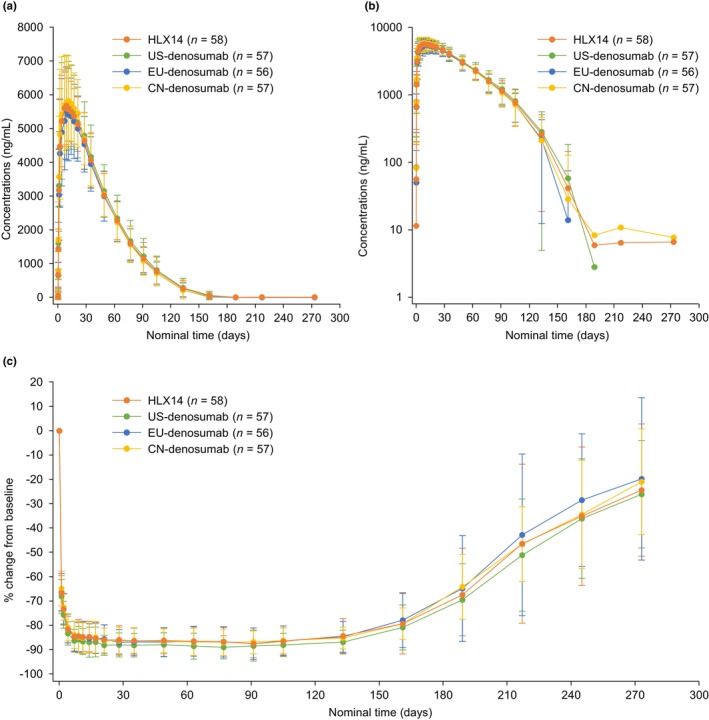
Pharmacokinetics and pharmacodynamics in Part 2. (a) Mean serum concentration–time profiles by treatment on a linear scale (PKCS). (b) Mean serum concentration–time profiles by treatment on a semi‐log scale (PKCS). (c) Mean percentage change from baseline in s‐CTX concentration (PDCS). Error bar represents standard deviation. PDCS, pharmacodynamic concentration set; PKCS, pharmacokinetic concentration set, s‐CTX, serum C‐terminal telopeptide of type I collagen.
TABLE 2.
Descriptive statistics of pharmacokinetic parameters in Part 2 (PKPS).
| Pharmacokinetic parameter | HLX14 (n = 57) | US‐denosumab (n = 56) | EU‐denosumab (n = 56) | CN‐denosumab (n = 56) |
|---|---|---|---|---|
| AUC0–t (day·μg/mL) | ||||
| Mean (SD) | 331.4 (72.3) | 343.0 (86.7) | 318.2 (76.5) | 331.2 (71.8) |
| GeoMean (CVb%) | 324.4 (20.8) | 332.6 (25.5) | 309.0 (25.2) | 323.7 (21.9) |
| C max (μg/mL) | ||||
| Mean (SD) | 6.0 (1.0) | 6.2 (1.4) | 5.8 (1.3) | 6.3 (1.5) |
| GeoMean (CVb%) | 6.0 (17.4) | 6.0 (24.1) | 5.6 (24.4) | 6.1 (23.7) |
| AUC0–inf (day·μg/mL) | ||||
| Mean (SD) | 342.1 (73.7) | 355.2 (87.9) | 330.3 (77.2) | 343.7 (73.4) |
| GeoMean (CVb%) | 335.0 (20.5) | 344.9 (25.1) | 321.3 (24.4) | 336.2 (21.5) |
| T max (day) | ||||
| Median (range) | 9.0 (2.0–22.0) | 11.0 (2.0–28.0) | 11.0 (2.0–28.0) | 9.1 (1.0–21.0) |
| CL/F (mL/day) | ||||
| Mean (SD) | 182.6 (35.4) | 179.4 (47.4) | 192.2 (48.3) | 182.5 (39.4) |
| GeoMean (CVb%) | 179.1 (20.5) | 174.0 (25.1) | 186.7 (24.4) | 178.5 (21.5) |
| λ z (1/day) | ||||
| Mean (SD) | 0.034 (0.008) | 0.031 (0.006) | 0.033 (0.007) | 0.033 (0.009) |
| GeoMean (CVb%) | 0.033 (21.7) | 0.031 (18.8) | 0.032 (20.8) | 0.032 (27.5) |
| t 1/2 (day) | ||||
| Mean (SD) | 21.5 (4.5) | 22.8 (4.3) | 22.2 (4.6) | 22.7 (6.9) |
| GeoMean (CVb%) | 21.0 (21.7) | 22.4 (18.8) | 21.7 (20.8) | 21.8 (27.5) |
| V d/F (L) | ||||
| Mean (SD) | 5.6 (1.3) | 5.8 (1.3) | 6.0 (1.6) | 5.8 (1.5) |
| GeoMean (CVb%) | 5.4 (22.3) | 5.6 (22.7) | 5.8 (26.1) | 5.6 (26.1) |
| %AUCex (%) | ||||
| Mean (SD) | 3.1 (1.7) | 3.5 (1.7) | 3.8 (2.3) | 3.7 (2.0) |
| GeoMean (CVb%) | 2.8 (53.2) | 3.1 (54.1) | 3.3 (58.5) | 3.2 (60.4) |
| MRT (day) | ||||
| Mean (SD) | 43.5 (6.3) | 44.6 (6.6) | 43.6 (5.4) | 43.3 (8.6) |
| GeoMean (CVb%) | 43.0 (15.1) | 44.1 (14.9) | 43.3 (12.6) | 42.7 (16.9) |
