

Abstract
Under physiological conditions in peptides or proteins, the -AsnGly- motif autonomously rearranges within hours/days to β-Asp and α-Asp containing sequence, via succinimide intermedier. The formation of the succinimide is the rate-limiting step, with a strong pH and temperature dependence. We found that Arg(+) at the (n + 2) position (relative to Asn in the nth position) favors isomerisation by forming a transition-state like structure, whereas Glu(-) disfavors isomerisation by adopting a β-turn like conformer. Four to six key intermediate structures (proton transfer, transition-state formation, ring-closure and ammonia-release steps) have been identified along the intrinsic reaction coordinate pathways. We explain how, under the right conditions, the N-atom of a backbone amide, hardly a potent nucleophile, can nevertheless initiate isomerisation. The new data are useful for the design of self-structuring motifs, more resistant protein backbones, antibodies, etc.
Subject terms: Peptides, Reaction mechanisms, Reaction kinetics and dynamics
AsnGly motif within peptides or proteins can rearrange to β-Asp and α-Asp by asparagine deamidation and isomerisation via a succinimide intermediate, however, the mechanism underlying this transition remains underexplored. Here, the authors present a quantitative kinetic model for the overall isomerisation reaction, show that for peptides containing both charged and neutral (n + 2) amino acid residues, geometry plays a more important role in their isomerisation reaction rates, and they also identify key reaction sub-steps within succinimide formation.
Introduction
It has been shown that a protein would remain stable for years under sterile and neutral conditions in water, with the typical half-life of uncatalysed amide hydrolysis reaching hundreds of years1. However, this is half the truth, as spontaneous isomerisation of Asn (N or asparagine) is a threat to backbone integrity that occurs on a much faster timescale. The amide side chain of Asn triggers the isomerisation reaction of α-Asn to form α-Asp and β-Asp (D or aspartic acid and β-D or β-aspartic acid), changing the primary sequence and thus, the constitution of the protein. In addition, the α-Asp → α-Asp/β-Asp-like reorganisation is complemented by racemisation of (L)-Asp to (L)-Asp/(D)-Asp, which can be completed within days or even hours, as observed for (L)-Asn-Gly segments2. The explanation for why all this occurs, and some of the factors that enhance or limit these reactions, remain unclear, despite the fact that these primary sequence ‘hot spots’ were identified decades ago3–6. Deamidation of the asparagine side chain via the formation of a five-membered substituted succinimide ring (Suc) and its subsequent hydrolysis to give both α-Asp and its isopeptide analogue β-Asp has been reported (Fig. 1)2,3. This natural, nonenzymatic intramolecular reaction results in a mixture of main-chain aspartyl (Asp or α-Asp) and isoaspartyl (isoAsp or β-Asp) residues in a ratio of ~1:3, depending on pH, temperature, solvent dielectric constant, etc.2,7. By studying the formation of the succinimide intermediate and its subsequent hydrolysis by quantum chemical methods, some have concluded that, contrary to previous findings in the literature, the hydrolysis of the succinimide is the rate-determining step rather than its formation. The succinimide intermediate was found to be formed by the tautomerization of asparagine after several reaction pathways were investigated. The effect of catalytic water on the reaction has also been described8–10.
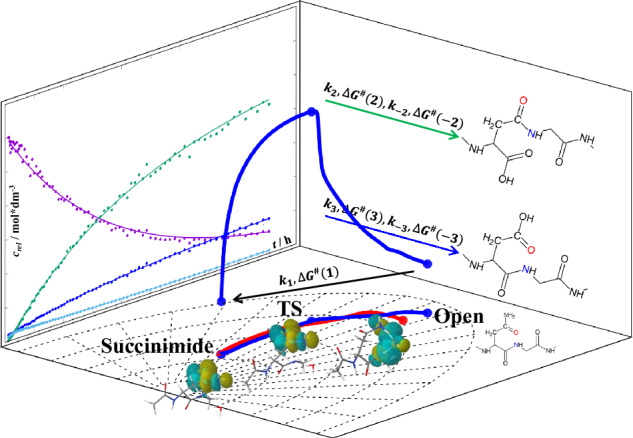
Fig. 1. A typical isomerisation of Asn-Gly sequence motif.

The isomerisation of the Asn-Gly sequence motif proceeds by N-nucleophilic attack of the glycine N-atom on the asparagine side chain carbonyl carbon, resulting in the formation of a five-membered succinimide followed by the release of ammonia. Subsequent hydrolysis leads to two different products, a segment containing β-Asp and a segment containing α-Asp. On the one hand, the side chain amide -CONH2 is replaced by a -COOH group and, on the other hand, the β-Asp nonproteinogenic residue appears within the primary sequence, in which the backbone is extended but the side chain is reduced by a -CH2- group with respect to the α-Asp residue.
Furthermore, in addition to the external parameters, the primary, secondary and tertiary structure of the polypeptide chain itself can accelerate or retard the rate of isomerisation2. First the neighbouring residues have a critical effect on the integrity of asparagine11–13. Glycine, following asparagine with a small side chain, promotes isomerisation, probably due to the increased flexibility of the backbone atoms2,4,5. Comparison of the average chemical shifts of the different residue types shows that the smallest residues typically have smaller 15N shifts at the amide bond, suggesting that these backbone N atoms have lower electron densities and their protons dissociate more readily, a prerequisite for nucleophilic attack. The isomerisation rate of asparagine within folded structural segments such as helices, β-turns, β-strands is slower2,14,15. For example, asparagine in native ribonuclease A is ~30 times less prone to isomerisation than in its reduced and unfolded forms16. Similarly, asparagine residues in intrinsically disordered proteins are expected to have an increased rate of isomerisation (compared to those in globular proteins), as enhanced internal dynamics contribute to faster and easier adoption of the ideal asparagine conformation for successful intramolecular nucleophilic attack2.
In contrast to the NG sequences found in mobile structures, 68Ga-NOTA-c(NGR) and 68Ga-NODAGA-c(NGR) molecules containing the cyclic NGR units have been successfully used as diagnostic contrast agents for positron emission tomography, as the NGR motif can selectively bind to aminopeptidase N/CD13 receptors, allowing precise detection of tumours expressing this receptor17. Furthermore, the tumour therapeutic effect of cytokines (e.g. tumour necrosis factor alpha - TNFα) is also greatly enhanced by their binding to target molecules (e.g. S-CNGRC cyclic peptide)18. In addition, the β-DGR sequence can be used as a tumour vasculature targeting motif19. Besides peptides many proteins, ranging from transport molecules (Cu: ceruloplasmin, Fe: haemoglobin β)20,21, apoptotic signalling proteins (Bcl-xL, p53, calmodulin)22–25 to neuronal maintenance proteins (α-synuclein, tau)26,27 or chromatin integrity, and thus involved in epigenetics (histone H4, histone H2B)28,29 or translational activity (4E-BP)30 were shown to undergo isomerisation. Some proteins have been studied by Nuclear Magnetic Resonance (NMR) spectroscopy, showing instability at Asn-Gly sites, which in some cases can also lead to protein aggregation31–33. These examples show that deamidation/isomerisation has not only structural, but also functional effects on proteins. Despite the fact that both the succinimide intermediate and/or the β-aspartate-containing products have been successfully crystallized for a half a dozen proteins (e.g., chicken egg white lysozymes PDB (Protein Data Bank) codes are 4QEQ, 1AT5 and 1AT6)34, the molecular explanation of how and why this transformation occurs in peptides or proteins in such a natural way is still incomplete. First, an amide N-atom in the backbone is not expected to behave as a nucleophile in water, which happens to be the prerequisite initial step in this isomerisation reaction. Here, an NMR-based kinetic analysis and a QM (quantum mechanical) derived reaction pathway with key intermediates provide reasonably but sufficiently detailed explanation for this reaction.
