

Abstract
The bioorthogonal tetrazine-triggered cleavage of trans-cyclooctene(TCO)-linked payloads has strong potential for widespread use in drug delivery and in particular in click-cleavable antibody-drug conjugates (ADCs). However, clinical translation is hampered by an inverse correlation between click reactivity and payload release yield, requiring high doses of less reactive tetrazines to drive in vivo TCO reactions and payload release to completion. Herein we report that the cause for the low release when using the highly reactive bis-(2-pyridinyl)-tetrazine is the stability of the initially formed 4,5-dihydropyridazine product, precluding tautomerization to the releasing 1,4-dihydropyridazine tautomer. We demonstrate that efficient tautomerization and payload elimination can be achieved by ortho-substituting bis-pyridinyl-tetrazines with hydrogen-bonding hydroxyl or amido groups, achieving a.o. release yields of 96% with 18-fold more reactive tetrazines. Applied to on-tumor activation of a click-cleavable ADC in mice, these tetrazines afforded near-quantitative ADC conversion at a ca. 10- to 20-fold lower dose than what was previously needed, resulting in a strong therapeutic response.
Subject terms: Drug delivery, Reaction mechanisms, Chemical tools, Drug delivery
The bioorthogonal tetrazine-triggered cleavage of trans-cyclooctene-linked payloads has strong potential in click-to-release drug delivery, however, an inverse correlation between click reactivity and payload release yield is hampering their clinical translation. Here, the authors develop ortho-substituted bis-pyridinyl-tetrazines with hydrogen-bonding hydroxyl or amido groups for efficient tautomerization and payload elimination, achieving release yields of 96% with 18-fold more reactive tetrazines.
Introduction
The ability to directly control drug activity in vivo is a highly sought after goal. Following the advent of bioorthogonal conjugation chemistry, bioorthogonal cleavage reactions were developed to amongst others control therapies in vivo1. The most reactive and very selective bioorthogonal conjugation reaction, the widely used inverse electron-demand Diels–Alder (IEDDA) cycloaddition between trans-cyclooctene (TCO) and tetrazine, led to the development of the IEDDA pyridazine elimination reaction wherein a TCO-linked payload is cleaved by tetrazine (Fig. 1)2–4, or vice versa, wherein a tetrazine-linked payload is cleaved by TCO5. The so-called click-to-release reaction of TCO-linked payloads has led to many in vivo applications, such as tetrazine-triggered on-target cleavage and activation of TCO-containing antibody-drug conjugates (ADCs), prodrugs and proteins6–14, as well as the off-target deactivation of radioimmunoconjugates15.
Fig. 1. Triggered drug release using “click-to-release” chemistry.

Previous results2,11,12 and results of the enhanced “click-to-release” presented in this work.
The TCO-tetrazine IEDDA cycloaddition initially forms a 4,5-dihydropyridazine, which can then tautomerize to a 1,4- or a 2,5-dihydropyridazine16–18. The IEDDA pyridazine elimination between TCO-payload conjugate and tetrazine retains the favorable properties of the parent reaction regarding stability and selectivity, and in addition affords the release of a carbamate, ester or ether-linked payload from the allylic position of the TCO upon tautomerization to the 1,4-dihydropyridazine2–4 (Fig. 1). However, whereas in vivo IEDDA conjugation applications can make use of highly reactive electron-deficient tetrazines such as the bis-(2-pyridinyl)-tetrazine motif19–22, when applied to IEDDA pyridazine elimination these compounds only afford ca. 10–15% payload release after quantitative cycloaddition adduct formation2. So far, the IEDDA pyridazine elimination has been hampered by an inverse correlation between tetrazine reactivity and payload release yield, with the 100-fold less reactive alkyl- or phenyl-substituted tetrazines affording ca. 80% release (Fig. 1)2,9,11,12.
An increase of the reaction rate with maintained high release yield would be beneficial for several applications. For example, complete in vivo activation of a target-localized protein or ADC requires the intravenous administration of an excess of tetrazine activator. A higher click reaction rate will allow a lower dose of the activator, facilitating clinical translation. Recently, the groups of Mikula, Rutjes and our group reported approaches to increase reaction kinetics of systems using the less reactive tetrazines by increasing the ring strain of the TCO linker through cis-fusion of a 3- or 5-membered ring23–25. However, further reactivity increases using this approach would likely impact TCO stability and thereby the application scope26. Alternatively, higher click conjugation rates of the IEDDA pyridazine elimination could become within reach by increasing the release yield afforded by the highly reactive bis-pyridinyl-tetrazine motif.
