

Abstract
Cancer remains a significant global challenge, and despite the numerous strategies developed to advance cancer therapy, an effective cure for metastatic cancer remains elusive. A major hurdle in treatment success is the ability of cancer cells, particularly cancer stem cells (CSCs), to resist therapy. These CSCs possess unique abilities, including self-renewal, differentiation, and repair, which drive tumor progression and chemotherapy resistance. The resilience of CSCs is linked to certain signaling pathways. Tumors with pathway-dependent CSCs often develop genetic resistance, whereas those with pathway-independent CSCs undergo epigenetic changes that affect gene regulation. CSCs can evade cytotoxic drugs, radiation, and apoptosis by increasing drug efflux transporter activity and activating survival mechanisms. Future research should prioritize the identification of new biomarkers and signaling molecules to better understand drug resistance. The use of cutting-edge approaches, such as bioinformatics, genomics, proteomics, and nanotechnology, offers potential solutions to this challenge. Key strategies include developing targeted therapies, employing nanocarriers for precise drug delivery, and focusing on CSC-targeted pathways such as the Wnt, Notch, and Hedgehog pathways. Additionally, investigating multitarget inhibitors, immunotherapy, and nanodrug delivery systems is critical for overcoming drug resistance in cancer cells.
Keywords: Cancer cells, Cancer stem cells, Drug resistance, Mammalian cancer stem cells
Introduction
Cancer, a heterogeneous and intricate collection of diseases, presents substantial challenges to the health of both humans and animals on a global scale. This condition is typified by a variety of pathological phenomena, including unregulated cellular proliferation, transformations at both the cellular and morphological levels, angiogenesis (the development of new vasculature to nourish neoplastic tissues), dysregulation of apoptosis (the programmed mechanism for cellular demise), metastasis (the dissemination of malignant cells to distant anatomical sites), and neoplasia, which refers to the abnormal proliferation or division of cells. On a global scale, cancer constitutes a predominant cause of mortality, accounting for over eight million fatalities annually [1, 2]. This alarming statistic highlights its importance as a paramount public health concern. The disease is characterized by a multifactorial etiology, encompassing intricate genomic modifications that arise from the interplay between inherited genetic susceptibilities and various environmental determinants, such as lifestyle choices, exposure to carcinogenic agents, and infectious agents [3]. These complex interactions significantly contribute to the pathogenesis and progression of diverse cancer types, rendering it a highly individualized condition that manifests uniquely in each patient [4].
The management of cancer continues to face substantial obstacles, particularly due to the challenges posed by drug resistance and immune evasion. These elements significantly impede the effectiveness of existing therapeutic interventions, resulting in treatment failure and exacerbation of the disease. A prominent concern in oncological treatment is the phenomenon of drug resistance, which can be delineated into intrinsic and acquired categories. Intrinsic resistance pertains to the inherent capacity of cancer cells to endure the effects of pharmacological agents, often attributable to preexisting genetic mutations or the activation of survival pathways [5, 6]. For example, numerous tumors overexpress ATP-binding cassette (ABC) transporters, which actively eliminate chemotherapeutic agents, consequently diminishing their intracellular concentrations and therapeutic efficacy [7]. Acquired resistance emerges as a consequence of therapeutic interventions, wherein cancer cells evolve mechanisms to survive in the presence of pharmacological agents. This occurrence is frequently driven by genetic and epigenetic modifications, such as mutations in drug targets or alterations in signaling pathways [8]. The tumor microenvironment (TME) is integral to the phenomenon of drug resistance. Elements such as hypoxia, acidity, and nutrient scarcity create an inhospitable milieu for drug action and may increase the survival of cancer stem cells (CSCs), which are characteristically resistant to standard therapeutic modalities [9, 10]. The presence of stromal cells, immune cells, and components of the extracellular matrix can also sequester drugs, thereby limiting their therapeutic effectiveness [11]. For example, the hypoxic TME may result in the activation of hypoxia-inducible factors (HIFs), which facilitate resistance mechanisms such as upregulated glycolytic metabolism and amplified survival signaling [12]. Another significant obstacle is immune evasion, whereby tumors devise mechanisms to elude detection and eradication by the host immune system. Cancer cells may modify their antigen expression, downregulate major histocompatibility complex (MHC) molecules, or secrete immunosuppressive factors, all of which contribute to immune tolerance [10, 13–15]. For example, numerous tumors express programmed cell death-ligand 1 (PD-L1), which interacts with PD-1 on T cells, inhibiting their activation and facilitating immune checkpoint evasion [10, 16]. This immune evasion not only permits unimpeded tumor proliferation but also constrains the efficacy of immune-based therapeutic strategies, such as immune checkpoint inhibitors. The introduction of immunotherapy, while groundbreaking, has further highlighted the complexities associated with immune evasion. Although therapies that target immune checkpoints have demonstrated potential, their effectiveness may be curtailed by the heterogeneity of tumors and the differential expression of immune checkpoints across various cancer types [17]. Furthermore, the emergence of resistance to immunotherapy can transpire, with tumors discovering alternative pathways to circumvent immune detection even subsequent to initial responsiveness [18]. The integration of targeted therapies with immunotherapy represents a prospective strategy to address both drug resistance and immune evasion. By targeting specific pathways that facilitate survival and resistance in tumor cells, increasing the efficacy of immunotherapeutic agents may be feasible. For example, the concomitant use of inhibitors of the PI3K/AKT/mTOR pathway with immune checkpoint inhibitors has exhibited promise in preclinical investigations ([19, 20]. However, the complexity of cancer biology necessitates careful consideration of combination strategies to avoid exacerbating resistance mechanisms. In conclusion, the challenges of drug resistance and immune evasion significantly complicate cancer treatment. A deeper understanding of the underlying mechanisms and interactions within the tumor microenvironment is crucial for developing more effective therapeutic strategies. Research aimed at identifying novel biomarkers and elucidating the intricacies of tumor biology is essential for improving patient outcomes in the fight against cancer [21, 22].
In recent years, the recognition of cancer as a heterogeneous entity characterized by tumors that comprise a variety of cellular populations exhibiting distinct characteristics and behaviors has increased [23, 24]. This heterogeneity is evidenced by the diverse differentiation phenotypes that can be observed within neoplastic tissues. While the predominant cellular constituents of a tumor may be classified as nontumorigenic, indicating that they do not facilitate cancer progression, a small yet pivotal subpopulation of cells, referred to as "cancer stem cells" (CSCs), is integral to the initiation, growth, and recurrence of neoplasms. These CSCs are believed to drive tumorigenesis because of their capacity for self-renewal and differentiation, thereby establishing them as critical focal points for cancer research and therapeutic intervention [23–26]. As our comprehension of the complexities inherent in cancer continues to evolve, it becomes increasingly apparent that the effective treatment and management of this disease necessitate a multifaceted strategy that addresses not only the predominant tumor cell populations but also the specialized subpopulations, such as CSCs, that are implicated in the most aggressive and enduring manifestations of cancer. This advancing knowledge underscores the persistent challenges and emerging opportunities in the global endeavor to combat cancer.
CSCs are a specific population of cells within tumors that have the ability to self-renew and reproduce the original tumor phenotype. They play crucial roles in tumor proliferation, differentiation, recurrence, metastasis, and chemoresistance. They are considered targets for anticancer therapy, and ongoing clinical trials are testing the efficacy of various drugs against CSCs [27]. Understanding cancer stem cells is important in the context of clonal genetic heterogeneity, as they possess clonogenicity and self-renewal capabilities. They have been identified in various malignancies, including leukemia, and advances in the field have been made through techniques such as lineage tracing [28].
Currently, mammalian cancer cells are the focus of several studies [29]. For example, many studies have demonstrated the integration of metabolic, signaling, and gene regulatory networks in these cells via support vector regression (SVR)-based three timescale models [4, 30–32]. Similarly, there are currently mechanisms of rapid turnover of microRNAs (miRNAs) in cancer cells and the effects of target mRNAs on miRNA abundance [33, 34]. Moreover, a lectin called iNoL was identified as a slipper lobster that displays cytotoxic effects on human cancer cells [35]. There is compelling evidence that pharmaceutical compositions comprising engineered mammalian cells for cancer treatment, including immunomodulators and immunotherapies, have been researched [4].
Notably, stem cells are the building blocks of tissues and organs and have been studied for their potential in regenerative medicine and tissue engineering [4, 36, 37]. These cells can be classified on the basis of their differentiation potential, with totipotent stem cells being able to give rise to any cell type, pluripotent stem cells being limited to the three embryonic germ layers, and multipotent stem cells being able to differentiate into one germ line tissue [38, 39]. They can be extracted from various sources, including bone marrow, amniotic cells, adipose tissue, the umbilical cord, and placental tissue [36].
In the context of cancer medicine, stem cell transplantation, particularly bone marrow transplantation, is a procedure used to treat certain types of cancer and other diseases by replacing damaged or diseased bone marrow with healthy stem cells [40, 41]. The process of stem cell transplantation offers the potential to enhance the treatment of neurodegenerative disorders by replacing compromised neurons across a variety of neurological conditions, including multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer's disease, Parkinson's disease, and Huntington's disease [42]. These innovative therapies have undergone rigorous evaluation in clinical trials to determine their efficacy in addressing these incapacitating condition [43]. A diverse array of stem cell types, including embryonic stem cells, mesenchymal stem cells, and neural stem cells, has been scrutinized for their potential to differentiate into neurons and glial cells, which are fundamental components of the nervous system [44]. The overarching objective of stem cell-based interventions is to reinstate lost or compromised neuronal cells while concurrently fostering a neuroprotective milieu. This is accomplished not only through the direct substitution of cellular entities but also via the secretion of bioactive molecules that can increase cellular viability, mitigate inflammation, and facilitate tissue regeneration. These therapeutic modalities exhibit significant promise, as they represent a comprehensive strategy for the treatment of neurodegenerative diseases, effectively addressing both the manifestations and the foundational etiologies of neuronal impairment [45]. As scientific inquiry progresses, stem cell transplantation may emerge as a pivotal element in the management and prospective resolution of neurodegenerative disorders. These therapies have the potential to provide tissue restoration and alleviate the symptoms of neurodegenerative disorders [42]. However, in the context of technological advancements, there is a need to refine cell therapy and increase its effectiveness in the human body as well as other animal species. Stem cells have the potential to revolutionize clinical treatment and are being explored for their role in drug development and toxicity evaluation [43].
Currently, drug resistance in mammalian cancer stem cells is a major challenge in cancer therapy, as it is driven by several key features. CSCs possess several mechanisms that allow them to survive exposure to chemotherapy drugs, making tumors resistant to treatment [46, 47]. Of course, they are a subset of tumor cells that are capable of multilineage progenitor expansion, and they are known to be intrinsically resistant to anticancer treatments [48]. The eradication of a tumor mass requires the complete removal of both tumor cells and CSCs [46, 48]. Elucidating the key features underlying drug resistance in CSCs is crucial for developing effective treatment strategies [49, 50]. Common characteristics between cancer stem cells (CSCs) and normal cells, such as shared signaling pathways and markers, complicate the targeted treatment of CSCs, impacting the safety and specificity of chemotherapy. Overcoming drug resistance in CSCs is crucial for enhancing cancer treatment outcomes [50–52].
CSCs have unique signaling pathways and markers that are important for the development of new anticancer strategies. The Wnt, Notch, Hedgehog, TGFβ/SMAD, JAK-STAT, and VEGF pathways are associated with CSC regulation and are potential therapeutic targets [42, 50, 53]. Furthermore, several other markers of CSCs, including CD133, CD44, ABCG2, and ALDH, have been identified for the development of promising pain points in the search for cancer treatments [34, 52, 54–56]. However, there is still no consensus on the characterization of antigens for CSCs. In addition to these markers, the expression levels of therapeutic target proteins such as PD-L1 and phosphorylated EGFR can also be examined in CSCs [44]. Identifying new markers and gaining a deeper understanding of CSC-specific antigens are crucial for developing novel therapeutic strategies against cancer.
Importantly, there is currently an immense struggle to explore ideal therapeutic approaches for targeting cancer stem cells (CSCs), including the development of treatments that block CSC-related signaling pathways [52, 57, 58]. In a similar fashion, chemical antibodies have been used as ligands for CSC-targeted therapeutic strategies [59]. These aptamers have been conjugated with various therapeutic cargoes, such as chemotherapy drugs, small interfering RNAs, and microRNAs, to kill CSCs [56, 60]. Natural products (NPs) have also been shown to target and inhibit CSCs, but their clinical translation has been hindered by pharmacokinetic defects and off-target effects [46, 50, 52, 61]. By targeting CSC-specific survival factors and interfering with CSC-selective survival signaling, overcoming drug resistance and enhancing the effectiveness of anticancer agents may be possible. This knowledge can inform the development of novel therapeutic approaches for targeting CSCs and improving outcomes for cancer patients. Hence, this comprehensive review explores findings from the scientific literature and provides an update on drug resistance in mammalian cancer stem cells.
Concepts of drug resistance in mammalian cancer cells
One of the significant challenges in modern cancer treatment is increasing resistance to chemotherapy and molecularly targeted therapies, which poses a substantial barrier to the effectiveness of these treatments [51]. Certain neoplasms demonstrate innate insensitivity to therapeutic modalities due to preexisting resistance determinants, referred to as primary or intrinsic resistance, whereas other malignancies acquire resistance throughout the therapeutic regimen, a phenomenon known as acquired resistance [62]. The emergence of resistance to anticancer pharmacotherapies is modulated by an array of factors, including genetic mutations, epigenetic modifications, and the upregulation of drug efflux mechanisms, among various cellular and molecular phenomena. These alterations enable cancer cells to endure and proliferate in the presence of therapeutic measures, thereby diminishing the overall efficacy of treatment strategies [63]. The current foundational elements of oncological management include a diverse array of methodologies, such as surgical intervention, cytotoxic chemotherapy, targeted therapies, radiation therapy, endocrine therapy, and immunotherapy. Despite the considerable progress achieved in these therapeutic modalities over recent decades, resistance to both conventional chemotherapeutic agents and novel targeted therapies remains a pivotal concern. This resistance significantly contributes to the recurrence of cancer, which remains one of the predominant causes of mortality among cancer patients [64, 65]. The persistent battle against pharmacologic resistance highlights the imperative for ongoing inquiry and innovation within the realm of cancer therapy, as surmounting this obstacle is crucial for enhancing patient outcomes and mitigating the elevated rates of cancer-associated mortalit [66].
Mammalian cancer and its severity
Mammalian cancer involves the development of abnormal cell growth in mammals and is characterized by genetic alterations that affect cellular fitness and can lead to the formation of tumors. Various methods have been developed for detecting and diagnosing cancer, including the detection of specific nucleic acids and proteins in biological samples [67–69]. The presence, absence, or relative quantity of these molecules can indicate the presence or risk of cancer development. The spatial distribution of specific components in the cell nucleus has also been used to detect and grade tumors, providing a method for efficient diagnostics and monitoring of proliferation disorders. The evolution of cancer suppression mechanisms across mammalian species has been studied, revealing a positive correlation between cancer gene copy number and longevity, as well as a strong association between longevity and the number of germline and somatic tumor suppressor genes [70]. For example, chimeric nonhuman mammals have also been developed as inducible spontaneous cancer models, allowing for the study of different types of cancer through the introduction of genetically modified embryonic stem cells [71], as depicted in Table 1.
Table 1.
Relative severity of common mammalian cancer stem cells
| Cancer Type | CSC Characteristics | Relative Severity | References |
|---|---|---|---|
| Breast Cancer | CD44 + /CD24-, ALDH1 + | High | [74, 75] |
| Colon Cancer | CD133 + , Lgr5 + | High | [76, 77] |
| Lung Cancer | CD133 + , CD44 + | High | [78–81] |
| Pancreatic Cancer | CD133 + , CD24 + | Very High | [82, 83] |
| Brain Cancer (Glioblastoma) | CD133 + , CD44 + | Very High | [69, 84–86] |
The capacity of cancer stem cells (CSCs) to start and sustain tumor growth, evade therapy, and aid in metastasis is frequently linked to the severity of the tumors they support. However, the particular cancer type and the properties of the implicated CSCs can affect the relative severity of disease.
The severity of cancer in mammals varies across species and is not directly correlated with lifespan or body mass. However, there is a positive association between litter size and the prevalence of malignancy, suggesting a trade-off between reproduction and cancer defenses [72]. The prevalence of neoplasia and malignancy in mammals increases with increasing adult weight and somatic mutation rate but decreases with increasing gestation time [73]. Certain species, such as ferrets and opossums, have particularly high levels of cancer, whereas others, such as the common porpoise, the Rodrigues fruit bat, and the black-footed penguin, have low levels [2]. Considering the different factors that contribute to these differences in cancer incidence may lead to improved strategies for cancer management and prevention (Table 1).
Area of cancer stem cell drug resistance factors
Drug resistance in cancer represents a formidable obstacle to the successful administration of therapeutic interventions, frequently culminating in treatment failure and suboptimal patient prognoses. Among the myriad of factors that contribute to the phenomenon of drug resistance, the tumor microenvironment (TME) plays a pivotal role. This complex ecosystem consists of neoplastic cells, adjacent stromal constituents, immune cells, vascular structures, and the extracellular matrix (ECM), all of which engage in dynamic interactions that facilitate tumor progression and confer resistance against therapeutic modalities. Hypoxia, defined by an inadequate oxygen supply, is a prevalent condition in solid tumors and is attributable to rapid cellular proliferation and insufficient vascular development. This hypoxic milieu precipitates the stabilization and activation of hypoxia-inducible factors (HIFs), particularly HIF-1α, which orchestrates a diverse array of adaptive responses [87]. HIF-1α enhances the transcriptional activity of genes implicated in angiogenesis, glucose metabolism, and cell survival, thereby augmenting the aggressive characteristics of tumors. In the context of cancer stem cells (CSCs), hypoxia is particularly important because it promotes self-renewal and stemness attributes, enabling these cells to endure hostile environments and resist standard therapeutic approaches [88]. Empirical investigations have indicated that hypoxic CSCs exhibit increased expression of drug efflux transporters, such as P-glycoprotein, which actively extrude chemotherapeutic agents from the cellular interior, resulting in diminished drug accumulation and therapeutic efficacy [89, 90]. Furthermore, hypoxia can promote epithelial‒mesenchymal transition (EMT), a mechanism that further amplifies the migratory and invasive capacities of CSCs, thereby facilitating their evasion of targeted therapeutic interventions.
The stroma that envelops tumor cells is composed of a diverse array of cell types, including fibroblasts, immune constituents, and endothelial cells, which collectively influence tumor behavior and drug resistance. Cancer-associated fibroblasts (CAFs), a significant element of the TME, secrete a plethora of growth factors and cytokines that increase tumor survival, proliferation, and drug resistance [91]. For example, CAFs are capable of producing transforming growth factor-beta (TGF-β) and hepatocyte growth factor (HGF), which activate signaling cascades, such as the PI3K/AKT and MAPK cascades, which have been shown to confer resistance to therapeutic agents [92]. The complex interplay between tumor cells and CAFs may give rise to a protective niche that sustains CSC viability, often mediated by the secretion of extracellular matrix components that provide both physical and biochemical support to CSCs, which further complicates therapeutic strategies [93]. Targeting CAFs represents a novel therapeutic strategy aimed at overcoming drug resistance. Therapeutic agents that inhibit CAF activation or disrupt CAF interactions with tumor cells have exhibited potential in enhancing tumor sensitivity to chemotherapy [91, 92]. For example, the application of monoclonal antibodies that specifically target CAF markers has demonstrated efficacy in preclinical models, resulting in improved drug delivery and therapeutic response. Nevertheless, the heterogeneity of CAFs, coupled with their dual roles in facilitating tumor progression and promoting antitumor immunity, introduces challenges that necessitate careful consideration within clinical contexts.
The extracellular matrix (ECM) offers essential structural support to neoplasms and is pivotal in mediating cellular signaling processes. In the context of malignancy, ECM remodeling transpires as a consequence of the excessive synthesis of matrix constituents and modifications in their composition, which may affect drug resistance mechanisms [94]. Elevated ECM stiffness, which is frequently correlated with the accumulation of collagen and fibronectin, has been associated with improved survival rates and enhanced drug resistance across various malignancies, including breast and pancreatic cancers [89]. Furthermore, ECM remodeling may influence the bioavailability and spatial distribution of therapeutic agents within the tumor microenvironment. For example, modifications in the ECM can establish physical barriers that obstruct drug infiltration, resulting in inadequate therapeutic concentrations in specific tumor locations. Additionally, the interplay between tumor cells and ECM constituents can activate signaling cascades, such as integrin-mediated signaling pathways, that promote cellular survival and contribute to resistance against apoptosis-inducing pharmacological agents [94]. Approaches designed to target ECM components or restore the normal architecture of the ECM are increasingly recognized as viable strategies to combat drug resistance. The enzymatic degradation of particular ECM constituents, such as hyaluronic acid, may facilitate enhanced drug delivery and augment therapeutic efficacy [95, 96]. Moreover, the advent of ECM-targeting agents that disrupt the interactions between tumor cells and the ECM presents a promising avenue for overcoming resistance phenomena. Nevertheless, the intricate nature of ECM dynamics alongside the potential for unforeseen consequences necessitates a prudent approach in the implementation of these therapeutic strategies.
The immunological landscape within the tumor microenvironment (TME) strongly influences drug resistance mechanisms. Tumors can create an immunosuppressive microenvironment through the recruitment of diverse immune cell types, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which effectively inhibit antitumor immune responses and facilitate the survival of cancer stem cells (CSCs) [93]. Furthermore, tumor cells frequently upregulate immune checkpoint molecules, such as PD-L1, thereby enabling them to evade immune surveillance and contributing to resistance to immunotherapeutic interventions [97]. In addition, the presence of immune cells within the TME may modulate CSC characteristics. For example, cytokines secreted by tumor-associated macrophages (TAMs) can promote CSC self-renewal and enhance resistance to therapeutic modalities, including chemotherapy and targeted therapies [98]. Immunotherapy has emerged as a promising strategy to mitigate drug resistance by re-engaging the immune system to target malignant cells. The combination of immune checkpoint inhibitors with conventional therapeutic approaches has demonstrated potential in enhancing patient outcomes, particularly in tumors characterized by elevated T-cell infiltration [99]. However, the existence of an immunosuppressive TME may constrain the effectiveness of these treatments, underscoring the necessity for strategies that can alter the immune landscape in conjunction with immunotherapeutic measures.
The tumor microenvironment (TME) significantly influences the metabolic characteristics of neoplastic cells, facilitating adaptations that enhance drug resistance. Neoplastic cells, including cancer stem cells (CSCs), frequently depend on glycolytic pathways for ATP generation, even in oxygen-rich conditions, a phenomenon referred to as the Warburg effect [100]. This metabolic reconfiguration promotes rapid cellular proliferation and sustains viability under conditions of nutrient deprivation. Moreover, the acidic milieu produced by metabolic byproducts, such as lactate, can promote the maintenance of CSCs and confer protection against the cytotoxicity of chemotherapeutic agents [101]. Metabolic modifications also impact the susceptibility of tumor cells to therapeutic interventions. For example, CSCs characterized by altered lipid metabolism may circumvent apoptosis triggered by specific pharmacological agents, resulting in heightened treatment resistance [102, 103]. Targeting metabolic pathways represents an innovative approach to mitigate drug resistance in oncological contexts. Inhibitors of glycolytic enzymes, such as hexokinase and lactate dehydrogenase, have demonstrated promise in preclinical investigations because they preferentially target cancer cells that have undergone metabolic adaptation [102, 104]. Furthermore, strategies aimed at metabolic reprogramming to restore oxidative phosphorylation could increase the sensitivity of tumors to chemotherapeutic agents. Nevertheless, the ability of tumor cells to adapt to metabolic stressors and the possibility of off-target effects warrant further exploration prior to clinical application, as presented in Table 2.