Aims
Quantitative NMR data were obtained to construct a coherent and quantitative kinetic and thermodynamic model of the -NGX- isomerisation reaction as a function of pH, temperature and chemical composition: X = K(+) (Lys(+): Lysine), R(+) (Arg(+): Arginine), A (Ala, Alanine) or E(-) (Glu(-): Glutamate). The characterisation of the model systems Ac-NGKA-NH2 (shorthand -NGKA-), -NGRA-, -NGAA- and -NGEA- thus required the acquisition of hundreds of time-dependent 1H NMR data sets. The quantitative analysis of these data helped to establish the key kinetic and thermodynamic parameters of a consistent model, to provide a better insight into the -NGX- isomerisation. Under different conditions, the data allowed to explain and characterize all coupled reactions of isomerisations k1, k2, k–2, k3, k–3, ∆G1, ∆G2, ∆G−2, ∆G3, ∆G−3 (Figs. 1–2). In addition in order to explain how an amide N-atom in water can be a sufficiently good nucleophile to effectively initiate isomerisation, the first irreversible step was studied by ab initio conformational analysis and intrinsic reaction coordinate (IRC) reaction path calculations, together with the determination of local electrostatic potential (ESP) charges and natural bond orbital analysis (NBO) data. As a result of the NBO analysis, we have identified a series of microsteps - and their evolution - on the IRC pathways from the „open” form, through the ring closure, to deamidation. Finally, we aimed to synchronise the NMR-driven kinetic data on the positively and a negatively charged (n + 2) residue (relative to Asn in the nth position), which enhances or retards isomerisation, with the QM data obtained for the relevant models.
Fig. 2. The time course of the AsnGly isomerisation reaction and the resulting activation energies.

a The decomposition of -α-NGAA- via isomerisation and the formation of the products β-Asp (β-DGAA) and α-Asp (α-DGAA), respectively (pH=7.4, T = 46 °C), via the intermediate succinimide. The vertical axis is scaled with arbitrary units proportional to selected non-overlapping NMR signal integrals plotted as a function of the time. b ΔG‡ profile of the -α-NGAA- isomerisation reaction at pH 7.4, based on selected non-overlapping NMR signal integral data from 1H-NMR experiments.
Kinetic inference of NMR data
The integrals of selected 1H-NMR resonances of the fine structures of the uncoupled spins were considered to be proportional to the respective concentrations of the molecules in the solution. Literature data and preliminary simulations2 compared with measured time-dependent 1D 1H-NMR signals suggested a first-order reaction network as the kinetic mechanism of isomerisation, as follows:
| 1 |
This mechanism proved to be a good fit to the experimental data obtained (Table 1, Supplementary Figs. 2–20 within Supplementary Information, Supplementary Data 4). In this mechanism asparagine is irreversibly converted to succinimide which is in equilibrium with both α-Asp and β-Asp (Fig. 1). The equilibrium constant of the latter two coupled reactions can also be described by first order rate coefficients:
| 2 |
Table 1.
Selected reaction kinetic parametersa of the different -NGXA-b model peptides
| peptide | pH | T, °C | # of data pointsc | k1, h−1 | k2, h−1 | k–2, h−1 | k3, h−1 | k–3, h−1 | t1/2, h | |
|---|---|---|---|---|---|---|---|---|---|---|
| -NGAA- | 5.1 | 55 | 980 | estimated value a | 0.00341 | 0.0531 | 0.0099 | 0.068 | 0.059 | 203.0 |
| error a | 0.00004 | 0.0017 | 0.0007 | 0.010 | 0.009 | |||||
| -NGAA- | 6.3 | 55 | 436 | estimated value | 0.0235 | 0.324 | 0.0123 | 0.077 | 0.0126 | 29.5 |
| error | 0.0012 | 0.030 | 0.0020 | 0.014 | 0.0043 | |||||
| -NGAA- | 7.4 | 55 | 376 | estimated value | 0.2192 | 5.211 | 0.0938 | 0.68 | 0.0346 | 3.2 |
| error | 0.0039 | 0.163 | 0.0044 | 0.08 | 0.0088 | |||||
| -NGAA- | 7.8 | 55 | 264 | estimated value | 0.267 | 8.06 | 0.119 | 2.08 | 0.172 | 2.6 |
| error | 0.004 | 0.19 | 0.007 | 0.13 | 0.017 | |||||
| -NGAA- | 7.4 | 28 | 16 | estimated value | 0.0047 | 0.120 | n. d.d | 0.055 | 0.017 | 147.5 |
| error | 0.00047 | 0.023 | n. d. | 0.033 | 0.011 | |||||
| -NGAA- | 7.4 | 37 | 476 | estimated value | 0.027 | 0.714 | n. d. | n. d. | n. d. | 25.7 |
| error | 0.008 | 0.32 | n. d. | n. d. | n. d. | |||||
| -NGAA- | 7.4 | 46 | 324 | estimated value | 0.0498 | 1.079 | 0.0224 | 0.2285 | 0.0237 | 13.9 |
| error | 0.0005 | 0.015 | 0.0012 | 0.0085 | 0.003 | |||||
| -NGAA- | 7.4 | 55 | 376 | estimated value | 0.2192 | 5.211 | 0.0938 | 0.68 | 0.0346 | 3.2 |
| error | 0.0039 | 0.163 | 0.0044 | 0.08 | 0.0088 | |||||
| -NGEA- | 7.4 | 28 | 68 | estimated value | 0.0028 | 0.4116 | 0.00007 | 0.0855 | 0.0007 | 248.5 |
| error | 0.0003 | 0.0732 | n. d. | 0.0522 | n. d. | |||||
| -NGEA- | 7.4 | 37 | 120 | estimated value | 0.0075 | 0.327 | 0.0052 | 0.058 | n. d. | 92.4 |
| error | 0.0003 | 0.022 | 0.0023 | 0.005 | n. d. | |||||
| -NGEA- | 7.4 | 46 | 272 | estimated value | 0.0269 | 1.993 | 0.011 | 0.2767 | 0.0038 | 25.8 |
| error | 0.0004 | 0.056 | n. d. | 0.0707 | n. d. | |||||
| -NGEA- | 7.4 | 55 | 24 | estimated value | 0.057 | 2.03 | 0.043 | 0.385 | n. d. | 12.2 |
| error | 0.007 | 0.44 | 0.044 | 0.24 | n. d. | |||||
| -NGKA- | 7.4 | 28 | 192 | estimated value | 0.0090 | 0.334 | n. d. | 0.174 | 0.0063 | 76.6 |
| error | 0.0003 | 0.015 | n. d. | 0.008 | 0.0012 | |||||
| -NGKA- | 7.4 | 37 | 128 | estimated value | 0.0362 | 0.643 | 0.0018 | 0.255 | n. d. | 19.1 |
| error | 0.0003 | 0.010 | 0.0008 | 0.004 | n. d. | |||||
| -NGKA- | 7.4 | 46 | 256 | estimated value | 0.074 | 2.30 | 0.0242 | 1.150 | n. d. | 9.4 |
| error | 0.000 | 0.018 | 0.0022 | 0.022 | n. d. | |||||
| -NGKA- | 7.4 | 55 | 141 | estimated value | 0.232 | 4.00 | 0.0121 | 0.134 | n. d. | 3.0 |
| error | 0.003 | 0.053 | 0.0014 | 0.026 | n. d. | |||||
| -NGRA- | 7.4 | 28 | 288 | estimated value | 0.0079 | 0.107 | 0.0005 | 0.086 | 0.0046 | 87.2 |
| error | 0.00005 | 0.0013 | 0.0000 | 0.0016 | 0.0002 | |||||
| -NGRA- | 7.4 | 37 | 400 | estimated value | 0.0318 | 1.290 | n. d. | n. d. | n. d. | 21.8 |
| error | 0.0002 | 0.0082 | n. d. | n. d. | n. d. | |||||
| -NGRA- | 7.4 | 46 | 272 | estimated value | 0.0535 | 1.056 | 0,059 | 0.66 | 0.205 | 13.0 |
| error | 0.0003 | 0.010 | 0.001 | 0.010 | 0.007 | |||||
| -NGRA- | 7.4 | 55 | 320 | estimated value | 0.235 | 5.83 | 0.0153 | 2.202 | 0.077 | 3.0 |
| error | 0.003 | 0.10 | 0.0016 | 0.048 | 0.011 |
aFor an explanation of rate coefficients, see subsection “Kinetic inference from NMR data”. t1/2 is the half-life of the Asn species. The term ‘estimated value’ refers to the parameter estimation result based on the fit of mechanism (1) to the temporal evolution of all four species while ‘error’ refers to the half-widths of their 95% confidence intervals.
bNGXA- stands for Ac-Asn-Gly-Xxx-Ala-NH2, where Xxx: Ala, Lys, Arg, Glu.
c# of data’ represents the number of the relative integral values used to determine all reaction kinetic parameters at a given pH, T and chemical composition.
dDue to larger uncertainties in the time-dependent concentrations of the peptides, some of the activation energies associated with the rate constants k−2, k3 and k−3 could not be determined.