To this aim, we set out to elucidate the cause for the poor elimination yields with bis-(2-pyridinyl)-tetrazine as activator, and to develop derivatives that would overcome this hurdle (Figs. 1 and 2). Herein we report that the main cause for the low release when using bis-(2-pyridinyl)-tetrazine is the stability of the initially formed 4,5-dihydropyridazine tautomer upon IEDDA cycloaddition with the TCO-payload conjugate, precluding formation of the releasing 1,4-dihydropyridazine tautomer. We demonstrate that 4,5-to-1,4-tautomerization and payload elimination can be achieved by ortho-substituting bis-pyridinyl-tetrazines with groups capable of hydrogen bonding, thereby enabling the use of more reactive tetrazines in this click-to-release reaction. We subsequently apply these compounds in the cleavage of an ADC, demonstrating strong therapeutic effect in tumor-bearing mice at 10- to 20-fold lower tetrazine doses than used in previous studies with a less reactive bis-alkyl-tetrazine activator.
Fig. 2. Compounds used in this study.
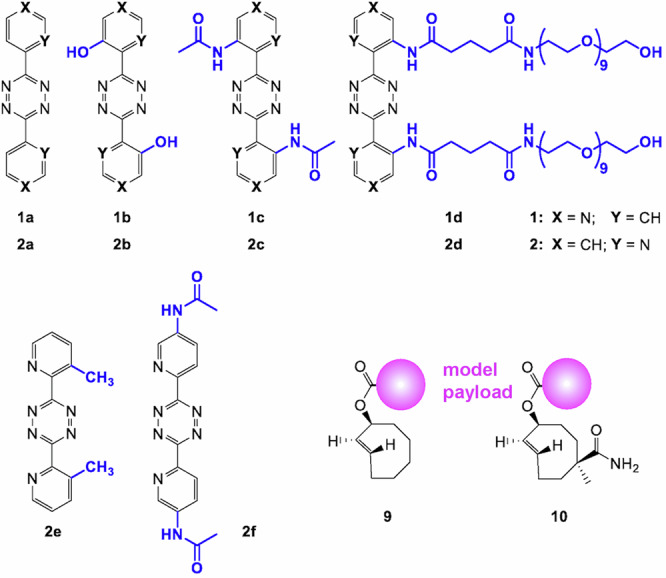
Ortho-substituted bis-(4-pyridinyl)-1,2,4,5-tetrazines 1b–d, ortho-substituted bis-(2-pyridinyl)-1,2,4,5-tetrazines 2b–d, control tetrazines 2e-f, parent compounds 1a and 2a, and TCO derivatives 9 and 10.
Results and discussion
We hypothesized that the poor release with bis-(2-pyridinyl)-tetrazine is either caused by the IEDDA adduct with TCO-payload being trapped in the 4,5-dihydropyridazine tautomeric form, or by the inability of the 1,4-dihydropyridazine tautomer to release the payload via the 1,4-elimination. To examine this, we studied the reaction of bis-(2-pyridinyl)-tetrazine 2a with TCO compounds 9 and 10, models for TCO-masked or linked payloads (Fig. 2). Both had dimethylamine as model payload to facilitate NMR analysis and to ensure sufficient aqueous solubility, while the tertiary carbamate was chosen to prevent potential side-product formation upon release4.
Detailed analysis (Fig. 3 and Supplementary Figs. 9–13) of the IEDDA reaction products with TCOs 9 and 10 showed the expected initial formation of 4,5-tautomers. For example, for the product derived from 9, 4,5-tautomer 19a, FT-IR analysis showed the absence of an NH band around 3300 cm−1 excluding the presence of either 1,4- or 2,5-tautomer (19b and 19c). And the aromatic region in 1H-NMR aligned well with that of 4,5-dihydropyridazine 17a (Fig. 4)16,27. However, in contrast to the IEDDA adduct with aliphatic tetrazines, these 4,5-tautomers did not readily tautomerize to either 1,4- and/or 2,5-tautomers. Even after prolonged times in aqueous solution, the 4,5-tautomer 19a was stable, and hence incapable of payload elimination. Addition of a small amount of formic acid to the aqueous solution resulted in slow tautomerization and ca. 10% payload release, and in this way it was possible to isolate a species that was characterized as the non-releasing 2,5-dihydropyridazine tautomer 19c (Supplementary Section 9). Incubation of this species in PBS did indeed not result in elimination (Supplementary Section 6).
Fig. 3. IEDDA reaction between bis-(2-pyridinyl)-tetrazine 2a and TCO-payload model 9.