Table 2.
Drug resistance factors for cancer stem cell areas
| Area | Factors | References |
|---|---|---|
| Intrinsic Properties of CSCs | ||
| Self-renewal and differentiation | Ability to maintain stem cell phenotype and generate differentiated cells | [107–109] |
| Altered metabolism | Metabolic adaptations that support survival and proliferation | [110, 111] |
| DNA repair mechanisms | Efficient DNA repair pathways | [112–114] |
| Drug efflux | Expression of efflux pumps (e.g., ABC transporters) that remove drugs from cells | [115–117] |
| Tumor Microenvironment | ||
| Hypoxia | Low oxygen conditions | [118, 119] |
| Immune suppression | Suppression of immune response | [110, 120] |
| Extracellular matrix | Physical and biochemical properties of the tumor microenvironment | [110, 121] |
| Therapeutic Interventions | ||
| Drug combinations | Synergistic or additive effects of multiple drugs | [122, 123] |
| Drug repurposing | Using existing drugs for new indications | [124–126] |
| Targeted therapies | Targeting specific molecular pathways involved in cancer development and progression | [127–129] |
An overview of the main regions and variables influencing cancer stem cell resistance is given in this table. Depending on the type of cancer, the properties of the cancer stem cells, and the therapeutic approaches employed, several pathways may be involved
In conclusion, the tumor microenvironment serves as a pivotal factor in determining drug resistance within cancer, affecting multiple facets of tumor biology, including cellular survival, proliferation, and therapeutic responsiveness. Variables such as hypoxic conditions, stromal interactions, extracellular matrix (ECM) remodeling, immune cell dynamics, and metabolic changes engender a complex landscape that promotes resilience against therapeutic interventions, particularly in the context of CSCs. A comprehensive understanding of these interactions is crucial for the formulation of effective therapeutic strategies designed to overcome drug resistance. As research advances, concurrently targeting the TME alongside conventional therapeutic modalities may augment treatment efficacy and enhance patient outcomes in the ongoing struggle against cancer.
Drug resistance in cancer stem cells is influenced by a myriad of factors that affect the responsiveness to therapeutic interventions. These factors encompass both elements that impede the delivery of therapeutic agents to the neoplasm and those that modify the characteristics of the tumor microenvironment. The manifestation of treatment resistance, arising from a combination of intrinsic and extrinsic influences, constitutes a considerable impediment in oncological treatment [105]. A pivotal factor that contributes to resistance is the presence of genetic mutations, which can significantly impair the drug's capacity to effectively bind to its designated target. Moreover, neoplastic cells can implement various strategies to circumvent the effects of chemotherapeutic agents. Such strategies include a decrease in the influx of drugs into the cells, an increase in efflux pumps that extrude drugs from the cellular interior, the activation of detoxifying proteins that neutralize the pharmacological agent, and the enhancement of DNA repair systems that mitigate the damage inflicted by cytotoxic chemotherapies. Additionally, abnormalities in signaling pathways that normally react to damage induced by chemotherapy can also significantly contribute to resistance [106]. Comprehending and addressing these factors that confer resistance is imperative for enhancing cancer therapeutic efficacy. Confronting these issues necessitates an in-depth study of the sophisticated interactions within the tumor microenvironment and the cellular mechanisms that allow for the survival and growth of cancer cells in defiance of therapeutic efforts. As research advances, the targeting of these resistance pathways may pave the way for the development of more effective therapeutic strategies and reduce the likelihood of relapse and improve outcomes for cancer patients, as presented in Table 2.
Off-target effects of nanodrugs: Targeting cancer stem cells (CSCs) in particular has made it possible to improve drug delivery and overcome drug resistance owing to the introduction of nanotechnology in cancer therapy. Although the use of nanodrugs has many benefits, there are also several hazards, particularly with respect to off-target effects, that could compromise the safety and effectiveness of treatment. The potential for unexpected interactions with nontarget cells and tissues to result in negative side effects that could make treatment outcomes more difficult is one of the main concerns with nanodrugs [130, 131]. In the tumor microenvironment (TME), for example, nanoparticles intended to specifically target CSCs may unintentionally impact normal stem cells or other cell types, leading to toxicity and possibly worsening treatment resistance [132].
A number of variables, such as the size, shape, composition, surface charge, and physicochemical characteristics of nanoparticles, might result in off-target effects. These features have a major impact on the cellular absorption and biodistribution of nanomedicines. For example, whereas larger particles might not effectively penetrate tumor tissues, excessively small nanoparticles might quickly accumulate in the liver and spleen, decreasing their availability to target tumors [133]. Furthermore, the surface functionalization of nanoparticles, which is meant to improve targeted specificity, may inadvertently increase binding to cells that are not the intended target, which could result in cytotoxicity. Because immune cells may be stimulated to attack both the tumor cells and the surrounding healthy tissue, this unintentional activation of immune responses might further complicate the treatment landscape and increase the risk of systemic adverse effects [134].
The possibility of changing drug release profiles from nanodrugs, which may affect their safety and therapeutic efficacy, is another major concern. Drug stability and release kinetics can be affected by changes in environmental factors, including pH and temperature; however, regulated release is essential for reducing toxicity and optimizing therapeutic effects [135]. A nanodrug may cause elevated off-target toxicity if its payload is released too quickly or incorrectly, which could result in high local concentrations of the drug in nontargeted locations.
Furthermore, persistent nanoparticle buildup in the body can lead to new hazards, especially in regard to compounds that cannot be easily eliminated. The long-term presence of these substances may cause chronic inflammatory reactions, which may result in additional issues such as fibrogenesis or cancer [132, 136]. For example, certain metal-based nanoparticles can cause oxidative stress in tissues that are not their intended targets, leading to cellular damage that may promote the growth of tumors or the establishment of drug-resistant cell populations [137, 138].
The possible hazards associated with the use of nanoparticles are also significantly influenced by their immunological response. According to Xiong et al. [139], nanoparticles can trigger an immunological response and activate the complement system, which can either improve antitumor immunity or cause severe allergic reactions and anaphylaxis in susceptible people. The production of antibodies against the nanoparticles due to the immunogenicity of the nanocarriers may eventually diminish their effectiveness and encourage the emergence of drug resistance.
Careful assessment of the safety and effectiveness characteristics of nanodrugs in preclinical and clinical settings is crucial to reduce these hazards. To evaluate how nanoparticles interact with biological systems and find any possible off-target effects, advanced characterization techniques should be used [39, 132]. Furthermore, techniques such as employing stimuli-responsive release mechanisms or covering nanoparticles with biocompatible materials might improve selectivity and lower the possibility of off-target interactions [132, 139]. Creating predictive models to comprehend the biodistribution and pharmacokinetics of nanomedicines can also help with design optimization for increased safety and efficacy in targeting CSCs.
In summary, although nanodrugs have the potential to prevent drug resistance in mammalian cancer stem cells, there are several hazards associated with their use, especially in regard to off-target effects. Optimizing the design of nanoparticles and enhancing therapeutic results require an understanding of how they interact with biological systems. To fully utilize nanotechnology in cancer treatment, it will be necessary to optimize nanodrug compositions and conduct thorough safety assessments as research advances.
Extrinsic factors
With respect to mammalian cancer stem cell (CSC) resistance, extrinsic variables are crucial. One significant extrinsic element influencing CSC survival and function is the tumor microenvironment (TME) [47], which is an ecosystem in which cancer cells interact with various components, such as immune cells, stromal cells and the extracellular matrix [140, 141]. Factors within the TME, such as the complex network of the tumor stroma and epidermal microenvironment, stimulate tumor progression via CSC plasticity [142]. Furthermore, microenvironmental cues regulate the biological phenotypes of CSCs and impact their ability to self-renew, differentiate, and respond to therapy [143].
These interactions influence tumor survival, growth and response to therapies. Additionally, factors such as hypoxia, nutrient deficiency, and acidic pH in the TME contribute to cancer malignancy and drug resistance [144]. Metabolic analysis constitutes a fundamental component in elucidating the complexities associated with the tumor microenvironment (TME) and its consequent effects on tumor metabolism and therapeutic responsiveness [145]. This analytical approach serves to uncover the mechanisms by which alterations within the TME can modulate the efficacy of cancer treatments. A prominent indicator of these modifications is the progressive decline in the anticancer potency of a pharmacological agent postadministration. This reduction in treatment efficacy may arise from a multitude of factors. A particularly salient factor is the activation of a secondary proto-oncogene, which has the potential to evolve into a novel driver gene that catalyzes tumor proliferation, thereby diminishing the drug's overall effectiveness. Furthermore, mutations or variations in the expression levels of drug targets can significantly erode therapeutic outcomes, consequently diminishing treatment effectiveness [61]. Additionally, alterations in the TME itself, which may ensue as a consequence of initial therapeutic intervention, can further exacerbate this attenuation in drug efficacy. Such alterations may encompass variations in nutrient availability, fluctuations in oxygen concentrations, and modifications in the overall cellular architecture of the TME, all of which can foster a more conducive milieu for tumor resilience and therapeutic resistance [66]. A thorough comprehension of these metabolic and microenvironmental factors is essential for the development of more effective cancer treatment strategies. By elucidating and targeting these mechanisms of resistance, researchers can strive to surmount the obstacles that impede the effectiveness of existing therapies, potentially culminating in improved clinical outcomes for patients. Efforts continue to target TME components, inhibit tumor matrix remodeling, and explore metabolic interactions between cells and organelles to develop innovative therapeutic strategies to address the challenges posed by the TME [146]. Understanding how external factors influence cancer stem cell (CSC) behavior is crucial for developing strategies that effectively counter therapeutic resistance and improve cancer treatment outcomes.
Tumor heterogeneity: As a distinguishing feature of mammalian cells, tumor heterogeneity reflects the heterogeneous nature of malignant cells and is visible both in vitro and in vivo. Intertumoral and intratumoral heterogeneity are the two primary categories into which heterogeneity can generally be divided [147]. The term "intertumoral heterogeneity" describes the variations that occur when tumors from several tissues and cell types are combined. Tumors originating from various tissues, tumors of the same tissue type from different patients, and even different tumors within the same person exhibit these differences. This type of heterogeneity emphasizes the individuality of every tumor by showing how malignancies can act significantly differently on the basis of their particular genetic and environmental circumstances, even when they are found in the same organ [148]. Conversely, intratumoral heterogeneity describes the variety that exists inside a single tumor. A combination of noncell-autonomous factors, such as the various components of the tumor stroma, and cell-autonomous factors, such as genomic and epigenomic variations, are responsible for this internal variability. The characteristics of tumor cell populations within a single tumor might differ greatly, including growth rates, genetic stability, cell surface indicators, growth rates, and responses to treatments. Tumor cell populations are heterogeneous, as numerous studies have confirmed their presence [32, 39, 149]. Because it affects how each patient responds to treatment and hastens the emergence of resistance to targeted medicines, this variation has significant therapeutic ramifications. For this reason, comprehending and treating tumor heterogeneity is essential for the development of more specialized, targeted cancer treatments.
Even under optimal culture conditions, human and mouse embryonic stem cells are heterogeneous and include both partially committed and pluripotent cells. Adult organ somatic stem cells are also diverse, with numerous subpopulations of self-renewing cells with unique regeneration capacities [147]. Extrinsic factors that contribute to tumor heterogeneity include microenvironmental factors that directly impact the cell of origin and can potentially lead to differentiation [150]. Furthermore, the tumor immune signature, which is influenced by the mutational landscape and microbiome components, plays a crucial role in determining the different cellular components of the tumor microenvironment (TME) in different cancer types and influences patient prognosis and drug responses [151]. These external factors, such as the TME and cellular components, are essential for understanding the variability of drug responses between tumor cells, even within clonal sublineages, highlighting the importance of both genetic and nongenetic factors in tumor relapse and acquired resistance [152].
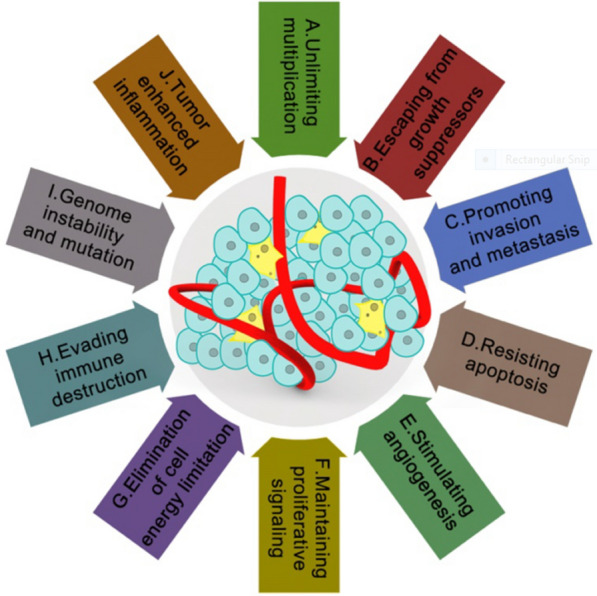
Tumor microenvironment: The tumor microenvironment (TME) is a complex ecosystem that encompasses not only cancer cells but also a diverse array of stromal cells, vascular cells, immune cells, and extracellular matrix components. These noncancerous elements actively participate in the cellular and molecular processes that drive tumor invasion, metastasis, and overall progression [153]. Although the importance of the TME in cancer development and progression has been recognized for many years, its influence on the tumor response to therapies is becoming increasingly clear. The interactions occurring within the TME can profoundly affect the effectiveness of cancer treatments, either by fostering tumor growth and survival or by contributing to therapeutic resistance [39, 149]. Moreover, the TME is shaped by a variety of factors closely linked to the hallmarks of cancer—ten key characteristics that define the behavior of cancer cells. These hallmarks include sustained proliferative signaling, evasion of growth suppressors, resistance to cell death, enabling replicative immortality, induction of angiogenesis, activation of invasion and metastasis, genome instability and mutation, tumor-promoting inflammation, deregulation of cellular energetics, and avoidance of immune destruction. Each of these characteristics directly influences the TME, creating a dynamic environment that can either support or hinder the success of cancer therapies [149]. Therefore, understanding the role of the TME in cancer biology is crucial for developing more effective treatment strategies. By targeting various components of the TME, researchers aim to overcome some of the barriers that currently limit the effectiveness of cancer treatments, ultimately leading to better outcomes for patients.
, as demonstrated in Fig. 1.
Fig. 1.
The tumor microenvironment and characteristics of cancer patients from Abadjian et al. [164]
The tumor microenvironment (TME) is integral to the emergence of drug resistance in mammalian neoplastic cells. A significant element within the TME that exacerbates this resistance is hypoxia, characterized by diminished oxygen levels. The aberrant formation of blood vessels within tumors frequently leads to the establishment of hypoxic regions, subsequently triggering the activation of the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α). HIF-1α facilitates the transcription of genes that enable cancer cells to endure adverse conditions, including the acquisition of resistance to chemotherapeutic agents. Furthermore, it catalyzes metabolic reprogramming, promotes glycolysis and increases the expression of drug efflux transporters such as P-glycoprotein, which diminishes intracellular drug retention and therapeutic efficacy [154], and further stated in Fig. 5.
Fig. 5.

An review of the ABC transporter, LRP, Bcl-2, and Topo ll drug resistance pathways in cancer cells. An ATP-activated transporter is the ATP-binding cassette (ABC) transporter. To remove foreign molecules (such as antibiotics and anticancer drugs) from the intracellular environment, cells express ABC transporters during general chemotherapy. The three main members of the ABC transporter family are P-glycoprotein (P-gp), multidrug-resistant protein 1 (MRP-1), and breast cancer resistance protein (BCRP). Lung resistance protein (LRP) is found in cytoplasmic vaults and aids in the exocytosis of foreign substances, such as anticancer medications. Additionally, studies have shown that p53 loss of function, downregulation of topoisomerase II (Topo-II), and upregulation of bcl-2 (an antiapoptotic factor acted upon by anticancer agents that activate the normal apoptotic process) also decrease cell apoptosis to increase the resistance of cancer cells to anticancer drugs [353, 354]
Another critical dimension of the TME involves the participation of cancer-associated fibroblasts (CAFs) and supplementary stromal cells. CAFs secrete a myriad of growth factors and cytokines, such as interleukin-6 (IL-6), which activate essential signaling cascades, including JAK/STAT3, pivotal for tumor proliferation and drug resistance. These fibroblasts further accumulate extracellular matrix (ECM) constituents, such as collagen, thereby establishing a robust, fibrotic framework that constitutes a physical impediment, hindering the infiltration of pharmacological agents into the neoplasm [155, 156]. The interplay between cancerous cells and stromal cells constitutes a fundamental mechanism through which the TME bolsters cancer cell viability and diminishes the effectiveness of therapeutic interventions. The immune system is similarly influenced by the TME to facilitate the evasion of therapeutic strategies by cancer cells. Immune evasion and immunosuppression are characteristic features of the TME. Regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) are frequently recruited by tumors, where they attenuate the activity of cytotoxic T cells and natural killer (NK) cells through the secretion of immunosuppressive cytokines, including transforming growth factor-beta (TGF-β) and interleukin-10 (IL-10). Moreover, numerous tumors express immune checkpoint molecules such as PD-L1, which interact with PD-1 receptors on T cells, effectively inhibiting immune responses. This immunosuppressive environment poses a considerable challenge to the efficacy of immunotherapeutic strategies, such as immune checkpoint inhibitors [157].
In addition to forming a physical barrier, the extracellular matrix (ECM) actively contributes to the promotion of drug resistance. Compared with proteins such as collagen and fibronectin, the ECM provides structural integrity for the tumor and facilitates interactions between cancer cells and stromal cells that activate survival signaling pathways, notably the PI3K/Akt pathway. These interactions increase the resilience of cancer cells and impede the effectiveness of therapeutic modalities. The dense and rigid characteristics of the ECM further obstruct the efficient delivery of chemotherapeutic agents, thereby diminishing their therapeutic potential [158–160].
Another mechanism through which the tumor microenvironment (TME) facilitates drug resistance is via exosomes. These diminutive extracellular vesicles, which are secreted by both neoplastic cells and stromal cells, transport a variety of molecular constituents, including proteins, lipids, and microRNAs (miRNAs), which possess the capacity to modify the behavior of recipient cells. Exosomes have the ability to convey miRNAs that inhibit proapoptotic pathways in malignant cells, thereby diminishing their sensitivity to chemotherapy-induced apoptosis. In certain instances, exosomes are responsible for transferring proteins associated with drug resistance, such as drug efflux transporters, to adjacent cells, thereby further disseminating resistance mechanisms [161].
The TME additionally cultivates tumor heterogeneity and clonal evolution, which are essential catalysts of drug resistance. As neoplastic cells engage with various elements of the TME, selective pressures such as hypoxia, nutrient scarcity, and therapeutic interventions instigate genetic and phenotypic modifications. Over an extended period, these pressures culminate in the emergence of drug-resistant clones of cancer cells, which subsequently become predominant within the tumor microenvironment, thereby diminishing the efficacy of treatment. This dynamic characteristic of tumors, shaped by the microenvironment, presents a considerable obstacle for therapies aimed at specific populations of cancer cells [162].
Finally, autophagy, a cellular process that facilitates the recycling of damaged or superfluous components, is frequently upregulated in neoplastic cells situated within the TME. Autophagy confers a survival advantage to cancer cells under adverse conditions such as nutrient deprivation or exposure to chemotherapy. Within the TME, the activation of autophagy assists neoplastic cells in resisting therapeutic interventions by obstructing cell death, thereby establishing autophagy as a potential therapeutic target for increasing the efficacy of cancer treatment modalities [163].
The notion of a "niche" pertaining to cancer stem cells (CSCs) has attracted considerable scholarly attention, as it signifies a distinct microenvironment that is instrumental in preserving the unique characteristics of CSCs [102, 165]. This protective niche constitutes a pivotal element that facilitates the retention of CSCs in a quiescent state within various tissues, thereby effectively safeguarding them from the deleterious impacts of chemotherapy and increasing their longevity. The niche delineates the particular microenvironment in which CSCs exist and engage with adjacent cells, encompassing stromal, immune, and vascular components. In the case of normal stem cells, the niche is generally localized within specific vascular territories that govern stem cell functionality during critical processes such as tissue regeneration, maintenance, and repair. This vasculature-associated niche offers both physical and physiological safeguarding, which is essential for the preservation of the stem cell population and the prevention of its depletion [166]. Within the framework of cancer, the tumor microenvironment (TME) assumes a comparable function in bolstering CSCs. It provides indispensable signals that modulate CSC maintenance and self-renewal, as well as homeostatic mechanisms vital for tumor survival, including angiogenesis, hypoxia, and the maintenance of a mildly acidic pH. These conditions within the TME not only facilitate CSC survival and proliferation but also increase resistance to standard cancer therapeutic modalities [167]. Understanding the dynamics of the CSC niche and its interactions within the TME is imperative for the formulation of more efficacious cancer treatment strategies. Targeting the specific components of the niche that underpin CSCs may undermine their capacity to circumvent therapeutic interventions, thereby leading to more successful cancer eradication and mitigating the risk of recurrence.
Intrinsic factors
Intrinsic factors that contribute to tumor heterogeneity include the genetic/epigenetic mutation profile of cells and the specific “cell of origin” within an organ[168]. Genomic instability caused by different mutational processes plays a significant role in heterogeneity within tumors, with tumors harboring high numbers of mutations and copy number alterations exhibiting the most intrinsic heterogeneity [169]. Furthermore, cell-to-cell heterogeneity in gene expression enables flexible cell fate decisions within cancer cells, with tumor-propagating and tumor-initiating cells adopting different molecular states for growth and survival [170]. The influence of intrinsic and extrinsic factors on cancer stem cells contributes to tumor complexity, treatment resistance and invasive ability, highlighting the importance of understanding and targeting these cells for effective therapies [143]. The integration of these intrinsic factors is crucial for the comprehensive treatment of tumor heterogeneity and the development of personalized treatment strategies.
Understanding the intrinsic factors involved in tumor heterogeneity, such as master transcription factors (mTFs) and cancer stem cells (CSCs), is crucial for cancer research and treatment [143, 171, 172]. These factors play important roles in the development of cancer complexity, treatment resistance and disease progression. Insights into the dysregulated gene expression programs controlled by mTFs and the modulation of cancer stem cells by the tumor microenvironment provide valuable information for the development of targeted therapies.
Intrinsic resistance refers to the innate, preexisting resistance to drug treatments observed in patients even before they receive any medication. This particular type of resistance frequently results in diminished efficacy of the treatment from the beginning [66]. Moreover, it is predominantly derived from the spontaneous mutation rate, which is characteristic of all proliferating cells and occurs independently of any external influences. These mutations emerge as a standard aspect of cellular activities and represent a significant factor contributing to the initial resistance noted in certain malignancies. Biologically intrinsic elements, which are responsible for the induction of DNA mutations in humans, are immutable. These elements are intrinsic to the genetic constitution and cellular milieu of an individual and lie beyond the scope of contemporary medical intervention. Consequently, addressing intrinsic resistance poses a considerable challenge in the development of efficacious oncological therapies [173]. Gaining insight into the mechanisms underpinning these intrinsic factors is essential for formulating strategies aimed at augmenting the effectiveness of treatments for patients who display this form of resistance from the outset.