| AUC0–28d (day·μg/mL) | ||||
| Mean (SD) | 141.6 (24.5) | 145.0 (32.4) | 136.8 (30.9) | 147.9 (31.7) |
| GeoMean (CVb%) | 139.6 (17.2) | 141.3 (23.6) | 133.2 (24.5) | 144.6 (22.1) |
| AUC0–112d (day·μg/mL) | ||||
| Mean (SD) | 322.8 (62.5) | 333.1 (77.0) | 312.3 (69.4) | 323.8 (61.3) |
| GeoMean (CVb%) | 317.3 (18.5) | 324.6 (23.4) | 304.6 (23.3) | 318.1 (19.5) |
Note: Given that %AUCex of two participants in EU‐denosumab group and one in CN‐ denosumab group were greater than 20%, the PK parameters of AUC0–inf, AUC0–t , %AUCex, V d/F, CL/F, λ z, t 1/2, MRT and AUC0–112d were not included in summary.
Abbreviations: %AUCex, area extrapolated from time t to infinity as a percentage of total AUC0–inf; AUC0–112d, area under the serum drug concentration–time curve from day 0 to day 112; AUC0–28d, area under the serum drug concentration–time curve from day 0 to day 28; AUC0–inf, area under the serum drug concentration–time curve from time 0 to infinity; AUC0–t , area under the serum drug concentration–time curve from time 0 to the last concentration‐quantifiable time t; CL/F, total clearance; C max, maximum serum drug concentration; CN, China; CVb, geometric coefficient of variation; EU, European Union; GeoMean, geometric mean; MRT, mean residence time; PKPS, pharmacokinetic parameter set; SD, standard deviation; t 1/2, elimination half‐life; T max, time to reach maximum serum drug concentration; US, United States; V d/F, apparent volume of distribution; λ z apparent terminal elimination rate constant.
PK similarity analysis for Part 2 was conducted in the PKPS. After a single dose of subcutaneous injection, the GMR (90% CI) for AUC0–t between HLX14 versus US‐denosumab, HLX14 versus EU‐denosumab, and HLX14 versus CN‐denosumab was 0.98 (0.91–1.05), 1.05 (0.98–1.13), and 1.00 (0.94–1.07), respectively (Table 3). The GMR (90% CI) for C max was 0.99 (0.93–1.06), 1.05 (0.99–1.13), and 0.97 (0.91–1.04) for the respective pairs, and for AUC0–inf was 0.97 (0.91–1.04), 1.04 (0.97–1.12), and 1.00 (0.93–1.06), respectively. The above GMRs were all close to 1, and all associated 90% CIs fell entirely within the pre‐specified similarity margins of 0.80–1.25, indicating that PK similarity was established between HLX14 and denosumab sourced from the US, the EU, and CN (Table 3).
TABLE 3.
