

Abstract
Tau is a microtubule‐associated protein implicated in Alzheimer's disease (AD) and other neurodegenerative disorders termed tauopathies. Pathological, aggregated forms of tau form neurofibrillary tangles (NFTs), impairing its ability to stabilize microtubules and promoting neurotoxicity. Indeed, NFTs correlate with neuronal loss and cognitive impairment. Hyperphosphorylation of tau is seen in all tauopathies and mirrors disease progression, suggesting an essential role in pathogenesis. However, hyperphosphorylation remains a generic and ill‐defined term, obscuring the functional importance of specific sites in different physiological or pathological settings. Here, we focused on global mapping of tau phosphorylation specifically during conditions of neuronal hyperexcitation. Hyperexcitation is a property of AD and other tauopathies linked to human cognitive deficits and increased risk of developing seizures and epilepsy. Moreover, hyperexcitation promotes extracellular secretion and trans‐synaptic propagation of tau. Using unbiased mass spectrometry, we identified a novel phosphorylation signature in the C‐terminal domain of tau detectable only during neuronal hyperactivity in primary cultured rat hippocampal neurons. These sites influenced tau localization to dendrites as well as the size of excitatory postsynaptic sites. These results demonstrate novel physiological tau functions at synapses and the utility of comprehensive analysis of tau phosphorylation during specific signaling contexts.
Keywords: Alzheimer’s disease; excitatory synapse; hyperphosphorylation; phosphorylation code; hippocampal neurons, MAPT
Tau is a microtubule‐associated protein implicated in neurodegenerative tauopathies. Tau hyperphosphorylation is essential for pathogenesis but remains ill defined. Here, global mapping of tau phosphorylation was conducted specifically during neuronal hyperexcitation, a condition which promotes trans‐synaptic tau propagation. Using unbiased mass spectrometry, we identified a novel phosphorylation signature in the tau C‐terminal domain detectable only during neuronal hyperactivity in primary cultured rat hippocampal neurons. These sites influenced tau dendritic localization and excitatory postsynapse size. These results demonstrate novel synaptic functions of tau and the importance of comprehensive analysis of tau phosphorylation to elucidate the role of specific phosphosites in different contexts.
Abbreviations
- AD
Alzheimer's disease
- AMPA
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid
- APP
amyloid precursor protein
- DIV
days in vitro
- GABA
gamma‐amino butyric acid
- HPLC
high‐performance liquid chromatography
- ICC
immunocytochemistry
- IP
immunoprecipitation
- MS
mass spectrometry
- NFT
neurofibrillary tangle
- PHF
paired helical filament
- PLK2
polo like kinase 2
- PSD‐95
postsynaptic density 95
- PTX
picrotoxin
- SDS‐PAGE
sodium dodecyl‐sulfate polyacrylamide gel electrophoresis
- WB
western blot
- WT
wild type
1. INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disorder marked by progressive memory impairment and loss of brain tissue throughout the cortex and hippocampus (Goedert & Spillantini, 2006). Hallmark pathologies of AD are amyloid‐beta (Aβ) plaques and neurofibrillary tangles (NFTs) composed of the microtubule‐associated protein tau (Sanabria‐Castro et al., 2017). Tau promotes stabilization and polymerization of microtubules, which are essential for the survival of neurons (Franker & Hoogenraad, 2013). In AD, tau is hyperphosphorylated and loses its ability to stabilize microtubules, yielding disrupted axonal transport, synaptic dysfunction, and eventual neuronal death (Murphy, 2018). Hyperphosphorylated tau also promotes aggregation into NFTs, which have originally been reported to correlate with neurodegeneration and cognitive impairment (Arriagada et al., 1992; Braak & Braak, 1991, 1995; Nelson et al., 2012; Schoenheit et al., 2004). However, it is currently thought that early and intermediate tau oligomeric species are likely the most toxic forms of tau (Lasagna‐Reeves et al., 2010, 2011; Maeda et al., 2006; Tian et al., 2013). It is thus essential to unveil the etiology and patterns of phosphorylated tau.
In AD and other tauopathies, hyperexcitation is an established characteristic (Bakker et al., 2012; Busche et al., 2008; Crimins et al., 2012; Hafkemeijer et al., 2012; Holth et al., 2013; Hunsberger et al., 2016; Minkeviciene et al., 2009; Palop et al., 2007; Palop & Mucke, 2009; Putcha et al., 2011; Rocher et al., 2010), a condition linked to human cognitive deficits (Bakker et al., 2012; Putcha et al., 2011; Verret et al., 2012). Moreover, AD patients present increased risk of developing seizures and epilepsy (Kazim et al., 2017). The origin of this aberrant overactivity is unclear, but studies point to GABAergic dysfunction and loss of inhibitory tone (Abbas et al., 2016; Busche et al., 2008, 2012) and/or excessive glutamate (Hunsberger et al., 2016; Zott et al., 2019). Convincingly, hyperexcitation exacerbates extracellular secretion of both Aβ (Kamenetz et al., 2003) and tau (Pooler et al., 2013; Wu et al., 2016; Yamada et al., 2014), inducing the spread of pathology. Moreover, combating CA3 hyperactivity in humans has resulted in memory and cognitive benefits (Bakker et al., 2012). It is plausible that tau plays an important role in hyperexcitation as it has been shown that mice overexpressing hAPP displayed increased resistance to excitotoxicity and seizures when tau was deleted (Roberson et al., 2007).
Here, we focused on using mass spectrometry (MS) to characterize site‐specific tau phosphorylation during chronic hyperexcitation. Such conditions are known to induce homeostatic synaptic plasticity, a class of compensatory negative feedback mechanism which stabilizes the neural network from runaway excitation or inhibition (Queenan et al., 2012). We showed that distinct sites at the C‐terminus of tau are phosphorylated during homeostatic adaptation to hyperexcitation and regulate both tau dendritic localization and the intensity of the postsynaptic marker PSD‐95. These findings shed light on novel tau functions and potential new targets for AD therapies.
2. METHODS
2.1. Animals
All experimental procedures using rodents were approved and performed per regulations of the Georgetown University Institutional Animal Care and Use Committee (protocol #2016–1145). Pregnant Sprague–Dawley rats (8‐ to 10‐week‐old females) were obtained from Charles River (Raleigh, NC, USA). Timed‐pregnant Sprague–Dawley dams were singly housed for 2 days in individually ventilated cages, with ad libitum access to food and water. This study was not pre‐registered. No exclusion criteria were predetermined and no animals were excluded. At embryonic day 18 (E18), pregnant Sprague–Dawley dams were euthanized using a flow‐regulated carbon dioxide chamber and death was verified by toe pinch and cervical dislocation. 30 dams were used for this study.
2.2. Primary hippocampal culture
Hippocampal neurons were prepared from embryonic day (E18) rat embryos (Lee et al., 2017) Neurons were plated on coverslips (for microscopy) or well plates (for biochemistry) coated with poly‐D‐lysine/laminin (Sigma, cat. #P8099, Sigma, cat. #L2020) and grown in neurobasal media (Invitrogen) supplemented with SM1 (STEMCELL Technologies), penicillin/streptomycin, 0.5 mM glutamine and 12.5 mM glutamate. Neurons used were plated on a 12‐well plate at a density of 400 000 cells/well (for biochemistry) or 150 000 cells/well (on coverslips). Cell media were changed at day in vitro (DIV)5 and 12. Neurons were collected between DIV20‐22. Between DIV15‐16, neurons were transfected with 2 μg of DNA using Lipofectamine 2000 (Invitrogen cat. #11688019) for 3 days. One dam was used for each plating for the immunochemistry and immunostaining experiments (combining hippocampi from ~10–12 embryos per culture). Four dams (~48 embryos) were used and pooled for each mass spectrometry (MS) experiment.