Moreover, recognizing distinct cell populations within tumors and comprehending their interactions and evolution under treatment pressure can aid in the development of more effective combination therapies and ideal treatment strategies. In general, addressing intrinsic factors related to tumor heterogeneity presents promising opportunities to improve cancer treatment outcomes and patient survival.
Epithelial–mesenchymal transition (EMT)-induced multidrug resistance (MDR) in CSCs: Epithelial‒mesenchymal transition (EMT) is a developmental process that regulates embryonic development and involves significant morphological and molecular changes in cells [174]. EMT is a cellular process involved in cancer progression and metastasis and is associated with multidrug resistance (MDR) in cancer stem cells (CSCs) [175, 176].
Epithelial‒Mesenchymal Transition (EMT): The process of epithelial‒mesenchymal transition (EMT) constitutes a critical biological event essential for tissue differentiation during embryonic development, wherein epithelial cells undergo a phenotypic shift to a mesenchymal state. This transition is characterized by a comprehensive array of biochemical alterations that equip cells with increased migratory potential, increased invasiveness, and augmented resistance to programmed cell death, commonly referred to as apoptosis [177]. The orchestration of EMT is mediated by a multitude of signaling cascades, including Wnt/β-catenin, Notch, and TGF-β, which are frequently disrupted throughout the progression of tumor apoptosis [89].
The transformation from an epithelial to a mesenchymal phenotype during EMT is associated with the upregulation of mesenchymal markers and the downregulation of epithelial markers. This phenomenon is correlated with the development of drug resistance and metastatic characteristics within malignant cells [178]. Furthermore, EMT activates signaling pathways that are linked to stem cell-like properties, which further increase resistance to therapeutic interventions. Despite these advancements in understanding, the specific molecular mechanisms that govern EMT and its contribution to multidrug resistance in cancer stem cells (CSCs) continue to represent a significant domain of ongoing investigation. Elucidating these mechanisms is imperative for the formulation of targeted therapeutic strategies aimed at overcoming treatment resistance and enhancing patient prognoses.
ATP-Binding Cassette (ABC) Transporters: ATP-binding cassette (ABC) transporters constitute a family of membrane-associated proteins that are pivotal in the establishment of multidrug resistance (MDR) in neoplastic cells. These transporters operate by actively extruding a diverse array of substrates, including pharmacological agents, toxins, and metabolic byproducts, from the intracellular environment, consequently diminishing the intracellular concentrations of therapeutic compounds [106].
ABC transporters are implicated in several fundamental biological functions, such as the uptake of nutrients, lipid translocation, drug excretion, and the modulation of cellular volume. They play crucial roles in the efflux of various entities from the cell, a process driven by ATP hydrolysis [179, 180]. This family of transporters, encompassing 49 distinct protein variants, is expressed at elevated levels on the surface of cancer stem cells, thereby contributing to their therapeutic resistance. The designation "ABC transporters" originates from the ATP-binding domains that are integral to their functional capacity. Biochemical investigations of these transporters, including P-glycoproteins, have revealed conserved motifs within their ATP-binding regions, indicating their evolutionary conservation across prokaryotic and higher eukaryotic organisms, including Homo sapiens [106]. ABC transporters utilize the energy derived from ATP hydrolysis to facilitate the translocation of molecules across cellular membranes, whether by effluxing them from the cell or translocating them to the outer leaflet of the membrane. Empirical studies have demonstrated that the expression and functional activity of ABC transporters are highly relevant at the blood–brain and blood–spinal cord barriers, where they play a role in the efflux of drugs and other bioactive substances [181], and represented in Fig. 5.
Hypoxia and Reactive Oxygen Species: Oxygen is indispensable for an array of physiological functions, including metabolism, respiration, and cellular proliferation. Nonetheless, oxygen concentrations within tissues frequently fall below atmospheric levels, resulting in hypoxia during extended durations of inadequate oxygen supply. This hypoxic state leads to diminished ATP levels and modified intracellular pH as a consequence of anaerobic metabolic processes and lactate accumulation, ultimately precipitating cell death through diverse mechanisms, including autophagy, apoptosis, necrosis, and necroptosis [182]. Hypoxia represents a salient characteristic of the tumor microenvironment and strongly influences cancer progression [106]. Through the modulation of hypoxia-inducible factors (HIFs), a milieu that is conducive to increased tumorigenicity and the sustenance of cancer stem cells is cultivated. Hypoxia is a significant characteristic of the tumor microenvironment and profoundly influences the progression of cancer. It cultivates an atmosphere that is favorable for increased tumorigenicity and the preservation of cancer stem cells (CSCs) by modulating hypoxia-inducible factors (HIFs). HIFs serve as transcription factors that orchestrate cellular responses to diminished oxygen availability by inhibiting cell differentiation, facilitating angiogenesis, and adjusting apoptotic processes [166].
The tumor microenvironment is defined by hypoxia, increased glycolytic activity, and elevated levels of reactive oxygen species (ROS). Tumors rapidly exhaust local oxygen resources, engendering a hypoxic niche that promotes tumor invasion and metastasis while simultaneously imparting resistance to radiation and chemotherapy modalities [168]. Furthermore, compared with their normal counterparts, malignant cells exhibit increased generation of ROS, which can further exacerbate tumor progression and contribute to therapeutic resistance. ROS play a critical role in the regulation of various signaling pathways and are elevated by hypoxic conditions across numerous cancer cell models. The presence of tumor hypoxia is frequently correlated with adverse patient outcomes because of its detrimental effect on the efficacy of radiation therapy, which predominantly relies on ROS to facilitate the apoptosis of tumor cells [106].
High survival capacity of cancer stem cells: Cancer stem cells (CSCs) exhibit remarkable survival capacities, contributing to drug resistance in cancer treatment. These cells possess self-renewal abilities, strong repair mechanisms, and resistance to apoptosis, making them resilient to chemotherapy drugs [183]. CSCs, which are found in various cancers, including lung and bone sarcomas, are responsible for therapy resistance, relapse, and tumor dissemination. These cells express specific markers and exhibit unique biological characteristics similar to those of normal stem cells, enabling them to evade the effects of chemotherapy and survive treatment, leading to cancer relapse. Cancer stem cells possess many mechanisms to avoid cell death, and some drugs circumvent all of these barriers and successfully damage CSC DNA. However, CSCs have other ways to overcome this damage. CSCs of the lung, pancreas, glioma and breast possess highly active DNA damage response systems [106, 183].
MicroRNAs in the Acquisition of Cancer Stem Cell Phenotypes: RNA plays a critical role in the development and maintenance of cancer stem cells (CSCs) by regulating gene expression and cellular processes [184, 185]. Various RNA modifications, such as inosine, 5-methylcytosine and N6-methyladenosine, have been identified as essential for the control of spatiotemporal gene expression during CSC fate transitions [186]. In addition, long noncoding RNAs (lncRNAs), such as H19, are involved in the regulation of CSC division, reprogramming, metastasis, and drug resistance. The interplay between microRNAs (miRNAs) and signaling pathways such as Wnt/β-catenin and EGFR/IGF1R also influences CSC properties and therapeutic efficacy [185–187]. An understanding of these RNA-mediated pathways is essential for the development of specific treatments aimed at eradicating CSCs and enhancing the efficacy of cancer therapy.
Moreover, RNA expression patterns differ significantly between cancer stem cells (CSCs) and their differentiated progeny. Studies have shown that, compared with nonstem cancer cells, CSCs have distinct miRNA signatures, suggesting a role in tumorigenesis and therapy resistance [44, 187]. In particular, miRNAs such as miR-21, miR-34 and miR-155 are differentially expressed between CSCs and their differentiated counterparts, suggesting that a unique miRNA profile in CSCs may contribute to tumor progression and prognosis [188]. Furthermore, the expression levels of certain miRNAs in CSCs are associated with self-renewal, tumorigenesis, and resistance to chemotherapy, highlighting the importance of understanding these RNA expression patterns for the development of targeted cancer therapies [187–189].
In addition, RNAs, especially noncoding RNAs such as long noncoding RNAs (lncRNAs) and short noncoding RNAs (miRNAs), play crucial roles in regulating the phenotypic acquisition of cancer stem cells (CSCs). For example, lncRNAs such as HOTAIR, H19, and LncTCF7 and miRNAs such as miR-324-5p are considered key regulators of stemness in cancer and enable CSCs to acquire their characteristic properties [190–192]. These RNA molecules modulate CSC initiation, self-renewal, metastasis, and chemoresistance by targeting lineage-related factors and influencing signaling pathways such as the Wnt/β-catenin, NF-κB, and EMT signaling pathways [185, 193, 194]. Furthermore, miRNAs regulate the tumor microenvironment and cellular plasticity, contributing to therapy resistance and poor clinical outcomes in cancers such as head and neck squamous cell carcinomas (HNSCCs) [193, 195]. Gaining knowledge of these biological mechanisms can help develop therapeutic approaches that target CSCs to improve cancer treatment outcomes.
Drug resistance mechanisms in cancer and cancer stem cells
The phenomenon of drug resistance presents a formidable obstacle in oncological treatment, wherein cancer stem cells (CSCs) assume a central role in this dilemma. CSCs possess unique abilities for self-renewal, differentiation, and tumor initiation, thereby significantly contributing to the ineffectiveness of cancer therapies. Their inherent resistance to chemotherapeutic agents and radiation frequently culminates in treatment failure, precipitating in tumor recurrence and metastasis, which considerably adversely influence patient prognosis and increase cancer-related mortality [106]. The prevalent issue of drug resistance in neoplastic cells is a fundamental determinant of the unfavorable prognostic outcomes observed in numerous patients. The survival of cancer cells after therapy often results in recurrence and metastasis, which are the principal contributors to mortality among individuals afflicted with malignant death [196]. Drug resistance can be classified into two categories: primary resistance, which is established prior to therapeutic intervention, and acquired resistance, which emerges throughout the therapeutic regimen. These mechanisms of resistance may arise from modifications in drug metabolism, including enhanced detoxification or sequestration of pharmacological agents, as well as alterations in drug targets [197].
Among CSCs, the mechanisms underlying drug resistance exhibit a particularly intricate and multifarious nature. A notable mechanism is the heightened expression of ATP-binding cassette (ABC) transporters. CSCs frequently overexpress these transporters, which facilitate the active efflux of chemotherapeutic agents from the cells, thereby diminishing the intracellular concentrations of these drugs and their therapeutic efficacy. Furthermore, CSCs have the capacity to activate cytoprotective and survival signaling pathways, which further bolster their resistance to therapeutic interventions. The dysregulation of signaling pathways associated with stemness also plays a role in drug resistance by perpetuating CSC characteristics that confer treatment resistance [102].
Moreover, CSCs are distinguished by their robust DNA repair mechanisms, which mitigate the damage inflicted by chemotherapeutic agents. They may also demonstrate increased quiescence, existing in a dormant state that renders them less vulnerable to drugs that target rapidly proliferating cells. The elevated levels of autophagy observed in CSCs facilitate their survival under conditions of drug-induced stress. Additionally, CSCs possess the ability to evade immune surveillance, thereby diminishing the efficacy of immunotherapeutic approaches. Deficiencies in mitochondria-mediated apoptotic pathways and the upregulation of antiapoptotic proteins within CSCs further exacerbate their therapeutic resistance. Finally, the activation of the PI3K/AKT signaling pathway in CSCs is integral to promoting cell survival and resistance to therapeutic modalities [102, 194]. A comprehensive understanding and targeted intervention of these diverse resistance mechanisms are imperative for the advancement of more efficacious cancer therapies and the enhancement of patient outcomes. By concentrating on the specific pathways and processes that contribute to CSC drug resistance, researchers aspire to surmount some of the considerable challenges posed by these resilient neoplastic cells.
Other mechanisms contributing to drug resistance in CSCs include the expression of CSC markers, epithelial–mesenchymal transition, hypoxia, intercellular communication in the tumor microenvironment, and inflammation [40, 194, 198]. CSCs also exhibit a high level of plasticity, quickly adapt to changes in the tumor microenvironment and are intrinsically resistant to current chemotherapies and radiotherapies [199]. The molecular mechanism of resistance in individual tumors is determined by the dependence of CSCs on the targeted pathway. Genetically based resistance occurs in tumors that contain CSCs through specific pathways, whereas tumors with bulk tumor cells but independent of CSCs acquire resistance through epigenetic reprogramming [26, 200]. Multiple cellular and molecular mechanisms have been shown to contribute to multidrug resistance (MDR) in CSCs [200, 201]. Understanding these mechanisms is crucial for developing strategies to overcome chemoresistance in CSCs, improve cancer treatment outcomes [201] and possibly eliminate CSCs, which are responsible for tumor initiation, maintenance, and metastasis (Fig. 2).
Fig. 2.

Depiction of the primary mechanisms that enable cancer cells to become drug resistant; source: Housman et al. [202]
Drug inactivation
The activation of pharmaceuticals in vivo represents a multifaceted process characterized by interactions between various chemical entities and an array of proteins. These interactions can alter the pharmacological properties of drugs, partially degrade them, or facilitate their binding with additional molecules or proteins, thereby culminating in their activation. For a considerable number of anticancer agents, the phenomenon of metabolic activation is paramount in attaining their therapeutic efficacy. Nonetheless, tumor cells have the potential to evolve mechanisms of resistance against these therapeutic modalities by attenuating the degree of drug activation [202].
In addition to anticancer agents, drug inactivation is a thoroughly documented mechanism of resistance in various other contexts. For example, the efficacy of penicillin G is compromised by the production of β-lactamases by penicillin-resistant bacterial strains; the therapeutic utility of chloramphenicol is significantly reduced by the action of chloramphenicol acetyltransferase in resistant organisms; and aminoglycosides undergo inactivation through enzymatic modifications such as phosphorylation, adenylation, or acetylation by aminoglycoside-modifying enzymes [203]. This exemplifies the critical role that drug inactivation assumes within the spectrum of resistance mechanisms applicable across diverse treatment modalities. In the context of cancer cells, resistance may manifest through an array of mechanisms, which include the augmented efflux of drugs from the cellular interior, the diminished influx of drugs into the cellular environment, and the direct inactivation of drugs themselves [202, 204, 205]. A comprehensive understanding of these mechanisms is vital for the formulation of strategies aimed at overcoming drug resistance and enhancing the efficacy of therapeutic interventions.
Inactivation of drugs can render them ineffective in fighting cancer cells, allowing the cells to survive and proliferate despite treatment attempts. Understanding the mechanisms underlying drug inactivation is crucial for developing strategies to overcome resistance and improve treatment outcomes. Different studies highlight the importance of exploring novel therapies and targeted agents to combat drug resistance and improve the effectiveness of cancer treatments [204, 206]. By studying drug inactivation and other resistance mechanisms, researchers have aimed to develop more effective treatment options and prevent the development of drug-resistant cancer cells.
Alterations in multidrug resistance (MDR) drug targets in CSCs
The therapeutic efficacy of pharmacological agents is profoundly affected by their interactions with designated molecular targets, and modifications to these targets can lead to drug resistance. Within the sphere of oncological treatment, modifications in drug targets—such as genetic mutations or variations in expression levels—can drastically diminish the effectiveness of therapeutic interventions. For example, many anticancer pharmacotherapies are engineered to interact with topoisomerase II, an enzyme that plays a pivotal role in the regulation of DNA supercoiling during cellular replication. Typically, the interaction between DNA and topoisomerase II is ephemeral; however, certain pharmacological agents stabilize this interaction, which may precipitate enduring DNA damage, disrupt DNA synthesis, and cessation of cellular division [202]. In neoplastic cells, particularly in cancer stem cells (CSCs), these alterations in molecular targets can lead to the development of multidrug resistance (MDR). Genetic mutations in the genes encoding these pharmacological targets or fluctuations in their expression levels can undermine the efficacy of agents that depend on these targets for their therapeutic action. For example, if the expression of topoisomerase II is modified or if the enzyme itself experiences mutations that impair drug binding, the drugs intended to inhibit this enzyme may exhibit diminished effectiveness. This situation exacerbates the overarching issue of MDR, wherein cancer cells, including CSCs, acquire resistance to an array of pharmacological agents as a result of such target alterations. Comprehending these modifications is imperative for the development of more efficacious therapeutic strategies and for overcoming the obstacles to successful oncological treatment.
As a result, drug target modification is a key mechanism contributing to drug resistance in mammalian cancer cells. This change can be caused by various factors, such as genetic, epigenetic and microenvironmental influences [204]. Furthermore, the RNA modification of N6-methyladenosine (m6A) plays a crucial role in drug resistance by affecting drug efficacy through the restructuring of multidrug efflux transporters, drug-metabolizing enzymes, and anticancer drug targets [207]. Furthermore, changes in the transcriptome, transcription factors, and DNA and chromatin regulatory proteins have been identified as key factors driving drug resistance in cancer cells, highlighting the complexity of this phenomenon [208]. Understanding and targeting these changes in drug targets is critical for developing effective strategies to overcome drug resistance in mammalian cancer cells.
Alterations in drug targets have been found to contribute to multidrug resistance (MDR) in cancer stem cells (CSCs) [209]. Epigenetic modifications play crucial roles in the initiation, formation, and maintenance of CSCs; aberrant epigenetic reprogramming has been implicated in the tumorigenesis of pediatric and early-stage adult cancers; and the overexpression of ATP-binding cassette (ABC) transporters, such as P-glycoprotein (P-gp)[179], was initially considered the main mechanism for drug resistance [210, 211]. Targeting CSCs and MDR has become an important focus in cancer research, with efforts to develop drugs and therapies specifically aimed at overcoming MDR and eradicating CSCs [31], and refer Fig. 5. Studies have identified potential MDR genes and epigenetic alterations associated with resistance to therapy, providing potential therapeutic targets for treatment-resistant patients [212]. Additionally, combination therapies involving CSC-targeting vaccines and conventional chemotherapies have shown promise in suppressing tumor growth, circumventing MDR, and increasing the efficacy of treatment [213]. These alterations lead to temporal intratumoral heterogeneity and clonal evolutionary processes, resulting in the emergence of MDR. Additionally, metabolic alterations in CSCs affect extracellular vesicle (EV) cargo and release, contributing to the acquisition of MDR. Understanding these alterations and their role in MDR may provide potential therapeutic targets for overcoming drug resistance in CSCs and would be indispensable for developing targeted therapies to overcome resistance, which are important areas of research in the field of oncology.
Existing drugs have shown promise in targeting cancer stem cells (CSCs) in mammals. CSCs are subpopulations of cancer cells that have self-renewal and differentiation capacity and are thought to be key drivers of tumor growth and recurrence. Several intracellular signaling pathways, including protein kinase signaling pathways, have been identified as critical for CSC regulation and drug resistance [214]. Efforts are being made to target CSCs with kinase inhibitors, and preclinical and clinical studies have shown potential for the use of kinase inhibitors alone or in combination with current therapies for effective cancer treatment [215]. Additionally, repurposed drugs, such as those used for diabetes, parasitic diseases, and inflammatory diseases, have shown efficacy in targeting CSCs [216]. However, further investigations are needed to evaluate the efficacy of these drugs without adverse effects on normal stem cells. Overall, while existing drugs have shown promise in targeting CSCs, more research is needed to optimize their effectiveness and minimize side effects.
3.3. Epigenetic alterations induced by drugs.
Epigenetic modifications play a pivotal role in the emergence of drug resistance within mammalian neoplastic cells [208, 217]. These modifications can profoundly influence the responsiveness of cancer cells to therapeutic interventions and contribute to the intricate process of carcinogenesis. Among the principal epigenetic alterations that affect drug resistance are DNA methylation and histone modifications, including acetylation and methylation [202]. DNA methylation involves the covalent addition of methyl groups to the DNA molecule, which may result in the silencing of gene expression and subsequent alterations in cellular functionality. This mechanism can precipitate the activation of resistance pathways or the suppression of genes that are critical for the efficacy of pharmacological agents. Conversely, histone modifications modulate the accessibility of DNA for transcriptional processes. Acetylation of histones typically results in relaxation of the chromatin structure, thereby increasing DNA accessibility and potentially affecting drug sensitivity, whereas histone methylation can lead to more condensed packaging of DNA, diminishing gene expression and altering the cellular response to therapeutic strategies. These epigenetic alterations are implicated not only in resistance to existing therapeutic modalities but also in the evolution and advancement of malignancies. By modulating the expression of genes that are integral to drug metabolism, DNA repair mechanisms, and cellular survival, epigenetic modifications can foster a more robust cancer cell phenotype. A comprehensive understanding of these mechanisms is imperative for the advancement of targeted therapeutic strategies that can circumvent epigenetically mediated resistance and enhance overall treatment efficacy.
Moreover, epigenetic changes include alterations in the expression of noncoding RNAs, which play a significant role in the development of mechanisms that confer resistance. Modifications in the epigenome can result in the upregulation of particular genes, such as ABCB1, which facilitates resistance to multiple drugs [218].
Epigenetic dysregulation represents a prevalent feature observed in nearly all forms of human malignancies. Neoplasms exhibit a dynamic epigenetic landscape characterized by alterations in the modifications of DNA promoter regions, irregular patterns of histone protein acetylation or methylation, and aberrant expression of repetitive DNA sequences. Collectively, these modifications significantly influence tumor biology and facilitate the onset and progression of cancer. The importance of epigenetic mechanisms in the context of drug resistance becomes particularly pronounced when resistance manifests swiftly, is ephemeral, and demonstrates functional heterogeneity. In contrast to genetic mutations, which tend to be stable and enduring, epigenetic modifications can exhibit greater fluidity and reversibility. This variability in epigenetic states may result in the rapid emergence of drug-resistant phenotypes that vary among tumor cells. Comprehending these epigenetic alterations is imperative for elucidating the mechanisms underlying resistance evolution and for formulating strategies to mitigate these processes in cancer therapeutics [38].
The use of epigenetic medications, such as inhibitors of DNA methyltransferase and histone deacetylase, has the potential to reverse drug resistance and increase sensitivity to conventional therapies [219]. It is imperative to understand the epigenetic profile of cells that are tolerant to drugs, as they have the ability to endure treatment and reestablish the tumor, thereby establishing enduring features of resistance to drugs. In general, manipulation of the epigenome has emerged as a potential therapeutic approach for sensitizing drug-resistant cells and increasing the effectiveness of treatment in cancer cells of mammalian origin.
3.4. Repairs of DNA damage and multidrug resistance (MDR) in CSCs.
DNA repair mechanisms are pivotal for preserving genomic integrity, especially within cancer stem cells (CSCs), which are often characterized by their propensity for multidrug resistance (MDR). During the processes of DNA replication and repair, DNA polymerases may inadvertently introduce errors that culminate in potentially deleterious mutations. To mitigate these events, cells initiate extensive DNA damage response (DDR) pathways, which provide the requisite temporal and mechanistic resources for substrate-specific repair processes to mitigate damage [210, 220]. Five principal DNA repair pathways operate at distinct phases of the cell cycle to rectify various forms of DNA damage. These pathways include base excision repair (BER), which involves minor, nonhelix-distorting base lesions induced by oxidative stress or spontaneous hydrolysis; nucleotide excision repair (NER), which involves substantial, helix-distorting lesions resulting from ultraviolet radiation or chemical exposure; mismatch repair (MMR), which corrects replication inaccuracies such as base–base mismatches and insertion–deletion loops; homologous recombination (HR), which accurately restores double-strand breaks employing a homologous template; and nonhomologous end joining (NHEJ), which directly ligates double-strand breaks, frequently in a manner that lacks the precision characteristic of HR [220].