Similarity of primary pharmacokinetic parameters among treatment groups in Part 2 (PKPS).
| Treatment groups for comparison | Geometric least squares mean ratio (90% CI) | ||
|---|---|---|---|
| AUC0–t (day μg/mL) | C max (μg/mL) | AUC0–inf (day μg/mL) | |
| HLX14 vs. US‐denosumab | 0.98 (0.91–1.05) | 0.99 (0.93–1.06) | 0.97 (0.91–1.04) |
| HLX14 vs. EU‐denosumab | 1.05 (0.98–1.13) | 1.05 (0.99–1.13) | 1.04 (0.97–1.12) |
| HLX14 vs. CN‐denosumab | 1.00 (0.94–1.07) | 0.97 (0.91–1.04) | 1.00 (0.93–1.06) |
| US‐denosumab vs. EU‐denosumab | 1.08 (0.99–1.16) | 1.06 (0.98–1.14) | 1.07 (0.99–1.16) |
| CN‐denosumab vs. EU‐denosumab | 1.05 (0.97–1.13) | 1.09 (1.01–1.17) | 1.05 (0.97–1.12) |
| CN‐denosumab vs. US‐denosumab | 0.97 (0.90–1.05) | 1.02 (0.95–1.10) | 0.97 (0.91–1.05) |
Note: Due to %AUCex of two participants in EU‐denosumab group and one in CN‐denosumab group were greater than 20%, the PK parameters AUC0–inf and AUC0–t were not included in similarity evaluation.
Abbreviations: AUC0–inf, area under the serum drug concentration–time curve from time 0 to infinity; AUC0–t , area under the serum drug concentration–time curve from time 0 to the last concentration‐quantifiable time t; CI, confidence interval; C max, maximum serum drug concentration; CN, China; EU, European Union; PKPS, pharmacokinetic parameter set; US, United States.
Pharmacodynamics
In the PDCS, after a single dose of HLX14 or denosumab, the profiles of percentage change from baseline in s‐CTX concentration were largely consistent among treatment groups, as shown in the mean percentage change from baseline in s‐CTX concentration–time curves (Figure 2c and Figure S2C). In Part 2 of the study, s‐CTX concentration was reduced by approximately 85% at day 5 post‐dose, with the maximal reduction observed at around 1‐month post‐dose. The inhibition of s‐CTX concentration was maintained at a high level for nearly six months, followed by a slow decrease, and eventually, the s‐CTX concentration was recovered to around 75% of baseline level at day 274 post‐dose.
Descriptive statistics of PD parameters in the PDPS showed comparable AUEC0–t , I max, I min, and T min among treatment groups in both Part 1 and Part 2 of the study (Table S6). In Part 2, the geometric mean (CVb) of AUEC0–t in the HLX14, US‐, EU‐, and CN‐denosumab groups was 18742.0 (16.7%), 19303.1 (15.7%), 18013.9 (26.2%), and 18372.1 (18.8%) day·% inhibition, respectively. The geometric mean (CVb) of I max and I min was 89.5 (5.7%) versus 90.9 (4.2%) versus 89.8 (5.6%) versus 89.7 (5.0%) % inhibition, and 0.051 (51.3%) versus 0.052 (48.6%) versus 0.053 (46.7%) versus 0.054 (44.7%) ng/mL. The median T min was 28.0 days (range 4.0–105.0), 42.0 days (range 4.0–105.1), 42.0 days (range 7.0–139.0), and 28.0 days (range 4.0–134.0) in the respective groups.
Safety
All participants (24 [100%]) enrolled in Part 1 of the study experienced treatment‐emergent AEs (TEAEs), and all these TEAEs were grade 1–2 (Table S7). The most common TEAEs are provided in Table S8. Three (25.0%) and two (16.7%) participants in the respective groups experienced AESIs, all of which were hypocalcemia (Table S7). No TEAE leading to death or injection site reaction was reported.
All participants in Part 2 experienced TEAEs (Table 4). TEAEs with an incidence of ≥20% in all participants were COVID‐19 (HLX14 vs. US‐denosumab vs. EU‐denosumab vs. CN‐denosumab, 62.1% vs. 56.1% vs. 60.7% vs. 59.6%), blood calcium increased (29.3% vs. 28.1% vs. 28.6% vs. 12.3%), blood phosphorus decreased (22.4% vs. 22.8% vs. 19.6% vs. 26.3%), blood triglycerides increased (24.1% vs. 21.1% vs. 21.4% vs. 17.5%), and alanine aminotransferase increased (19.0% vs. 15.8% vs. 28.6% vs. 19.3%). Grade ≥3 TEAEs were reported in three (5.2%), six (10.5%), six (10.7%), and two (3.5%) participants in the respective groups, most commonly (≥1% in all participants) blood triglycerides increased (3.4% vs. 3.5% vs. 5.4% vs. 3.5%). Treatment‐related AEs (TRAEs) occurred in a similar proportion of patients in the four treatment groups (82.8% vs. 84.2% vs. 85.7% vs. 80.7%), among whom two (3.4%), four (7.0%), one (1.8%), and zero reported grade ≥3 TRAEs, respectively (Table S7). Four (6.9%), nine (15.8%), nine (16.1%), and nine (15.8%) participants in the respective groups experienced AESIs, including hypocalcemia (6.9% vs. 14.0% vs. 16.1% vs. 15.8%, all grade 1–2) and serious infection (one [1.8%] grade 3 tuberculous arthritis in the US‐denosumab group). Two (3.5%) participants in the US‐denosumab group reported serious TRAEs, one (1.8%) each of arthritis infective and synovitis. Only one (1.8%) participant in the US‐denosumab group had an injection site reaction. No subject experienced TEAE leading to death.