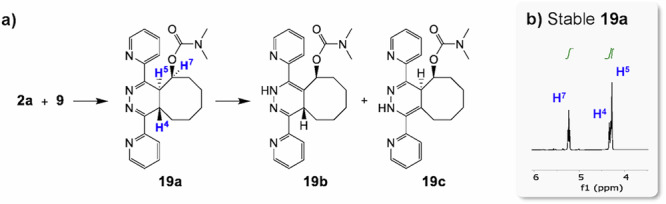
a Reaction scheme; b 1H-NMR of the IEDDA reaction product, the 4,5-tautomer 19a.
Fig. 4. IEDDA reaction between tetrazines 2a-c and TCO.
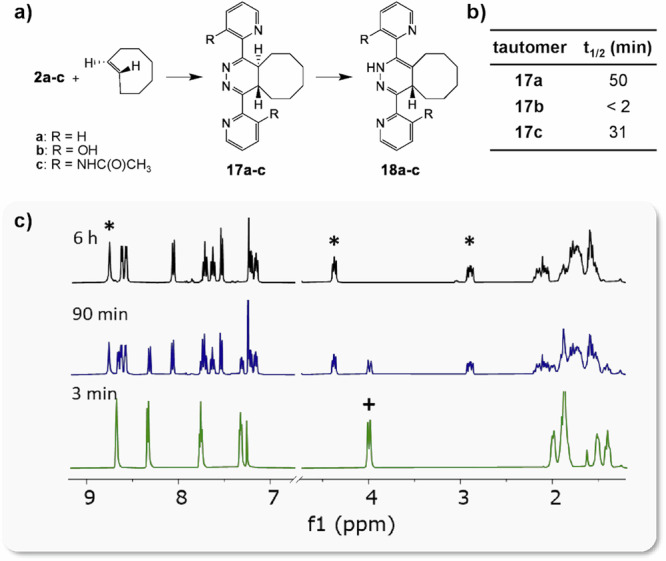
a Reaction scheme; b table with half-life of 4,5-tautomers 17a–c in organic solution at 20 °C; c 1H-NMR spectra (CDCl3) of reaction of 2a and TCO, resulting in 4,5-tautomer 17a, and its tautomerization to 1,4-tautomer 18a (legend: + 17a; * 18a).
Apparently, the activation energy for tautomerization of IEDDA adducts with 2a is too high for spontaneous formation of the releasing 1,4-tautomer and the non-releasing 2,5-tautomer. Having learned the possible cause for the low release with 2a, we questioned whether an ortho-positioned (vs. tetrazine core) protic or hydrogen-bonding group on the pyridine ring could induce the tautomerization of a corresponding 4,5-dihydropyridazine IEDDA adduct due to its close vicinity to the central ring nitrogens (Fig. 1). While functionalization of aliphatic tetrazines with quaternary ammonium or carboxylic acid substituents was previously shown to accelerate tautomerization and elimination4,28, IEDDA products from aliphatic tetrazines are already prone to tautomerization, while the 4,5-dihydropyridazine derived from bis-pyridinyl-tetrazine is in itself stable. Also, as shown above, addition of acid to the medium with 19a does not lead to substantial release. Nevertheless, we set out to study the effect of protic or hydrogen-bonding groups on the tautomerization and, in addition, to learn whether these could also facilitate elimination by deprotonating the 1,4-dihydropyridazine and/or by protonating the releasing carbamate. To this end, several 2- and 4-bis-pyridinyl-tetrazine derivatives containing hydroxyl or amide groups at the ortho position were prepared (Fig. 2).
First, the suitability of these compounds was determined in terms of stability and reactivity. The stability of 1b, 1d, 2b, and 2d varied with half-lives between 4 and 38 h in PBS at room temperature, and between 1 and 2 h in plasma at 37 °C, which is stable enough for application as a fast-clearing activator since small tetrazines typically have a blood clearance half-life of 1–5 min (Table 1, Supplementary Figs. 1 and 2 and Supplementary Table 1)12,29. Dissolution of 2b in PBS revealed a remarkable color change from pink to yellow due to deprotonation of a hydroxyl group (Supplementary Fig. 7). In contrast, 2a, 2c, and 2d did not show such a pH dependency, underlining the relative acidity of the hydroxyl group of 2b. The second-order rate constants of the IEDDA reaction of these activators with TCO 9 in 25% MeCN/PBS varied between 270 and 748 M−1 s−1, which is up to 26-fold faster than the highly releasing but less reactive bis-alkyl-tetrazines such as 3,6-dimethyl-1,2,4,5-tetrazine (Table 1, Supplementary Figs. 2–6, Supplementary Table 2)2,4,11. Bis-(2-pyridinyl)-tetrazines 2 were more reactive and stable than the 4-pyridinyl analogues 1, most likely due to greater out-of-plane rotation of the pyridinyl due to lone pair interactions combined with the slightly reduced electron withdrawing nature of the 2- vs. -4-pyridinyl group30–32. And the tetrazines with the more electron donating hydroxyl groups were less reactive compared to those with amide groups, which is in agreement with previous observations27,33.