2.3. Pharmacology
For synaptic overactivity modulation, neurons were treated between DIV20‐21 with 100 μM picrotoxin (PTX) (Abdel‐Ghani et al., 2023; Seeburg & Sheng, 2008) dissolved in 0.1 N NaOH, or with vehicle control (0.1 N NaOH), for 24 h. During time course experiments, 100 μM of PTX was added for 30 min, 8 h, 24 h, and 48 h. For kinase inhibitor studies, 50 nM BI‐6727 (BI‐6727, Chemietek) or 50 nM TC‐S 7005 (Tocris cat. #4459) were applied during the final 4 h of PTX administration.
2.4. Antibodies
The following antibodies were purchased from the indicated supplier and used at the indicated dilution listed: Total tau (Agilent Cat# A0024, RRID:AB_10013724, western blot (WB) 1/100000, immunoprecipitation (IP) 2 μg antibody/mg of total protein); phospho‐serine 422 (Abcam Cat# ab79415, RRID:AB_1603345, WB 1/1000, immunocytochemistry (ICC) 1/300); Flag (Sigma‐Aldrich Cat# F3165, RRID:AB_259529, ICC 1/80000); MAP2 (PhosphoSolutions Cat# 1100‐MAP2, RRID:AB_2492141, ICC 1/500); Alexa Fluor anti‐chicken 647 (Thermo Fisher Scientific Cat# A‐21449, RRID:AB_2535866, ICC 1/300); Alexa Fluor anti‐mouse 555 (Thermo Fisher Scientific Cat# A28180, RRID:AB_2536164, ICC 1/300).
2.5. Immunocytochemistry
Primary hippocampal cultured neurons were fixed at room temperature (1% paraformaldehyde for 7 min followed by ice‐cold methanol for 7 min), washed 3 × 5 min in phosphate buffered saline (PBS), and incubated with primary antibodies diluted in GDB (0.1% gelatin (wt/vol), 0.3% Triton X‐100 (vol/vol), and 450 mM NaCl in PBS). Cells were then incubated in Alexa‐555 or Alexa‐647 tagged secondary antibodies diluted in GDB buffer.
2.6. IP and immunoblotting
Cells were removed from the incubator and lysed immediately at room temperature in RIPA buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.5% deoxycholate, 1% nonidet P40, 1% SDS) with phosphatase inhibitors (NaF and Na2VO4) and protease inhibitors (AEBSF and mini cOmplete cocktail (Thermo Scientific cat# 78431, Millipore Sigma Cat# 11836153001)). Using a BCA Assay (Thermo, Cat#23225) to quantify protein loads, equal amounts of protein were prepared for each sample. For western blotting (WB), 4x NuPage LDS Buffer (Invitrogen #NP0007) was combined with beta‐mercaptoethanol at a ratio of 4:1. The solution was loaded at a 1x dilution with protein onto an SDS‐PAGE gel. For phosphospecific antibody westerns, phosphospecific antibody was probed first and then stripped (62.5 mM Tris, 20% SDS, 0.1% β–mercaptoethanol) and reprobed for loading control (beta‐actin or total tau, as neither were affected by activity stimulation). Total tau signal was also normalized to quantified protein load. SDS‐PAGE gel was soaked in 20% ethanol for 7 min and transferred to a nitrocellulose membrane (Invitrogen cat. #IB23001) using Invitrogen iBlot 2 Gel Transfer Device (IB21001). Following transfer, membranes were washed with Tris‐buffered saline with 0.1% Tween‐20 (TBST) and incubated in blocking solution (5% skim milk/TBST) for 1 h at room temperature on a shaker. Blots were incubated overnight at 4C in primary antibody in blocking solution on a shaker. The next day, blots were washed 3× for 10 min each in TBST on a shaker. The blot incubated in either mouse or rabbit horseradish peroxidase‐conjugated secondary antibodies (Sigma) in blocking solution for 1 h at room temperature on a shaker. Finally, the blots were washed for 30 min in a container of TBST (>200 mL) at room temperature on a shaker. Blots were incubated for 1 min with enhanced chemiluminescence solution (Thermo Scientific Pierce cat #P132106). For immunoprecipitation (IP), Dynabeads Antibody Coupling Kit (Invitrogen, Cat#14311D) was used to conjugate Total Tau antibody (Agilent #A0024) to beads (approximately 0.5 mg beads/1 mg protein). Beads were incubated with samples overnight at 4°C and eluted in elution buffer (Laemmli sample buffer, 10% beta‐mercaptoethanol) for 10 min at 50°C. Samples were run on a 10%–12.5% SDS‐PAGE gel. The gel was then fixed for 45 min at 25°C in fix solution (10% acetic acid, 25% isopropyl alcohol), then incubated overnight, shaking at 25°C in Coomassie stain. The gel was destained (40% methanol, 10% acetic acid) and bands were excise at ~55 kDa for MS.
2.7. Mass spectrometry
2.7.1. Sample preparation
The gel bands were destained with 50% ACN followed by the addition of 100 μL of 10 mM DTT (Sigma) in 50 mM bicarbonate buffer and incubation at 37°C for 0.5 h. After removal of DTT solution, 100 μL of 30 mM iodoacetamide (Thermo Fisher Scientific) in 50 mM bicarbonate buffer was added and incubated in dark for 30 min. Proteins were then digested with the addition of sequencing‐grade trypsin/LysC mixture (Promega) followed by incubation at 37°C overnight. Yielding peptides were extracted and desalted with C18 Ziptip columns (Millipore), with elutes dried down with a SpeedVac NanoUPLC‐MS/MS. Dried peptides were dissolved into 0.1% formic acid and loaded onto a C18 Trap column (Waters Acquity UPLC Symmetry C18 NanoAcquity 10 K 2G V/M, 100 A, 5 μm, 180 μm × 20 mm) at 15 μL/min for 2 min. Peptides were then separated with an analytical column (Waters Acquity UPLC M‐Class, peptide BEH C18 column, 300 A, 1.7 μm, 75 μm × 150 mm) which was temperature controlled at 40°C. The flow rate was set as 400 nL/min. A 60‐min gradient of buffer A (2% ACN, 0.1% formic acid) and buffer B (0.1% formic acid in ACN) was used for separation: 1% buffer B at 0 min, 5% buffer B at 1 min, 45% buffer B at 35 min, 99% buffer B at 37 min, 99% buffer B at 40 min, 1% buffer B at 40.1 min, and 1% buffer B at 60 min. Data were acquired with the TripleTOF 6600 mass spectrometer using an ion spray voltage of 2.3 kV, GS1 5 psi, GS2 0, CUR 30 psi and an interface heater temperature of 150°C. Mass spectra was recorded with Analyst TF 1.7 software in the IDA mode. Each cycle consisted of a full scan (m/z 400–1600) and 50 information dependent acquisitions (IDAs) (m/z 100–1800) in the high sensitivity mode with a 2+ to 5+ charge state. Rolling collision energy was used.