DNA damage may originate from both intrinsic and extrinsic factors. Endogenous damage predominantly arises from internal cellular mechanisms, including oxidative stress attributable to reactive oxygen species (ROS) and hydrolytic reactions that naturally transpire within cellular environments [40, 220]. In the context of CSCs, these repair mechanisms assume a vital function in their capacity to withstand therapeutic measures. Augmented DNA repair ability in CSCs can significantly underpin their resistance to a multitude of pharmacological agents, as these cells can effectively rectify damage induced by chemotherapeutic agents and radiation therapy. A comprehensive understanding of the complexities inherent in these repair pathways is essential for the formulation of strategies aimed at overcoming MDR in oncological treatment and enhancing patient prognoses [28, 220].
In cancer stem cells (CSCs), DNA damage repair systems serve two distinct functions. Aggressive CSC characteristics are driven by genomic instability caused by mutations in DNA repair genes, even though improved DNA repair in CSCs leads to therapeutic resistance and tumor progression [221]. Furthermore, genetic alterations that affect tissue homeostasis and increase the risk of cancer can be caused by DNA damage in stem cells, including CSCs [222]. Inducing synthetic lethality and sensitizing CSCs to anticancer medicines requires targeting their DNA repair mechanisms. To successfully target CSCs and enhance patient outcomes, tailored cancer treatments must consider the complex interplay between DNA damage repair and CSC maintenance. Furthermore, CSCs are known to exhibit multidrug resistance (MDR) via various mechanisms, including increased DNA repair efficiency [221, 223]. CSCs possess properties that enable them to repair DNA damage more effectively than other cancer cells do, which contributes to their resistance to chemotherapy and radiotherapy [113]. This enhanced DNA repair can occur through increased expression and splicing fidelity of DNA repair genes, robust activation of cell cycle checkpoints, and increased homologous recombination-mediated DNA repair [224]. Additionally, there is a relationship between MDR and autophagy in CSCs [200]. Understanding the DNA damage response (DDR) in CSCs and targeting this pathway could lead to novel therapeutic approaches to overcome MDR and improve treatment efficacy in cancer patients.
Drug efflux
A fundamental mechanism through which neoplastic cells acquire resistance to chemotherapeutic agents is the augmented efflux of pharmacological compounds. This phenomenon significantly curtails the intracellular concentration of therapeutic agents, thereby reducing their pharmacological effectiveness. Integral to this mechanism are proteins belonging to the ATP-binding cassette (ABC) transporter family. These transporters are extensively characterized regulatory entities situated in the plasma membranes of both normal and malignant cells, and they play pivotal roles in the translocation of various substances across cellular membranes [202].
ABC transporters are transmembrane proteins that are present not only in human cells but also throughout all identified biological kingdoms. Their primary role is to facilitate the transport of a heterogeneous array of molecules, including pharmaceuticals, from the intracellular environment to the extracellular space. This process is driven by the hydrolysis of ATP, which supplies the requisite energy for the active translocation of substrates against their concentration gradients. In cancer cells exhibiting drug resistance, particular drug efflux transporters are frequently overexpressed. Prominent among these proteins are P-glycoprotein, breast cancer resistance protein (BCRP), and multidrug resistance-associated proteins (MRPs). P-glycoprotein, in particular, is recognized as an ABC transporter characterized by its extensive substrate specificity and is acknowledged as a significant contributor to therapeutic resistance in the treatment of leukemia and various solid tumors [166], and demonstrated in Fig. 5. The heightened expression levels of these transporters in cancer stem cells (CSCs) further complicate the challenge of effective treatment by actively expelling chemotherapeutic agents prior to their ability to manifest therapeutic effects. Comprehending the function and regulatory mechanisms of these efflux transporters is imperative for the formulation of strategies aimed at mitigating drug resistance. By targeting these transporters or inhibiting their activity, it may be feasible to augment drug retention within neoplastic cells and enhance the overall effectiveness of anticancer therapeutic interventions.
Furthermore, drug efflux, particularly that mediated by proteins such as MDR1, is a significant risk factor for drug resistance in cancer stem cells (CSCs) [102, 225]. CSCs, owing to their unique biological characteristics and resemblance to stem cells, possess mechanisms such as ABC protein drug pump function, which aids in coping with chemotherapy drug invasion. This drug efflux capability, along with other properties, such as quiescence and evasion of apoptosis, contributes to therapy resistance in CSCs. Resistance to protein-degrading drugs, including those targeting key oncogenes, can be mediated by MDR1 overexpression, but this resistance can be overcome by combination treatment with MDR1 inhibitors such as lapatinib. Therefore, understanding and targeting drug efflux mechanisms are crucial for combating CSC-mediated drug resistance in cancer therapy.
Cancer cell inhibition of drug resistance (DR) in CSCs
Autophagy plays a significant role in multidrug resistance (MDR) in cancer stem cells (CSCs) [226]. It helps CSCs maintain stemness, adapt to changes in the tumor microenvironment, and promote tumor survival. However, autophagy can also act as a double-edged sword, with both protumorigenic and antitumorigenic properties depending on the context [227]. Autophagy-related proteins, such as Beclin1 and MAPLC3-II, are upregulated in CSCs with MDR, and inhibiting autophagy can sensitize drug-resistant CSCs to treatment [41, 58, 223, 228]. Similarly, resistance to apoptosis and multidrug resistance (MDR) are common characteristics of cancer stem cells (CSCs) [198, 229]. The development of MDR in CSCs involves dysregulation of apoptotic pathways, including the Bcl-2 superfamily, as well as overactivation of the PI3K/AKT pathway, and inhibitors of apoptosis (IAPs) family members contribute to the development of MDR in CSCs (Fig. 3 and Table 3). Cancer stem cells (CSCs) frequently exhibit increased expression of ATP-binding cassette (ABC) membrane transporters, which significantly contributes to their resistance to chemotherapeutic agents. These transporters promote the efflux of pharmacological agents from the cellular interior, thereby diminishing their therapeutic efficacy. Moreover, CSCs often display the activation of survival signaling cascades, dysregulation of stemness-related signaling pathways, and anomalies in DNA repair processes, all of which are implicated in their resistance to therapeutic interventions. Additionally, factors secreted by the tumor microenvironment, including those produced by cancer-associated fibroblasts, can further impede CSC apoptosis and reduce the effectiveness of anticancer pharmacotherapy, consequently contributing to multidrug resistance (MDR) [224, 230]. In the context of cellular demise mechanisms, autophagy and apoptosis frequently exert opposing influences; however, both are indispensable in the overarching process of cellular death. Apoptosis, also referred to as programmed cell death, transpires via two principal pathways: the intrinsic pathway, which is governed by mitochondrial dynamics and involves B-cell lymphoma 2 (BCL-2) family proteins, and the extrinsic pathway, which is instigated by death receptors present on the cell surface [202], and depicted in Fig. 5. While these pathways are inherently distinct, they ultimately converge upon the common endpoint of cellular death. Cancer is an extraordinarily intricate disease characterized by multiple hallmarks, and it represents a substantial threat to public health. The predominant cause of mortality associated with cancer is not typically the primary tumor itself but rather metastatic dissemination to distant organs, which complicates both treatment and management strategies [231].
Fig. 3.

Schematic representation of multidrug resistance (MDR) mechanisms in cancer cells. This is a schematic representation of the different mechanisms that lead to the development of multidrug resistance (MDR) in cancer cells. Arrows illustrating their interactions and pathways are used to demonstrate the essential mechanisms, which include drug efflux, changes in drug levels, drug inactivation, EMT, the tumor microenvironment, DNA damage repair, cancer stem cells, and immune evasion. A mechanism is represented by each colored ellipse, and the linkages or signaling exchanges between these mechanisms are shown by arrows. This streamlined illustration aids in illustrating the intricacy of MDR in cancer [135]
Table 3.
Important molecular roles and processes of resistance gene expression in mammalian cancer stem cells
| Resistance Gene | Key Molecular Function | Mechanism of Gene Expression | References |
|---|---|---|---|
| ABC Transporters (e.g., ABCB1, ABCG2) | Efflux of chemotherapeutic drugs | Overexpression or altered expression due to genetic mutations, epigenetic modifications (e.g., DNA methylation, histone modifications), or transcriptional regulation by signaling pathways (e.g., Notch, Wnt) | [115, 265, 266] |
| DNA Repair Genes (e.g., BRCA1, BRCA2, ATM) | Repair of DNA damage caused by chemotherapy or radiation | Overexpression or enhanced activity due to genetic mutations, epigenetic modifications, or transcriptional regulation by signaling pathways (e.g., PI3K/AKT, NF-κB) | [114, 267, 268] |
| Anti-Apoptotic Genes (e.g., Bcl-2, Bcl-xL) | Inhibition of apoptosis | Overexpression or altered expression due to genetic mutations, epigenetic modifications, or transcriptional regulation by signaling pathways (e.g., PI3K/AKT, NF-κB) | [110, 138, 269] |
| Stem Cell Markers (e.g., CD44, CD133, ALDH1) | Maintenance of stem cell phenotype and self-renewal | Overexpression or altered expression due to genetic mutations, epigenetic modifications, or transcriptional regulation by signaling pathways (e.g., Notch, Wnt, Hedgehog) | [22, 107, 194, 270–273] |
| Epithelial–Mesenchymal Transition (EMT) Markers (e.g., vimentin, N-cadherin, Twist) | Acquisition of migratory and invasive properties | Overexpression or altered expression due to genetic mutations, epigenetic modifications, or transcriptional regulation by signaling pathways (e.g., TGF-β, Wnt, Notch) | [121, 274, 275] |
| Metabolic Enzymes (e.g., GLUT1, LDHA) | Altered metabolism to support survival and proliferation under stress conditions | Overexpression or altered expression due to genetic mutations, epigenetic modifications, or transcriptional regulation by signaling pathways (e.g., HIF-1α, MYC) | [110, 111, 276] |
Several strategies have been investigated in the field of cancer research to suppress cancer cells and overcome treatment resistance. Research has indicated the need to focus on particular molecules, such as USP45, which can be blocked by natural substances such as α-mangostin and promote drug resistance, stemness, and cancer formation [64]. Furthermore, the ability of PGC1α, or peroxisome proliferator-activated receptor-γ coactivator-1α, to control the viability of cancer cells under metabolic stress has been clarified, providing information about the survival strategies of resistant cells [232]. How nongenetic adaptations, such as epithelial‒to-mesenchymal transition (EMT), contribute to drug resistance and how to successfully fight resistance, which requires a focus on several signaling pathways, has also been explored [180]. Understanding the complex interplay between autophagy and MDR and between apoptosis and MDR in CSCs may lead to the development of targeted therapeutics to overcome MDR in CSCs and is crucial for developing effective cancer therapies. Together, these results support current initiatives to suppress cancer cells and address medication resistance in cancer cell treatment.
Heterogeneity of cancer cells
Cancer stem cells (CSCs) are distinguished by their capacity to undergo asymmetric cell division, a mechanism that produces both CSCs and differentiated progeny. This phenomenon leads to a diverse array of cell types within neoplasms, thereby contributing to significant phenotypic and functional heterogeneity. This intrinsic variability serves as a pivotal element in the complexity and treatment resistance frequently observed in CSC-enriched metastases. Traditionally, the concept of heterogeneity within a CSC population has been underappreciated, as it is often more convenient to conceptualize CSCs as a homogeneous subset of cells. Nonetheless, emerging empirical evidence indicates that CSC populations can exhibit substantial heterogeneity, which complicates clinical therapeutic approaches. This internal heterogeneity among CSCs plays a critical role in treatment failure and relapse by facilitating the emergence of various drug-resistant and immune-evasive clones, thereby rendering the disease more challenging to manage and eradicate [106].
In particular, drug resistance is significantly influenced by the heterogeneity of cancer stem cells (CSCs) [29, 233]. Owing to their capacity for self-renewal and differentiation, CSCs frequently elude the effects of immunological, chemo-, and radiotherapy treatments used in conventional cancer therapy [40]. Tumor recurrence and the negation of the effects of chemotherapy are greatly aided by the existence of diverse subpopulations of cancer cells, such as CSCs [234]. Research indicates that CSCs have a condition akin to a plastic stem, resulting in phenotypic variability, which is a crucial element in resistance to therapy [39]. Furthermore, increasing tumor heterogeneity, which affects therapy resistance and promotes malignant characteristics in recurring tumors, is a result of the hierarchical organization of cancer cell populations, especially in a scenario involving CSCs [38, 39]. To combat drug resistance in cancer treatment, comprehending the mechanisms underlying CSC heterogeneity and designing more potent therapeutic approaches are imperative.
The term "niche" describes specific microenvironments observed in different tissues that are home to stem cells, including cancer stem cells (CSCs). The control of stem cell processes such as self-renewal, differentiation, and carcinogenesis depends on these habitats. They accomplish this by means of an intricate interaction between growth factors, cytokines, short RNAs, and extracellular matrix constituents, all of which are involved in the maintenance and operation of stem cells [106]. The CSC niche in cancer includes a variety of stromal cells, such as immune cells, fibroblasts, mesenchymal cells, and endothelial cells. These cells work together to form a milieu that promotes CSC proliferation and survival [235].
The CSC niche contributes significantly to treatment resistance in addition to helping to maintain CSCs. Stem-related signaling pathways that lead to drug resistance are maintained in part by the distinct morphological and molecular features of the CSC niche [106]. For example, the niche of cancer-associated fibroblasts (CAFs) can secrete cytokines such as interleukin-17A (IL-17A), which encourage CSC self-renewal and invasiveness to produce a chemoresistant environment [235]. Furthermore, mesenchymal stem cells (MSCs) in the niche can promote angiogenesis and provide an immunosuppressive milieu, which can accelerate the growth of tumors. Because they play crucial roles in angiogenesis—the process by which new blood arteries emerge from existing arteries—endothelial cells (ECs) stand out in particular [236]. In a similar vein, MSCs stimulate the growth of cancer by interacting with CSCs and creating an environment that supports the survival and spread of tumors. The development of ways to overcome therapeutic resistance and enhance the outcomes of cancer treatment requires an understanding of the dynamic interactions between CSCs and their habitat [106].
Signaling pathways for multidrug resistance (MDR) in CSCs
The phenomenon of drug resistance associated with cancer stem cells (CSCs) necessitates an in-depth examination of the intricate molecular pathways that govern CSC behavior, the therapeutic ramifications of targeting these pathways, and the distinctive attributes of CSCs across diverse malignancies. A comprehensive understanding of these elements may enhance the formulation of innovative therapeutic strategies designed to overcome drug resistance and improve patient prognosis.
Molecular Pathways in the CSC-Mediated Drug Resistance Notch Signaling Pathway: The Notch signaling pathway serves as a pivotal modulator of cell fate determination, thereby influencing the preservation of CSC characteristics. In the context of breast cancer, for example, Notch signaling is markedly upregulated within CSCs, thereby fostering their survival and self-renewal capabilities [237, 238]. This pathway not only augments the proliferation of CSCs but also plays a significant role in their resistance to therapeutic interventions such as chemotherapy and radiation, as it facilitates the evasion of apoptosis and supports the survival of a quiescent population capable of repopulating the tumor after treatment [239, 240].
Hedgehog Signaling Pathway: In a similar vein, the Hedgehog signaling pathway is crucial for augmenting CSC features, particularly in malignancies such as pancreatic cancer and glioblastoma. This pathway governs cellular growth and differentiation, with its activation being correlated with an aggressive tumor phenotype and heightened drug resistance [241, 242]. Inhibitors directed at the Hedgehog pathway, such as vismodegib, have demonstrated potential efficacy in preclinical models, suggesting that disruption of this pathway may increase the vulnerability of CSCs to conventional therapeutic modalities [243].
Wnt/β-catenin pathway: The Wnt/β-catenin signaling cascade represents another essential pathway implicated in CSC-mediated drug resistance. The activation of this pathway fosters self-renewal and amplifies the tumor-initiating potential of CSCs, thereby increasing the probability of tumor recurrence following therapeutic intervention [99, 244, 245]. Empirical investigations have indicated that inhibition of the Wnt pathway can diminish the CSC population across a spectrum of cancers, suggesting that this pathway is a promising target for therapeutic exploitation [246, 247].
PI3K/AKT/mTOR pathway: The PI3K/AKT/mTOR signaling pathway is frequently activated in CSCs and is associated with cellular growth, survival, and resistance to programmed cell death. This pathway has been implicated in various cancer types, including ovarian and colorectal cancers, where it promotes CSC survival in the face of therapeutic challenges [248, 249]. Through the inhibition of components within this pathway, investigators have observed a reduction in CSC viability alongside an increase in sensitivity to chemotherapeutic agents [250].
CSC characteristics and drug resistance in various types of cancer.
In the context of breast cancer, cancer stem cells (CSCs) present elevated expression levels of the aldehyde dehydrogenase (ALDH) enzyme, which correlates with stem cell-like characteristics and facilitates resistance to chemotherapeutic agents such as doxorubicin and cyclophosphamide [251, 252]. The detection of ALDH + cellular populations within breast tumors has led to the development of targeted therapeutic strategies aimed at eliminating these resistant cell subsets.
Colorectal cancer: In colorectal cancer, CSCs are distinguished by the expression of particular surface markers, including CD44 and CD133. These markers are associated with increased self-renewal capacity, tumor initiation, and resistance to standard chemotherapy regimens [47, 253]. The strategic targeting of these CSC markers has shown promise in preclinical investigations, indicating that an integrated therapeutic approach that addresses both the bulk tumor and CSC populations may augment treatment efficacy.
Lung cancer: In lung cancer, the proliferation of CSCs characterized by ATP-binding cassette (ABC) transporters significantly contributes to the phenomenon of multidrug resistance. These transporters actively extrude chemotherapeutic compounds, thereby diminishing their therapeutic effectiveness and enabling the survival of the CSC population posttreatment [254]. Investigative efforts are currently focused on strategies that inhibit the functionality of ABC transporters to increase the therapeutic impact of existing treatment modalities, and more explained in Fig. 5.
Glioblastoma: Glioblastoma is widely recognized for its resistance to conventional therapeutic interventions, which is primarily attributed to the existence of CSCs that flourish within a hypoxic tumor microenvironment. Hypoxia-inducible factors (HIFs) are instrumental in facilitating the survival and self-renewal of these CSCs under hypoxic conditions, culminating in treatment failure [88, 255]. Targeting the HIF signaling pathway or altering the tumor microenvironment to mitigate hypoxia may yield improved therapeutic outcomes.
Therapeutic implications and targeting of molecular pathways to CSCs
The therapeutic ramifications of comprehending the drug resistance associated with cancer stem cells (CSCs) are substantial. Focusing on the molecular pathways that are distinctive to those of CSCs offers an auspicious approach for overcoming resistance and preventing relapse. For example, gamma-secretase inhibitors that specifically target the Notch signaling pathway have demonstrated efficacy in diminishing CSC populations in both breast and pancreatic neoplasms [256, 257]. This methodology holds significant potential for augmenting patient responses to traditional therapeutic interventions. In parallel, the application of Hedgehog inhibitors, such as vismodegib, has been investigated across numerous malignancies characterized by a high abundance of CSCs, including pancreatic and glioblastoma cancers [258, 259]. Through the effective targeting of CSCs within neoplastic masses, these therapeutic modalities may contribute to enhanced overall tumor management and mitigate the probability of disease recurrence.
Combination therapies: The integration of conventional therapeutic modalities with CSC-targeted interventions constitutes an avant-garde strategy aimed at optimizing treatment efficacy. By amalgamating Wnt pathway inhibitors or PI3K/AKT/mTOR inhibitors with standard chemotherapeutic regimens, investigators aspire to eradicate both CSCs and their non-CSC counterparts within tumors, thereby addressing the challenge of tumor heterogeneity [260, 261]. This combinatorial approach has the potential not only to increase response rates but also to prolong survival and diminish the likelihood of relapse. Furthermore, immunotherapeutic strategies that leverage the host immune system to specifically target CSCs are currently under investigation. Immune checkpoint inhibitors, for example, have exhibited promise in amplifying the immune response directed against CSCs, culminating in improved therapeutic outcomes [262, 263]. The synergy of immunotherapy with targeted CSC interventions could yield a more holistic treatment paradigm. Drug resistance associated with CSCs is a complex phenomenon governed by an intricate interplay of molecular pathways that modulate CSC survival, self-renewal, and proliferation. The variability in CSC characteristics across different cancer types further complicates the development of treatment strategies. Nevertheless, by focusing on these pathways and incorporating innovative therapeutic methodologies, there is a promising trajectory for overcoming resistance and enhancing the efficacy of oncological therapies. Ongoing research in this domain is imperative to translate these discoveries into clinical applications, ultimately improving patient outcomes in cancer management, as described in Table 3, Figs. 3, 4.
Fig. 4.

Schematic representation of drug resistance pathways in cancer cells that are both pathway-dependent and pathway-independent. For example, the kinase domain and ectodomain mutation of the epidermal growth factor receptor (EGFR) or the overexpression of a truncated version of the target receptor can both activate a putative target receptor. These pathway-dependent (black) mechanisms work similarly. Furthermore, downstream pathways can multiply due to loss-of-function mutations (PTEN, a well-known downstream pathway inhibitor) or gain-of-function mutations in downstream components (e.g., PIK3CA, BRAF, and KRAS). Other plausible molecular mechanisms that rely on pathways include bypass activation, which can result in the amplification of different isoforms. Epigenetic modifications are typically involved in pathway-independent (red) processes. The development of resistance to cancer treatment is largely dependent on epithelial–mesenchymal transition (EMT) in cancer tissues and the tumor microenvironment. (M, methylation; dM, demethylation; TKI, tyrosine kinase inhibitor; RTK, receptor tyrosine kinase) [135, 353]
In general, signaling pathways play crucial roles in multidrug resistance (MDR) in cancer stem cells (CSCs) [200]. Dysregulation of developmental and stem cell-specific signaling pathways, such as the Notch, Wnt/β-catenin, Hedgehog, and STAT3 pathways, has been observed in CSCs [55, 165]. These pathways can regulate both intrinsic and extrinsic factors involved in the maintenance of CSCs, including drug transporter pumps, autophagy, apoptosis, DNA repair, and inflammation [264]. Targeting pathway molecules that regulate CSCs, either through small molecule inhibitors, monoclonal antibodies, or immunotherapies, provides a potential approach to overcome therapeutic resistance and improve patient outcomes. Further research is needed to fully elucidate the mechanisms underlying MDR in CSCs and to develop novel strategies for reversing resistance, as explicitly demonstrated in Fig. 5, Tables 3 and 4.
Table 4.