As and , it follows that
However, it is important to note that this equilibrium is not maintained during the reaction due to kinetic couplings. In fact, equilibrium is only reached at the “end” of the reaction, i.e. at infinite reaction time, when the reaction is complete. Until then, the following coupled differential equations describe the time evolution of the different concentrations:
| 3 |
| 4 |
| 5 |
| 6 |
Note that Eqs. (4) to (6) are responsible for the coupling, which complicates the temporal evolution of the concentrations. One of the consequences is that the ratio is not constant during the reaction even if the initial concentration of the two aspartic acid species, namely and are zero; the ratio of their (time-dependent) instantaneous formation rate can be expressed as follows:
| 7 |
Thus, the ratio is time-dependent with a limiting value at infinite time (or practically, after the reaction is completed) of .
The kinetic inference of the time-dependent measurements was performed numerically using the COPASI software application, version 4.27 (http://copasi.org/). The estimated parameters of the mechanism (3)-(6) together with their standard deviations were the rate coefficients k1, k2, k3 and the equilibrium constants K2 and K3. The rate coefficients k−2 and k−3 were calculated from Eq. (2) using the error propagation formula including the estimated covariances to calculate their standard deviations. All errors reported are half-widths of 95% confidence intervals based on the Student distribution of the estimated values.
The activation Gibbs free energies for the forward reactions were estimated using the Arrhenius equation from the slope of the linear function ln ki vs. 1/T; their error was also calculated using the error propagation formula (see Supplementary Note 2/A. within Supplementary Information, Supplementary Eqs. 1–4 within Supplementary Information, Supplementary Table 1 within Supplementary Information).
Results and discussion
Kinetic model
In agreement with most literature data, we found that in the isomerisation of NG-peptides, the rate-determining step is, in almost all cases, the first step of the reaction with a reaction rate coefficient k1 the value of k1 is in most cases smaller than those of k2 or k3 (see Eq. (1) and Table 1). The first step involves the formation of succinimide and is accompanied by the release of NH3, which makes the step irreversible. The height of the Gibbs free energy barrier ΔG‡ for the whole pseudo-first order reaction is thus, related to the rate coefficient of the first step (“bottleneck” step: Eq. (1)). The succinimide is then rapidly hydrolyzed to a mixture of β-Asp and α-Asp products, in two alternative steps that are measured by k2 and k3, respectively.
However, the concentration of the intermediate product succinimide is never zero during the overall reaction (Figs. 1–2), as both β- and α-Asp products are in equilibrium via this species. These equilibrium steps are quantitatively described by the temperature and pH dependent rate coefficients, k2 and k−2, and k3 and k−3, respectively. With the help of our detailed kinetic model, in addition to k1, if the spectra allowed, we were able to determine the most of the k2, k–2, k3, k–3, as well as the ∆G2, ∆G−2, ∆G3, ∆G−3 values.
Using the -NGAA- sequence and our kinetic model, the analysis of asparagine isomerisation revealed the following: (i) Regardless of pH or temperature, k1 is usually an order of magnitude smaller than k2 or k3 (Table 1), specifying that the succinimide formation is the slowest reaction of all. (ii) The isomerisation rate limited by k1 shows a strong pH dependence, i.e. under acidic conditions (e.g. pH= 5.1) the reaction is slow, but increases by about one order of magnitude when moving towards the alkaline range and shifting the pH by one unit (Table 1), confirming previous findings in the literature2. As a function of the increasing pH (pH = 5.1 → 7.8), the ratio k2/k1 doubles, indicating that β-Asp formation is increasingly faster at higher pH, than succinimide formation. (iii) Due to equilibrium, both products, β- and α-Asp are converted back to succinimide, with rate coefficients k−2 and k−3, respectively. Since k−2 << k2 and k−3 << k3 (Table 1), respectively, β- and α-Asp are both formed at a much faster rate than they are degraded (back-cyclized). The ratio of both k2/k−2 and k3/k−3 increases significantly with increasing pH, but moderately with increasing temperature. Thus, both β- and α-Asp are formed more rapidly from succinimide and are relatively slower to be degraded to succinimide at elevated pH. iv By increasing the temperature all the reaction rates increase (Table 1), as has been written in the literature2. (v) The determination of the change in Gibbs free energy of each step gave the expected results. In most cases, ΔG‡(1) of the first, rate-determining and non-equilibrium step, leading to succinimide, is the largest (Fig. 2b, Supplementary Table 1 within Supplementary Information). In the case of the equilibrium steps, the two backward reactions that convert either β-Asp or α-Asp to succinimide are slower, than the forward reactions (hydrolysis), so their respective ΔG‡s are larger: ΔG‡(2) < ΔG‡(−2) and ΔG‡(3) < ΔG‡(−3) (Fig. 2b, Supplementary Table 1 within Supplementary Information).
Comparing the kinetic parameters of the reference system -NGAA- with those in which the side chains carry an explicit charge in the (n + 2) position, -NGEA-, -NGKA- and -NGRA-, we showed, in agreement with previous work2, that the rate of NG isomerisation is influenced. When the side chain of the amino acid residue following glycine has a positive charge: i.e. Arg(+)(n+2) and Lys(+)(n+2) at pH = 7.4, the isomerisation rate increases. The current quantitative kinetic model provided for the isomerisation of -NGAA- remains valid, for -NGKA-, -NGRA- and even -NGEA-, so that both rate constants and t1/2 are directly comparable. At pH = 7.4, the -NGEA- peptide has a net negative charge because the glutamic acid’s side chain is fully deprotonated, and according to the measurements the rate of isomerisation decreases. All rate constants are influenced by the charged side chains of the (n + 2) residue as described above. In fact, the increase in the rate coefficient k1 quantitatively illustrates the extent to which the reaction rate increases for positively charged residues (k1-NGAA- < k1-NGRA- ~ k1-NGKA-), and decreases for negatively charged (n + 2) residues, such as glutamic acid (k1-NGEA- < k1-NGAA-) at all four temperatures, at pH = 7.4 (Table 1). In other words, the asparagine half-lives become shorter for Arg(+) and Lys(+) and longer for Glu(-) compared to the reference value of the neutral Ala. Also for the charged models, -NGE(-)A-, -NGK(+)A-, -NGR(+)A-, k1 is significantly smaller than k2 or k3 indicating that the formed succinimide immediately evolves to either β- or α-Asp. Furthermore, k2/k1 ranges from 13.5 to 147.0, while k3/k1 ranges from 0.6 to 30.5. This indicates that in most cases not only β-Asp but also α-Asp forms faster, than the transient succinimide is formed (Table 1). As found for the reference model -NGAA-, charged peptides form β- or α-Asp derivatives from succinimide and back cyclise to succinimide, due to the equilibrium reaction, with k-2 << k2 and k-3 << k3, respectively. The activation Gibbs free energies of the -NGAA-, -NGE(-)A-, -NGK(+)A- and -NGR(+)A- peptides (110.8, 94.7, 87.3 and 97.3 kJ mol−1, respectively) show that the activation energy of the rate-determining initial step decreases for -NGRA-, -NGEA- and -NGKA- when compared to -NGAA- (Supplementary Table 1 within Supplementary Information). In other words, longer side chains facilitate isomerisation, both because they can carry explicit charge, and because they can adopt special conformers (see below), making isomerisation more likely to occur. In addition, the change in Gibbs free energy of the β-Asp formation steps, ∆G‡(2), for the -NGE(-)A- and -NGK(+)A- peptides (61.2 and 85.9 kJ mol−1 respectively) is lower than the change in Gibbs free energy of the succinimide formation, ∆G‡(1) (94.7 and 87.3 kJ mol−1 respectively), suggesting that in these cases succinimide facilitates the formation of β-peptides even more (Supplementary Table 1 within Supplementary Information).