2.8. Data analysis
Data files were submitted for simultaneous searches using Protein Pilot version 5.0 software (Sciex) utilizing the Paragon and Progroup algortihms and the integrated false discovery rate (FDR) analysis function. MS/MS data were searched against the customized Tau protein database (rat). Trypsin/LysC was selected as the enzyme. Carbamidomethylation was set as a fixed modification on cysteine. Phosphorylation emphasis was chosen as a special factor. Other search parameters include instrument (TripleTOF 6600), ID Focus (Biological modifications), search effort (Thorough), false discovery rate (FDR) analysis (Yes), and user modified parameter files (No). Proteins were inferred based on the ProGroupTM algorithm associated with the ProteinPilot software. The detected protein threshold in the software was set to the value which corresponded to 5% FDR. Peptides were defined as redundant if they had identical cleavage site(s), amino acid sequence, and modification. All peptides were filtered with confidence to 5% FDR, with the confidence of phosphorylation sites such as phosphor serine (p‐Ser), phospho‐threonine (p‐Thr), and phospho‐tyrosine (p‐Tyr) automatically calculated. Data from the workflow provided from the Georgetown University Proteomics Shared Resource were compiled using a custom program written in Julia. Julia v1.7 was used with BioSequences package, DataFrames package, XSLX package, Plots package, and PyPlots package.
2.9. DNA constructs
Control neurons for experiments were transfected with Flag tagged‐Variant 2 (tau 441) human Tau from Genescript (#OHu28029D). This plasmid was used as template to create constitutively phosphorylated (S422D) or phospho‐null (S422A) mutants. The following oligonucleotides were used for site mutagenesis of S422D and S422A, respectively: 5′‐catcgacatggtagacgacccccagctcgccacgc‐3′ (sense), 5′‐gcgtggcgagctgggggtcgtctaccatgtcgatg‐3′ (antisense) and 5′‐cgacatggtagacgcgccccagctcgc‐3′ (sense) 5′‐gcgagctggggcgcgtctaccatgtcg‐3′ (antisense). The bulk mutations of S412A/S413A/T414A, S412D/S413D/T414D, S409A/S412A/S413A/T414A/S416A/S422A/T427A, and S409D/S412D/S413D/T414D/S416D/S422D/T427D, were created using a mammalian gene expression vector, CMV promotor, and C‐terminal Flag epitope‐tagged MAPT open reading frame (ORF). Bulk mutations were made using Vector Builder. The coding sequence used for the ORF can be found under NM_005910.6 CDS and changes in codons were made according to mutations desired.
2.10. Quantification and image analysis
Images were obtained using Axiovert 200 M (Zeiss) epifluorescence at 40× magnification coupled with Zen software for primary hippocampal neurons. Exposure time was determined by ensuring all non‐transfected neurons were not visible in the image taken. Once the exposure time was found, it was kept consistent between control and treatment groups per antibody per cell culture preparation. FIJI software was used for image intensity analysis. ROIs with were drawn around secondary dendrites just after the bifurcation. Average intensity measurements were taken and background intensity was subtracted from this number. For psd95 intensity measurements, a length of 10 microns was taken just after the bifurcation, and ROIs were drawn around all puncta within the 10 microns on the secondary dendrite. Area and intensity were multiplied for each puncta which resulted in the integrated intensity. At least four secondary dendrites were measured per neuron; if there were not four secondary dendrites to measure, the maximum amount of secondary dendrites on the neuron imaged were measured. Measurements from the secondary dendrites were averaged to create a value per neuron (n = number or neurons). All analysis was done blinded; images were excluded if dendrites were overlapping in such a way that we could not measure only a single, isolated dendrite.
2.11. Statistical analyses
All data were analyzed using Graphpad Prism 9. All values are expressed as ± SEM representing at least two culture groups and two coverslips. Sample number (n) represents the number of neurons or culture groups as indicated in the figure legends. For the analysis of cultured hippocampal neurons, secondary dendritic segments were measured and averaged per neuron. Two‐tailed unpaired t‐tests were used for comparison between two independent groups, and one‐way analysis of variance (ANOVA) coupled with post‐hoc Tukey test were used for comparisons between multiple groups. Statistical significance was determined at p < 0.05. All image quantification was performed blind with respect to experimental condition to avoid bias.
3. RESULTS
To understand the acute, dynamic modification of tau specifically as a result of hyperexcitation, we used a pharmacological approach in primary cultured hippocampal neurons coupled with MS. Cultures were treated at day in vitro (DIV) 20 with picrotoxin (PTX), an antagonist of the inhibitory GABAA receptor, for 24 h to model persistent hyperexcitability (Lee et al., 2019). Cell lysates were collected, and tau was isolated via IP, SDS‐PAGE, and excision from Coomassie‐stained gels for subsequent nanoHPLC‐MS/MS analysis. To visualize the landscape of tau phosphorylation holistically, we developed a schematic tau “phosphomap” that includes all possible phosphorylation sites in rodent and human tau (Figure 1a). When we input the peptide hits obtained from MS into the phosphomap, we found that even under basal conditions tau displayed phosphorylation on multiple sites (Figure 1b). The overall sequence coverage for rat tau isoform A (ortholog to canonical human tau 441aa variant 2) was 77%. These basal hits overlapped with tau sites previously identified to be phosphorylated in unstimulated conditions, supporting the validity of our method (Funk et al., 2014; Wesseling et al., 2020). Interestingly, with PTX the most striking difference was the stimulated appearance of robust phosphorylation on sites S409, S412, S413, T414, and S416 (S = serine, T = threonine) that was absent in control cultures (Figure 1b). Because this tau phosphorylation cluster was uniquely present following PTX and has not been previously reported, this novel motif was termed a “signature” of hyperexcitation.
FIGURE 1.
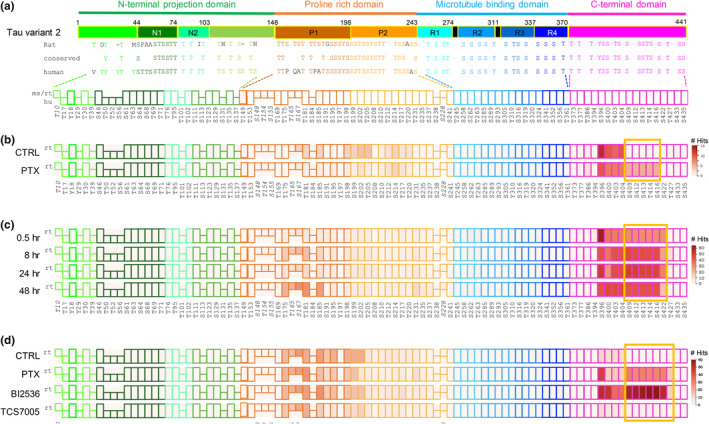
Tau phosphorylation during hyperexcitation. (a) Top, tau domain structure is depicted schematically with amino acid numbering at domain boundaries. Domains and subdomains are color coded as shown. Black bars represent PHF6* and PHF6 motifs. Middle, all 85 serine (S), threonine (t), and tyrosine (Y) residues in tau are shown in the approximate domain locations for mouse MAPT isoform a, 430aa (NP_001033698.1) (identical to rat) and human MAPT variant 2, 441aa (NP_005901). Conserved sites are displayed in between the rodent and human sequences. Bottom, schematic of phosphomap. Each phosphomap is a line of colored boxes representing all possible tau phosphorylation sites. Tau domains are color coded as shown at top. Sites conserved between rodent and human are depicted as rectangles. Phosphoresidues unique to rodent (ms/rt) or human (hu) tau are represented as squares on upper or lower rows, respectively. Numbering at bottom refers to the human MAPT 441 aa isoform, except italicized numbers which denote sites in mouse sequence not found in human tau; for these sites the mouse numbering is given. (b–d) Phosphomapping of tau phosphorylation with hyperexcitation. Following stimulation, tau was immunopurified and MS performed. Phosphorylation cluster induced by PTX condition (hyperexcitation) is highlighted with yellow box. Legend at right indicates number of peptide hits at that site. (b) Primary rat hippocampal neurons were stimulated with 100 μM PTX for 24 h or vehicle control (CTRL) as indicated. Phosphomap represents one MS run consisting of two rat cultures. (c) Time course of PTX administration. Primary rat hippocampal neurons were stimulated with 100 μM PTX for 30 min, 8 h, 24 h, and 48 h or with vehicle control as indicated. Phosphomap represents one MS run consisting of four rat cultures. (d) Plk2 inhibitor blocks hyperexcitation‐induced C‐terminal phosphorylation. Primary rat hippocampal neurons were treated with 100 μM PTX for 24 h or with vehicle control as indicated. During the final 4 h of PTX administration, 50 nM BI‐6727 or 50 nM TC‐S7005 were applied. Phosphomap represents one MS run consisting of four rat cultures.