Table 1.
Stability and reactivity data of tetrazines 1b, 2b, 1d, and 2d
| Tetrazine | Stability, plasma 37°Ca t1/2 (h) (95% CI) | Reactivityb with 9 k2 (M−1 s−1) | |
|---|---|---|---|
| 1b | 2.04 (1.66-2.66) | 270 ± 11 | |
| 2b | 2.00 (1.77-2.30) | 397 ± 4 | |
| 1d | 0.92 (0.72-1.30) | 519 ± 12 | |
| 2d | 1.98 (1.78-2.24) | 748 ± 20 | |
| dimethyl-Tz | 29.0 ± 0.4 | ||
CI confidence interval.
a50% mouse plasma, n = 3.
bIn 25% MeCN/PBS at 20 °C, n = 5.
Reaction with TCO and tautomerization
To examine the effect of the ortho-substituted pyridinyl groups of the tetrazines on the rate of tautomerization of their IEDDA adducts, an NMR study was performed in which 2a, 2b, and 2c were reacted with unfunctionalized TCO in organic solvent (Fig. 4, Supplementary Fig. 8, Supplementary Table 3). Due to the fast kinetics of the IEDDA reaction, the initial 4,5-tautomer 17 was formed within minutes16. Subsequently, this tautomerized to the asymmetric 1,4-tautomer 18, which is thermodynamically more stable. For the unfunctionalized bis-(2-pyridinyl)-tetrazine 2a, the half-life of 4,5-tautomer 17a was about 60 min. Gratifyingly, the hydroxyl groups in 2b resulted in a significant increase of the tautomerization rate, reducing the half-life of the 4,5-tautomer to less than 2 min, possibly by hydrogen bonding between the hydroxyl group and the nitrogen in the 4,5-dihydropyridazine, aided by the relative acidity of the hydroxyls. Although slower than 2b, the acetamide groups in 2c also afford marked acceleration of the tautomerization rate compared to unsubstituted tetrazine 2a.
Release with model TCO linker
Now that we succeeded in enhancing the tautomerization of the bis-(pyridinyl)-tetrazine IEDDA adduct with unmodified TCO by introducing hydrogen-bonding and/or protic groups at the ortho position of the pyridinyl substituents, we examined tetrazines 2b and 2c for their capability as activators of the pyridazine elimination with TCO linker-payload model 10. The reaction of 2b and 10 was performed in a mixture of DMSO-d6 and deuterated phosphate buffer (pD 7.4) and monitored by 1H-NMR and HPLC-MS/PDA (Fig. 5, Supplementary Figs. 14 and 15). To our delight, we observed the quantitative liberation of N,N-dimethylamine within minutes, accompanied by the formation of an elimination product that slowly precipitated from solution, which was surprisingly shown to be cyclic ether 14 (Fig. 5). We questioned whether this product originated from direct substitution of the neighboring hydroxyl onto the carbon adjacent to the carbamate group driving the N,N-dimethylamine release or if it resulted from Michael addition onto the exocyclic double bond in the intermediate elimination product (14-exo). To get an indication, we oxidized the IEDDA adduct of tetrazine 2b and TCO 9 in situ, forming pyridazine 22 (Fig. 5), which cannot undergo an 1,4-elimination but could possibly still release the payload via hydroxyl substitution. This species, however, did not release N,N-dimethylamine, supporting the Michael addition mechanism for the formation of 14 (Supplementary Information Section 6). Furthermore, reaction of ether-linked TCO-payload conjugate 23 with 2b afforded near quantitative formation of comparable elimination product 26 within 20 min., again supporting the Micheal addition mechanism, as alkoxyl is a poorer leaving group than carbamate and should have led to a much slower release, if any, in the case of direct substitution (Supplementary Information Section 6). The quantitative release yield with 2b suggests that the ortho-hydroxyl groups also catalyze the tautomerization from non-releasing 2,5-tautomer 13c to releasing tautomer 13b, likely via the 4,5-tautomer.
Fig. 5. Reaction of tetrazine 2b with TCO 10.

Reaction of tetrazine 2b and TCO 10, resulting in instantaneous formation of cyclized elimination product 14, formed either through pathway A or B. Control experiment 1: Reaction of 2b with 9 in presence of sodium nitrite to in situ oxidize 4,5-tautomer to pyridazine, preventing 1,4-elimination; lack of release supports A. Control experiment 2: Reaction of 2b with TCO ether 23, leading to rapid and quantitative release of hydroxyl, supporting A.