In conclusion, it is clear that NG isomerisation is spontaneous, and can be controlled by adjusting pH, temperature and primary sequences. In other words, the control and fine-tuning this reaction appears to be straightforward. From a kinetic point of view, the rate-determining step - in almost all cases - (marked by k1) is the formation of succinimide. At the molecular level, however, this step is complex and its details are not yet fully understood. It is unclear where the starting point of the mechanism is, and how it can be interpreted in the light of the extremely poor nucleophilicity of the backbone amide N-atom. It is doubtful that the NH proton is “simply” released from the backbone amide group (Fig. 3a/III), since the deprotonation of an amide group in N-methylacetamide is estimated to be rather unfavourable, with pKa=1835. Due to (C = O)N conjugation, the N-atom of an amide bond is a poor nucleophile at neutral pH. This raises the fundamental question: how does this AdN reaction occur? It is reasonable to assume that the very first step is the deprotonation of the amide bond by a water molecule, allowing the short-lived, electron-paired neutral N-species to attack the carbonyl of the asparagine side chain (Fig. 3b). This consideration is consistent with the experimental observation that this nucleophilic attack and subsequent succinimide ring closure becomes less likely as the pH becomes more acidic36 because the proton exchange between the backbone amide and the catalytic water molecule is hindered (Table 1).
Fig. 3. The isomerisation reaction requires the formation of an N-nucleophile.

a First, the two non-equivalent resonance hybrids of the amide bond (a/I) must be considered. Secondly, the imino form, the zwitterion, can bind a water molecule in two different ways (a/II). The left one can be more stable (marked in green), while the right one is more reactive, producing an N- and O-nucleophile (marked in red) (a/III). b A way to explain how the Asn-Gly isomerisation is initiated using the latter type of nucleophile, which is simply formed with the aid of a water molecule (b/II). The ring closure of the Asn-Gly subunit (b/III) to form succinimide, shown as the rate-limiting step of the isomerisation reaction.
Computational aspects
As shown above, the rate-limiting step in NG isomerisation, the succinimide ring closure, is achieved by an effective AdN reaction. This reaction step can best be described in the conformational subspace defined by the Bürgi-Dunitz (BD) distance (d), and angle (θ) variables37,38 (Fig. 4). With increasing temperature and/or increasing internal dynamics of the polypeptide chain, the ideal d and θ structural constraint is more frequently satisfied, thus facilitating succinimide formation more easily due to the spatial proximity of CγAsn and NGly atoms. This desired increase in backbone plasticity is better achieved when the side-chainless and thus more flexible glycine is in the (n + 1) position, as described earlier2.
Fig. 4. The geometric description of the succinimide formation.

a The Bürgi-Dunitz distance d (Å), NGly-CγAsn and the angle θ (°), NGly-CγAsn-OδAsn are two variables describing the intramolecular conformational changes associated with the backbone-side chain conformational restructuring, required for succinimide ring closure. b In a polar coordinate system, both the waterless and the water assisted IRC paths are plotted, as calculated at the B3LYP/6-31+G(d,p) level of theory.
Recently, the Ac-Asn-NH-CH3 model - based on previous literature8–10 - has been studied by QM methods to determine the reaction mechanism of asparagine isomerisation, considering two possible scenarios. According to their waterless concept the direct intramolecular attack between the asparagine side chain and the NH of the (n + 1) residue, denoted -NH-CH3, occurs in the absence of water. In line with the second alternative, a water molecule assists the proton transfer from the backbone amide group to the -NH2 group of the side chain, referred to as the water assisted mechanism8–10. These three seminal studies focus on the energetics and structural changes associated with the isomerisation reaction.
We have identified 4 (for the waterless) and 6 (for the water assisted) microsteps based on the NBO analysis over the IRC pathway calculated between the „open” and the succinimide forms. Information on the stabilising interactions were obtained from the NBO interaction energies (i.e. second perturbation energies, denoted as E(2) kJ mol−1) of the donor-acceptor orbitals. The stronger the interaction between two natural bond orbitals, the higher the energy value. In the waterless mechanism the 4 main steps along the reaction pathway are as follows. (i) The first one is the proton transfer step, where the amide N-H bond is broken and the proton is attached to the -NH2 group of the asparagine side chain. (ii) The formation of the transition state is followed by (iii) the ring closure and (iv) the subsequent irreversible step of the NH3 release (Supplementary Tables 1–6 within Supplementary Data 2, Supplementary Figs. 22–26 within Supplementary Information, Supplementary Tables 2–5 within Supplementary Information). Similar steps as described for the waterless ring closure can also be observed in the water assisted reaction, but in this case the reaction is carried out with the aid of a water molecule. The inclusion of an explicit water molecule can provide a better insight into a more realistic reaction mechanism, as the isomerisation reaction is completed in an aqueous medium. In the case of the water assisted reaction, the atomic labelling is as follows: NGly: the (n + 1) amide N-atom of the backbone, HGly: the leaving amide proton / the attaching proton to the water, NδAsn: the leaving N-atom of the asparagine side chain, CγAsn: the carbonyl C-atom of the asparagine side chain, OδAsn: the carbonyl O-atom of the asparagine side chain, OH2O is the O-atom of the explicit water molecule and HH2O: the leaving proton of the water / the attaching proton to the nitrogen of the asparagine side chain (Supplementary Tables 7–12 within Supplementary Data 2). The initial structure based on the literature data10 and transition state reoptimized by us (Fig. 5a) and the 6 main steps of the water assisted reaction mechanism identified by us from (i) to (vi) are as follows.
Fig. 5. Reaction sub-steps of the water assisted reaction.

a The initial structure, known as the „open” form of the water assisted reaction. b Schematic and NBO computational structure of the first water assisted proton transfer step, which is the result of orbital overlapping between the LP(3):OH2O and the σ*(1):NδAsn-HH2O orbitals. c Schematic and NBO computational structure of the water assisted proton transfer. In this mechanism a significant orbital overlapping occur between the LP(3):OH2O and the σ*(1):NδAsn-HH2O orbitals and between the LP(3):OH2O and the σ*(1):NGly-HGly orbitals. d A representative schematic and NBO computational molecular structure of the second phase of the water assisted proton transfer step. Overlap occurs between the LP(2) NGly and the σ*(1) OH2O-HGly orbitals. e The transition state formed by overlapping the LP(2):OH2O and the σ*(1):NδAsn-HH2O orbitals and the LP(2):OδAsn and the σ*(1):CγAsn-NδAsn orbitals. f The ring-closure step takes place with orbital overlapping between the LP(3):OδAsn and the σ*(1):CγAsn-NGly orbitals. g Schematic and NBO computational structure of the final ammonia release step. Orbital overlapping occur between the LP(3):OδAsn and the σ*(1):CγAsn-NδAsn orbitals.
In the water assisted mechanism, the proton transfer occurs in 3 consecutive microsteps or phases. (i) The first is when the water molecule begins to protonate the side chain N-atom. This can be inferred from the significant overlap of the LP(3):OH2O orbitals, which denote the lone pair of electrons of the oxygen in water, and the σ*(1):NδAsn-HH2O orbitals, which denotes an anti-bonding orbital between the NδAsn and HH2O atoms, with an overlap energy of 639.86 kJ mol−1 (Fig. 5b and Supplementary Table 6 within Supplementary Information).