We next examined whether the tau C‐terminal phosphorylation pattern changed over the course of stimulation, and specifically if there existed a temporal sequence to the modification of different sites. We therefore performed a time‐course experiment in which primary rat hippocampal cultures were treated at DIV19‐20 with PTX for 0.5, 8, 24, and 48 h. We found that the hyperexcitation signature appears to occur in a coordinated fashion, as all sites were phosphorylated as a group as early as 30 min (Figure 1c). Alternatively, it is possible that some sites may precede others but on a faster timescale than was examined here. In addition, there was some variability in the phosphorylation cluster size between preparations, as we found in this experiment the C‐terminal phosphorylation extended further to include residue S422.
To identify kinases responsible for this C‐terminal phosphorylation, we tested the hypothesis that polo‐like kinase 2 (Plk2) may be involved. Plk2 is a member of the polo‐like kinase family that is highly upregulated during hyperexcitation and promotes decreased spine density as a homeostatic protective measure against excitotoxicity (Evers et al., 2010; Kauselmann et al., 1999; Seeburg & Sheng, 2008). To test the role of Plk2, primary rat hippocampal cultures were treated at DIV20 with PTX for 24 h as before, except that during the final 4 h of stimulation we added either a pan‐Plk family inhibitor (BI‐6727), or a more Plk2‐selective inhibitor (TC‐S 7005) prior to harvesting. In this experiment, PTX again induced phosphorylation of the hyperexcitation signature, but included more extensive C‐terminal phosphorylation out to site T427 (Figure 1d). Although the pan‐specific BI‐6727 did not reduce phosphorylation levels at the tau C‐terminus following PTX, administration of the Plk2‐specific TC‐S 7005 completely blocked hyperexcitation‐induced phosphorylation, bringing levels to baseline control (Figure 1d). Thus, Plk2 activity is critical for hyperexcitation‐dependent tau C‐terminal phosphorylation.
Over the course of these experiments, we noted that there was variability in the C‐terminal sites of phosphorylation. The sites from S409‐S416 were consistent, while the downstream boundary fluctuated with regard to S422 and T427. When we compared peptide hit results from triplicate experiments, we observed that indeed the core motif induced by PTX included residues 409–427, although the further downstream the site was situated, the weaker the response became (Figure 2a, hyperexcitation‐inducible domain “H”). In this region, basal phosphorylation was very low, with only rare peptide hits in the three runs.
FIGURE 2.
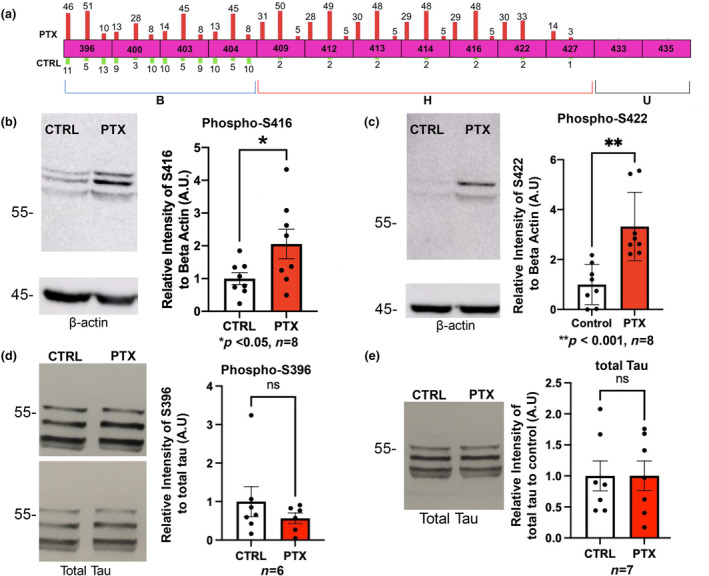
Replication and validation of MS findings at selected sites. (a) Top, MS was performed in triplicate and number of phosphopeptide hits per run are shown as bars on each residue. No number is shown if zero hits were obtained in that run. Hits obtained with PTX treatment are shown as red bars above the sites (scaled in height to the number of hits), and those observed in control condition are green bars below each site. Each PTX bar is paired with its cognate control bar beneath. Bottom, C‐terminus is divided into 3 phosphosite subdomains: B, basally phosphorylated (aa396‐404); H, hyperexcitation inducible (aa409‐427); and U, unmodified (aa433‐435) regions. (b–e) Primary rat hippocampal neurons were stimulated with 100 μM PTX for 24 h or vehicle control as indicated. Cells were collected and immunoblotted for phospho‐S416 (ROUT outlier test (Q = 1%)). D'Afostino and Pearson test (alpha = 0.05) was used to test for normality and passed; t‐test used (p = 0.0469, t = 2.179, df = 14) (b), phospho‐S422 (ROUT outlier test (Q = 1%)). D'Afostino and Pearson test (alpha = 0.05) was used to test for normality and did not pass; Mann–Whitney test used (p = 0.0007). (c), phospho‐S396 (ROUT outlier test (Q = 1%)). D'Afostino and Pearson test (alpha = 0.05) was used to test for normality and did not pass; Mann–Whitney test used (p = 0.5338) (d), and total tau (ROUT outlier test (Q = 1%)). D'Afostino and Pearson test (alpha = 0.05) was used to test for normality and did not pass; Mann–Whitney test used (p > 0.9999) (e). Quantifications are shown to the right of panels and represent relative intensity to the control. Loading controls were beta‐Actin (b, c) and total tau (d). For (e) the loading was controlled by quantified protein amount. n = 6–8 independent cell culture preparations as indicated below each graph.
The sites immediately upstream from the hyperexcitation signature, residues 396–404, showed variability as well but were generally phosphorylated under both basal conditions and during PTX (Figure 2a, basally phosphorylated region “B”). Prior studies have observed basal phosphorylation in this B region as well (Funk et al., 2014; Wesseling et al., 2020), supporting the idea that these sites were not part of the hyperexcitation‐responsive group. The last 2 phosphosites of the C‐terminus, S433 and S435, were never observed to be phosphorylated under any conditions we used (Figure 2a, unmodified group “U”).