In a comparable manner, activator 2c was reacted with 10 (Fig. 6, Supplementary Figs. 16–18). After the fast formation of the IEDDA adduct, 4,5-tautomer 13a, the liberation of N,N-dimethylamine was slower, affording 50% in 3 days, which is still a pronounced increase compared to the 10–15% maximum yield achieved with unsubstituted tetrazine 2a. The disappearance of 4,5-tautomer 13a yielded two species, which were isolated and characterized as the non-releasing 2,5-tautomer 15c and aromatized elimination product 162.
Fig. 6. Reaction of tetrazine 2c and TCO 10, resulting in tautomerization of the initial IEDDA adduct and elimination.
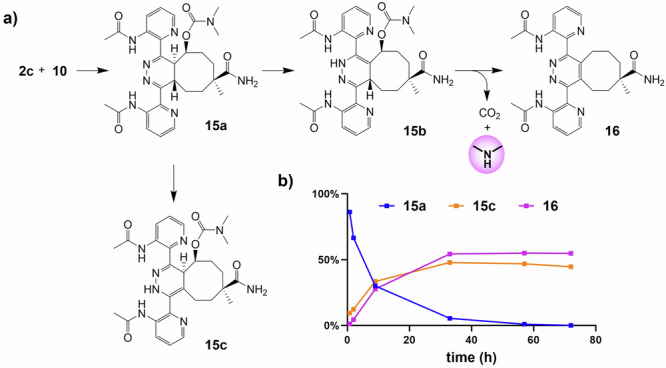
a Reaction scheme; b distribution plot of the reaction products.
These model studies demonstrate that the ortho-functionalization of bis-pyridinyl-tetrazines results in an improved release by these activators compared to unsubstituted 2a, which is at least in part due to the increased ability to tautomerize to the desired 1,4-tautomer. This is further supported by indicative DFT calculations of the 4,5-dihydropyridazine structures derived from TCO 9 and tetrazines 1a-c and 2a-c, showing hydrogen bonding between the N1 and the hydroxyl and amido groups of the top pyridine ring adjacent to N1 (Fig. 7, Supplementary Fig. 25, Supplementary Data 1). Furthermore, these groups likely make the pyridinyl rings less electron-withdrawing as result of electron-donation and possibly due to out-of-plane rotation of the top pyridinyl through steric hindrance with the H1 in the 1,4-dihydropyridazine, disturbing π-system conjugation. While the present study does not inform whether increased electron density in the 1,4-dihydropyridazine ring (i.e. after the tautomerization has occurred) facilitates 1,4-elimination, this would be in line with structure−activity relationships established for the widely used self-immolative para-aminobenzyloxycarbonyl linker wherein an electron-donating group on the aromatic core leads to faster fragmentation due to stabilization of the increasing positive charge upon release of the carbamate34,35.
Fig. 7. DFT calculations.
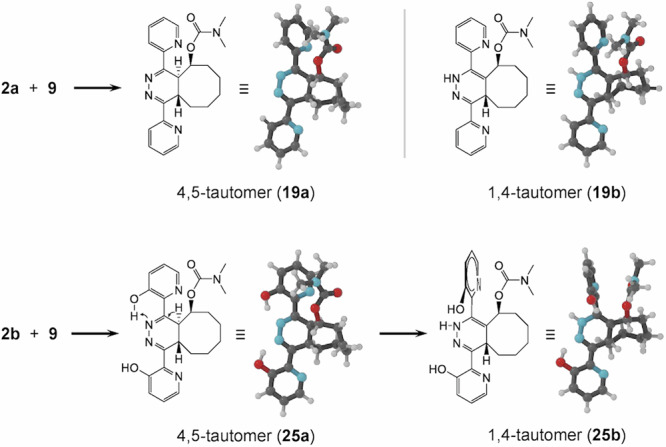
DFT calculations of lowest energy structures of 4,5- and 1,4-dihydropyridazine tautomer products derived from 2a and 2b with TCO 9. DFT calculations demonstrate hydrogen bonding in 25a that is postulated to lead to 4,5-1,4 dihydropyridazine tautomerization and subsequent out-of-plane rotation of the top ring in 25b, while this is not observed for 19a and 19b.