(ii) Beside the LP(3):OH2O and the σ*(1):NδAsn-HH2O orbitals overlap with a perturbation energy of 343.38 kJ mol−1. The amide proton is involved in the proton transfer, with the help of the lone pair electrons of the O-atom of the water molecule, by the overlap of the LP(3):OH2O and the σ*(1) NGly-HGly orbitals, with a perturbation energy of 343.09 kJ mol−1 (Fig. 5c and Supplementary Table 7 within Supplementary Information). (iii) Finally, when the orbitals LP(2):NGly and σ*(1):OH2O-HGly interact with a perturbation energy of 379.07 kJ mol−1, the proton transfer step is completed (Fig. 5d and Supplementary Table 8 within Supplementary Information). (iv). In the transition state, as before, two significant interactions take place simultaneously, namely the LP(2):OH2O and the σ*(1):NδAsn-HH2O orbitals and the LP(2):OδAsn and the σ*(1):CγAsn-NδAsn orbitals interact, with a perturbation energy of 202.05 kJ mol−1 and 193.97 kJ mol−1, respectively (Fig. 5e and Supplementary Table 9 within Supplementary Information). (v) Ring closure is aided by the donation of electrons to the O-atom of the side chain, which is indicated by the overlap of the LP(3):OδAsn and the σ*(1):CγAsn-NGly orbitals, with an overlap energy of 311.42 kJ mol−1 (Fig. 5f and Supplementary Table 10 within Supplementary Information). (vi) Finally, in the deamidation step, the O-atom of the side chain plays a decisive role, in addition to the CγAsn and NδAsn atoms, since the overlap of the LP(3):OδAsn and the σ*(1):CγAsn-NδAsn orbitals has the most significant perturbation energy, with a value of 391.83 kJ mol−1. (Fig. 5g and Supplementary Table 11 within Supplementary Information).
From these microsteps we conclude that the amide N-atom alone is acidic enough to initiate the isomerisation reaction. At each step of the IRC pathways, our quantum mechanical study yields calculated ESP charges and orbital shapes. Starting from an „open” form of asparagine e.g. [[d, θ]= ~3 Å, 83°] (Fig. 4), d does not decrease significantly in the initial phase of the ring closure reaction to complete succinimide formation. However, θ shifts to the ideal 109.5° as approaching the transition state (Fig. 4). In the presence of a catalytic water molecule, θ gets closer to the ideal (~110°), while for the waterless system, it is close to ~116.0° (Fig. 4). The barrier heights calculated at the B3LYP/6-311++G(d,p) level of theory are 210.13 kJ mol−1 and 243.70 kJ mol−1 in the presence and absence of catalytic water, respectively (Fig. 6c, d). When the implicit solvent model of the IEFPCM is used, the barrier heights are significantly lower: 179.15 and 196.76 kJ mol−1, respectively. Not only their absolute values, but also the differences between the transition state energies of the explicit system, with and without water, decrease from 33.57 to 17.61 kJ mol−1. In order to gain a better understanding of this part of the reaction, IRC steps in both directions were determined, starting from the above transition states, calculated at the B3LYP/6-31+G(d,p) level of theory. Along the IRC paths, selected geometric parameters were monitored, namely: the torsional angles of the asparagine (φ, ψ, χ1, χ2), the H-NGly distance, in the waterless reaction the HGly-NδAsn distance and in the water assisted case from the HGly-OH2O distance, and the distance of the nearest proton (HH2O) from the NδAsn atoms.
Fig. 6. Changes of the interatomic distance and relative energy during succinimide formation.

Panels (a), (c) for the waterless, and (b), (d) for the water assisted reactions on the basis of NBO data (for NBO data see Supplementary Figs. 22–26 within Supplementary Information, Supplementary Tables 2–12 within Supplementary Information, and Supplementary Note 1 within Supplementary Data 2, Supplementary Tables 1–18 within Supplementary Data 2). The IRC paths were calculated in vacuum at the B3LYP/6-31+G(d,p) level of theory. The molecular energy reference point is the optimised „open” conformer of asparagine, calculated at the same level of theory. e, f Schematic mechanism of the succinimide formation for the waterless and for the water assisted reactions, respectively.
We found that the torsion angles of the asparagine backbone, (φ,ψ)Asn, change little. For example, the variation of φ throughout the IRC path is about 5-7°, and φ returns to its original value at the end of the reaction (Fig. 7 and Supplementary Fig. 21 within Supplementary Information). The same is true for ψ. Although it changes more during the first phase of the reaction; e.g. in the waterless model, from the „open” form to the transition state, ψ changes more (Δψ ~ 25°), while towards the succinimide formation, ψ changes only ~5°. In the water assisted reaction, ψ remains practically unchanged (~10°) during the first phase, as does the succinimide closure. In order to achieve the ideal transition state structure, both χ1 and χ2 had to be adjusted. As the first step of the reaction proceeds, there is a disrotational motion for side chain torsion angles: with χ1 decreasing from ~198 ± 7° to ~150°, while χ2 completes a rotation of ~145°, namely from 35° to 155°, or 5° to 150°, respectively (Fig. 7).
Fig. 7. Variation of asparagine dihedral angles (φ, ψ, χ1, χ2) and the Bürgi-Dunitz distance (d) along the calculated IRC pathway.
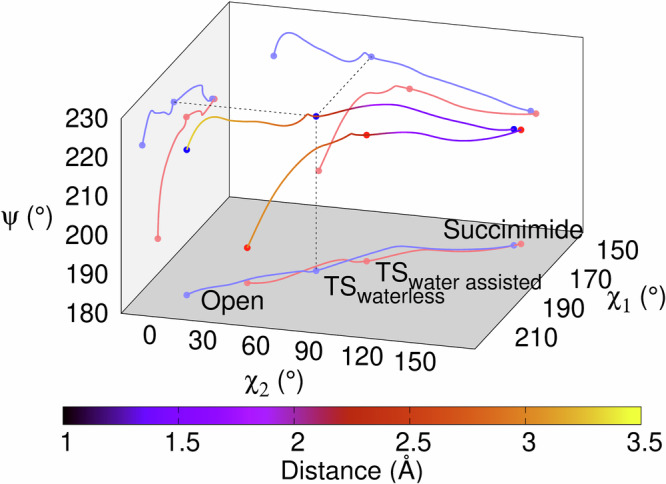
Pathways from the „open” form of asparagine, towards succinimide formation, for both the waterless (red lines and dots) and the water assisted (blue lines and dots) reactions. The combined change of the side chain dihedral angles creates the spatial arrangement required for the successful reaction. There is no significant change in the peptide backbone, but the χ1 and χ2 torsional angles change simultaneously but in the opposite direction.
On an IRC pathway, by definition, the transition state belongs to the highest energy molecular form that connects two neighbouring and stable (low energy) molecular structures called conformers (Fig. 6c, d). Until the proton transfer takes place, the HGly-NGly bond distance changes very little: it remains ~1 Å (Fig. 6a, b / green). The rotation of both χ1 and χ2 allows the formation of the key molecular “packing”, that leads to the isomerisation reaction. Both CγAsn and NGly atoms are involved in the ring closure and thus both gradually approach to each other (Fig. 6a, b / magenta). Similarly, NδAsn and NGly must be in close proximity to each other, to prepare for the proton transfer. In the presence of a catalytic water molecule, both the OH2O-NGly and the OH2O-NδAsn distances are the shortest at the moment of the proton transfer (Fig. 6b / yellow and red). From this point on, the reaction partners begin to move away from each other. The NGly–HGly and the OH2O–HH2O distances change in parallel with each other during the reaction, which means that the proton transfer in the NGly-H2O–NδAsn system occurs in a synchronized mode (Fig. 6b / green and black).