To validate these MS data and quantify the responses, we used immunoblotting with selected phosphospecific tau antibodies. Primary cultured hippocampal neurons were treated with PTX or control vehicle for 24 h and cell lysates harvested for SDS‐PAGE and western analysis. We confirmed that S416 and S422, residues within the H region, were indeed significantly phosphorylated during PTX stimulation (Figure 2b,c). In contrast, S396, residing in the B region, exhibited strong levels of basal phosphorylation not significantly different from PTX (Figure 2d). Moreover, levels of total tau did not change with neuronal overactivity (Figure 2d,e), indicating that alterations in phosphorylation were not simply because of the differences in tau expression. These results using an independent methodology supported the semi‐quantitative MS findings and validated our approach.
To probe the functional role of the tau C‐terminal phosphorylation pattern we identified, we used site‐directed mutagenesis. Because the C‐terminal sites were observed to be regulated as a single unit, we first generated a “bulk” mutant in which all 7 phosphosites in the H region (409–427) were changed to either non‐phosphorylatable alanine (bulk‐Ala, or BA) or phosphomimetic aspartic acid (bulk‐Asp, or BD) within the Flag epitope‐tagged wild‐type (WT) human tau variant 2 (V2) (Figure 3a). When transfected into hippocampal neuron cultures, both WT tau and bulk‐Ala exhibited a much higher expression in dendrites following PTX treatment, compared to vehicle control (Figure 3b,c). Interestingly, however, we observed no significant change in bulk‐Asp expression upon PTX administration (Figure 3b,c). Furthermore, we observed that PTX upregulated WT tau significantly more than it did with bulk‐Ala (Figure S2a). These expression changes in the dendrite paralleled those in the soma as well for each construct (Figure S2b–d).
FIGURE 3.
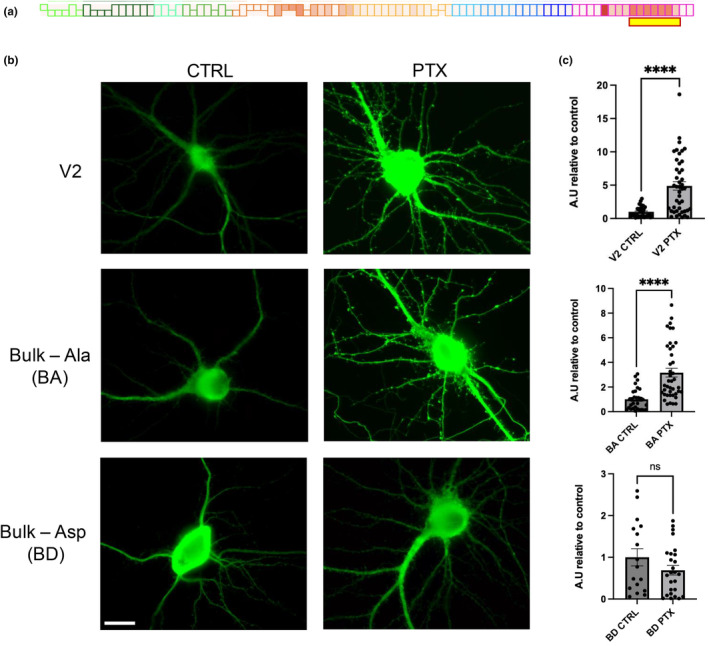
Effects of C‐terminal phosphosite mutations on tau localization in secondary dendrites. (a) Schematic of the hyperexcitation‐responsive amino acid residues used for mutagenesis (highlighted with yellow box underneath representative PTX stimulation phosphomap). (b) Hippocampal neuron localization and expression of Flag‐tau control and mutant constructs as indicated left, treated with vehicle (CTRL) or PTX as indicated on top. Green, Flag tag antibody. (c) Quantification of Flag intensity in secondary dendrites from (b); n = 14–43 neurons, from at least 6 separate coverslip plantings of 2 independent culture groups. Data are means ± SEM. ****p < 0.0001, ns = not significant; ROUT outlier test (Q = 1%). D'Afostino and Pearson test (alpha = 0.05) was used to test for normality. V2 did not pass normality, therefore Mann–Whitney test used (p < 0.0001). BA and BD passed normality (BA ctrl, BA ptx, BD ctrl, BD ptx, p = 0.0959, 0.0641, 0.2911, and 0.2908, respectively). Two‐tailed Student's t‐test used for BA (p < 0.0001, t = 4.968, df = 69) and BD (p = 0.1623, t = 1.423, df = 41). Scale bar represents 20 μm.
To understand the physiological relevance of an increase in WT Flag‐tau dendritic expression upon PTX administration, we examined the expression of endogenous tau under similar conditions. We treated primary cultured hippocampal neurons with PTX or control vehicle for 24 h, fixed, and labeled with total tau antibodies. Indeed, endogenous tau levels in secondary dendrites treated with PTX were significantly increased compared to control vehicle (Figure 4a,b). These data corroborate the activity‐dependent increase in tau expression seen with transfected constructs.
FIGURE 4.
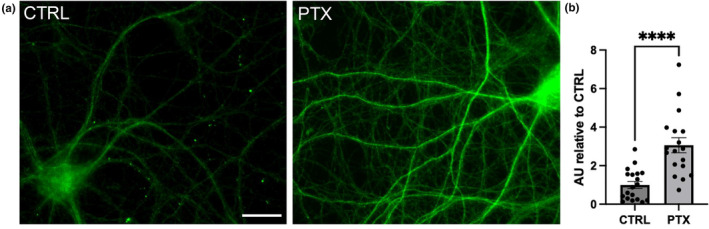
Endogenous total tau levels increase in secondary dendrites after PTX administration. (a) Hippocampal neuron expression of total endogenous tau (green, Tau Dako antibody) under basal control and 24 h. PTX conditions. (b) Quantification of endogenous tau intensity in secondary dendrites from (a); n = 18–19 neurons from at least 12 separate coverslip plantings of 2 independent culture preparations. ROUT outlier test (Q = 1%). D'Afostino and Pearson test (alpha = 0.05) was used to test for normality and passed; V2 and PTX, p = 0.4075 and p = 0.0848, respectively. Data are means ± SEM. ****p < 0.0001, two‐tailed Student's t‐test. Scale bars represent 20 μm.
The expression of tau in dendrites implied that tau may have normal functions at postsynaptic sites. To analyze the effect of tau on excitatory synapses, we transfected hippocampal neuron cultures with WT or mutant bulk‐Ala/Asp constructs, followed by immunostaining for the postsynaptic marker PSD‐95. We observed that non‐phosphorylatable Bulk‐Ala expression greatly increased the integrated intensity of individual PSD‐95 puncta, compared to both control tau and bulk‐D transfected neurons (Figure 5b,c). Because PSD‐95 intensity correlates with synaptic strength and postsynaptic size (Melander et al., 2021), these results suggest that C‐terminal phosphorylation of tau may normally function to restrain the growth of the postsynaptic specialization.
FIGURE 5.