Overview of Drug Resistance Mechanisms and Genes in Cancer Stem Cells
| Drug | Resistance Gene(s) | Mechanism of Resistance | Cancer Type | Associated Cancer Stem Cell Characteristics | References |
|---|---|---|---|---|---|
| Doxorubicin | ABCB1 (P-glycoprotein) | Efflux pump expels drug from cells, reducing intracellular drug concentration | Breast, Ovarian, Leukemia | CSCs overexpress ABCB1, leading to multidrug resistance (MDR) | [39, 277–279] |
| Cisplatin | ERCC1, XPF | Enhanced DNA repair capacity through NER (nucleotide excision repair) mechanism | Ovarian, Lung | CSCs exhibit high DNA repair and survival capabilities | [280–284] |
| Imatinib | BCR-ABL | Point mutations in the kinase domain prevent drug binding | Chronic Myeloid Leukemia (CML) | CSCs harbor mutations in the BCR-ABL fusion protein | [285–289] |
| Methotrexate | DHFR (Dihydrofolate Reductase) | Gene amplification increases DHFR enzyme levels, bypassing methotrexate inhibition | Breast, Leukemia | CSCs show survival due to DHFR amplification | [290–294] |
| 5-Fluorouracil (5-FU) | TYMS (Thymidylate Synthase) | Overexpression or mutations reduce the drug’s ability to inhibit DNA synthesis | Colorectal, Gastric | CSCs upregulate TYMS, resulting in enhanced survival | [295–298] |
| Paclitaxel | TUBB (Beta-tubulin) | Beta-tubulin mutations alter microtubule stability, reducing drug binding | Breast, Lung, Ovarian | CSCs show alterations in tubulin that lead to resistance | [299–304] |
| Vincristine | ABCC1 (MRP1) | Efflux pump reduces intracellular concentration of the drug | Leukemia, Neuroblastoma | CSCs overexpress ABCC1 leading to efflux-mediated resistance | [248, 305–310] |
| Temozolomide | MGMT (O6-Methylguanine-DNA Methyltransferase) | MGMT repairs DNA by removing drug-induced lesions | Glioblastoma | CSCs exhibit high MGMT levels, contributing to therapy resistance | [112, 311–317] |
| Gefitinib/Erlotinib | EGFR (Epidermal Growth Factor Receptor) | Mutations (e.g., T790M) reduce drug-binding efficacy | Non-small cell lung cancer (NSCLC) | CSCs harbor EGFR mutations that prevent efficient drug inhibition | [318–322] |
| Sorafenib | RAF, MEK, ERK (MAPK Pathway) | Activation of MAPK signaling pathway leads to compensatory survival pathways | Hepatocellular carcinoma | CSCs bypass drug effect via alternative signaling pathway activations | [323–327] |
| Oxaliplatin | MLH1, MSH2 (Mismatch Repair Genes) | Deficient mismatch repair reduces apoptosis triggered by DNA damage | Colorectal | CSCs exhibit mismatch repair deficiency (MSI-high), evading apoptosis | [22, 328–333] |
| Carboplatin | BRCA1/2 (Breast Cancer Genes) | Secondary mutations restore BRCA function, enabling homologous recombination repair | Ovarian, Breast | CSCs restore BRCA functionality, promoting drug resistance | [334–339] |
| Docetaxel | TUBB3 (Class III Beta-tubulin) | Overexpression of TUBB3 decreases microtubule binding affinity, leading to resistance | Prostate, Breast, Lung | CSCs show altered microtubule dynamics via TUBB3 expression | [340–343] |
| Tamoxifen | ESR1 (Estrogen Receptor) | ESR1 mutations alter estrogen receptor function, reducing the efficacy of anti-estrogen drugs | Breast | CSCs harbor ESR1 mutations, leading to tamoxifen resistance | [244, 344–348] |
| Trastuzumab | HER2 (Human Epidermal Growth Factor Receptor 2) | Mutations in HER2 prevent effective trastuzumab binding | HER2-positive Breast Cancer | CSCs expressing altered HER2 maintain resistance to trastuzumab | [349–352] |
Extracellular matrix in the development of drug resistance in cancer cells and cancer stem cells
The extracellular matrix (ECM) has garnered increasing recognition as a pivotal factor influencing the emergence of drug resistance in both cancer cells and cancer stem cells (CSCs). Compared with a sophisticated network of proteins, glycoproteins, and polysaccharides, the ECM not only provides structural integrity to tissues but also plays a fundamental role in modulating cellular behavior, affecting various processes ranging from cell survival to responses to pharmacological agents. A primary mechanism by which the ECM facilitates drug resistance is through mechanical signaling. The biomechanical properties of the ECM, including stiffness and density, can profoundly affect the behavior of cancer cells. For example, in solid tumors, the ECM frequently becomes rigid due to the excessive synthesis of collagen and other matrix proteins. This augmented stiffness can activate mechanotransductive pathways, particularly the Rho/ROCK pathway, which has been demonstrated to increase survival signaling in CSCs and foster resistance to chemotherapy [355–357]. As these mechanical alterations transpire, they may also initiate epithelial-to-mesenchymal transition (EMT), a process whereby epithelial cells acquire mesenchymal traits, thereby increasing their invasiveness and therapeutic resistance [356, 358–360]. The rigidity of the ECM not only affects cellular responses but also shapes the overall architecture of the tumor, rendering it a dynamic player in the biology of the tumor.
The composition of the ECM is an essential determinant of drug resistance. In numerous tumors, significant modifications in ECM components are observed, characterized by increased deposition of collagen fibers and the presence of glycoproteins such as fibronectin. These alterations can establish a dense, fibrous milieu that physically sequesters therapeutic agents, thereby diminishing their accessibility to target cancer cells [357, 360, 361]. Furthermore, changes in ECM composition can precipitate the upregulation of drug efflux pumps, particularly ATP-binding cassette (ABC) transporters, which actively extrude chemotherapeutic agents from cells [362–364], and verified in Fig. 5. Consequently, the modified ECM not only provides a protective barrier for cancer cells against the effects of pharmacological agents but also endows them with augmented mechanisms for survival under therapeutic conditions.
Moreover, the ECM exerts substantial influence on the tumor microenvironment by mediating interactions between cancer cells and stromal cells. These interactions frequently culminate in the secretion of diverse growth factors and cytokines, which can increase cell survival and confer drug resistance [103]. For example, cancer-associated fibroblasts (CAFs) within the tumor microenvironment (TME) are capable of releasing factors such as transforming growth factor-beta (TGF-β) and hepatocyte growth factor (HGF). These factors are associated with the preservation of CSC characteristics and are recognized for their role in activating survival pathways that assist cancer cells in resisting treatment [103, 365, 366]. This interaction engenders a supportive niche for CSCs, wherein they can flourish despite the administration of therapeutic agents intended to eradicate them.
The existence of hypoxic zones within neoplasms, which are frequently associated with modifications in the extracellular matrix (ECM) architecture, presents an additional obstacle in the pursuit of mitigating drug resistance. Under conditions of low oxygen availability, hypoxia-inducible factors (HIFs) undergo stabilization, leading to the activation of diverse genetic programs that promote cellular survival and increase the stemness attributes of cancer stem cells (CSCs) [103, 367, 368]. The ECM plays a significant role in the establishment of these hypoxic microenvironments, thereby further consolidating the protective milieu that supports CSC persistence and drug resistance [360, 367, 369]. Hypoxic conditions not only diminish the effectiveness of numerous conventional therapeutic interventions but also foster a more aggressive tumor phenotype.
In relation to CSCs, the ECM is particularly important for preserving their stem-like traits. Interactions between CSCs and ECM constituents, including hyaluronic acid, laminin, and fibronectin, have been demonstrated to increase self-renewal potential and confer resistance to therapeutic modalities [20, 357, 366, 368]. For example, the affinity of CSCs for specific ECM proteins can initiate signaling cascades, such as integrin signaling, which promotes cellular proliferation and survival. This underscores the imperative for therapeutic strategies that not only directly target CSCs but also modify their ECM interactions to disrupt their survival mechanisms.
In light of these findings, therapeutic modalities that specifically target the ECM or its associated signaling pathways present promising prospects for overcoming drug resistance in oncological contexts. For example, therapies aimed at ECM modulation, such as those that facilitate collagen degradation or alter the mechanical properties of the ECM, could augment the infiltration and efficacy of chemotherapeutic agents. Likewise, strategies designed to interfere with the signaling pathways activated by ECM components, such as Rho/ROCK or integrin signaling, may sensitize resistant neoplastic cells to therapeutic intervention [366, 370–372].
In summary, the extracellular matrix is integral to the manifestation of drug resistance in both cancer cells and cancer stem cells. Via mechanical signaling, compositional alterations, interactions within the tumor microenvironment, and adaptations to hypoxic conditions, the ECM not only shields tumor cells from therapeutic agents but also reinforces the aggressive and stem-like attributes of CSCs. Addressing these ECM-related mechanisms may yield novel therapeutic strategies that enhance the efficacy of existing treatment modalities and ultimately improve patient outcomes in cancer therapy.
Off-target effects of nanodrugs: Targeting cancer stem cells (CSCs), in particular, has made it possible to improve drug delivery and overcome drug resistance owing to the introduction of nanotechnology in cancer therapy. Although the use of nanodrugs has many benefits, there are also several hazards, particularly with respect to off-target effects, that could compromise the safety and effectiveness of treatment. The potential for unexpected interactions with nontarget cells and tissues to result in negative side effects that could make treatment outcomes more difficult is one of the main concerns with nanodrugs [130, 131]. In the tumor microenvironment (TME), for example, nanoparticles intended to specifically target CSCs may unintentionally impact normal stem cells or other cell types, leading to toxicity and possibly worsening treatment resistance [132].
A number of variables, such as the size, shape, composition, surface charge, and physicochemical characteristics of nanoparticles, might result in off-target effects. These features have a major impact on the cellular absorption and biodistribution of nanomedicines. For example, whereas larger particles might not effectively penetrate tumor tissues, excessively small nanoparticles might quickly accumulate in the liver and spleen, decreasing their availability to target tumors [133]. Furthermore, the surface functionalization of nanoparticles, which is meant to improve targeted specificity, may inadvertently increase binding to cells that are not the intended target, which could result in cytotoxicity. Because immune cells may be stimulated to attack both the tumor cells and the surrounding healthy tissue, this unintentional activation of immune responses might further complicate the treatment landscape and increase the risk of systemic adverse effects [134].
The possibility of changing drug release profiles from nanodrugs, which may affect their safety and therapeutic efficacy, is another major concern. Drug stability and release kinetics can be affected by changes in environmental factors, including pH and temperature; however, regulated release is essential for reducing toxicity and optimizing therapeutic effects [135]. A nanodrug may cause elevated off-target toxicity if its payload is released too quickly or incorrectly, which could result in high local concentrations of the drug in nontargeted locations.
Furthermore, persistent nanoparticle buildup in the body can lead to new hazards, especially in regard to compounds that cannot be easily eliminated. The long-term presence of these substances may cause chronic inflammatory reactions, which may result in additional issues such as fibrogenesis or cancer [136]. For example, certain metal-based nanoparticles can cause oxidative stress in tissues that are not their intended targets, leading to cellular damage that may promote the growth of tumors or the establishment of drug-resistant cell populations [137, 138].
The possible hazards associated with the use of nanoparticles are also significantly influenced by their immunological response. According to Xiong et al. [139], nanoparticles can trigger an immunological response and activate the complement system, which can either improve antitumor immunity or cause severe allergic reactions and anaphylaxis in susceptible people. The production of antibodies against the nanoparticles due to the immunogenicity of the nanocarriers may eventually diminish their effectiveness and encourage the emergence of drug resistance.
Careful assessment of the safety and effectiveness characteristics of nanodrugs in preclinical and clinical settings is crucial to reduce these hazards. To evaluate how nanoparticles interact with biological systems and find any possible off-target effects, advanced characterization techniques should be used [39]. Furthermore, techniques such as employing stimuli-responsive release mechanisms or covering nanoparticles with biocompatible materials might improve selectivity and lower the possibility of off-target interactions [132]. Creating predictive models to comprehend the biodistribution and pharmacokinetics of nanomedicines can also help with design optimization for increased safety and efficacy in targeting CSCs, as described in Fig. 6 A and B.
Fig. 6.

Diagrams A and B depict schematic representations of the off-target effects of nanodrugs on drug resistance in mammalian cancer cells
In summary, although nanodrugs have the potential to prevent drug resistance in mammalian cancer stem cells, there are several hazards associated with their use, especially in regard to off-target effects. Optimizing the design of nanoparticles and enhancing therapeutic results require an understanding of how they interact with biological systems. To fully utilize nanotechnology in cancer treatment, it will be necessary to optimize nanodrug compositions and conduct thorough safety assessments as research advances.
Therapeutic modalities for targeting cancer stem cells and future directions
Chemotherapy continues to serve as a fundamental component of oncological management; however, the emergence of resistance to these pharmacological agents constitutes a substantial obstacle to achieving therapeutic efficacy [373]. The enduring presence of drug resistance within neoplastic cells complicates therapeutic outcomes and highlights the need for alternative intervention strategies. Contemporary oncological treatment protocols frequently encompass a multimodal approach, integrating surgical intervention, radiotherapy, and chemotherapy, whether administered independently or in succession. Surgical resection is often the primary intervention, particularly in the context of early-stage malignancies, where it has the potential for curative outcomes. Radiotherapy is routinely utilized in localized disease scenarios and is frequently combined with surgical procedures to increase therapeutic effectiveness [106]. Despite its established status as a treatment modality for more than a century, the efficacy of radiation therapy is constrained by the incomplete comprehension of the comprehensive genetic response necessary for cellular protection against radiation-induced injury [106]. This lack of knowledge impedes the formulation of strategies aimed at mitigating normal tissue toxicity while simultaneously enhancing neoplastic control.
Recent innovations in oncological therapy have given rise to a novel class of treatments explicitly designed to target cancer stem cells (CSCs). CSCs are distinguishable from non-CSCs and bulk tumor cells by virtue of their unique molecular profiles. To address this differentiation, high-throughput screening methodologies have been utilized to discover small molecules that are capable of selectively targeting and diminishing CSC populations [374]. This strategy is particularly vital for the management of malignancies that are resistant to conventional treatment modalities and underscores the dynamic evolution of cancer therapy paradigms. As research advances, a deeper comprehension of CSC biology and interactions with therapeutic agents will be imperative for formulating more effective and targeted oncological therapies.
As a result, various therapeutic approaches have been developed for targeting cancer stem cells (CSCs). These approaches include targeting intracellular signaling pathways related to CSC functions [57], using aptamers as targeting ligands for CSC-specific therapies [59], and inhibiting CSCs through monoclonal antibodies, signaling pathway inhibitors, and energy metabolism enzymes inhibitors, induce differentiation therapy [375], and utilize natural products (NPs) and NP-based nanoformulations (NPNs) to target and inhibit CSCs [60]. The further development of multifunctional aptamer-based therapeutic strategies has shown promising results in recent years [59]. Additionally, immunotherapy with immune cells engineered with a chimeric antigen receptor (CAR) has demonstrated favorable outcomes in CSC therapy [376]. However, challenges such as pharmacokinetic defects and off-target effects need to be addressed for effective clinical translation of these approaches [377]. Overall, targeting CSCs through these therapeutic strategies holds great potential for comprehensive cancer therapy in the future.
In fact, targeting cancer stem cells (CSCs) is a promising approach for comprehensive cancer therapy. Several therapeutic approaches have been developed to eliminate CSCs and reduce cancer recurrence. Natural products (NPs) derived from various sources have shown significant antitumor effects and have been shown to target and inhibit CSCs [57, 378, 379]. These NPs can inhibit crucial signaling pathways involved in the maintenance of CSC stemness and sensitize CSCs to standard chemotherapeutic treatments [60]. Additionally, NP-based nanoformulations (NPNs) have been developed to overcome the pharmacokinetic limitations of NPs and improve their targeting capabilities [60]. These NPNs have the potential to deliver NPs specifically to CSCs, increasing their efficacy in eliminating CSCs and reducing cancer recurrence [60]. Furthermore, bispecific chemically self-assembled nanorings (CSANs) have been developed to selectively target CSCs and bulk tumor cells simultaneously, resulting in synergistic effects and complete tumor eradication [379]. These findings suggest that therapeutic approaches targeting CSCs, such as blocking signaling pathways and using NPs or NPNs, can be effective in eliminating CSCs and reducing cancer recurrence.
Current approaches to understanding the mechanisms of drug resistance in mammalian cancer stem cells and their impact on cancer recurrence involve studying genetic alterations such as point mutations and gene amplification [380].
To address the intricate challenge of drug resistance in mammalian cancer stem cells (CSCs), investigators are concentrating on the genetic modifications that contribute to this phenomenon. Significant alterations currently under scrutiny include point mutations and gene amplifications, which are pivotal in facilitating drug resistance and the ensuing recurrence of cancer [380]. Sophisticated methodologies, such as stochastic multitype branching process models, are utilized to investigate the dynamics associated with tumor recurrence. These models assist in estimating the probabilities of tumor extinction and offer valuable insights into the temporal aspects of potential recurrences [102, 207]. By simulating a range of scenarios, researchers are able to ascertain how genetic modifications affect tumor behavior over time. A fundamental component of this research involves elucidating the relative influence of various genetic alterations on drug resistance. In particular, the ratio of recurrence intervals attributed to gene amplification in comparison to point mutations is of significant interest. This ratio is contingent upon the quantity of amplification events necessary to attain an equivalent level of resistance as that conferred by a single mutation event. The linear correlation between amplification events and mutation events underscores the disparate contributions of these genetic mechanisms to drug resistance and the relapse of cancer [381].
Through the integration of these methodologies, researchers aspire to enhance the understanding of the fundamental mechanisms underlying drug resistance in CSCs and their consequential implications for cancer recurrence. This comprehension is crucial for the formulation of more efficacious treatment strategies and the enhancement of patient prognoses. Cancer stem cells (CSCs), which have unique biological characteristics and drug resistance mechanisms, such as ABC protein drug pump function and microenvironmental factors, have been identified as important factors in drug resistance in tumors. Understanding the role of specific markers, such as HMGA2, in the development and invasion of cancer cells, as well as their function in drug resistance mechanisms in CSCs, is crucial for overcoming drug resistance and recurrence. Advances in technology, such as single-cell RNA sequencing and CRISPR gene editing, have greatly impacted our understanding of drug resistance in mammalian cancer stem cells. Single-cell RNA sequencing allows for the identification and characterization of individual cancer stem cells, providing insights into their gene expression profiles and heterogeneity, which can contribute to drug resistance [382]. On the other hand, CRISPR gene editing has emerged as a powerful tool for studying the mechanisms of drug resistance in cancer cells. It enables the targeted manipulation of specific genes involved in drug resistance pathways, allowing researchers to investigate their role in conferring resistance and potentially developing strategies to overcome it [383]. These technological advancements have the potential to revolutionize our understanding of drug resistance in cancer stem cells and pave the way for the development of more effective therapies.
Achievements and constraints in mitigating the drug resistance of mammalian cancer stem cells (CSCs) in oncological therapeutics
Cancer stem cells (CSCs) have attracted considerable scholarly attention in oncological research owing to their pivotal involvement in tumor initiation, advancement, and recurrence. Their intrinsic characteristics, which include self-renewal and differentiation capabilities, significantly contribute to drug resistance, thereby posing substantial obstacles in cancer treatment. Although various methodologies have been formulated to specifically target CSCs and overcome drug resistance, these approaches are not devoid of limitations. This study seeks to thoroughly investigate the achievements and constraints of diverse strategies aimed at addressing drug resistance in mammalian CSCs.
Molecular Targeting of CSC Pathways Achievements: The strategic targeting of critical signaling pathways that sustain CSC attributes has demonstrated considerable promise in mitigating drug resistance. For example, Notch signaling is essential for the survival and self-renewal of CSCs. Inhibitors of the Notch pathway, such as gamma-secretase inhibitors, have proven effective in preclinical models involving breast and pancreatic cancers, resulting in diminished CSC populations and increased sensitivity to chemotherapy [252, 257]. Similarly, the Hedgehog signaling pathway has been effectively targeted. Investigations have revealed that Hedgehog inhibitors, such as vismodegib, can significantly reduce CSC populations in basal cell carcinoma and pancreatic cancer, thereby increasing the efficacy of conventional therapies [43, 259]. These findings underscore the potential of pathway-specific targeting in overcoming drug resistance. Constraints: Notwithstanding these accomplishments, the targeting of such pathways may be confounded by compensatory mechanisms that neoplastic cells exploit. For example, upon inhibition of the Notch pathway, CSCs may engage alternative pathways, such as the Wnt/β-catenin signaling cascade, which can preserve their stem-like characteristics and foster resistance [245]. Moreover, prolonged inhibition of these pathways could precipitate the emergence of resistant cell populations, thereby limiting the long-term efficacy of these targeted interventions [62].
Combination therapeutic achievements: Combination therapies that amalgamate conventional chemotherapeutics with CSC-targeted agents have exhibited significant promise in overcoming drug resistance. For example, the integration of standard chemotherapeutics with PI3K/AKT/mTOR inhibitors has proven successful in diminishing the viability of CSCs in ovarian and colorectal cancers, thereby improving treatment outcomes [89, 384]. This approach aims to concurrently target both bulk tumor cells and CSC populations, thereby mitigating the likelihood of relapse. Furthermore, the integration of immunotherapy with CSC-targeted strategies has yielded encouraging results. Immune checkpoint inhibitors have demonstrated efficacy in specifically targeting CSCs, thereby enhancing the immune response against tumors [245]. These combinatorial strategies offer a more holistic approach to cancer intervention. Constraints: Nonetheless, combination therapies encounter a myriad of challenges, including heightened toxicity and the potential for exacerbated side effects, which may restrict the feasibility of treatment. The synergistic interactions between agents could engender unforeseen complications that complicate treatment protocols [261]. Additionally, the inherent heterogeneity of cancer may result in disparate responses to combination therapies, complicating the predictability of outcomes across various patient cohorts.
Targeting CSC Markers Successes: Elucidating how to target specific cancer stem cell (CSC) markers, including CD44, CD133, and ALDH, has emerged as a pivotal area in the formulation of CSC-targeted therapeutic interventions. For example, therapeutic strategies directed at ALDH + CSCs within the context of breast cancer have demonstrated promise in mitigating drug resistance and augmenting treatment efficacy [251]. The use of antibodies or small molecules to selectively engage these markers facilitates more refined interventions aimed at resistant CSC populations. Despite the promise associated with targeting CSC markers, several challenges persist in the clinical implementation of these strategies. CSCs frequently exhibit a degree of plasticity, which enables them to alter their marker expression in response to therapeutic interventions, thereby circumventing targeted treatments [271]. This inherent plasticity complicates the accurate identification of CSCs and may necessitate the formulation of novel strategies to dynamically monitor and adapt to these evolving changes.
Hypoxia and the Tumor Microenvironment Successes: The tumor microenvironment is instrumental in mediating CSC drug resistance. Hypoxic conditions prevalent within tumors can amplify CSC characteristics and confer resistance against therapeutic agents. Consequently, targeting the hypoxia-inducible factor (HIF) pathway has emerged as a compelling therapeutic strategy. Inhibitors of HIF have been shown to decrease the viability of CSCs under hypoxic conditions, thereby increasing the efficacy of concomitant therapeutic modalities [88]. Nevertheless, efforts to target the tumor microenvironment are fraught with considerable challenges. The inherently dynamic nature of the tumor microenvironment suggests that therapeutic interventions may not consistently produce the anticipated outcomes, given that CSCs can adapt to hypoxic environments through a variety of mechanisms [241]. Moreover, HIF inhibitors may inadvertently impact normal stem cells and adjacent healthy tissues, resulting in potential adverse effects.