Prior to the proton transfer, the water molecule should be properly oriented, causing a slight increase in the NδAsn – HH2O distance. The nature of the above motions can be followed in detail, by the changes in the orbits of the NBO (Figs. 4, 5, 6c, d). In addition to the structural constraints outlined above (e.g. d & θ), changes in electrostatic properties are also characteristic of NG isomerisation, which is followed by monitoring the ESP atomic charges along the IRC paths. The electronic changes associated with succinimide formation are similar for both the waterless and water assisted ring closure reactions, although there are subtle differences (Fig. 8a–d). The ESP charges of the two key atoms have a minimum as a function of the reaction coordinate. The NGly is more negative during proton transfer and transition state (Figs. 8a, b, orange), while the CγAsn-atom is more negative after succinimide ring formation (Figs. 8a, b, purple). The partial charges of the leaving HGly (+0.3–0.4) and OδAsn (−0.6-(−0.5)) atoms are nearly parallel throughout the path (Figs. 8a, b, yellow and green). The charge of the NδAsn has a local minimum (−0.8 and −0.6) near the proton transfer point, and reaches its most negative partial charge (−0.95) after leaving as NH3 (Figs. 8a, b, light blue). The charge difference between CγAsn- and the NGly-atoms, ΔQ(ξ), compared in the waterless and in the water assisted cases in the first step of the reaction, is higher by ~0.25 in the absence of water (Fig. 8c, d). High values (~0.8 and 1.1) are reached during the proton transfer-transition state segment of the reaction, representing a significant attractive force to close the ring. Thereafter, the value of ΔQ(ξ) decreases continuously as the reaction proceeds along the reaction coordinate.
Fig. 8. Changes of the ESP atomic charge during succinimide formation.

a For the waterless, and (b) for the water assisted reactions on the basis of NBO data (for NBO data see Supplementary Figs. 22–26 within Supplementary Information, Supplementary Tables 2–12 within Supplementary Information, and Supplementary Note 1 within Supplementary Data 2, Supplementary Tables 1–18 within Supplementary Data 2). The IRC paths were calculated in vacuum at the B3LYP/6-31+G(d,p) level of theory. The molecular energy reference point is the optimised „open” conformer of asparagine, calculated at the same level of theory. c The „open” conformer of the QM determined asparagine model with the incorporated water molecule assisting the succinimide formation. d ESP atomic charge differences ΔQ(ξ)= QCγ Asn(ξ) – QNGly(ξ) along the reaction path, during succinimide formation. Charge differences vs. reaction coordinate, ξ, as calculated in vacuum at the B3LYP/6-31+G(d,p) level of theory.
A positively charged amino acid residue of the (n + 2) sequential position (e.g. Lys(+), Arg(+)) increases the isomerisation rate, as described previously2. However, the possible mechanism by which this occurs is not yet known. The structures of both Ac-Asn-Gly-Arg(+)-NH-CH3 and Ac-Asn-Gly-Glu(-)-NH-CH3 were optimised, in addition to the reference Ac-Asn-Gly-Ala-NH-CH3, which has no charge at the (n + 2) position, in order to identify the possible roles of the positively and negatively charged side chains of residue (n + 2) (Supplementary Tables 13–18 within Supplementary Data 2). Different orientations of the side chains were investigated in different models, and the following results were obtained.
The driving force of a reaction can be revealed by following the change in dipole moment along the reaction path from the reactant to the rate-determining transition state39. Therefore we evaluated the dipole moments of the water assisted Ac-Asn-NH-CH3 model along the IRC path (Supplementary Fig. 27 within Supplementary Information). Comparing the change in dipole moment with the change in ESP charge differences between the CγAsn and NGly atoms (Fig. 8d), we see that they follow a very similar path, but with a slight phase shift. The dipole moment values of the Ac-Asn-Gly-Arg(+)-NH-CH3, Ac-Asn-Gly-Ala-NH-CH3 and Ac-Asn-Gly-Glu(-)-NH-CH3 one-water systems are 19.7472 Debye, 10.7260 Debye and 6.2423 Debye, respectively. Meanwhile, the local ESP charge and other geometry parameters (see Supplementary Table 12 in the Supplementary Information) are similar. It can be concluded that although the global electrostatic environment around the reaction centre is different, no significant local dipole moment difference is expected, since these parameters are basically the same.
From a geometric perspective, in the case of the positively charged Ac-Asn-Gly-Arg(+)-NH-CH3, the most ‘sensible’ structural orientation is that, where the Arg(+) side chain is in focus over the backbone amide, while the leaving NδAsn-atom is oriented via an H-bond to the amide at position (n + 3), providing electrostatic stabilisation of the two carbonyl oxygens (Fig. 9b and Supplementary Tables 13–14 within Supplementary Data 2). In contrast to the positively charged Arg(+) model, the negatively charged molecular structure of the Glu(-) model has the key atoms fixed furthest from the transition state (Fig. 9a, d, Supplementary Tables 9–10 within Supplementary Data 2, Supplementary Tables 17–18 within Supplementary Data 2 and Supplementary Table 9 within Supplementary Information), hindering isomerisation. In fact Ac-Asn-Gly-Glu(-)-NH-CH3 is locked in a β-turn, making the “condensation” of the NGly- and the CγAsn-atoms (Supplementary Table 12 within Supplementary Information) less likely to occur. Finally, the uncharged Ala residue behaves in a neutral way, Ac-Asn-Gly-Ala-NH-CH3 is intermediate between the two models mentioned above, as it remains more flexible (Fig. 9c and Supplementary Tables 15–16 within Supplementary Data 2).
Fig. 9. Comparison of the transition state structure of the isomerisation reaction with various optimised oligopeptide structures in the presence of explicit water.

The transition state structure of the water assisted isomerisation reaction associated with (a) Ac-Asn-NH-CH3. The „open” form of the peptide model (b) Ac-Asn-Gly-Arg-NH-CH3, (c) Ac-Asn-Gly-Ala-NH-CH3 and (d) Ac-Asn-Gly-Glu-NH-CH3, calculated with IEFPCM at the B3LYP/6-31+G(d,p) level of theory. (For structural parameters of the molecules see Fig. 9, Supplementary Tables 9–10 within Supplementary Data 2, Supplementary Tables 13–18 within Supplementary Data 2, Supplementary Table 9 within Supplementary Information, Supplementary Table 12 within Supplementary Information).
The comprehensive analysis of the optimised water assisted oligopeptide structures (containing Ala, Arg(+) or Glu(–) residues) with the water assisted transition state of succinimide formation (Fig. 9, Supplementary Tables 9–10 within Supplementary Data 2, Supplementary Tables 13–18 within Supplementary Data 2, Supplementary Table 9 within Supplementary Information, Supplementary Table 12 within Supplementary Information) reveals the following. Both structural and charge parameters for the Arg(+) oligopeptide are very similar to the water assisted transition state of the parent model (Fig. 9a, b and Supplementary Table 12(1)-(2) within Supplementary Information). Despite of the lower polarisation of the key atoms (NGly and CγAsn), the correct orientation of the reaction partners characterized by d and θ remains the measure of success. Thus, for the Arg(+) derivative, a stable transition state-like molecular arrangement was found and an increased reaction rate is expected to occur. In contrast, the negatively charged Glu(–) derivative, which has very different ψ and χ1 torsion angles compared to the transition state, and thus both d or θ are very different from the ideal values for ring closure and succinimide formation, is expected to have a reduced reaction rate (Fig. 9a, d and Supplementary Tables 12(1), (4) within Supplementary Information). In the case of both the Arg(+) and Glu(–) models, we have found a QM conformer with a higher degree of rigidity compared to that of the Ala model (Fig. 9c and Supplementary Table 12(3) within Supplementary Information). This can either be a favourable conformer, as it was found for the Arg(+) model, since its fixed backbone conformer brings the key atoms in close proximity to the transition state. However, backbone fixation can also be unfavourable, as is the case for the Glu(-) model, where the resulting β-turn structure keeps the key atoms away from the expected transition state molecular structure. Therefore, the stabilisation of a given molecular conformer can be either positive, or negative in terms of succinimide formation.