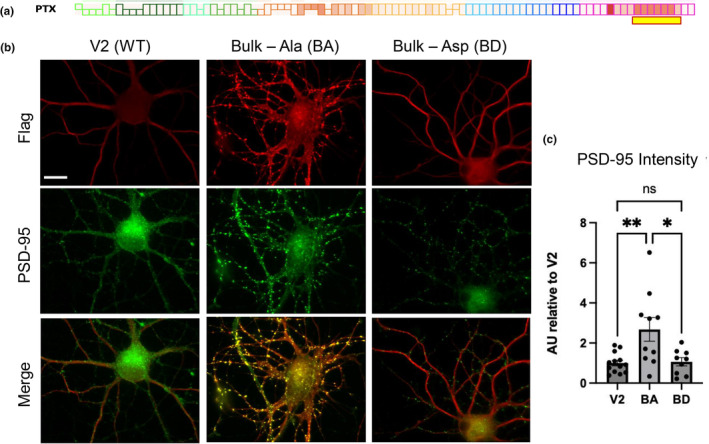
Effects of C‐terminal tau phosphosite mutations on excitatory postsynapses. (a) Schematic of the hyperexcitation‐responsive amino acid residues used for mutagenesis (highlighted with yellow box underneath representative PTX stimulation phosphomap). (b) Hippocampal neuron localization and expression of Flag‐tau control and mutant constructs as indicated at top. Flag‐tau constructs were transfected into hippocampal neurons and co‐stained for excitatory synaptic marker PSD‐95 (green) and Flag tag (red). (c) Quantification of PSD‐95 integrated intensity from (b); n = 9–13 transfected neurons from at least 8 separate coverslip plantings per 2 independent culture preparations. Data are means ± SEM. **p < 0.001, *p < 0.01, ns = not significant; D'Afostino and Pearson test (alpha = 0.05) was used to test for normality (passed; for V2, BA, and BD, p = 0.5061, 0.3728, and 0.07327, respectively); ROUT outlier test (Q = 1%). ANOVA (F(2, 29) = 7.263, p = 0.0028) with Tukey's post‐hoc test. Scale bar represents 20 μm.
4. DISCUSSION
Previously published work has described profound effects of neuronal hyperexcitation on tau function, including acceleration of tau cell‐to‐cell propagation (Wu et al., 2016). Nevertheless, the precise signaling pathways and residues of tau which are phosphorylated during this condition remained unknown. Prior studies of tau phosphorylation have frequently employed candidate phosphospecific antibodies, which is a biased approach, or MS using postmortem brain tissue in which activity manipulation is not possible (Figure S1) (Buée et al., 2000; Hanger et al., 2002; Funk et al., 2014; Wesseling et al., 2020). Here, using pharmacological stimulation of hippocampal neurons, we uncovered for the first time a unique tau C‐terminal phosphorylation motif that was dynamically responsive to neuronal hyperactivity. The extent of phosphorylation was variable in the downstream boundary, suggesting that different degrees of activity may cause more or less extensive C‐terminal tau phosphorylation. In this way, tau may act as a sensor or “thermostat” of neuronal activity level.
Intriguingly, some of the C‐terminal hyperexcitation‐induced sites we identified have been previously associated with paired helical filaments (PHFs) from AD (Buée et al., 2000) compared to normal brain (Funk et al., 2014), specifically sites S409, S412, S413, and S422, with pS422 representing the epitope for PHF‐selective phosphospecific antibody AP422 (Figure S1a–c). Additionally, a recent MS study identified multiple phosphorylation events in AD brain compared to asymptomatic control brain, but sites residing within the hyperexcitation‐responsive region studied here were observed to be phosphorylated only in AD brain and in sarkosyl‐insoluble tau fibrils (Wesseling et al., 2020). Notably, AD and other tauopathy mouse models are well established to exhibit hyperexcitation (Bakker et al., 2012; Busche et al., 2008; Crimins et al., 2012; Hafkemeijer et al., 2012; Holth et al., 2013; Hunsberger et al., 2016; Minkeviciene et al., 2009; Palop et al., 2007; Palop & Mucke, 2009; Putcha et al., 2011; Rocher et al., 2010), suggesting that tau C‐terminal hyperphosphorylation could be associated with pathogenesis if occurring in an uncontrolled manner. Although it has been shown that temperature can influence tau phosphorylation (Canet et al., 2023), temperature would affect both control and PTX conditions equally and could not explain the differences in phosphorylation signature we observed here.
Our study also used pharmacological inhibition to identify a novel tau kinase, the homeostatic regulator Plk2 (Evers et al., 2010; Lee et al., 2017; Seeburg & Sheng, 2008), as being critical for hyperexcitation‐induced tau C‐terminal phosphorylation. However, because the pan‐Plk family inhibitor did not block tau phosphorylation with PTX (but does inhibit Plk2), there may be opposing functions of different Plk family members in this process, which will be interesting for future studies. Selective inhibitors of Plk2 could provide a potential therapeutic option to reduce tau hyperphosphorylation at the C‐terminus, as Plk2 is present at low levels basally (Seeburg & Sheng, 2008) and its inhibition could produce low off‐target effects under basal conditions.
An additional observation was that PTX application caused a dramatic increase in tau expression in dendrites. As this phenomenon occurred with both endogenous and transfected WT tau, this effect may represent a physiological mechanism to recruit dendritic tau during persistent neuronal hyperactivity. The tau Bulk‐Ala mutant also increased in dendrites with PTX, suggesting that these phosphorylation sites are not strictly required for dendritic localization/upregulation and there may be additional mechanisms that can mediate this process. However, the Bulk‐Asp mutant did not show any upregulation with PTX, likely because of the already high level of dendritic expression under control basal conditions. Thus, the bulk C‐terminal sites seem to be sufficient, but not necessary, for tau dendritic recruitment. Because levels of tau were increased to similar degree in both dendrites and soma, an alternative possibility is that PTX induced a phosphorylation‐dependent increase in tau levels in the whole cell rather than a phosphorylation‐dependent recruitment of tau from the soma to the dendrites, although the two possibilities are not mutually exclusive. The function of increased tau in the soma may be simply to supply additional tau to the dendrites, or there may be additional roles of tau not explored here that require cell‐wide augmentation.
These findings implicate tau in postsynaptic function, and we found that inability to become phosphorylated in the C‐terminus significantly increased PSD‐95 puncta intensity and size. These data suggest that tau C‐terminal phosphorylation may have a role in the downregulation of PSD‐95 during conditions of hyperexcitation. Tau could therefore potentially have a function in a negative feedback mechanism to protect from excitotoxicity, regulated by Plk2 phosphorylation. This function would be in concordance with the known role of Plk2 in homeostatic dampening of excitatory synapses. Intriguingly, Plk2 is also involved in the downscaling of glutamatergic AMPA receptors, in part via phosphorylation of amyloid precursor protein (APP), the precursor of the AD pathological hallmark Aβ (Lee et al., 2017). It is thus possible that tau and APP physiologically collaborate to exert effects on postsynapses, which may help explain their intimate relationship in AD pathology as well.
Although the rat hippocampal neurons utilized here are an imperfect model for human disease, this preparation can nevertheless reveal insights into normal physiological functions of tau. In this regard, we note that the sites we identified are all conserved from rodent to human tau, suggesting a shared signaling apparatus. In terms of disease relevance, previous tau phosphorylation and MS studies utilizing postmortem AD brain tissue focus on relatively late‐occurring events, whereas hyperexcitation is likely a characteristic of early, asymptomatic stages (Crimins et al., 2012; Kazim et al., 2017; Putcha et al., 2011). Comprehensive analysis of tau phosphorylation, including potential isoform‐specific events (Liu et al., 2016), under acute signaling contexts as well as disease types and stages will help disentangle the functions of the multitude of tau phosphosites and eventually allow the compilation of an “atlas” of modifications to decipher the tau phosphorylation code in physiology and pathology.