TABLE 4.
Treatment‐emergent adverse events in Part 2 (SS).
| n (%) | HLX14 (n = 58) | US‐denosumab (n = 57) | EU‐denosumab (n = 56) | CN‐denosumab (n = 57) | ||||
|---|---|---|---|---|---|---|---|---|
| Any | Grade ≥ 3 | Any | Grade ≥ 3 | Any | Grade ≥ 3 | Any | Grade ≥ 3 | |
| Any treatment‐emergent adverse event | 58 (100) | 3 (5.2) | 57 (100) | 6 (10.5) | 56 (100) | 6 (10.7) | 57 (100) | 2 (3.5) |
| COVID‐19 | 36 (62.1) | 0 | 32 (56.1) | 0 | 34 (60.7) | 0 | 34 (59.6) | 0 |
| Blood calcium increased | 17 (29.3) | 0 | 16 (28.1) | 1 (1.8) | 16 (28.6) | 1 (1.8) | 7 (12.3) | 0 |
| Blood triglycerides increased | 14 (24.1) | 2 (3.4) | 12 (21.1) | 2 (3.5) | 12 (21.4) | 3 (5.4) | 10 (17.5) | 2 (3.5) |
| Blood phosphorus decreased | 13 (22.4) | 0 | 13 (22.8) | 0 | 11 (19.6) | 0 | 15 (26.3) | 0 |
| Hypophosphatemia | 11 (19.0) | 0 | 10 (17.5) | 0 | 4 (7.1) | 0 | 12 (21.1) | 0 |
| Alanine aminotransferase increased | 11 (19.0) | 0 | 9 (15.8) | 0 | 16 (28.6) | 0 | 11 (19.3) | 0 |
| Protein urine present | 11 (19.0) | 0 | 5 (8.8) | 0 | 7 (12.5) | 0 | 8 (14.0) | 0 |
| Blood cholesterol increased | 10 (17.2) | 0 | 12 (21.1) | 0 | 10 (17.9) | 0 | 5 (8.8) | 0 |
| Neutrophil count increased | 10 (17.2) | 0 | 7 (12.3) | 0 | 3 (5.4) | 0 | 8 (14.0) | 0 |
| Aspartate aminotransferase increased | 8 (13.8) | 0 | 8 (14.0) | 0 | 8 (14.3) | 0 | 10 (17.5) | 0 |
| Blood creatinine increased | 8 (13.8) | 0 | 5 (8.8) | 0 | 5 (8.9) | 0 | 8 (14.0) | 0 |
| White blood cell count increased | 8 (13.8) | 0 | 4 (7.0) | 0 | 2 (3.6) | 0 | 2 (3.5) | 0 |
| Neutrophil count decreased | 7 (12.1) | 0 | 7 (12.3) | 0 | 9 (16.1) | 2 (3.6) | 9 (15.8) | 0 |
| Upper respiratory tract infection | 6 (10.3) | 0 | 12 (21.1) | 0 | 6 (10.7) | 0 | 10 (17.5) | 0 |
| White blood cell count decreased | 6 (10.3) | 0 | 9 (15.8) | 0 | 6 (10.7) | 0 | 6 (10.5) | 0 |
| Blood uric acid increased | 6 (10.3) | 0 | 6 (10.5) | 0 | 9 (16.1) | 0 | 7 (12.3) | 0 |
| Hypokalemia | 5 (8.6) | 0 | 5 (8.8) | 0 | 5 (8.9) | 0 | 8 (14.0) | 0 |
| Arthralgia | 4 (6.9) | 0 | 7 (12.3) | 0 | 7 (12.5) | 0 | 0 | 0 |
| Blood calcium decreased | 3 (5.2) | 0 | 7 (12.3) | 0 | 7 (12.5) | 0 | 8 (14.0) | 0 |
| Blood magnesium increased | 1 (1.7) | 0 | 2 (3.5) | 0 | 3 (5.4) | 1 (1.8) | 1 (1.8) | 0 |
| Blood potassium increased | 1 (1.7) | 1 (1.7) | 1 (1.8) | 0 | 3 (5.4) | 0 | 0 | 0 |
| Neutropenia | 0 | 0 | 1 (1.8) | 1 (1.8) | 1 (1.8) | 0 | 1 (1.8) | 0 |
| Arthritis infective | 0 | 0 | 1 (1.8) | 1 (1.8) | 0 | 0 | 0 | 0 |
| Synovitis | 0 | 0 | 1 (1.8) | 1 (1.8) | 0 | 0 | 0 | 0 |
Note: Treatment‐emergent adverse events were reported in ≥10% of participants in any treatment group and all grade ≥3 treatment‐emergent adverse events were shown in the table.