Conclusions
In order to improve our understanding of the spontaneous and fundamental process of asparagine deamidation coupled with isomerisation reactions, we have undertaken a detailed study of the mechanism of -Asn-Gly- isomerisation, that affect the overall stability of the host polypeptide and / or protein. We have shown that succinimide formation is the rate-limiting step, with a strong pH and temperature dependence. The most complete quantitative kinetic model to date was established, providing values for k1, k2, k−2, k3, k−3, ∆G1, ∆G2, ∆G−2, ∆G3, and ∆G−3 based on quantitative NMR data. We found that Arg(+) at the (n + 2) position enhances the reaction by forming a transition state-like structure, whereas Glu(-) at the same position, fixes a β-turn-like conformer, which on the other hand disfavours the isomerisation. We have identified the four or six key reaction sub-steps of the succinimide formation, based on a detailed NBO interaction analysis, which provides insight into the structural and electronic backgrounds of the key structures of proton transfer, transition state formation, ring closure and NH3 release. Based on these results, we can now explain how, under the right conditions, the N atom of a backbone amide, which is hardly a strong nucleophile, can however be the initiator of the isomerisation reaction.
In addition to the -NG- and -β-DG- sequence elements, which can be used in targeted chemotherapy in NGR/β-DGR targeting molecules40,41, isomerisation of asparagine may also be responsible for the loss of protein function. Furthermore, many biological processes are based on the recognition of sites containing either aspartic acid or asparagine, so our observations could be used to design self-structuring, more resistant protein backbones, for drug or antibody design, etc., such as the integrin receptor binding β-DGR/RGD motif41,42.
Methods
Synthesis of acetylated linear peptides
Linear tetrapeptides were synthesised as previously described2 using Rink-Amide MBHA (methylbenzhydryl amine) resin (0.67 mmol g−1 capacity) and Fmoc/tBu (fluorenylmethoxycarbonyl protecting group/tert-butyl) protecting group strategy. The N-terminal Fmoc group was cleaved prior to acetylation. Acetylation was performed by adding a mixture of acetic anhydride, N,N-diisopropylethylamine (DIPEA) and dimethylformamide (DMF) in a 1:1.2:3 ratio. For ~1 g Rink-Amide MBHA resin, 1728 µl acetic anhydride, 2032 µl DIPEA and 5072 µl DMF were mixed and stirred for 1 h. The products were purified by reversed-phase high-performance liquid chromatography (RP-HPLC) using a semi-preparative Phenomenex Luna C18 column with eluent A (0.1% trifluoroacetic acid/H2O) and B (0.1% trifluoroacetic acid in acetonitrile/H2O (80/20)) and identified by electrospray ionization mass spectrometry (Bruker Esquire 3000+ ion-trap) and NMR (Bruker 700 MHz).
1H-NMR experiments and resonance assignment
To follow the isomerisation reaction, time-dependent 1D 1H-NMR measurements of Ac-Asn-Gly-Xxx-Ala-NH2 peptides were completed (where Xxx was used as an arbitrary three-letter abbreviated amino acid). Typically, 1 mg of tetrapeptide was dissolved in 500 μl of Na2HPO4 buffer (50 mM with respect to the final volume) to which 45 µl of D2O and 5 µl of sodium 3-(trimethylsilyl)propane-1-sulphonate (DSS)/NaN3 were added. The pHs of the NMR samples was adjusted to 5.1, 6.3, 7.4 and 7.8 for the -NGAA- peptide, and to 7.4 for the -NGKA-, -NGRA-, -NGAA- and -NGEA- peptides with 0.1 M HCl and/or NaOH solutions. If the samples were heated to 55 °C, they were ultrasonicated for 1 min prior to measurement to avoid bubble formation. Both 1D (Supplementary Notes 1–5 within Supplementary Data 1, Supplementary Figs. 1–12 within Supplementary Data 1) and 2D 1H-NMR (COSY, TOCSY) experiments were used for resonance assignment. To follow the isomerisation and to provide quantitative data for kinetic analysis, time-dependent 1D 1H-NMR signals (t = 5, 10, 15, 20, 25, 30, 45, 60, 90, 120 min, etc.) were recorded, in several cases until the reactant concentration dropped below ~10%. Further 1H-NMR spectra recording, processing, and analysis procedures are detailed in the Supplementary Information Supplementary Note 1 chapter.
QM calculation and NBO representations
The IRC path and NBO 5.9 calculations were performed at the B3LYP/6-31+G(d,p) level of theory in vacuum using the Gaussian 09 B01 software package. All other calculations were performed using the Gaussian 16 C01 software. Individual points of the IRC paths were collected and a frequency calculation was performed at each point to obtain both ∆G values and electrostatic potential atomic charges. IRC path endpoints and transition states were further optimised at the DFT B3LYP/6-31++G(d,p) level of theory both in vacuum and using the IEFPCM water model43. The NBO analysis of the various key structures of the isomerisation reaction along the IRC path was completed (Figs. 4–9 and additional data are available in the Supplementary Note 3 within Supplementary Information, Supplementary Tables 1–18 within Supplementary Data 2, Supplementary Tables 2–12 within Supplementary Information and Supplementary Figs. 21–27 within Supplementary Information). All NBO interactions were visualised with open source Jmol 3D chemical structure viewer (http://www.jmol.org/).
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We thank to Gábor Mező for the oligopeptide synthesis. The authors thank the help in NMR work to Andrea Bodor, DSc and Dr. Fanni Sebák. This work was supported by the National Research, Development and Innovation Office, Hungary (grant number NKFI K137940; PD 146764). This work was completed in the ELTE Thematic Excellence Programme (Szint+) supported by the Hungarian Ministry for Innovation and Technology. ELTE Institutional Excellence Programme (1783-3/2018/FEKUTSTRAT) supported by the Hungarian Ministry of Human Capacities and grants from the European Union and the State of Hungary, co-financed by the European Regional Development Fund (VEKOP-2.3.3-15-2017-00018, VEKOP-2.3.3-15-2017-00020, VEKOP-2.3.2-16-2017-00014). Project no. 2018-1.2.1-NKP-2018-00005 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the 2018-1.2.1-NKP funding scheme. Project number RRF-2.3.1-21-2022-00015 is implemented with the support of the European Union’s Recovery and Resilience Instrument. Supported by the Ministry for Innovation and Technology from the Hungarian NRDI Fund (2020-1.1.6-JÖVŐ-2021-00010).
Abbreviations
- A or Ala
alanine
- β-D or β-Asp
β-aspartate
- BD
Bürgi-Dunitz
- CγAsn
the carbonyl C-atom of the asparagine side chain
- D(–) or Asp(–)
aspartate (with its negative charge emphasized)
- DSS
sodium 3-(trimethylsilyl)propane-1-sulfonate
- E(–) or Glu(–)
glutamate (side chain negatively charged)
- ESP
electrostatic potential
- G or Gly
glycine
- HGly
the leaving amide proton / the attaching proton to the water
- HH2O
the leaving proton of the water / the attaching proton to the nitrogen of the asparagine side chain
- IRC
intrinsic reaction coordinate
- K(+) or Lys(+)
lysine (side chain positively charged)
- N or Asn
asparagine
- NBO
natural bond orbitals
- NδAsn
the leaving N-atom of the asparagine side chain
- NGly
the (n+1)th amide N-atom of the backbone
- NMR
Nuclear Magnetic Resonance
- OδAsn
the carbonyl O-atom of the asparagine side chain
- OH2O
is the O-atom of the explicit water molecule
- QM
quantum mechanical
- R(+) or Arg(+)
arginine (side chain positively charged)
- Suc
succinimide
Author contributions
F.P. contributed with peptide synthesis, NMR measurements, theoretical calculations, kinetic and theoretical data analysis and visualization, and manuscript writing. I.J. contributed with theoretical calculations supervision, data analysis and visualization, and paper preparation. E.K. contributed with the kinetic data analysis and model establishment, and paper preparation. A.L. contributed with the peptide selection and NMR experiment planning and acquisition, and manuscript writing. A.P. contributed with general supervision and manuscript conceptual elaboration and writing. A.P. also provided financial support and instrumentation for the research.
Peer review
Peer review information
Communications Chemistry thanks Binju Wang and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information, Supplementary Data 1–4 files. All other relevant data are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-024-01374-1.