Indicative DFT calculations of the optimal dihedral angle between the pyridinyl rings vs. the dihydropyridazine of the 1,4-tautomers of the IEDDA adducts of TCO 9 and tetrazines 2a-c afforded an angle close to 90° for the top pyridinyl ring (adjacent to the H1) for 2b and 2c, while the less sterically hindered pyridinyl in 2a could still adopt a π-conjugated near-planar configuration just like the bottom rings for 2a-c (Fig. 7, and Supplementary Table 8 and Supplementary Fig. 25, Supplementary Data 1). The strong out-of-plane rotation of the top ring in the 1,4-tautomer suggest it is unlikely that the pyridinyl substituents are accelerating the release through hydrogen bonding with the carbamate or the 1,4-dihydropyridazine H1. To get an indication of the relative importance, if any, of electron donation vs. top ring rotation on the 1,4-elimination, we prepared control tetrazine 2e (Fig. 2), wherein the pyridinyl rings are ortho-substituted with the sterically demanding but less electron donating methyl group. Reaction with TCO 9 did not result in the formation of elimination product, even after forced tautomerization by the addition of acid (Supplementary Fig. 19), suggesting that out-of-plane rotation does not lead to tautomerization and indicating that rotation would not be the dominant factor in the case that increased electron density facilitates elimination from these pyridinyl-substituted 1,4-dihydropyridazines.
Release with TCO in ADC in vitro
Next the new tetrazines were tested as activators of a click-cleavable ADC (Fig. 8). Current ADCs rely on binding to an internalizing cancer receptor and subsequent intracellular drug release mediated by e.g. proteases36. However, the number of suitable cancer-specific receptors in solid cancers is limited, while there are non-internalizing receptors and extracellular markers in the tumor microenvironment that would be good ADC targets if cleavage can be achieved without relying on intracellular process. Click-cleavable ADCs are designed to expand the application scope of ADC therapy by allowing targeting of such targets, followed by administration of a tetrazine activator to cleave the drug from the tumor-bound ADC, allowing it to distribute into surrounding cancer cells11,12. We previously developed such an ADC against the non-internalizing tumor marker Tumor Associated Glycoprotein 72 (TAG-72), comprising a diabody with four copies of TCO-linked monomethyl auristatin E (MMAE) (Fig. 8a), affording strong antimitotic therapy in mice with colorectal and ovarian xenografts12.
Fig. 8. Tetrazine-triggered ADC cleavage in vitro.
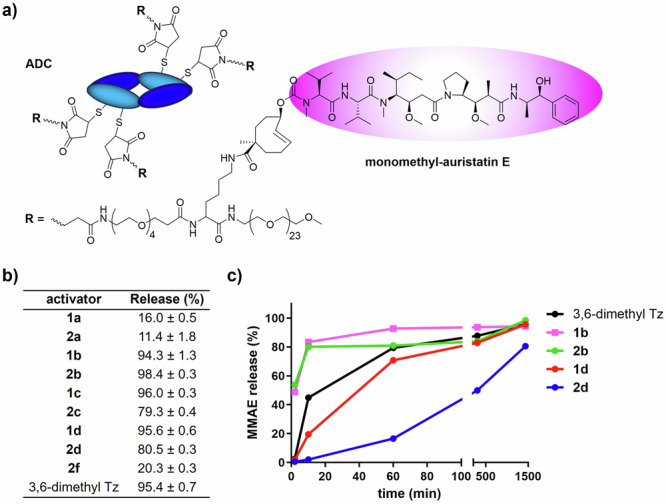
a Structure of the diabody-based anti-TAG-72 ADC used in this work12; b MMAE release from ADC in reaction with ortho-substituted tetrazines in 50% mouse plasma at 37 °C after 24 h (mean ± SD, n = 3) and; c time-dependent MMAE release from ADC in the presence of selected tetrazines in 50% mouse plasma at 37 °C up to 24 h.
Incubation of unmodified 1a and 2a with ADC in 50% plasma at 37 °C gave expected low release yields of 16 and 11%, respectively. As hoped, the new series of ortho-functionalized bis-pyridinyl-tetrazines was highly effective in releasing MMAE, finally departing from the inverse reactivity-release correlation, with yields up to 95%, equaling 3,6-dimethyl tetrazine and surpassing the 80–85% yield of the previously developed bis-alkyl-tetrazine activator designed for in vivo use12. We were especially encouraged to see that now also bisamides 1c, 2c, 1d and 2d produced high payload release, likely due to the increased water content, and possibly other factors such as increased temperature and an altered TCO environment, which may have favored the formation of the 1,4-dihydropyridazine tautomer. However, as with the model studies, following rapid IEDDA conjugate formation, products derived from the hydroxyl tetrazines 1b and 2b afforded near instantaneous release while those derived from the amido tetrazines required several hours, with 2d being the slowest. Nevertheless, for an ADC activation application release speed is less important than release yield. Importantly, control tetrazine 2f, an analog of 2c with para-positioned amides afforded much less release than 2c, underlining that the tautomerization effected by the ortho-substituents is not due to electron donation but due to hydrogen bonding with the 4,5-tautomer N1.