Abbreviations: CN, China; EU, European Union; SS, safety set; US, United States.
Immunogenicity
Immunogenicity was analyzed in the SS. In Part 1 of the study, no ADA‐positive sample was detected for any subject at any timepoint. In Part 2, six (10.3%) participants in the HLX14 group, 10 (17.5%) in the US‐denosumab group, 13 (23.2%) in the EU‐denosumab group, and 12 (21.1%) in the CN‐denosumab group tested positive for ADAs at least once after dosing. Among the ADA‐positive participants, three participants in the US‐denosumab group, three in the EU‐denosumab group, and one in the CN‐denosumab group only had negative titers, indicating weak ADA positivity in these participants. Additionally, one participant in the US‐denosumab group, one in the EU‐denosumab group, and two in the CN‐denosumab group was ADA‐positive at baseline with their post‐dose ADA titers not exceeding four times the baseline levels. Therefore, ADAs detected in these four participants were considered non‐treatment‐boosted and unrelated to study drugs, as they did not meet the criterion for treatment‐boosted ADAs. Throughout the study, only one (1.8%) participant in the US‐denosumab group tested positive for NAbs.
DISCUSSION
In this randomized phase I study, the primary PK parameters have met the predefined similarity criteria, with the 90% CIs of GMR for AUC0–t , C max, and AUC0–inf being entirely within the pre‐specified similarity margins of 0.80–1.25, thereby demonstrating similarity between HLX14 and reference denosumab from the EU, UK, and CN. A single dose of HLX14, US‐denosumab, EU‐denosumab, and CN‐denosumab also produced similar PD characteristics, safety, and immunogenicity in Chinese healthy adult male participants.
The US FDA approved its first denosumab biosimilar GP2411 (denosumab‐bddz) in March 2024. 18 Three additional denosumab biosimilars have been approved by the CN NMPA, namely LY06006, MW031, and QL1206. 19 , 20 , 21 The discrepancy in regulatory approvals of biosimilars across geographical regions have limited treatment accessibility for patients globally. In the current phase I study, the similarity between HLX14 and denosumab from three different sources (US, EU, and CN) was demonstrated. In the meantime, there is an ongoing international randomized phase III clinical study comparing HLX14 with EU‐denosumab as a reference in women with postmenopausal osteoporosis at high risk for fracture (NCT05352516). The development of HLX14 is expected to increase the accessibility of patients to effective treatment both in China and internationally.
The serum concentration–time curves as well as the PK parameters of HLX14 in our study shared the same pattern as those of other denosumab biosimilars. 22 , 23 , 24 The change in s‐CTX concentration after a single dose of HLX14 was also similar to the change with other denosumab biosimilars; the maximal inhibition of bone resorption appeared at about 1‐month post‐dose, and was maintained for almost six months. 22 , 23 , 24 , 25 As treatment effects of denosumab rapidly diminish after treatment discontinuation, a marked rise of s‐CTX in the 7–9 months post‐injection was commonly observed, making the repeated injections at 6‐month intervals necessary. 8 In our trial, s‐CTX concentrations increased to ≥50% of baseline levels at around 7‐month post‐dose, which is consistent with this unique property of denosumab.