References
- 1.Radzicka, A. & Wolfenden, R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J. Am. Chem. Soc.118, 6105–6109 (1996). [Google Scholar]
- 2.Láng, A. et al. Off-pathway 3D-structure provides protection against spontaneous Asn/Asp isomerization: shielding proteins Achilles heel. Quart. Rev. Biophys.53, e2 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Geiger, T. & Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem.262, 785–794 (1987). [PubMed] [Google Scholar]
- 4.Aswad, D. W., Paranandi, M. V. & Schurter, B. T. Isoaspartate in peptides and proteins: formation, significance, and analysis. J. Pharm. Biomed. Anal.21, 1129–1136 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Stephenson, R. C. & Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem.264, 6164–6170 (1989). [PubMed] [Google Scholar]
- 6.Oliyai, C. & Borchardt, R. T. Chemical pathways of peptide degradation. IV. Pathways, kinetics, and mechanism of degradation of an aspartyl residue in a model hexapeptide. Pharm. Res.10, 95–102 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Patel, K. & Borchardt, R. T. Chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm. Res.07, 703–711 (1990). [DOI] [PubMed] [Google Scholar]
- 8.Catak, S., Monard, G., Aviyente, V. & Ruiz-López, M. F. Reaction mechanism of deamidation of asparaginyl residues in peptides: Effect of solvent molecules. J. Phys. Chem. A110, 8354–8365 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Catak, S., Monard, G., Aviyente, V. & Ruiz-López, M. F. Computational study on nonenzymatic peptide bond cleavage at asparagine and aspartic acid. J. Phys. Chem. A112, 8752–8761 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Catak, S., Monard, G., Aviyente, V. & Ruiz-López, M. F. Deamidation of asparagine residues: Direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A113, 1111–1120 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Wright, H. T. Sequence and structure determinants of the nonenzymatic deamidation of asparagine and glutamine residues in proteins. Protein Eng. Des. Sel.4, 283–294 (1991). [DOI] [PubMed] [Google Scholar]
- 12.Tonie Wright, H. & Urry, D. W. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in protein. Crit. Rev. Biochem. Mol. Biol.26, 1–52 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Patel, K. & Borchardt, R. T. Chemical pathways of peptide degradation. III. Effect of primary sequence on the pathways of deamidation of asparaginyl residues in hexapeptides. Pharm. Res.07, 787–793 (1990). [DOI] [PubMed] [Google Scholar]
- 14.Xie, M. & Schowen, R. L. Secondary structure and protein deamidation. J. Pharm. Sci.88, 8–13 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Sinha, S. et al. Effect of protein structure on deamidation rate in the Fc fragment of an IgG1 monoclonal antibody. Protein Sci.18, 1573–1584 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wearne, S. J. & Creighton, T. E. Effect of protein conformation on rate of deamidation: ribonuclease A. Proteins5, 8–12 (1989). [DOI] [PubMed] [Google Scholar]
- 17.Kis, A. et al. In vivo assessment of aminopeptidase N (APN/CD13) specificity of different 68Ga-labelled NGR derivatives using PET/MRI imaging. Int. J. Pharm.589, 119881 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Corti, A. et al. NGR-TNF engineering with an N-terminal serine reduces degradation and post-translational modifications and improves its tumor-targeting activity. Mol. Pharm.17, 3813–3824 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Curnis, F. et al. Isoaspartate-glycine-arginine: A new tumor vasculature–targeting motif. Cancer Res.68, 7073–7082 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Barbariga, M. et al. Oxidation-induced structural changes of ceruloplasmin foster NGR motif deamidation that promotes integrin binding and signaling. J. Biol. Chem.289, 3736–3748 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Charache, S. et al. Postsynthetic deamidation of hemoglobin Providence (beta 82 Lys replaced by Asn, Asp) and its effect on oxygen transport. J. Clin. Invest.59, 652–658 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deverman, B. E. et al. Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell111, 51–62 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Dho, S. H. et al. Control of cellular Bcl-xL levels by deamidation-regulated degradation. PLoS Biol.11, e1001588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee, J.-C. et al. Protein L-isoaspartyl methyltransferase regulates p53 activity. Nat. Commun.3, 927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potter, S. M., Henzel, W. J. & Aswad, D. W. In vitro aging of calmodulin generates isoaspartate at multiple Asn–Gly and Asp–Gly sites in calcium‐binding domains II, III, and IV. Protein Sci.2, 1648–1663 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson, N. E., Robinson, M. L., Schulze, S. E. S., Lai, B. T. & Gray, H. B. Deamidation of α‐synuclein. Protein Sci.18, 1766–1773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe, A., Takio, K. & Ihara, Y. Deamidation and isoaspartate formation in smeared tau in paired helical filaments. J. Biol. Chem.274, 7368–7378 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Hayashi, T., Ohe, Y., Hayashi, H. & Iwai, K. Human spleen histone H4. Isolation and amino acid sequence1. J. Biochem.92, 1995–2000 (1982). [DOI] [PubMed] [Google Scholar]
- 29.Young, A. L., Carter, W. G., Doyle, H. A., Mamula, M. J. & Aswad, D. W. Structural integrity of histone H2B in vivo requires the activity of protein L-isoaspartate O-methyltransferase, a putative protein repair enzyme. J. Biol. Chem.276, 37161–37165 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Bidinosti, M., Martineau, Y., Frank, F. & Sonenberg, N. Repair of isoaspartate formation modulates the interaction of deamidated 4E-BP2 with mTORC1 in brain. J. Biol. Chem.285, 19402–19408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwasa, H., Meshitsuka, S., Hongo, K., Mizobata, T. & Kawata, Y. Covalent structural changes in unfolded GroES that lead to amyloid fibril formation detected by NMR: insight into intrinsically disordered proteins. J. Biol. Chem.286, 21796–21805 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krause, M. E., Martin, T. T. & Laurence, J. S. Mapping site-specific changes that affect stability of the N-terminal domain of calmodulin. Mol. Pharm.9, 734–743 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murciano-Calles, J., Corbi-Verge, C., Candel, A. M., Luque, I. & Martinez, J. C. Post-translational modifications modulate ligand recognition by the third PDZ domain of the MAGUK protein PSD-95. PLoS ONE9, e90030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noguchi, S., Miyawaki, K. & Satow, Y. Succinimide and isoaspartate residues in the crystal structures of hen egg-white lysozyme complexed with tri-N-acetylchitotriose. J. Mol. Biol.278, 231–238 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Molday, R. S., Englander, S. W. & Kallen, R. G. Primary structure effects on peptide group hydrogen exchange. Biochemistry11, 150–158 (1972). [DOI] [PubMed] [Google Scholar]
- 36.Perrin, C. L. & Lollo, C. P. Mechanism of NH proton exchange in amides and proteins: solvent effects and solvent accessibility. J. Am. Chem. Soc.106, 2754–2757 (1984). [Google Scholar]
- 37.Bürgi, H. B., Dunitz, J. D., Lehn, J. M. & Wipff, G. Stereochemistry of reaction paths at carbonyl centres. Tetrahedron30, 1563–1572 (1974). [Google Scholar]
- 38.Ospina, E. & Villaveces, J. L. Theoretical calculation of the reaction mechanism between ammonia and formaldehyde. J. Mol. Structure: THEOCHEM287, 201–209 (1993). [Google Scholar]
- 39.Yan, S., Ji, X., Peng, W. & Wang, B. Evaluating the transition state stabilization/destabilization effects of the electric fields from scaffold residues by a QM/MM approach. J. Phys. Chem. B127, 4245–4253 (2023). [DOI] [PubMed] [Google Scholar]
- 40.Zou, M., Zhang, L., Xie, Y. & Xu, W. NGR-based strategies for targeting delivery of chemotherapeutics to tumor vasculature. ACAMC12, 239–246 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Höltke, C. isoDGR-peptides for integrin targeting: is the time up for RGD? J. Med. Chem.61, 7471–7473 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Plow, E. F., Haas, T. A., Zhang, L., Loftus, J. & Smith, J. W. Ligand binding to integrins. J. Biol. Chem.275, 21785–21788 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Tomasi, J., Mennucci, B. & Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Structure: THEOCHEM464, 211–226 (1999). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information, Supplementary Data 1–4 files. All other relevant data are available from the corresponding author upon reasonable request.