Gene Editing Technology Successes: The advent of innovative gene-editing technologies, such as CRISPR-Cas9, has paved new paths for the targeted manipulation of CSCs. By directly altering genes that are implicated in CSC characteristics, researchers have effectively diminished CSC populations and improved sensitivity to chemotherapy in preclinical models [53, 385] Nevertheless, the implementation of gene editing within clinical contexts is beset with numerous challenges. Ethical dilemmas associated with gene editing, the potential for off-target effects, and the technical complexities involved in delivering these therapies to tumor sites must be thoroughly addressed prior to the widespread adoption of these methodologies in cancer treatment. By and large, substantial advancements have been achieved in the comprehension and targeting of CSCs to overcome drug resistance in cancer therapy. Methods such as targeting specific molecular pathways, employing combination therapies, and concentrating on CSC markers have exhibited efficacy in both preclinical and clinical investigations. However, limitations, including compensatory mechanisms, treatment-related toxicity, and the plasticity of CSCs, present formidable challenges that require resolution. Research into the complex biology of CSCs and their interactions with the tumor microenvironment is imperative for the development of effective strategies to combat drug resistance. Personalized treatment modalities that consider the heterogeneity of tumors and the distinctive characteristics of CSCs may ultimately lead to more efficacious cancer therapies.
Clinical implications of the current review
The clinical implications of examining drug resistance in mammalian cancer stem cells (CSCs) are profound and significantly influence the trajectory of cancer therapeutics in various essential dimensions. An in-depth comprehension of the mechanisms underlying CSC-mediated drug resistance is imperative for the formulation of more efficacious treatment methodologies.
A primary consequence is the imperative to directly target CSCs. CSCs frequently contribute to the failure of treatment and the recurrence of tumors, as conventional therapeutic modalities predominantly focus on rapidly proliferating bulk tumor cells while leaving CSCs unscathed. Consequently, approaches specifically aimed at the eradication of CSCs have the potential to markedly improve treatment outcomes [386, 387]. For example, inhibitors that specifically target critical signaling pathways associated with the maintenance of CSC characteristics, such as the Notch and Wnt pathways, have shown promise in preclinical investigations and may yield improved therapeutic efficacy [12, 92, 388, 389]. Combination therapies have other vital clinical implications. Insights into drug resistance linked to CSCs underscore the potential benefits of amalgamating traditional therapies with agents that specifically inhibit CSCs. Empirical evidence indicates that the coadministration of chemotherapeutic agents alongside immunotherapeutics or targeted therapies that obstruct CSC pathways can effectively diminish the probability of resistance while increasing overall treatment effectiveness [19]. For example, investigations have demonstrated that the combination of immune checkpoint inhibitors with agents that target the PI3K/AKT/mTOR pathway results in enhanced therapeutic outcomes in models of breast and pancreatic malignancies [388, 390–393]
Personalized medicine constitutes another significant dimension influenced by the degree of CSC drug resistance. The profiling of tumors for distinctive CSC markers and resistance mechanisms facilitates the development of tailored treatment strategies that are congruent with a patient’s specific tumor attributes. For example, genomic analyses have revealed mutations within CSC populations that can guide the selection of targeted therapies, thereby increasing response rates [7]. This individualized approach is becoming increasingly attainable due to advancements in precision medicine technologies. The importance of the tumor microenvironment (TME) in sustaining CSC viability and fostering drug resistance further highlights its relevance in therapeutic planning. The modulation of the TME, including the normalization of hypoxic conditions or the modification of immune cell interactions, may increase the efficacy of therapies directed at CSCs. Research has indicated that therapies targeting both CSCs and the TME can yield superior outcomes by disrupting the protective niches inhabited by CSCs [255].
The identification of predictive biomarkers associated with CSCs and their mechanisms of resistance is crucial for enhancing treatment strategies. Biomarkers linked to CSCs can assist clinicians in stratifying patients and selecting the most effective therapeutic interventions. For example, elevated levels of aldehyde dehydrogenase (ALDH) activity in CSCs have been correlated with suboptimal responses to conventional chemotherapy, implying that evaluating ALDH expression could inform treatment decisions [251, 394]. Finally, the review of the mechanisms underlying drug resistance in CSCs underscores the need for persistent research and development. As our understanding of CSC biology advances, new therapeutic targets and innovative strategies are likely to arise. Ongoing investigations into the characteristics of CSCs and their interactions with the TME are vital for the progression of cancer therapy and for overcoming the challenges posed by drug resistance [239].
Views on overcoming mammalian cancer stem cell drug resistance
Advances in cancer therapy necessitate a comprehensive understanding of drug resistance in cancer stem cells (CSCs) and their involvement in tumor recurrence. Crucial pathways such as the Wnt/β-catenin, Notch, and Hedgehog pathways play significant roles in CSC self-renewal and tumorigenesis, rendering them attractive targets for therapeutic interventions. Ongoing efforts by researchers involve the development of multitarget inhibitors and the exploration of immunotherapy, autophagy, hyperthermia, nanodrug delivery systems, mitochondria targeting, and disruption of the CSC microenvironment. The interplay of epigenetic modifications and molecular interactions plays a pivotal role in resistance mechanisms, with reactive oxygen species (ROS) and microRNAs (miRNAs) being implicated in the proliferation of CSCs. Therapeutic strategies based on epidrugs can render resistant cells more susceptible to conventional treatments. Various factors, including genetic and nongenetic determinants, cellular diversity, and alterations in target molecules, collectively contribute to the development of resistance. Addressing this challenge requires the development of drugs with enhanced efficacy, reduced efflux mechanisms, and targeted actions, alongside the utilization of nanocarriers to improve specificity. Intensive research endeavors are imperative for the identification of novel biomarkers and the formulation of precise therapeutic approaches aimed at managing tumorigenesis and preventing relapse.
Acknowledgements
All the authors listed in this manuscript are appreciated for their independent contributions to the further development of the paper.
Abbreviations
- TME
Tumor Microenvironment
- ABC-ATP
Binding cassette
- CSCs
Cancer stem cells
- HIFs
Hypoxia-inducible factors
- MHC
Major histocompatibility complex
- PD
L1-programmed cell death: ligand 1
- VEGF
Vascular endothelial growth factor
- SMAD3
Suppressor of mothers against decapentaplegic homolog3
- JAK–STAT
The Janus kinase and signal transducer of activation pathway
- EGFR
Endothelial growth factors receptors
- CAFs
Cancer-associated fibroblasts
- TGF-β
Transforming growth factor-beta and HGF-hepatocyte growth factor
- ECM
Extracellular matrix
- MDSCs
Myeloid-derived suppressor cells
- TAMs
Tumor-associated macrophages
- MDR
Multidrug-induced multidrug resistance
- EMT
Epithelial‒mesenchymal transition
- ROS
Reactive oxygen species
- HNSCCs
Head and neck squamous cell carcinomas
- P-gp
P-glycoprotein
Author contributions
In the preparation of this review paper, all active members of the working group contributed to the article search, drafting, writing, editing, and overall conception and design of the study. Material preparation, article collection, and the development of key points and analyses were carried out by Bemrew Admassu, Tirunesh Tsegaw, Yitayew Demssie, Kalkidan Getnet, Melkie Dagnaw Fenta, Yesuneh Tefera Mekasha, Melaku Getahun, Abebe Belete, Mebrie Zemene, Asnakew Mulaw, and Atsede Solomon. Bemrew Admassu Mengistu and Tirunesh Tsegaw wrote the initial draft of the manuscript, with all the authors providing feedback on earlier versions. The entire working group reviewed and approved the final manuscript.
Funding
Funding was not applicable, as this manuscript is a review article, and as such, no specific funding or financial support was allocated for its development. The work was completed without any external grants, sponsorships, or designated budgets. All efforts involved in researching, drafting, and editing the manuscript were carried out without the provision of dedicated financial resources.
Data availability
For this comprehensive review, data were obtained from previous research/publication works.
Declarations
Ethics approval and consent to participate
This manuscript is a concise review. No ethical approval is needed.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Abbas Z, Rehman S, An overview of cancer treatment modalities. 2018.
- 2.Compton, Z.T., et al., Cancer prevalence across vertebrates. Research Square, 2023.
- 3.Das A, et al. Cancer stem cells, their origin and niche: a search for the therapeutic target. J Stem Cell Res Med. 2017;2(1):1. [Google Scholar]
- 4.Fan X, et al., Engineered mammalian cells for cancer therapy. 2019, Google Patents.
- 5.Gottesman MM, Robey RW, Ambudkar SV. New mechanisms of multidrug resistance: an introduction to the Cancer Drug Resistance special collection. Cancer Drug Resistance. 2023;6(3):590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veeraraghavan VP, et al. Emerging targeted therapies in oral oncology: focus on EGFR inhibition. Oral Oncol Rep. 2024;1:100592. [Google Scholar]
- 7.López-Lázaro M. Opium, street opium, and cancer risk. Curr Pharm Des. 2022;28(25):2039–42. [DOI] [PubMed] [Google Scholar]
- 8.Lord SR, Harris AL. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br J Cancer. 2023;128(6):958–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J, et al. Tumor microenvironment characterization identifies two lung adenocarcinoma subtypes with specific immune and metabolic state. Cancer Sci. 2020;111(6):1876–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibrahim B, et al. Stem cell marker aldehyde dehydrogenase 1A1 expression in triple-negative breast carcinoma. Open Access Maced J Med Sci. 2022;20(10):287–94. [Google Scholar]
- 11.Qin Y, et al. Autophagy and cancer drug resistance in dialog: preclinical and clinical evidence. Cancer Lett. 2023;570:216307. [DOI] [PubMed] [Google Scholar]
- 12.Zhou S, et al. Acid and hypoxia tandem-activatable deep near-infrared nanoprobe for two-step signal amplification and early detection of cancer. Adv Mater. 2023;35(36):2212231. [DOI] [PubMed] [Google Scholar]
- 13.Itahashi K, et al. BATF epigenetically and transcriptionally controls the activation program of regulatory T cells in human tumors. Sci Immunol. 2022;7(76):0957. [DOI] [PubMed] [Google Scholar]
- 14.Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. 2023;41(3):450–65. [DOI] [PubMed] [Google Scholar]
- 15.Lu Q, et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J Hematol Oncol. 2024;17(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma P, Allison JP. Immune checkpoint therapy: forging ahead. Am Assoc Adv Sci. 2022;14:2947. [DOI] [PubMed] [Google Scholar]
- 17.Sharma P, et al. Immune checkpoint therapy—current perspectives and future directions. Cell. 2023;186(8):1652–69. [DOI] [PubMed] [Google Scholar]
- 18.Chalabi M, et al. Neoadjuvant Immunotherapy in Locally Advanced Mismatch Repair-Deficient Colon Cancer. N Engl J Med. 2024;390(21):1949–58. [DOI] [PubMed] [Google Scholar]
- 19.Zhao L, et al. Strategies to synergize PD-1/PD-L1 targeted cancer immunotherapies to enhance antitumor responses in ovarian cancer. Biochem Pharmacol. 2023;215:115724. [DOI] [PubMed] [Google Scholar]
- 20.Zhao M, et al. Breaking the mold: overcoming resistance to immune checkpoint inhibitors. Antiviral Res. 2023;219: 105720. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, et al. Autophagy-driven regulation of cisplatin response in human cancers: exploring molecular and cell death dynamics. Cancer Lett. 2024;587: 216659. [DOI] [PubMed] [Google Scholar]
- 22.Hao J, et al. A novel TOX3-WDR5-ABCG2 signaling axis regulates the progression of colorectal cancer by accelerating stem-like traits and chemoresistance. PLoS Biol. 2023;21(9): e3002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, et al. Cancer stem cell theory: therapeutic implications for nanomedicine. Int J Nanomedicine. 2013;8:899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, et al. Viscoelastic rheology of polymer solution probed by resonant thermal capillary fluctuation. Phys Fluids. 2023;35(12):10. [Google Scholar]
- 25.Devilakshmi S, Madhumathi J, Verma RS. Immunotoxins, resistance and cancer stem cells: future perspective. Resist Immun Cancer Ther. 2015;39:33–56. [Google Scholar]
- 26.George J, et al. Cancer stem cells, not bulk tumor cells, determine mechanisms of resistance to SMO inhibitors. Cancer Res Commun. 2022;2(6):402–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaikh MA, et al. Introduction to cancer cell biology. In: Shaikh MA, editor., et al., Advanced drug delivery systems in the management of cancer. Amsterdam: Elsevier; 2021. p. 1–7. [Google Scholar]
- 28.Wilczyński JR. Cancer stem cells: an ever-hiding foe. In: Wilczyński JR, editor. Interaction of immune and cancer cells. Cham: Springer; 2022. p. 219–51. [DOI] [PubMed] [Google Scholar]
- 29.Beshiri ML, et al., The origin and dynamics of cellular heterogeneity vary across lineage subtypes of castrate resistant prostate cancer. bioRxiv, 2022: p. 2022.03. 24.484651. [DOI] [PMC free article] [PubMed]
- 30.Dasgupta A, et al. A control theoretic three timescale model for analyzing energy management in mammalian cancer cells. Comput Struct Biotechnol J. 2021;19:477–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho Y, Kim YK. Cancer stem cells as a potential target to overcome multidrug resistance. Front Oncol. 2020;10:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eid RA, et al. Targeting cancer stem cells as the key driver of carcinogenesis and therapeutic resistance. Int J Mol Sci. 2023;24(2):1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh S, et al., GW182 proteins restrict extracellular vesicle mediated selective export of used miRNAs in mammalian cancer cells. bioRxiv, 2020: p. 2020.09. 11.294488.
- 34.Guo Y, et al. P-glycoprotein (P-gp)-driven cancer drug resistance: biological profile, noncoding RNAs, drugs and nanomodulators. Drug Dis Today. 2024;29:104161. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh S, et al. Gw182 proteins restrict extracellular vesicle-mediated export of micrornas in mammalian cancer cells. Mol Cell Biol. 2021;41(5):e00483-e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poliwoda S, et al. Stem cells: a comprehensive review of origins and emerging clinical roles in medical practice. Orthopedic Rev. 2022;14(3):37498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirabelli P, Coppola L, Salvatore M. Cancer cell lines are useful model systems for medical research. Cancers. 2019;11(8):1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Angelis ML, et al. Stem cell plasticity and dormancy in the development of cancer therapy resistance. Front Oncol. 2019;9:626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang T, et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics. 2020;10(19):8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zahra MH, et al. Cancer stem cells contribute to drug resistance in multiple different ways. In: Zahra MH, editor., et al., Cancer stem cell markers and related network pathways. Cham: Springer; 2023. p. 125–39. [Google Scholar]
- 41.Wang X, Lee J, Xie C. Autophagy regulation on cancer stem cell maintenance, metastasis, and therapy resistance. Cancers. 2022;14(2):381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonaventura G, et al. Stem cells: innovative therapeutic options for neurodegenerative diseases? Cells. 2021;10(8):1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cecerska-Heryć E, et al. The use of stem cells as a potential treatment method for selected neurodegenerative diseases. Cell Mol Neurobio. 2023;43:1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Comertpay B, et al. Cancer stem cell transcriptome profiling reveals seed genes of tumorigenesis: new avenues for cancer precision medicine. OMIC J Integr Biol. 2021;25(6):372–88. [DOI] [PubMed] [Google Scholar]
- 45.Puranik N, et al. Exploring the role of stem cell therapy in treating neurodegenerative diseases: challenges and current perspectives. Curr Stem Cell Res Ther. 2022;17(2):113–25. [DOI] [PubMed] [Google Scholar]
- 46.Telang N. Drug-resistant stem cells: Novel approach for colon cancer therapy. Int J Mol Sci. 2022;23(5):2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, et al. Drug resistance and cancer stem cells. Cell Commun Signal. 2021;19(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rezayatmand H, Razmkhah M, Razeghian-Jahromi I. Drug resistance in cancer therapy: the Pandora’s box of cancer stem cells. Stem Cell Res Ther. 2022;13(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarkadi B. Cancer stem cells in drug resistance: an introduction to the e-book covering the special issue on the “cancer stem cells and drug resistance.” Cancer Drug Resist. 2023;6(2):239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vaez A, et al. A bright horizon of intelligent targeted-cancer therapy: nanoparticles against breast cancer stem cells. Curr Stem Cell Res Ther. 2023;18(6):787–99. [DOI] [PubMed] [Google Scholar]
- 51.Fapohunda O, Ajayi D. Cancer cell metabolism resulting in multidrug resistance to chemotherapy and possible ways out. J Cancer Prev Curr Res. 2020;11(3):64–70. [Google Scholar]
- 52.Zhang L, et al. Targeting breast cancer stem cells. Int J Biol Sci. 2023;19(2):552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aramini B, et al. Targeting cancer stem cells: new perspectives for a cure to cancer. In: Aramini B, editor., et al., Handbook of stem cell therapy. Cham: Springer; 2022. p. 1303–31. [Google Scholar]
- 54.Staudte S, et al. Multiparametric phenotyping of circulating tumor cells for analysis of therapeutic targets, oncogenic signaling pathways and DNA repair markers. Cancers. 2022;14(11):2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manni W, Min W. Signaling pathways in the regulation of cancer stem cells and associated targeted therapy. MedComm. 2022;3(4): e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cierpikowski P, Lis-Nawara A, Bar J. Prognostic value of WNT1, NOTCH1, PDGFRβ, and CXCR4 in oral squamous cell carcinoma. Anticancer Res. 2023;43(2):591–602. [DOI] [PubMed] [Google Scholar]
- 57.Mukai S. Therapeutic approaches targeting cancer stem cells. In: Mukai S, editor. Possibilities and limitations in current translational stem cell research. London: IntechOpen; 2022. [Google Scholar]
- 58.Ma S, et al. p53-induced autophagy regulates chemotherapy and radiotherapy resistance in multidrug resistance cancer cells. Dose-Response. 2021;19(4):15593258211048046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu B, et al. Recent advances in aptamer-based therapeutic strategies for targeting cancer stem cells. Materials Today, Bio. 2023;19: 100605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao W, et al. Natural products-based nanoformulations: a new approach targeting CSCs to Cancer therapy. Int J Nanomed. 2022;17:4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esmaeili SA, et al. Selectively targeting cancer stem cells: Current and novel therapeutic strategies and approaches in the effective eradication of cancer. IUBMB Life. 2021;73(8):1045–59. [DOI] [PubMed] [Google Scholar]
- 62.Zhou H-M, et al. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal Transduct Target Ther. 2021;6(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–91. [DOI] [PubMed] [Google Scholar]
- 64.Tu X, et al. Suppression of cancer cell stemness and drug resistance via MYC destabilization by deubiquitinase USP45 inhibition with a natural small molecule. Cancers. 2023;15(3):930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Korde LA, et al. Neoadjuvant chemotherapy, endocrine therapy, and targeted therapy for breast cancer: ASCO guideline. J Clin Oncol. 2021;39(13):1485–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019;2:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim JW. Mammalian cancer cell and transgenic mammal carrying human protooncogene and kit for diagnosing cancer using said protooncogene. 2008, Google Patents.
- 68.Tollis M, Schneider-Utaka AK, Maley CC. The evolution of human cancer gene duplications across mammals. Mol Biol Evol. 2020;37(10):2875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L, et al. The application of peptides in Glioma: a novel tool for therapy. Curr Pharm Biotechnol. 2022;23(5):620–33. [DOI] [PubMed] [Google Scholar]
- 70.Zhou Y, et al. Chimeric mouse tumor models reveal differences in pathway activation between ERBB family–and KRAS-dependent lung adenocarcinomas. Nat Biotechnol. 2010;28(1):71–8. [DOI] [PubMed] [Google Scholar]
- 71.Lampreht T, Horvat US, Cemazar M. Transgenic mouse models in cancer research. Front Oncol. 2018;8:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raimondi F, et al. Insights into cancer severity from biomolecular interaction mechanisms. Sci Rep. 2016;6(1):34490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boddy AM, et al. Lifetime cancer prevalence and life history traits in mammals. Evolution, medicine, and public health. 2020;2020(1):187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al-Hajj M, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci. 2003;100(7):3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ricardo S, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64(11):937–46. [DOI] [PubMed] [Google Scholar]
- 76.O’Brien CA, et al. A human colon cancer cell capable of initiating tumor growth in immunodeficient mice. Nature. 2007;445(7123):106–10. [DOI] [PubMed] [Google Scholar]
- 77.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–7. [DOI] [PubMed] [Google Scholar]
- 78.D’Eramo F. Dark matter and Higgs boson physics. Phys Rev D Part Fields Gravitation Cosmol. 2007;76(8):083522. [Google Scholar]
- 79.Eramo A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15(3):504–14. [DOI] [PubMed] [Google Scholar]
- 80.Codony-Servat J, Verlicchi A, Rosell R. Cancer stem cells in small cell lung cancer. Translational lung cancer research. 2016;5(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao C, et al. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than nonstem cancer cells. PLoS ONE. 2016;11(5): e0154576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hermann PC, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–23. [DOI] [PubMed] [Google Scholar]
- 83.Neoptolemos JP, et al. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15(6):333–48. [DOI] [PubMed] [Google Scholar]
- 84.Singh SK, et al. Identification of human brain tumor initiating cells. Nature. 2004;432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 85.Yi Y, et al. Glioblastoma stem-like cells: characteristics, microenvironment, and therapy. Front Pharmacol. 2016;7:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garnier D, et al. Glioblastoma stem-like cells, metabolic strategy to kill a challenging target. Front Oncol. 2019;9:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brahimi-Horn MC, Chiche J, Pouysségur J. Hypoxia and cancer. J Mol Med. 2007;85:1301–7. [DOI] [PubMed] [Google Scholar]
- 88.Ren P, et al. A novel hypoxia-driven gene signature that can predict the prognosis and drug resistance of gliomas. Front Genet. 2022;13: 976356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhong W, Sun T. Epithelial–mesenchymal transition (EMT) as a therapeutic target in cancer. Front Oncol. 2023;13:1121416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shi T, et al. The role of hypoxia and cancer stem cells in development of glioblastoma. Cancers. 2023;15(9):2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. [DOI] [PubMed] [Google Scholar]
- 92.Zhou S, et al. Targeting tumor endothelial cells with methyltransferase inhibitors: mechanisms of action and the potential of combination therapy. Pharmacol Ther. 2023;247: 108434. [DOI] [PubMed] [Google Scholar]
- 93.Fridman WH, et al. The immune contexture in human tumors: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. [DOI] [PubMed] [Google Scholar]
- 94.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cao J, Chow L, Dow S. Strategies to overcome myeloid cell induced immune suppression in the tumor microenvironment. Front Oncol. 2023;13:1116016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morschhauser F, et al. A phase 2 study of venetoclax plus R-CHOP as first-line treatment for patients with diffuse large B-cell lymphoma. Blood J Am Soc Hematol. 2021;137(5):600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bengsch B, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T-cell exhaustion. Immunity. 2016;45(2):358–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Patel SA. Uncovering molecular targets to overcome immunosuppression in non-small cell lung cancer with acquired tki resistance. 2023.