No new safety signal was identified in the HLX14 group as compared to the denosumab groups in our study, or to other denosumab biosimilars. 22 , 23 , 24 , 25 Most TEAEs in the current trial were grade 1–2. As the study period (from November 2020 to September 2023) spanned over the years of COVID‐19 pandemic, it was not surprising to observe such a high incidence of COVID‐19 (56.1%–62.1%), and the cases were evenly distributed across treatment groups. Blood phosphorus decrease (all grade 1–2) was frequently observed in this study, which was consistent with known effects of denosumab on serum phosphorus levels. 25 , 26 Other TEAEs of relatively high incidence included changes in lipid profile (i.e., blood triglycerides increased and blood cholesterol increased) and upper respiratory tract infection, both of which were often reported during treatment with denosumab and denosumab biosimilars. 14 , 22 , 23 , 25 The most common AESI was hypocalcemia (6.9%–25.0%), a known common adverse event caused by denosumab. 8 , 14 In this trial, all participants received calcium and vitamin D supplements to ensure adequate intake, and all events of hypocalcemia were grade 1–2. One (1.8%) subject receiving US‐denosumab treatment experienced a grade 3 AESI of arthritis infective, which was resolved within three months. No TEAE leading to death occurred during the study.
Immunogenicity was similar between HLX14 and reference denosumab. With the exclusion of participants with negative tiers, the ADA‐positivity rate was thereby 10.5% (n = 6), 16.1% (n = 9), and 15.8% (n = 9) in the US‐, EU‐, and CN‐denosumab groups, respectively, while that for the HLX14 group remained at 10.3% (n = 6). The incidence of positive ADAs was nevertheless higher than that in the other trials of denosumab biosimilars. 22 , 23 , 24 , 25 However, various factors may have an impact on the ADA‐positivity rate, including but not limited to the sensitivity and specificity of the detection method and the study population. Furthermore, PK, PD, and safety profiles were generally not affected by the presence of ADAs in the current trial.
This study was designed so HLX14 was compared with reference denosumab sourced from the US, EU, and China, three major markets with large patient populations, in one trial. This design mitigates potential risks in regional differences in formulation, packaging, or manufacturing processes that could impact the trial outcomes. With comparison with the differently sourced references as a bridge, results from studies adopting this design may therefore facilitate greater scientific rigor and broader regulatory approval.
Clinical trials of denosumab enrolled East Asian participants from study sites in Korea (NCT01575873) along with participants from EU and US, and PK analysis concluded that was not affected by race. 14 Consistent with this finding, Chen et al. evaluated the PK, PD, safety and tolerability of denosumab in 46 healthy Chinese adults and demonstrated similar PK and PD (s‐CTX) profiles of the Chinese participants and those previously reported of Caucasians. 27 Given the high similarity between the pharmacokinetics of HLX14 and denosumab demonstrated in this study, it is therefore likely that PK of HLX14 is also not affected by race.
One potential limitation of the present study is that the study population consisted of only male participants. However, this enrolment criteria were selected to ensure the characterization of PK and PD in the most homogenous population with the lowest variability. Furthermore, the PK of denosumab is not affected by gender as reported in the summary of product characteristics. 13
The long‐term findings after repeated dosing are currently being investigated in the ongoing, international, randomized phase III clinical study of HLX14 versus reference denosumab in women with postmenopausal osteoporosis at high risk for fracture.
CONCLUSION
This phase I study demonstrated the PK similarity between HLX14 and denosumab (US‐, EU‐, and CN‐sourced) in Chinese healthy adult male participants. PD characteristics, safety, and immunogenicity were also comparable among treatment groups. Data from this study supports large‐scale phase III clinical trials of HLX14 as a potential denosumab biosimilar in the treatment of osteoporosis and bone loss.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. J.Z., W.Y., and K.H. designed the research; J.Z., N.L., N.C., L.Z., X.W., Q.W. performed the research; J.W., X.H., H.Y., and Q.W. analyzed the data. All authors read and approved the final manuscript.
FUNDING INFORMATION
This study was sponsored by Shanghai Henlius Biotech, Inc.
CONFLICT OF INTEREST STATEMENT
Jiahui Wang, Xuhui Hu, Haoyu Yu, and Qingyu Wang are employees of Shanghai Henlius Biotech, Inc. All other authors declared no competing interests for this work.