AUTHOR CONTRIBUTIONS
Amanda Schneeweis: Conceptualization; writing – original draft; writing – review and editing; formal analysis; data curation; funding acquisition; methodology. Dawson Hillyer: Formal analysis; data curation; methodology. Tsering Lama: Formal analysis; data curation; methodology. Daeun Kim: Data curation; formal analysis. Charles Palka: Formal analysis; data curation. Sarra Djemil: Data curation; formal analysis; conceptualization. Mai Abdel‐Ghani: Data curation. Kelly Mandella: Data curation; formal analysis; visualization. William Zhu: Formal analysis; data curation; visualization. Nicole Alvarez: Data curation. Lara Stefansson: Data curation. Robert Yasuda: Conceptualization. Junfeng Ma: Data curation; methodology; writing – original draft. Daniel T. S. Pak: Writing – original draft; funding acquisition; investigation; conceptualization; writing – review and editing; supervision.
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1111/jnc.16221.
Supporting information
Data S1.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (R21 AG070673‐01 to DTSP and 5T32AG071745‐02 to AS) and Momental Foundation (Mistletoe grant to AS).
Schneeweis, A. , Hillyer, D. , Lama, T. , Kim, D. , Palka, C. , Djemil, S. , Abdel‐Ghani, M. , Mandella, K. , Zhu, W. , Alvarez, N. , Stefansson, L. , Yasuda, R. , Ma, J. , & Pak, D. T. (2025). Mass spectrometry identifies tau C‐terminal phosphorylation cluster during neuronal hyperexcitation. Journal of Neurochemistry, 169, e16221. 10.1111/jnc.16221
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Abbas, G. , Mahmood, W. , & Kabir, N. (2016). Recent progress on the role of GABAergic neurotransmission in the pathogenesis of Alzheimer's disease. Reviews in the Neurosciences, 27, 449–455. [DOI] [PubMed] [Google Scholar]
- Abdel‐Ghani, M. , Lee, Y. , Akli, L. A. , Moran, M. , Schneeweis, A. , Djemil, S. , El Choueiry, R. , Murtadha, R. , & Pak, D. T. S. (2023). Plk2 promotes synaptic destabilization through disruption of N‐cadherin adhesion complexes during homeostatic adaptation to hyperexcitation. Journal of Neurochemistry, 167, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada, P. V. , Growdon, J. H. , Hedley‐Whyte, E. T. , & Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology, 42, 631–639. [DOI] [PubMed] [Google Scholar]
- Bakker, A. , Krauss, G. L. , Albert, M. S. , Speck, C. L. , Jones, L. R. , Stark, C. E. , Yassa, M. A. , Bassett, S. S. , Shelton, A. L. , & Gallagher, M. (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron, 74, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1991). Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathologica, 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1995). Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiology of Aging, 16, 278–284. [DOI] [PubMed] [Google Scholar]
- Buée, L. , Bussière, T. , Buée‐Scherrer, V. , Delacourte, A. , & Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Research Reviews, 39, 95–130. [DOI] [PubMed] [Google Scholar]
- Busche, M. A. , Chen, X. , Henning, H. A. , Reichwald, J. , Staufenbiel, M. , Sakmann, B. , & Konnerth, A. (2012). Critical role of soluble amyloid‐β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 109, 8740–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche, M. A. , Eichhoff, G. , Adelsberger, H. , Abramowski, D. , Wiederhold, K.‐H. , Haass, C. , Staufenbiel, M. , Konnerth, A. , & Garaschuk, O. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science, 321, 1686–1689. [DOI] [PubMed] [Google Scholar]
- Canet, G. , Rocaboy, E. , Laliberté, F. , Boscher, E. , Guisle, I. , Diego‐Diaz, S. , Fereydouni‐Forouzandeh, P. , Whittington, R. A. , Hébert, S. S. , Pernet, V. , & Planel, E. (2023). Temperature‐induced artifacts in tau phosphorylation: Implications for reliable Alzheimer's disease research. Exp Neurobiol, 32, 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimins, J. L. , Rocher, A. B. , & Luebke, J. I. (2012). Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathologica, 124, 777–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers, D. M. , Matta, J. A. , Hoe, H. S. , Zarkowsky, D. , Lee, S. H. , Isaac, J. T. , & Pak, D. T. S. (2010). Plk2 attachment to NSF induces homeostatic removal of GluA2 during chronic overexcitation. Nature Neuroscience, 13, 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franker, M. A. M. , & Hoogenraad, C. C. (2013). Microtubule‐based transport—Basic mechanisms, traffic rules and role in neurological pathogenesis. Journal of Cell Science, 126, 2319–2329. [DOI] [PubMed] [Google Scholar]
- Funk, K. E. , Thomas, S. N. , Schafer, K. N. , Cooper, G. L. , Liao, Z. , Clark, D. J. , Yang, A. J. , & Kuret, J. (2014). Lysine methylation is an endogenous post‐translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochemical Journal, 462, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert, M. , & Spillantini, M. G. (2006). A century of Alzheimer's disease. Science, 314, 777–781. [DOI] [PubMed] [Google Scholar]
- Hafkemeijer, A. , Grond, J. , Der, V. , & Rombouts, S. A. R. B. (2012). Imaging the default mode network in aging and dementia. Biochimica et Biophysica Acta, 1822, 431–441. [DOI] [PubMed] [Google Scholar]
- Hanger, D. P. , Betts, J. C. , Loviny, T. L. F. , Blackstock, W. P. , & Anderton, B. H. (2002). New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament‐tau) from Alzheimer's disease brain using Nanoelectrospray mass spectrometry. Journal of Neurochemistry, 71, 2465–2476. [DOI] [PubMed] [Google Scholar]
- Holth, J. K. , Bomben, V. C. , Reed, J. G. , Inoue, T. , Younkin, L. , & Younkin, S. G. (2013). Tau loss attenuates neuronal network hyperexcitability in mouse and drosophila genetic models of epilepsy. The Journal of Neuroscience, 33, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger, H. C. , Rudy, C. C. , Batten, S. R. , Gerhardt, G. A. , & Reed, N. (2016). P301L tau expression affects glutamate release and clearance in the hippocampal Trisynaptic pathway. Journal of Neurochemistry, 132, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz, F. , Tomita, T. , Hsieh, H. , Seabrook, G. , Borchelt, D. , Iwatsubo, T. , Sisodia, S. , & Malinow, R. (2003). APP processing and synaptic function. Neuron, 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Kauselmann, G. , Weiler, M. , Wulff, P. , Jessberger, S. , Konietzko, U. , Scafidi, J. , Staubli, U. , Bereiter‐Hahn, J. , Strebhardt, K. , & Kuhl, D. (1999). The polo‐like protein kinases Fnk and Snk associate with a Ca(2+)‐ and integrin‐binding protein and are regulated dynamically with synaptic plasticity. The EMBO Journal, 18, 5528–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazim, S. F. , Chuang, S. C. , Zhao, W. , Wong, R. K. S. , Bianchi, R. , & Iqbal, K. (2017). Early‐onset network hyperexcitability in presymptomatic Alzheimer's disease transgenic mice is suppressed by passive immunization with anti‐human APP/Aβ antibody and by mGluR5 blockade. Frontiers in Aging Neuroscience, 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna‐Reeves, C. A. , Castillo‐Carranza, D. L. , Guerrero‐Muñoz, M. J. , Jackson, G. R. , & Kayed, R. (2010). Preparation and characterization of neurotoxic tau oligomers. Biochemistry, 49, 10039–10041. [DOI] [PubMed] [Google Scholar]