Reactivity and release with TCO in ADC in vivo
We continued the evaluation with the relatively hydrophilic bis-pyridyl-tetrazine activators 1b,d and 2b,d. First, we examined the tetrazines in a cell proliferation assay and to amplify any differences between the compounds, instead of a pulse treatment they were incubated for 72 h, which is 3 orders of magnitude longer than their expected in vivo residence times. We observed a ca. 50, 18 and 5% cell growth inhibition at the highest tested concentration of 10 µM for, respectively, the cell permeable hydroxyl tetrazines (1b, 2b), the cell impermeable PEGylated amido tetrazines (1d, 2d), and the unmodified tetrazines (1a, 2a) (see Supplemental Information Section 7). This difference is likely caused by tetrazine core degradation induced by the ortho substituents on the pyridinyl augmented by cell permeability in the case of the hydroxyl tetrazines. However, overall, the values were largely comparable to previous 72 h results with bis-pyridyl- and bis-methyl-tetrazine3, of which the latter has been safely administered to mice up to very high doses of 140 mg/kg11,13. Considering the much shorter residence times of the tetrazines in vivo (minutes), the measured in vitro tolerability of the PEGylated amido and hydroxyl-substituted tetrazines was expected to be more than adequate.
We then set out to determine the required dose for complete on-tumor ADC activation in a mouse model of human colon carcinoma (LS174T). Previously, this ADC exhibited high tumor uptake in this model and fast clearance from blood (6-7 h half-life), allowing i.v. injection of activator and on-tumor MMAE release 48 h post-ADC injection12. However, due to the low reactivity of the previously used bis-alkyl-tetrazine activator, a 0.335 mmol/kg activator dose (ca. 450 mg/kg) was necessary to completely activate the ADC on-tumor12. We expected that the 9- to 26-fold higher reactivity (Table 1) of the present activators should allow for a markedly lower dose for complete on-tumor reaction.
As in our previous studies, to estimate the on-tumor reaction yield between the activators and the ADC we used a highly reactive 111In-labeled DOTA-bis-(2-pyridinyl)-tetrazine probe as a readout of the presence of unreacted TCO in the tumor after ADC activation11,12. Following this approach, mice bearing LS174T xenograft were first i.v. administered 125I-labeled ADC (2 mg/kg) followed 48 h later by ca. 3.35 µmol/kg (dose 1×) activator 1b, 1d, 2b or 2d, which were rapidly cleared as evidenced by the purple coloration of the urine only within the first 5 minutes. One hour post-activator administration, 0.33 µmol/kg [111In]In-DOTA-tetrazine probe (Fig. 9a, e) was injected i.v. followed by biodistribution measurements 24 h post-probe injection. We observed high ADC uptake in tumor (15-19%ID/g), combined with very low levels in non-target tissues, and reduced tumor uptake of 111In-labeled probe compared to mice that did not receive any activator, signifying partial on-tumor reaction between the ADC and the activators injected at 1x dose (Supplementary Tables 4 and 5, Supplementary Fig. 24). Furthermore, the 111In-probe exhibited only very low uptake in non-target tissue mirroring the high tumor-nontumor ratio of the ADC, with the exception of the kidney due to tetrazine excretion. Perusal of the tumor biodistribution data revealed a lower in vivo reaction yield (20–40%) for hydroxyl tetrazines 1b and 2b compared to amido tetrazines 1d and 2d (43–65%) (Fig. 9a and Supplementary Table 4). This higher TCO reaction yield is most likely due to a combination of higher reactivity (Table 1) and a lower volume of distribution (Vd) of the non-cell permeable PEGylated tetrazines with respect to the cell permeable hydroxyl tetrazines. Higher reactivity of the 2- vs. the 4-pyridinyl compounds resulted in a higher on-tumor reaction yield for 2d compared to 1d (Fig. 9a). However, due to the lower release yield afforded by 2d vs. 1d the free MMAE recovered in the tumors harvested from these two groups of mice 24 h after activator administration was comparable (Figs. 8b, 9b, Supplementary Table 6). Based on the foregoing and the higher in vitro tolerability observed for the amido tetrazines, we proceeded with 1d and 2d and increased their dose to 16.7 µmol/kg (dose 5×) to push the on-tumor ADC activation and MMAE release further. As expected, we found higher reaction yields and liberated MMAE levels, with 2d even achieving near quantitative on-tumor ADC reaction (94.2 ± 1.8%, Fig. 9a). Surprisingly, however, the amount of recovered free MMAE in tumor 24 h after ADC activation with 2d was significantly lower than found for the better releasing but slightly less reactive 1d (128 ± 14 nM and 226 ± 26 nM, respectively; P < 0.01, Fig. 9b, Supplementary Table 6), despite its somewhat lower tumor reaction yield (80.1 ± 0.8%; P > 0.05; Fig. 9a). While this difference is likely in part due to variation in MMAE recovery efficiencies from tumor homogenates, the high intrinsic release yield and speed of activator 1d (Fig. 8) led us to select 1d for a therapy study in combination with the click-cleavable ADC.