CONSENT TO PARTICIPATE
All participants provided written informed consent prior to study enrollment.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We would like to thank the participants who participated in the trial, their families, the principal investigators, clinicians, study coordinators, nurses, as well as the clinical study team (Clinical Operations: Juan Du, Jing Li; Clinical Pharmacology: Liang Zhou; Medical R&D: Lin Wang; Statistics: Yunling Chen, Mengkai Chen), Jun Zhu, and Wenjie Zhang from Shanghai Henlius Biotech, Inc. Medical writing support was provided by Xiao Zou PhD, Shiqi Zhong PhD and Chen Hu PhD of Shanghai Henlius Biotech, Inc., and funded by Shanghai Henlius Biotech, Inc.
Li N, Chu N, Zhu L, et al. Pharmacokinetics, pharmacodynamics, safety, and immunogenicity of HLX14 versus reference denosumab in healthy males: A randomized phase I study. Clin Transl Sci. 2024;17:e70089. doi: 10.1111/cts.70089
Nanyang Li, Nannan Chu and Leilei Zhu contributed equally as co‐first authors.
Trial registration: This study was registered with ClinicalTrials.gov (NCT04534582) and Chinadrugtrials.org.cn (CTR20201905).
Contributor Information
Wei'an Yuan, Email: weian_1980@163.com.
Kai Huang, Email: hk19820627@sina.com.
Jing Zhang, Email: zhang.j61@fudan.edu.cn, Email: zhang.j61@fudan.edu.cn.
DATA AVAILABILITY STATEMENT
Data is available from the corresponding author upon reasonable request.
REFERENCES
- 1. National Institute of Arthritis and Musculoskeletal and Skin Diseases . Osteoporosis. Reviewed December 2022. https://www.niams.nih.gov/health‐topics/osteoporosis. Accessed February 28, 2024. https://www.niams.nih.gov/health‐topics/osteoporosis
- 2. MSD MANUAL Professional Version . Osteoporosis. Reviewed/Revised September 2023. https://www.msdmanuals.com/professional/musculoskeletal‐and‐connective‐tissue‐disorders/osteoporosis/osteoporosis?query=osteoporosis. Accessed February 28, 2024.
- 3. Xiao PL, Cui AY, Hsu CJ, et al. Global, regional prevalence, and risk factors of osteoporosis according to the World Health Organization diagnostic criteria: a systematic review and meta‐analysis. Osteoporos Int. 2022;33:2137‐2153. doi: 10.1007/s00198-022-06454-3 [DOI] [PubMed] [Google Scholar]
- 4. Chinese Society of Osteoporosis and Bone Mineral Research . Guidelines for the diagnosis and treatment of primary osteoporosis (2022). Chinese Gen Pract. 2023;26:1671‐1691. doi: 10.12114/j.issn.1007-9572.2023.0121 [DOI] [Google Scholar]
- 5. LeBoff MS, Greenspan SL, Insogna KL, et al. The clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int. 2022;33:2049‐2102. doi: 10.1007/s00198-021-05900-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. National Osteoporosis Guideline Group, UK . Clinical guideline for the prevention and treatment of osteoporosis. Updated September 2021. https://www.nogg.org.uk/full‐guideline. Accessed February 28, 2024.
- 7. Lyu H, Jundi B, Xu C, et al. Comparison of denosumab and bisphosphonates in patients with osteoporosis: a meta‐analysis of randomized controlled trials. J Clin Endocrinol Metabol. 2018;104:1753‐1765. doi: 10.1210/jc.2018-02236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reid IR, Billington EO. Drug therapy for osteoporosis in older adults. Lancet. 2022;399:1080‐1092. doi: 10.1016/S0140-6736(21)02646-5 [DOI] [PubMed] [Google Scholar]
- 9. Zhang L, Zeng F, Jiang M, Han M, Huang B. Roles of osteoprotegerin in endocrine and metabolic disorders through receptor activator of nuclear factor kappa‐B ligand/receptor activator of nuclear factor kappa‐B signaling. Front Cell Dev Biol. 2022;10:1005681. doi: 10.3389/fcell.2022.1005681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prolia(R) (Denosumab) Granted Marketing Authorization in the European Union. 2010. https://www.amgen.com/newsroom/press‐releases/2010/05/proliar‐denosumab‐granted‐marketing‐authorization‐in‐the‐european‐union. Accessed March 12, 2024.
- 11. FDA Approves Amgen's Prolia(TM) (Denosumab) for Treatment of Postmenopausal Women With Osteoporosis at High Risk for Fracture. 2010. https://www.amgen.com/newsroom/press‐releases/2010/06/fda‐approves‐amgens‐proliatm‐denosumab‐for‐treatment‐of‐postmenopausal‐women‐with‐osteoporosis‐at‐high‐risk‐for‐fracture. Accessed March 12, 2024.