- 100.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–70. [PubMed] [Google Scholar]
- 101.Liu X, et al. Dysregulation of cholesterol metabolism in cancer progression. Oncogene. 2023;42(45):3289–302. [DOI] [PubMed] [Google Scholar]
- 102.Lu J. Cancer stem cells and their drug resistance mechanisms. Highlights Sci Eng Technol. 2023;36:1334–41. [Google Scholar]
- 103.Liu Q, et al. Cancer stem cells and their niche in cancer progression and therapy. Cancer Cell Int. 2023;23(1):305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y, et al. Targeting metabolic-redox nexus to regulate drug resistance: from mechanism to tumor therapy. Antioxidants. 2024;13(7):828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205(2):275–92. [DOI] [PubMed] [Google Scholar]
- 106.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–98. [DOI] [PubMed] [Google Scholar]
- 107.Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154(2):274–84. [DOI] [PubMed] [Google Scholar]
- 108.Yang Y, et al. Emerging agents that target signaling pathways in cancer stem cells. J Hematol Oncol. 2020;13:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang XJ, et al. Journal of hematology & oncology. J Hematol Oncol. 2010;3:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 111.Vander Heiden MG, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329(5998):1492–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen H, et al. miR-130a can predict response to temozolomide in patients with glioblastoma multiforme, independently of O6-methylguanine-DNA methyltransferase. J Transl Med. 2015;13:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nathansen J, et al. Beyond the double-strand breaks: the role of DNA repair proteins in cancer stem-cell regulation. Cancers. 2021;13(19):4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Torre LA, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. [DOI] [PubMed] [Google Scholar]
- 115.Patra M, Gasser G. The medicinal chemistry of ferrocene and its derivatives. Nat Rev Chem. 2017;1(9):0066. [Google Scholar]
- 116.Chang F-W, et al. Estrogen enhances the expression of the multidrug transporter gene ABCG2—increasing drug resistance of breast cancer cells through estrogen receptors. Int J Mol Sci. 2017;18(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wei YN, et al. The role and application of vesicles in triple-negative breast cancer: opportunities and challenges. Mol Ther Oncolytics. 2023;31:100752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24(2):97–106. [DOI] [PubMed] [Google Scholar]
- 119.Ruan Y, et al. Mechanisms of cell adhesion molecules in endocrine-related cancers: a concise outlook. Front Endocrinol. 2022;13: 865436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Investig. 2009;119(6):1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang S, et al. A review on curability of cancers: more efforts for novel therapeutic options are needed. Cancers. 2019;11(11):1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mokhtari RB, et al. Combination therapy in combating cancer. Oncotarget. 2017;8(23):38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Trajanoska K, et al. From target discovery to clinical drug development with human genetics. Nature. 2023;620(7975):737–45. [DOI] [PubMed] [Google Scholar]
- 125.Ma Z, et al. Repurposing artemisinin and its derivatives as anticancer drugs: a chance or challenge? Front Pharmacol. 2021;12: 828856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang G, et al. Improving the therapeutic efficacy of sorafenib for hepatocellular carcinoma by repurposing disulfiram. Front Oncol. 2022;12: 913736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19(56):6550–65. [DOI] [PubMed] [Google Scholar]
- 128.Pazarentzos E, Bivona TG. Adaptive stress signaling in targeted cancer therapy resistance. Oncogene. 2015;34(45):5599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Padma VV. An overview of targeted cancer therapy. Biomedicine. 2015;5:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brady J, Horie S, Laffey JG. Role of the adaptive immune response in sepsis. Intensive Care Med Exp. 2020;8(Suppl 1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brady RV, Thamm DH. Tumor-associated macrophages: prognostic and therapeutic targets for cancer in humans and dogs. Front Immunol. 2023;14:1176807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mura P. Advantages of the combined use of cyclodextrins and nanocarriers in drug delivery: a review. Int J Pharm. 2020;579: 119181. [DOI] [PubMed] [Google Scholar]
- 133.Huang J, Xiao K. Nanoparticles-based strategies to improve the delivery of therapeutic small interfering RNA in precision oncology. Pharmaceutics. 2022;14(8):1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xiong T, et al. Reactive oxygen species triggered cleavage of thioketal-containing supramolecular nanoparticles for inflammation-targeted oral therapy in ulcerative colitis. Adv Funct Mater. 2024. 10.1002/adfm.202411979. [Google Scholar]
- 135.Murayama T, Gotoh N. Drug resistance mechanisms of cancer stem-like cells and their therapeutic potential as drug targets. Cancer Drug Resist. 2019;2(3):457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Saptarshi SR, Duschl A, Lopata AL. Interaction of nanoparticles with proteins: relation to bioreactivity of the nanoparticle. J Nanobiotechnol. 2013;11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dreaden EC, et al. Size matters: gold nanoparticles in targeted cancer drug delivery. Ther Deliv. 2012;3(4):457–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Urabe F, et al. Extracellular vesicles as biomarkers and therapeutic targets for cancer. Am J Physiol Cell Physiol. 2019. 10.1152/ajpcell.00280.2019. [DOI] [PubMed] [Google Scholar]
- 139.Xiong Y-X, et al. Rhodiola rosea polysaccharides-based nanoparticles loaded with DOX boosts chemo-immunotherapy for triple-negative breast cancer by re-educating tumor-associated macrophages. Int J Biol Macromol. 2023;239: 124110. [DOI] [PubMed] [Google Scholar]
- 140.Peng X, et al. Tumor microenvironment heterogeneity, potential therapeutic avenues, and emerging therapies. Curr Cancer Drug Targets. 2024;24(3):288–307. [DOI] [PubMed] [Google Scholar]
- 141.Witz IP, Izraely S. Introduction to the tumor microenvironment. 2022.
- 142.Das PK, et al., The role of cancer stem cells in disease progression and therapy resistance. 2022
- 143.Ponomarev A, et al. Intrinsic and extrinsic factors impacting cancer stemness and tumor progression. Cancers. 2022;14(4):970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aki S, et al. Cancer metabolism within tumor microenvironments. Biochim Biophys Acta Gen Sub. 2023;1867(5):130330. [DOI] [PubMed] [Google Scholar]
- 145.Zeng Z, Chen C-X. Metabonomic analysis of tumor microenvironments: a mini-review. Front Oncol. 2023;13:1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wu P, et al. Adaptive mechanisms of tumor therapy resistance driven by tumor microenvironment. Front Cell Dev Biol. 2021;9: 641469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22(3):457–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rich JN. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine. 2016;95(1 Suppl 1):S2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cassidy JW, Caldas C, Bruna A. Maintaining tumor heterogeneity in patient-derived tumor xenografts. Cancer Res. 2015;75(15):2963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hayford CE, et al. An in vitro model of tumor heterogeneity resolves genetic, epigenetic, and stochastic sources of cell state variability. PLoS Biol. 2021;19(6): e3000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang W, et al. Genetic mutation and tumor microbiota determine heterogenicity of tumor immune signature: evidence from gastric and colorectal synchronous cancers. Front Immunol. 2022;13: 947080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rahman MS, Haffari G. Analyzing tumor heterogeneity by incorporating long-range mutational influences and multiple sample data into heterogeneity factorial hidden Markov model. J Comput Biol. 2019;26(9):985–1002. [DOI] [PubMed] [Google Scholar]
- 153.Wels J, et al. Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev. 2008;22(5):559–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brown JM, Wilson WR. Exploiting tumor hypoxia in cancer treatment. Nat Rev Cancer. 2004;4(6):437–47. [DOI] [PubMed] [Google Scholar]
- 155.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98. [DOI] [PubMed] [Google Scholar]
- 157.Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541(7637):321–30. [DOI] [PubMed] [Google Scholar]
- 158.Insua-Rodríguez J, Oskarsson T. The extracellular matrix in breast cancer. Adv Drug Deliv Rev. 2016;97:41–55. [DOI] [PubMed] [Google Scholar]
- 159.Han Y, et al. Mesenchymal stem cells for regenerative medicine. Cells. 2019;8(8):886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fang M, et al. Collagen as a double-edged sword in tumor progression. Tumor Biology. 2014;35:2871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32:623–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613–28. [DOI] [PubMed] [Google Scholar]
- 163.White E. The role for autophagy in cancer. J Clin Investig. 2015;125(1):42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Abadjian MCZ, Edwards WB, Anderson CJ. Imaging the tumor microenvironment. Tumor Immune Microenviron Cancer Progress Cancer Ther. 2017. 10.1007/978-3-319-67577-0_15. [Google Scholar]
- 165.Kilmister EJ, et al. Cancer metastasis and treatment resistance: mechanistic insights and therapeutic targeting of cancer stem cells and the tumor microenvironment. Biomedicines. 2022;10(11):2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond). 2012;7(4):597–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dragu DL, et al. Therapies targeting cancer stem cells: current trends and future challenges. World J Stem Cells. 2015;7(9):1185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sutherland KD, Visvader JE. Cellular mechanisms underlying intertumoral heterogeneity. Trends Cancer. 2015;1(1):15–23. [DOI] [PubMed] [Google Scholar]
- 169.Raynaud F, et al. Pancancer inference of intratumor heterogeneity reveals associations with different forms of genomic instability. PLoS Genet. 2018;14(9): e1007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang C-C, Janes KA. Nongenetic heterogeneity caused by differential single-cell adhesion. Cell Cycle. 2014;13(14):2149–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pruller J. Intratumoural heterogeneity as a major challenge for cancer modeling and successful treatment.
- 172.Goyette M-A, Lipsyc-Sharf M, Polyak K. Clinical and translational relevance of intratumor heterogeneity. Trends Cancer. 2023. 10.1016/j.trecan.2023.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wu S, et al. Evaluating intrinsic and nonintrinsic cancer risk factors. Nat Commun. 2018;9(1):3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sato M, Shames DS, Hasegawa Y. Emerging evidence of epithelial-to-mesenchymal transition in lung carcinogenesis. Respirology. 2012;17(7):1048–59. [DOI] [PubMed] [Google Scholar]
- 175.Manfioletti G, Fedele M. Epithelial–mesenchymal transition (EMT). 2023, MDPI. p. 11386. [DOI] [PMC free article] [PubMed]
- 176.Fonseca LMD, et al. Bittersweet sugars: how unusual glycan structures may connect epithelial-to-mesenchymal transition and multidrug resistance in cancer. Medicines. 2023;10(6):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sun, T. and W. Zhong, Editorial [epithelial–mesenchymal transition (EMT) as a therapeutic target in cancer. Front Oncol. 13: p. 1218855. [DOI] [PMC free article] [PubMed]
- 178.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chen Z, et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Cancer Lett. 2016;370(1):153–64. [DOI] [PubMed] [Google Scholar]
- 180.Engle K, Kumar G. Cancer multidrug-resistance reversal by ABCB1 inhibition: a recent update. Eur J Med Chem. 2022;239: 114542. [DOI] [PubMed] [Google Scholar]
- 181.Wilkens S. Structure and mechanism of ABC transporters. Prime Rep. 2015;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen Q, Kang J, Fu C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct Target Ther. 2018;3(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Aponte PM, Caicedo A. Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017;2017(1):5619472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hao L, et al. Epitranscriptomics in the development, functions, and disorders of cancer stem cells. Front Oncol. 2023;13:1145766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fitriana M, et al. Roles of micrornas in regulating cancer stemness in head and neck cancers. Cancers. 2021;13(7):1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liang W, et al. mRNA modification orchestrates cancer stem cell fate decisions. Mol Cancer. 2020;19(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Stark VA, et al. Differential miRNA expression contributes to emergence of multiple cancer stem cell subpopulations in human colorectal cancer. J Stem Cell Res Ther. 2023;13(2):582. [PMC free article] [PubMed] [Google Scholar]
- 188.Shoff M, et al. Differential exosome miRNA expression in oral cancer stem cells. ExRNA. 2020;2:1–9. [Google Scholar]
- 189.Khan AQ, et al. Role of miRNA-regulated cancer stem cells in the pathogenesis of human malignancies. Cells. 2019;8(8):840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gutierrez-Cruz JA, Maldonado V, Melendez-Zajgla J. Regulation of the cancer stem phenotype by long noncoding RNAs. Cells. 2022;11(15):2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhao Y, et al. Panoramic view of microRNAs in regulating cancer stem cells. Essays Biochem. 2022;66(4):345–58. [DOI] [PubMed] [Google Scholar]
- 192.Ghatak D, et al. MicroRNA-324-5p–CUEDC2 axis mediates gain-of-function mutant p53-driven cancer stemness. Mol Cancer Res. 2021;19(10):1635–50. [DOI] [PubMed] [Google Scholar]
- 193.Ayob AZ, Said NA, Ramasamy TS. Integrated miRNA‒mRNA analysis revealed distinct molecular signatures and regulatory networks in cancer stem-like cell models of hepatocellular carcinoma. 2022.
- 194.Dzobo K, et al. Cancer stem cell markers in relation to patient survival outcomes: lessons for integrative diagnostics and next-generation anticancer drug development. Omics. 2021;25(2):81–92. [DOI] [PubMed] [Google Scholar]
- 195.Lala M, et al. Clinical outcomes with therapies for previously treated recurrent/metastatic head-and-neck squamous cell carcinoma (R/M HNSCC): a systematic literature review. Oral Oncol. 2018;84:108–20. [DOI] [PubMed] [Google Scholar]
- 196.Murayama T, Gotoh N. Drug resistance mechanisms of cancer stem-like cells and their therapeutic potential as drug targets. Cancer Drug Resist. 2019;2:457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zahreddine H, Borden K. Mechanisms and insights into drug resistance in cancer. Front Pharmacol. 2013;4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Safa AR. Drug and apoptosis resistance in cancer stem cells: a puzzle with many pieces. Cancer Drug Resist. 2022;5(4):850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Siqueira JM, et al. Mechanisms involved in cancer stem cell resistance in head and neck squamous cell carcinoma. Cancer Drug Resist. 2023;6(1):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zahedipour F, Kesharwani P, Sahebkar A. Mechanisms of multidrug resistance in cancer. In: Zahedipour F, editor. Aptamers engineered nanocarriers for cancer therapy. Amsterdam: Elsevier; 2023. p. 51–83. [Google Scholar]
- 201.Kuo Y-C, et al. Identification and clinical significance of pancreatic cancer stem cells and their chemotherapeutic drug resistance. Int J Mol Sci. 2023;24(8):7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Housman G, et al. Drug resistance in cancer: an overview. Cancers. 2014;6(3):1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ughachukwu P, Unekwe P. Efflux pump-mediated resistance in chemotherapy. Ann Med Health Sci Res. 2012;2(2):191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Liu Y, et al. Circumventing drug resistance pathways with a nanoparticle-based photodynamic method. Nano Lett. 2021;21(21):9115–23. [DOI] [PubMed] [Google Scholar]
- 205.Upadhyay SK, et al. Drug resistance in cancer. In: Upadhyay SK, editor., et al., Drug resistance in bacteria, fungi, malaria, and cancer. Cham: Springer; 2017. p. 449–73. [Google Scholar]
- 206.Gough NR. Paths to resistance. Sci Signaling. 2009;2(89):ec311–ec311. [Google Scholar]
- 207.Lei ZN, et al. Understanding and targeting resistance mechanisms in cancer. MedComm. 2023;4(3): e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wajapeyee N, Gupta R. Epigenetic alterations and mechanisms that drive resistance to targeted cancer therapies. Can Res. 2021;81(22):5589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Harrold E, et al. Genomic landscape of acquired resistance to targeted therapies in metastatic colorectal cancer (mCRC). Am Soc Clin Oncol. 2023. 10.1158/1078-0432.CCR-23-4005. [Google Scholar]
- 210.VD P. Targeting epigenetic alterations in cancer stem cells. Front Mol Med, 2022. 2: 1011882. [DOI] [PMC free article] [PubMed]
- 211.Mohammad IS, He W, Yin L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed Pharmacother. 2018;100:335–48. [DOI] [PubMed] [Google Scholar]
- 212.Min Q, et al. Genomic and epigenomic evolution of acquired resistance to combination therapy in esophageal squamous cell carcinoma. JCI Insight. 2021. 10.1172/jci.insight.150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.El-Ashmawy N, et al. Dual-targeted therapeutic strategy combining CSC–DC-based vaccine and cisplatin overcomes chemo-resistance in experimental mice model. Clin Transl Oncol. 2020;22:1155–65. [DOI] [PubMed] [Google Scholar]
- 214.Chu CN, Lee TKW. Targeting protein kinases in cancer stem cells. Essays Biochem. 2022;66(4):399–412. [DOI] [PubMed] [Google Scholar]
- 215.Raghav PK, Mann Z, Mohanty S. Therapeutic potential of chemical compounds in targeting cancer stem cells. In: Raghav PK, editor. Handbook of oxidative stress in cancer: therapeutic aspects. Cham: Springer; 2022. p. 1–39. [Google Scholar]
- 216.Fong D, Christensen CT, Chan MM. Targeting cancer stem cells with repurposed drugs to improve current therapies. Recent Patents Anticancer Drug Discov. 2021;16(2):136–60. [DOI] [PubMed] [Google Scholar]
- 217.Mani N, et al. Epigenetic adaptations in drug-tolerant tumor cells. Adv Cancer Res. 2023;158:293–335. [DOI] [PubMed] [Google Scholar]
- 218.Chen KG, et al. Genetic and epigenetic modeling of the origins of multidrug-resistant cells in a human sarcoma cell line. Can Res. 2005;65(20):9388–97. [DOI] [PubMed] [Google Scholar]
- 219.Nallasamy P, et al. Tumor microenvironment enriches the stemness features: the architectural event of therapy resistance and metastasis. Mol Cancer. 2022;21(1):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gillespie MS, Ward CM, Davies CC. DNA repair and therapeutic strategies in cancer stem cells. Cancers. 2023;15(6):1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Telang N. Isolation and characterization of chemo-resistant stem cells from a mouse model of hereditary nonpolyposis colon cancer. Stem Cells Cloning Adv Appl. 2021;14:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bui KD, Van Pham P. The relationship between autophagy and multidrug resistance in cancer stem cells. Sci Technol Dev J. 2022;25(4):2625–36. [Google Scholar]
- 224.Mohammad IS, He W, Yin L. Insight on multidrug resistance and nanomedicine approaches to overcome MDR. Critic Rev Ther Drug Carrier Syst. 2020;37(5):473. [DOI] [PubMed] [Google Scholar]
- 225.Kurimchak AM, et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci Signaling. 2022;15(749):eabn2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Li D, et al. Crosstalk between autophagy and CSCs: molecular mechanisms and translational implications. Cell Death Dis. 2023;14(7):409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Das S, et al. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis. 2021;26:1–22. [DOI] [PubMed] [Google Scholar]
- 228.Hu X, et al. Relationship between autophagy and drug resistance in tumors. Mini Rev Med Chem. 2023;23(10):1072–8. [DOI] [PubMed] [Google Scholar]
- 229.Safa AR. Resistance to drugs and cell death in cancer stem cells (CSCs). J Transl Sci. 2020;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Safa AR. Cancer stem cells, apoptosis pathways and mechanisms of death resistance, in oncogenomics. Amsterdam: Elsevier; 2019. p. 89–101. [Google Scholar]
- 231.Zhou T, et al. Inhibition of cancer cell migration by gold nanorods: molecular mechanisms and implications for cancer therapy. Adv Func Mater. 2014;24(44):6922–32. [Google Scholar]
- 232.Park KC, et al. PMCA inhibition reverses drug resistance in clinically refractory cancer patient-derived models. BMC Med. 2023;21(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Maleki EH, Bahrami AR, Matin MM. Cancer cell cycle heterogeneity as a critical determinant of therapeutic resistance. Genes Dis. 2024;11(1):189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sottoriva A, et al. Cancer stem cell tumor model reveals invasive morphology and increased phenotypical heterogeneity. Can Res. 2010;70(1):46–56. [DOI] [PubMed] [Google Scholar]
- 235.Phi LTH, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yoder MC. Endothelial stem and progenitor cells (stem cells): (2017 Grover conference series). Pulm Circ. 2018;8(1):2045893217743950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhang Y, et al. Notch signaling pathway: a new target for neuropathic pain therapy. J Headache Pain. 2023;24(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Smith KG, et al. Suppression of the humoral immune response by mycophenolate mofetil. Nephrol Dial Transplant. 1998;13(1):160–4. [DOI] [PubMed] [Google Scholar]
- 239.Davies A, et al. The transcriptional and epigenetic landscape of cancer cell lineage plasticity. Cancer Discov. 2023;13(8):1771–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Davis A, et al. An update on emerging therapeutic options for malignant pleural mesothelioma. Lung Cancer Targets Ther. 2022;13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lee D, et al. Superenhancer activation of KLHDC8A drives glioma ciliation and hedgehog signaling. J Clin Investig. 2023;133(2):e163592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zhang L, et al. Hedgehog signaling and the glioma-associated oncogene in cancer radioresistance. Front Cell Dev Biol. 2023;11:1257173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Pedersen KK, et al. Topical delivery of hedgehog inhibitors: current status and perspectives. Int J Mol Sci. 2022;23(22):14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Patel NS, Lee M, Marti JL. Assessment of screening mammography recommendations by breast cancer centers in the US. JAMA Intern Med. 2021;181(5):717–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Patel P, et al. Nanotheranostics for diagnosis and treatment of breast cancer. Curr Pharm Des. 2023;29(10):732–47. [DOI] [PubMed] [Google Scholar]
- 246.Pinto MP, et al. Differentially expressed genes and signaling pathways potentially involved in primary resistance to chemo-immunotherapy in advanced-stage gastric cancer patients. Int J Mol Sci. 2022;24(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Moreno-Londoño AP, et al. Canonical Wnt pathway is involved in chemoresistance and cell cycle arrest induction in colon cancer cell line spheroids. Int J Mol Sci. 2023;24(6):5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zhou X, et al. Therapy resistance in neuroblastoma: mechanisms and reversal strategies. Front Pharmacol. 2023;14:1114295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhou Y, et al. The molecular biology of prostate cancer stem cells: from the past to the future. Int J Mol Sci. 2023;24(8):7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Tyagi K, Roy A, Mandal S. Protein kinase C iota promotes glycolysis via PI3K/AKT/mTOR signaling in high grade serous ovarian cancer. Mol Biol Rep. 2024;51(1):1–10. [DOI] [PubMed] [Google Scholar]
- 251.Singh P, Sahoo SK. Piperlongumine loaded PLGA nanoparticles inhibit cancer stem-like cells through modulation of STAT3 in mammosphere model of triple negative breast cancer. Int J Pharm. 2022;616: 121526. [DOI] [PubMed] [Google Scholar]
- 252.Singh D, Assaraf YG, Gacche RN. Long noncoding RNA mediated drug resistance in breast cancer. Drug Resist Updates. 2022;63: 100851. [DOI] [PubMed] [Google Scholar]
- 253.Li J, et al. Trends in disparities and transitions of treatment in patients with early breast cancer in China and the US, 2011 to 2021. JAMA Netw Open. 2023;6(6):e2321388–e2321388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee JM, et al. Real-world adjuvant chemotherapy patterns and outcomes among elderly patients with resected early non-small cell lung cancer in the USA. Future Oncol. 2023;19(1):37–47. [DOI] [PubMed] [Google Scholar]