- Lasagna‐Reeves, C. A. , Castillo‐Carranza, D. L. , Sengupta, U. , Clos, A. L. , Jackson, G. R. , & Kayed, R. (2011). Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild‐type mice. Molecular Neurodegeneration, 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. S. , Lee, Y. , André, E. A. , Lee, K. J. , Nguyen, T. , Feng, Y. , Jia, N. , Harris, B. T. , Burns, M. P. , & Pak, D. T. S. (2019). Inhibition of polo‐like kinase 2 ameliorates pathogenesis in Alzheimer's disease model mice. PLoS One, 14, e0219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. , Lee, J. S. , Lee, K. J. , Turner, R. S. , Hoe, H. S. , & Pak, D. T. S. (2017). Polo‐like kinase 2 phosphorylation of amyloid precursor protein regulates activity‐dependent amyloidogenic processing. Neuropharmacology, 117, 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Song, X. , Nisbet, R. , & Götz, J. (2016). Co‐immunoprecipitation with tau isoform‐specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. Journal of Biological Chemistry, 291, 8173–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, S. , Sahara, N. , Saito, Y. , Murayama, S. , Ikai, A. , & Takashima, A. (2006). Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer's disease. Neuroscience Research, 54, 197–201. [DOI] [PubMed] [Google Scholar]
- Melander, J. B. , Nayebi, A. , Jongbloets, B. C. , Ganguli, S. , Mao, T. , & Zhong, H. (2021). Distinct in vivo dynamics of excitatory synapses onto cortical pyramidal neurons and parvalbumin‐positive interneurons. Cell Reports, 37, 109972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene, R. , Rheims, S. , Dobszay, M. B. , Zilberter, M. , Hartikainen, J. , Fülöp, L. , Penke, B. , Zilberter, Y. , Harkany, T. , Pitkänen, A. , & Tanila, H. (2009). Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. The Journal of Neuroscience, 29, 3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, M. P. (2018). Amyloid‐Beta solubility in the treatment of Alzheimer's disease. The New England Journal of Medicine, 378, 391–392. [DOI] [PubMed] [Google Scholar]
- Nelson, P. T. , Alafuzoff, I. , Bigio, E. H. , Bouras, C. , Braak, H. , Cairns, N. J. , Castellani, R. J. , Crain, B. J. , Davies, P. , Tredici, K. D. , Duyckaerts, C. , Frosch, M. P. , Haroutunian, V. , Hof, P. R. , Hulette, C. M. , Hyman, B. T. , Iwatsubo, T. , Jellinger, K. A. , Jicha, G. A. , … Beach, T. G. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. Journal of Neuropathology and Experimental Neurology, 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop, J. J. , Chin, J. , Roberson, E. D. , Wang, J. , Thwin, M. T. , Bien‐Ly, N. , Yoo, J. , Ho, K. O. , Yu, G. Q. , Kreitzer, A. , Finkbeiner, S. , Noebels, J. L. , & Mucke, L. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron, 55, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop, J. J. , & Mucke, L. (2009). Epilepsy and cognitive impairments in Alzheimer disease. Archives of Neurology, 66, 435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler, A. M. , Phillips, E. C. , Lau, D. H. W. , Noble, W. , & Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Reports, 14, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha, D. , Brickhouse, M. , O'Keefe, K. , Sullivan, C. , Rentz, D. , Marshall, G. , Dickerson, B. , & Sperling, R. (2011). Hippocampal hyperactivation associated with cortical thinning in Alzheimer's disease signature regions in non‐demented elderly adults. The Journal of Neuroscience, 31, 17680–17688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan, B. N. , Lee, K. J. , & Pak, D. T. S. (2012). Wherefore art thou, homeo(stasis)? Functional diversity in homeostatic synaptic plasticity. Neural Plasticity, 2012, 718203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson, E. D. , Scearce‐Levie, K. , Palop, J. J. , Yan, F. , Cheng, I. H. , Wu, T. , Gerstein, H. , Yu, G.‐Q. , & Mucke, L. (2007). Reducing endogenous tau ameliorates amyloid ‐induced deficits in an Alzheimer's disease mouse model. Science, 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Rocher, A. B. , Crimins, J. L. , Amatrudo, J. M. , Kinson, M. S. , Todd‐Brown, M. A. , Lewis, J. , & Luebke, J. I. (2010). Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Experimental Neurology, 223, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanabria‐Castro, A. , Alvarado‐Echeverría, I. , & Monge‐Bonilla, C. (2017). Molecular pathogenesis of Alzheimer's disease: An update. Annals of Neurosciences, 24, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenheit, B. , Zarski, R. , & Ohm, T. G. (2004). Spatial and temporal relationships between plaques and tangles in Alzheimer‐pathology. Neurobiology of Aging, 25, 697–711. [DOI] [PubMed] [Google Scholar]
- Seeburg, D. P. , & Sheng, M. (2008). Activity‐induced polo‐like kinase 2 is required for homeostatic plasticity of hippocampal neurons during epileptiform activity. Journal of Neuroscience, 28, 6583–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, H. , Davidowitz, E. , Lopez, P. , Emadi, S. , Moe, J. , & Sierks, M. (2013). Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. International Journal of Cell Biology, 2013, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret, L. , Mann, E. O. , Hang, G. B. , Barth, A. M. I. , Cobos, I. , Ho, K. , Devidze, N. , Masliah, E. , Kreitzer, A. C. , Mody, I. , Mucke, L. , & Palop, J. J. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell, 149, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling, H. , Mair, W. , Kumar, M. , Schlaffner, C. N. , Tang, S. , Beerepoot, P. , Fatou, B. , Guise, A. J. , Cheng, L. , Takeda, S. , Muntel, J. , Rotunno, M. S. , Dujardin, S. , Davies, P. , Kosik, K. S. , Miller, B. L. , Berretta, S. , Hedreen, J. C. , Grinberg, L. T. , … Steen, J. A. (2020). Tau PTM profiles identify patient heterogeneity and stages of Alzheimer's disease. Cell, 183, 1699–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. W. , Hussaini, S. A. , Bastille, I. M. , Rodriguez, G. A. , Mrejeru, A. , Rilett, K. , Sanders, D. W. , Cook, C. , Fu, H. , Boonen, R. A. C. M. , Herman, M. , Nahmani, E. , Emrani, S. , Figueroa, Y. H. , Diamond, M. I. , Clelland, C. L. , Wray, S. , & Duff, K. E. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nature Neuroscience, 19, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K. , Holth, J. K. , Liao, F. , Stewart, F. R. , Mahan, T. E. , Jiang, H. , Cirrito, J. R. , Patel, T. K. , Hochgräfe, K. , Mandelkow, E. M. , & Holtzman, D. M. (2014). Neuronal activity regulates extracellular tau in vivo. The Journal of Experimental Medicine, 211, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zott, B. , Simon, M. M. , Hong, W. , Unger, F. , Chen‐Engerer, H. J. , Frosch, M. P. , Sakmann, B. , Walsh, D. M. , & Konnerth, A. (2019). A vicious cycle of β amyloid−dependent neuronal hyperactivation. Science, 365, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.