- 12. Amgen Receives National Medical Products Administration Approval For Prolia® (Denosumab) in China. 2020. https://www.amgen.cn/en/media/amgen_denosumab_china.html. Accessed March 12, 2024.
- 13. Prolia Summary of Product Characteristics. Latest renewal on January 16, 2020. https://www.ema.europa.eu/en/documents/product‐information/prolia‐epar‐product‐information_en.pdf. Accessed March 12, 2024.
- 14. Prolia® Prescribing Information . Recent major changes in March 2024. https://www.pi.amgen.com/united_states/prolia/prolia_pi.pdf. Accessed March 12, 2024.
- 15. Amgen Prolia® Receives Approval for New Indication for the Treatment of Osteoporosis in Men at High Risk for Fracture. 2023. https://www.amgen.cn/cn/media/amgen_Prolia.html. Accessed March 12, 2024.
- 16. Zhang J, Wu X, Zhang X, et al. A randomized phase 1 study evaluating the pharmacokinetic equivalence of proposed biosimilar HLX14 and denosumab in healthy Chinese volunteers. Clin Pharmacol Ther. 2022;111:S59. doi: 10.1002/cpt.2521 [DOI] [Google Scholar]
- 17. Morris HA, Eastell R, Jorgensen NR, et al. Clinical usefulness of bone turnover marker concentrations in osteoporosis. Clin Chim Acta. 2017;467:34‐41. doi: 10.1016/j.cca.2016.06.036 [DOI] [PubMed] [Google Scholar]
- 18.Sandoz receives FDA approval for first and only denosumab biosimilars. 2024. https://www.sandoz.com/sandoz‐receives‐fda‐approval‐first‐and‐only‐denosumab‐biosimilars/. Accessed March 12, 2024.
- 19.China's First Denosumab Biosimilar Boyoubei® Approved for Launch by NMPA. 2022. https://www.boan‐bio.com/en/info.php?id=164. Accessed March 13, 2024.
- 20. The World's Second Approved Biosimilars of Denosumab (MAILISHU) . 2023. https://www.mabwell.com/en/news_info/id‐98.html. Accessed March 13, 2024.
- 21. Qilu Pharmaceutical's Denosumab Injection (LUKEXIN) Approved for Marketing . 2023. https://www.qilu‐pharma.com/news_details/157.html. Accessed March 13, 2024.
- 22. Gu J, Zhang H, Xue Q, et al. Denosumab biosimilar (LY06006) in Chinese postmenopausal osteoporotic women: a randomized, double‐blind, placebo‐controlled, multicenter phase III study. J Orthopaed Transl. 2023;38:117‐125. doi: 10.1016/j.jot.2022.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo Y, Guo T, Di Y, et al. Pharmacokinetics, pharmacodynamics, safety and immunogenicity of recombinant, fully human anti‐RANKL monoclonal antibody (MW031) versus denosumab in Chinese healthy subjects: a single‐center, randomized, double‐blind, single‐dose, parallel‐controlled trial. Expert Opin Biol Ther. 2023;23:705‐715. doi: 10.1080/14712598.2023.2178298 [DOI] [PubMed] [Google Scholar]
- 24. Jeka S, Dokoupilová E, Kivitz A, et al. Equivalence trial of proposed denosumab biosimilar GP2411 and reference denosumab in postmenopausal osteoporosis: the ROSALIA study. J Bone Miner Res. 2024;39:202‐210. doi: 10.1093/jbmr/zjae016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang H, Gu J‐m, Chao A‐j, et al. A phase III randomized, double‐blind, placebo‐controlled trial of the denosumab biosimilar QL1206 in postmenopausal Chinese women with osteoporosis and high fracture risk. Acta Pharmacol Sin. 2023;44:446‐453. doi: 10.1038/s41401-022-00954-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakamura Y, Suzuki T, Kamimura M, et al. Vitamin D and calcium are required at the time of denosumab administration during osteoporosis treatment. Bone Research. 2017;5:17021. doi: 10.1038/boneres.2017.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Q, Hu C, Liu Y, et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of single‐dose denosumab in healthy Chinese volunteers: a randomized, single‐blind, placebo‐controlled study. PLoS One. 2018;13:e0197984. doi: 10.1371/journal.pone.0197984 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Data is available from the corresponding author upon reasonable request.