- 255.Ren Y, et al. Multifaceted role of redox pattern in the tumor immune microenvironment regarding autophagy and apoptosis. Mol Cancer. 2023;22(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Martínez-Pérez J, et al. Targeted treatment against cancer stem cells in colorectal cancer. Int J Mol Sci. 2024;25(11):6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Greco S, et al. High levels of hypusinated eIF5A in leiomyoma and leiomyosarcoma pathologies: a possible novel therapeutic target. Reprod Biomed Online. 2023;47(1):15–25. [DOI] [PubMed] [Google Scholar]
- 258.Banaszek N, et al. Hedgehog pathway in sarcoma: from preclinical mechanism to clinical application. J Cancer Res Clin Oncol. 2023;149(19):17635–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.de Oliveira VA, et al. Application of nanoformulations as a strategy to optimize chemotherapeutic treatment of glioblastoma: a systematic review. J Toxicol Environ Health Part B. 2024;27(4):131–52. [DOI] [PubMed] [Google Scholar]
- 260.Brown PD, et al. A prospective phase II randomized trial of proton radiotherapy vs intensity-modulated radiotherapy for patients with newly diagnosed glioblastoma. Neuro Oncol. 2021;23(8):1337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Brown NF, et al. Survival outcomes and prognostic factors in glioblastoma. Cancers. 2022;14(13):3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hiremath IS, et al. The multidimensional role of the Wnt/β-catenin signaling pathway in human malignancies. J Cell Physiol. 2022;237(1):199–238. [DOI] [PubMed] [Google Scholar]
- 263.Kumar L, et al. Therapeutic approaches in pancreatic cancer: recent updates. Biomedicines. 2023;11(6):1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yamanoi K, Mandai M. Signaling and drug resistance. Mol Diagn Targeting Gynecol Malignancy. 2021. 10.1007/978-981-33-6013-6_7. [Google Scholar]
- 265.Zhang Y, et al. Poziotinib inhibits the efflux activity of the ABCB1 and ABCG2 transporters and the expression of the ABCG2 transporter protein in multidrug resistant colon cancer cells. Cancers. 2020;12(11):3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Müller M, et al. Light-mediated discovery of surfaceome nanoscale organization and intercellular receptor interaction networks. Nat Commun. 2021;12(1):7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hollingsworth JS. ISGylation and EGFR signaling is modulated by BRCA1 expression and follicular fluid exposure in human fallopian tube epithelium. Toronto: University of Toronto; 2017. [Google Scholar]
- 268.Mouhri ZS. Overcoming resistance to targeted and nontargeted therapies with combi-molecules designed to target PARP. Canada: McGill University; 2018. [Google Scholar]
- 269.Shibata M, Hoque MO. Targeting cancer stem cells: a strategy for effective eradication of cancer. Cancers. 2019;11(5):732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Szlachcic WJ, et al. Endocrine pancreas development and dysfunction through the lens of single-cell RNA-sequencing. Front Cell Dev Biol. 2021;9: 629212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Li Y, et al. Drug resistance and Cancer stem cells. Cell Commun Signaling. 2021;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Zhang X, Powell K, Li L. Breast cancer stem cells: biomarkers, identification and isolation methods, regulating mechanisms, cellular origin, and beyond. Cancers. 2020;12(12):3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Liu H, et al. Immunomodulatory effects of mesenchymal stem cells and mesenchymal stem cell-derived extracellular vesicles in rheumatoid arthritis. Front Immunol. 2020;11:1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Thiery JP, et al. Epithelial–mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. [DOI] [PubMed] [Google Scholar]
- 275.Gerstberger S, Jiang Q, Ganesh K. Metastasis. Cell. 2023;186(8):1564–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Keibler MA, et al. Metabolic requirements for cancer cell proliferation. Cancer Metabol. 2016;4:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Jagust P, et al. Metabolism-based therapeutic strategies targeting cancer stem cells. Front Pharmacol. 2019;10:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Robey RW, et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18(7):452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. [DOI] [PubMed] [Google Scholar]
- 280.Lehto J. Finding synergies for cancer treatment: new ways to modulate DNA damage repair by CX3CR1 and PFKFB3 inhibition. Sweden: Karolinska Institutet; 2021. [Google Scholar]
- 281.Sun H, et al. Diet and ovarian cancer risk: an umbrella review of systematic reviews and meta-analyses of cohort studies. Clin Nutr. 2021;40(4):1682–90. [DOI] [PubMed] [Google Scholar]
- 282.Fucikova J, et al. TIM-3 dictates functional orientation of the immune infiltrate in ovarian cancer. Clin Cancer Res. 2019;25(15):4820–31. [DOI] [PubMed] [Google Scholar]
- 283.Galluzzi L, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–83. [DOI] [PubMed] [Google Scholar]
- 284.Olaussen KA, et al. DNA repair by ERCC1 in non–small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355(10):983–91. [DOI] [PubMed] [Google Scholar]
- 285.Brumatti G, et al. BH3 mimetics and TKI combined therapy for chronic myeloid leukemia. Biochem J. 2023;480(2):161–76. [DOI] [PubMed] [Google Scholar]
- 286.Pei X, Huang X. New approaches in allogenic transplantation in AML. Seminars in hematology. Amsterdam: Elsevier; 2019. [DOI] [PubMed] [Google Scholar]
- 287.Cortes JE, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33(2):379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Chereda B, Melo JV. The biology and pathogenesis of chronic myeloid leukemia. Chronic Myeloid Leukemia. 2016. 10.1007/978-3-319-33198-0_2. [Google Scholar]
- 289.Corbin AS, et al. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Investig. 2011;121(1):396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Tafani M, et al. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-κB. Carcinogenesis. 2011;32(8):1167–75. [DOI] [PubMed] [Google Scholar]
- 291.Matsushita Y, Nakagawa H, Koike K. Lipid metabolism in oncology: why it matters, how to research, and how to treat. Cancers. 2021;13(3):474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wang J-Q, et al. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist Updates. 2021;54: 100743. [DOI] [PubMed] [Google Scholar]
- 293.Malik JA, et al. Nanodrug delivery system: a promising approach against breast cancer. Ther Deliv. 2023;14(5):357–81. [DOI] [PubMed] [Google Scholar]
- 294.Assaraf YG. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007;26:153–81. [DOI] [PubMed] [Google Scholar]
- 295.Kumar A, et al. Regulation of thymidylate synthase: an approach to overcome 5-FU resistance in colorectal cancer. Med Oncol. 2022;40(1):3. [DOI] [PubMed] [Google Scholar]
- 296.Ortíz R, et al. Nanomedicine to overcome multidrug resistance mechanisms in colon and pancreatic cancer: recent progress. Cancers. 2021;13(9):2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Meisenberg C, et al. Epigenetic changes in histone acetylation underpin resistance to the topoisomerase I inhibitor irinotecan. Nucleic Acids Res. 2017;45(3):1159–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8. [DOI] [PubMed] [Google Scholar]
- 299.Maloney SM, et al. Mechanisms of taxane resistance. Cancers. 2020;12(11):3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Jurj A, et al. New insights in gene expression alteration as effect of paclitaxel drug resistance in triple negative breast cancer cells. Cell Physiol Biochem. 2020;54(4):648–64. [DOI] [PubMed] [Google Scholar]
- 301.Maniam S, Maniam S. Small molecules targeting programmed cell death in breast cancer cells. Int J Mol Sci. 2021;22(18):9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Vasconcelos IC, et al. A freeze-and-thaw-induced fragment of the microtubule-associated protein tau in rat brain extracts: implications for the biochemical assessment of neurotoxicity. Biosci Rep. 2021;41(3):BSR20203980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Guo Q, et al. Sequential release of pooled sirnas and paclitaxel by aptamer-functionalized shell-core nanoparticles to overcome paclitaxel resistance of prostate cancer. ACS Appl Mater Interfaces. 2021;13(12):13990–4003. [DOI] [PubMed] [Google Scholar]
- 304.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10(3):194–204. [DOI] [PubMed] [Google Scholar]
- 305.van Dijk A, et al. The multidrug resistance protein breast cancer resistance protein (BCRP) protects adipose-derived stem cells against ischemic damage. Cell Biol Toxicol. 2012;28:303–15. [DOI] [PubMed] [Google Scholar]
- 306.Shang Y, Cai X, Fan D. Roles of epithelial–mesenchymal transition in cancer drug resistance. Curr Cancer Drug Targets. 2013;13(9):915–29. [DOI] [PubMed] [Google Scholar]
- 307.Gao L, et al. Nanodrug delivery system for the treatment of multidrug-resistant breast cancer: Current status and future perspectives. Biomed Pharmacother. 2024;179: 117327. [DOI] [PubMed] [Google Scholar]
- 308.Fletcher JI, et al. Too many targets, not enough patients: rethinking neuroblastoma clinical trials. Nat Rev Cancer. 2018;18(6):389–400. [DOI] [PubMed] [Google Scholar]
- 309.Hanssen KM, Haber M, Fletcher JI. Targeting multidrug resistance-associated protein 1 (MRP1)-expressing cancers: Beyond pharmacological inhibition. Drug Resist Updates. 2021;59: 100795. [DOI] [PubMed] [Google Scholar]
- 310.Hipfner DR. Structural and functional analysis of the multidrug resistance protein, MRP, using monoclonal antibodies. London: Queen’s University; 1999. [Google Scholar]
- 311.Lan Z, Li X, Zhang X. Glioblastoma: an update in pathology, molecular mechanisms and biomarkers. Int J Mol Sci. 2024;25(5):3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Tarasov VV, et al. Feasibility of targeting glioblastoma stem cells: from concept to clinical trials. Curr Top Med Chem. 2019;19(32):2974–84. [DOI] [PubMed] [Google Scholar]
- 313.Lang F, et al. Genotoxic therapy and resistance mechanism in gliomas. Pharmacol Ther. 2021;228: 107922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Brennan CW, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Fu W, et al. Enhanced efficacy of temozolomide loaded by a tetrahedral framework DNA nanoparticle in the therapy for glioblastoma. ACS Appl Mater Int. 2019;11(43):39525–33. [DOI] [PubMed] [Google Scholar]
- 316.Zhang D, et al. Near infrared-activatable biomimetic nanogels enabling deep tumor drug penetration inhibit orthotopic glioblastoma. Nat Commun. 2022;13(1):6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Hegi ME, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 318.Yonesaka K, et al. Alternating therapy with osimertinib and afatinib blockades EGFR secondary mutation in EGFR-mutant lung cancer: a single-arm phase II trial. Clinical Lung Cancer. 2023;24(6):519-527. e4. [DOI] [PubMed] [Google Scholar]
- 319.Kobayashi H, et al. Chemoradiotherapy for limited-stage small-cell lung cancer and interstitial lung abnormalities. Radiat Oncol. 2021;16:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Taniguchi Y, et al. Predictive factors for poor progression-free survival in patients with non-small cell lung cancer treated with nivolumab. Anticancer Res. 2017;37(10):5857–62. [DOI] [PubMed] [Google Scholar]
- 321.Inoue T, et al. Analysis of early death in Japanese patients with advanced non–small-cell lung cancer treated with nivolumab. Clin Lung Cancer. 2018;19(2):e171–6. [DOI] [PubMed] [Google Scholar]
- 322.Kosaka T, et al. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Can Res. 2004;64(24):8919–23. [DOI] [PubMed] [Google Scholar]
- 323.Llovet JM, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151–72. [DOI] [PubMed] [Google Scholar]
- 324.Elkhenany H, et al. Stem cell therapy for hepatocellular carcinoma: future perspectives. Cell Biol Transl Med. 2020;7:97–119. [DOI] [PubMed] [Google Scholar]
- 325.Liu L, et al. Robotic versus laparoscopic major hepatectomy for hepatocellular carcinoma: short-term outcomes from a single institution. BMC Surg. 2022;22(1):432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Jiang Y, Han Q-J, Zhang J. Hepatocellular carcinoma: mechanisms of progression and immunotherapy. World J Gastroenterol. 2019;25(25):3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wilhelm SM, et al. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.You YN, et al. Detection of pathogenic germline variants among patients with advanced colorectal cancer undergoing tumor genomic profiling for precision medicine. Dis Colon Rectum. 2019;62(4):429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Mulet-Margalef N, et al. Challenges and therapeutic opportunities in the dMMR/MSI-H colorectal cancer landscape. Cancers. 2023;15(4):1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Gulubova M, et al. New approach in understanding colorectal cancer immunosuppression and immunotherapy-based strategies in the treatment of microsatellite stable colorectal cancer. Acta Med Bulgarica. 2024;51(2):65–72. [Google Scholar]
- 331.Tanaka T, et al. Pharmacokinetic evaluation of oxaliplatin combined with S-1 (SOX) chemotherapy in a rat model of colorectal cancer with acute kidney injury: predictive renal biomarkers for dose optimization. Xenobiotica. 2023;53(10–11):613–20. [DOI] [PubMed] [Google Scholar]
- 332.Tanaka S, et al. miR-33a-5p in small extracellular vesicles as noninvasive biomarker for oxaliplatin sensitivity in human colorectal cancer cells. Biochem Biophys Rep. 2021;26: 100996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Le DD, Van Vo T, Sarakarn P. Overall survival rate of Vietnamese patients with colorectal cancer: a hospital-based cohort study in the central region of Vietnam. Asian Pacific J Cancer Prevent. 2021;22(11):3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Telli ML, et al. Neoadjuvant talazoparib in patients with germline BRCA1/2 mutation-positive, early-stage triple-negative breast cancer: exploration of tumor BRCA mutational status. Breast Cancer. 2024;31:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Telli ML, et al. Phase 1b study of berzosertib and cisplatin in patients with advanced triple-negative breast cancer. NPJ Breast Cancer. 2022;8(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Zhu Y, et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat Commun. 2018;9(1):1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Yu K-D, et al. Effect of adjuvant paclitaxel and carboplatin on survival in women with triple-negative breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. 2020;6(9):1390–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20. [DOI] [PubMed] [Google Scholar]
- 339.Kwa M, et al. Ovarian cancer in BRCA mutation carriers: improved outcome after intraperitoneal (IP) cisplatin. Ann Surg Oncol. 2014;21:1468–73. [DOI] [PubMed] [Google Scholar]
- 340.Na HY, et al. Expression of class III beta-tubulin is associated with invasive potential and poor prognosis in thyroid carcinoma. J Clin Med. 2020;9(12):3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Mukhtar TK, et al. Protein-truncating and rare missense variants in ATM and CHEK2 and associations with cancer in UK biobank whole-exome sequence data. J Med Genet. 2024;61:6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Karihtala P, et al. Expression levels of microRNAs miR-93 and miR-200a in pancreatic adenocarcinoma with special reference to differentiation and relapse-free survival. Oncology. 2019;96(3):164–70. [DOI] [PubMed] [Google Scholar]
- 343.le Sève JD, et al. Risk factors for venous thromboembolic disease in cancer patients treated with immune checkpoint inhibitor. Thrombosis Hemostasis. 2023;123(11):1049–56. [DOI] [PubMed] [Google Scholar]
- 344.Grinshpun A, Sandusky ZM, Jeselsohn R. The clinical utility of ESR1 mutations in hormone receptor-positive, HER2-negative advanced breast cancer. Hematol Oncol Clin. 2023;37(1):169–81. [DOI] [PubMed] [Google Scholar]
- 345.Patel JM, Jeselsohn RM. Estrogen receptor alpha and ESR1 mutations in breast cancer, nuclear receptors in human health and disease. Amsterdam: Springer; 2022. p. 171–94. [DOI] [PubMed] [Google Scholar]
- 346.Lok SW, et al. A phase Ib dose-escalation and expansion study of the BCL2 inhibitor venetoclax combined with tamoxifen in ER and BCL2–positive metastatic breast cancer. Cancer Discov. 2019;9(3):354–69. [DOI] [PubMed] [Google Scholar]
- 347.Corti C, et al. Novel endocrine therapies: what is next in estrogen receptor positive, HER2 negative breast cancer? Cancer Treat Rev. 2023;117: 102569. [DOI] [PubMed] [Google Scholar]
- 348.Jeselsohn R, et al. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12(10):573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Esteva FJ, Katz E. Tailoring neoadjuvant therapy in human epidermal growth factor receptor 2–positive early breast cancer: recent advances and strategies. JCO Oncol Pract. 2024;20(8):1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Barot SV, Roesch E, Abraham J. Optimizing adjuvant and postneoadjuvant therapy in HER2-positive early breast cancer. Expert Rev Anticancer Ther. 2022;22(12):1289–99. [DOI] [PubMed] [Google Scholar]
- 351.Gonzalez-Conchas GA, et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: a systematic review and a meta-analysis. Cancer Treat Rev. 2018;62:1–8. [DOI] [PubMed] [Google Scholar]
- 352.Samsudin F, et al. Not all therapeutic antibody isotypes are equal: the case of IgM versus IgG in Pertuzumab and Trastuzumab. Chem Sci. 2020;11(10):2843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Emran TB, et al. Multidrug resistance in cancer: understanding molecular mechanisms, immunoprevention and therapeutic approaches. Front Oncol. 2022;12: 891652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDH hi CD44+ human breast cancer cells. Breast Cancer Res Treat. 2012;133:75–87. [DOI] [PubMed] [Google Scholar]
- 355.Jiang Y, et al. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Luthold C, et al. The extracellular matrix stiffening: a trigger of prostate cancer progression and castration resistance? Cancers. 2022;14(12):2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Levental KR, et al. Lipidomic and biophysical homeostasis of mammalian membranes counteracts dietary lipid perturbations to maintain cellular fitness. Nat Commun. 2020;11(1):1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Nonnast E, Mira E, Mañes S. Biomechanical properties of laminins and their impact on cancer progression. Biochim Biophys Acta Rev Cancer. 2024;1879:189181. [DOI] [PubMed] [Google Scholar]
- 359.Dzobo K, Dandara C. The extracellular matrix: its composition, function, remodeling, and role in tumorigenesis. Biomimetics. 2023;8(2):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Wullkopf L, et al. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol Biol Cell. 2018;29(20):2378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Bigos KJ, et al. Tumor response to hypoxia: understanding the hypoxic tumor microenvironment to improve treatment outcome in solid tumors. Front Oncol. 2024;14:1331355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Kazmierczak D, et al. The significance of MicroRNAs expression in regulation of extracellular matrix and other drug resistance genes in drug resistant ovarian cancer cell lines. Int J Mol Sci. 2020;21(7):2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. 2020;6:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Lv Y, et al. Three-dimensional decellularized tumor extracellular matrices with different stiffness as bioengineered tumor scaffolds. Bioactive Mater. 2021;6(9):2767–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Lin Z, et al. Cancer therapy resistance mediated by cancer-associated fibroblast-derived extracellular vesicles: biological mechanisms to clinical significance and implications. Mol Cancer. 2024. 10.1186/s12943-024-02106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Shu J, et al. Cancer cell response to extrinsic and intrinsic mechanical cue: opportunities for tumor apoptosis strategies. Regenerative Biomater. 2024;11:rbae016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Harris B, et al. Targeting hypoxia in solid and hematological malignancies. J Exp Clin Cancer Res. 2022;41(1):318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Gao Q, et al. Cancer stem cells and the tumor microenvironment in tumor drug resistance. Stem Cell Rev Rep. 2023;19(7):2141–54. [DOI] [PubMed] [Google Scholar]
- 369.Harrison H, et al. HIF1-alpha expressing cells induce a hypoxic-like response in neighboring cancer cells. BMC Cancer. 2018;18:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Borlongan MC, Wang H. Profiling and targeting cancer stem cell signaling pathways for cancer therapeutics. Front Cell Dev Biol. 2023;11:1125174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Mukherjee S, Chatterjee K. Targeting signaling pathways in solid tumors part A. Amsterdam: Elsevier; 2024. [Google Scholar]
- 372.Lin PK, Koller GM, Davis GE. Elucidating the morphogenic and signaling roles of defined growth factors controlling human endothelial cell lumen formation versus sprouting behavior. Am J Pathol. 2023;193(12):2203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Zheng T, et al. Role of microRNA in anticancer drug resistance. Int J Cancer. 2010;126(1):2–10. [DOI] [PubMed] [Google Scholar]
- 374.Takahashi RU, Miyazaki H, Ochiya T. The role of microRNAs in the regulation of cancer stem cells. Front Genet. 2014;4:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Huang B, et al. A promising antitumor method: targeting CSC with immune cells modified with CAR. Front Immunol. 2022;13: 937327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Alhabbab RY. Targeting cancer stem cells by genetically engineered chimeric antigen receptor T cells. Front Genet. 2020;11:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Gu W, et al. Actively targeted nanomedicines for precision cancer therapy: concept, construction, challenges and clinical translation. J Control Release. 2021;329:676–95. [DOI] [PubMed] [Google Scholar]
- 378.Lo Iacono M, et al. Destroying the shield of cancer stem cells: natural compounds as promising players in cancer therapy. J Clin Med. 2022;11(23):6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Petersburg J, Vallera DA, Wagner CR. Eradication of heterogeneous tumors by T cells targeted with combination bispecific chemically self-assembled nanorings. Mol Cancer Ther. 2023;22(3):371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Li A, Kibby D, Foo J. A comparison of mutation and amplification-driven resistance mechanisms and their impacts on tumor recurrence. J Math Biol. 2023;87(4):59. [DOI] [PubMed] [Google Scholar]
- 381.Huldani H, et al. Mechanisms of cancer stem cells drug resistance and the pivotal role of HMGA2. Pathol Res Pract. 2022;234: 153906. [DOI] [PubMed] [Google Scholar]
- 382.Zhang J. The therapeutic potential of CRISPR-Cas9 in drug resistance during cancer treatment. Highlights Sci Eng Technol. 2023;45:286–90. [Google Scholar]
- 383.McLean B, et al. A CRISPR path to finding vulnerabilities and solving drug resistance: targeting the diverse cancer landscape and its ecosystem. Adv Genet. 2022;3(4):2200014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Jenkins MA, et al. Assessment of a polygenic risk score for colorectal cancer to predict risk of Lynch syndrome colorectal cancer. JNCI Cancer Spectr. 2021;5(2):pkab022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Poghosyan H, et al. A brief report of lung cancer screening utilization before, during, and in the later stages of the COVID-19 pandemic in the United States. JTO Clin Res Rep. 2024;5(9): 100705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Xu X, et al. Application of mPEG-CS-cRGD/Bmi-1RNAi-PTX nanoparticles in suppression of laryngeal cancer by targeting cancer stem cells. Drug Delivery. 2023;30(1):2180112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Yin W, et al. Cancer and stem cells. Exp Biol Med. 2021;246(16):1791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Ebrahimi M, et al. PI3K/Akt/mTOR signaling pathway in cancer stem cells. Pathol Res Pract. 2022;237: 154010. [DOI] [PubMed] [Google Scholar]
- 389.Zhou W, Yan K, Xi Q. BMP signaling in cancer stemness and differentiation. Cell Regeneration. 2023;12(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Huang T, et al. The roles of extracellular vesicles in gastric cancer development, microenvironment, anticancer drug resistance, and therapy. Mol Cancer. 2019;18:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Huang H, Tsui Y-M, Ng IO-L. Fueling HCC dynamics: interplay between tumor microenvironment and tumor initiating cells. Cell Mol Gastroenterol Hepatol. 2023;15(5):1105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Huang S, et al. Cellular dynamics of tumor microenvironment driving immunotherapy resistance in non-small cell lung carcinoma. Cancer Lett. 2024;604:217272. [DOI] [PubMed] [Google Scholar]
- 393.Li B, et al. Immune checkpoint inhibitors combined with targeted therapy: the recent advances and future potentials. Cancers. 2023;15(10):2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Lavudi K, et al. ALDH and cancer stem cells: pathways, challenges, and future directions in targeted therapy. Life Sci. 2024;356:123033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
For this comprehensive review, data were obtained from previous research/publication works.