Fig. 9. Tetrazine-triggered ADC cleavage in tumor-bearing mice.

a On-tumor ADC reaction yields in mice pre-treated with ADC (2 mg/kg) followed 2 days later by activators 1b, 1d, 2b and 2d (dose 1× = 3.35 µmol/kg; dose 5× = 16.7 µmol/kg), estimated from the 125I-ADC and [111In]In-DOTA-tetrazine probe (0.33 µmol/kg, injected 1 h post-activation) biodistribution data (Supplementary Tables 4 and 5); b Intra-tumor free MMAE concentration 24 h post-ADC activation (therapy dose = 33.5 µmol/kg in mice pre-treated with a 4 mg/kg ADC dose). The data are the mean with SEM (n = 4; *P < 0.05; **P < 0.01; ***P < 0.001; one-way ANOVA with Newman–Keuls multiple-comparison test); c Mean tumor volume (with SEM) in groups of LS174T bearing mice (n = 10 at day 0) treated with four cycles of ADC (4 mg/kg) followed by activator 1d (33.5 µmol/kg) 48 h post-ADC, ADC only, activator only, and vehicle (***P < 0.001 at day 17; one-way ANOVA with Newman–Keuls multiple-comparison test); d Survival curves for the mice in c (P = 0.0005, Log-rank Mantel–Cox test); e Experimental timelines for a–d. Individual values for panels a–c are presented in Supplementary Tables 4 and 6 and in Supplementary Fig. 22. Created with Biorender.com with permission.
Therapy study
We embarked on a therapy study in LS174T-bearing mice to evaluate the performance of the new bis-pyridinyl-tetrazine 1d. To be able to compare to the earlier therapy study with the bis-alkyl-tetrazine activator we chose a 4 mg/kg ADC dose (average of the two highest doses used previously) combined with a 10-fold lower activator dose of 33.5 µmol/kg 1d12. This combination was found to produce a high concentration of free MMAE in tumor 24 h post-activation (339 ± 45 nM; Fig. 9b and Supplementary Table 6)12.
Mice bearing LS174T xenografts received four cycles (one every 4 days) of ADC followed by activator 1d 48 h post-ADC, or ADC alone, activator alone, or vehicle (injection scheme in Fig. 9e and Supplementary Fig. 21). For the group of mice that received the ADC and 1d combination, we were pleased to observe that the growth of the very aggressive LS174T tumors was inhibited for over 3 weeks affording a 42-day median overall survival and one complete response at the end of the 2 months observation period (Fig. 9c, d), Supplementary Fig. 22). On the contrary, all control mice were euthanized within 3 weeks due to large tumor size (>1.5 gr) or poor health (Supplementary Table 7) and had a 15.5–17.5 days median survival. The repeated treatments were tolerated well, with only minimal and reversible weight loss, mainly due to tumor toxicity. Importantly there were no overt signs of hematological or other toxicity that could be attributed to the use of ADC and activator, either in combination or alone, amongst others supporting the tolerability and in vivo applicability of the new PEGylated amido tetrazines (Supplementary Fig. 23a, b). Overall, these results were very similar to the outcome of our previous therapy study performed with the bis-alkyl-tetrazine activator given at a 10-fold higher dose, with an overall 34 days (3 mg/kg ADC) or 39 days (5 mg/kg ADC) median survival12.
Conclusion
It has been more than 10 years since the first report of the pyridazine elimination reaction2 between tetrazine and TCO conjugates, and the reaction has enjoyed growing and widespread use, covering diverse chemical, in vitro, and in vivo applications. However, the reaction is beset by an unrelenting inverse correlation between click reactivity and payload release yield, requiring high in vivo reagent concentrations that hamper clinical translation of systemic drug delivery applications. We show that the IEDDA product of the highly reactive bis-pyridinyl-tetrazines gets trapped in the 4,5-dihydropyridazine tautomer and therefore cannot give substantial release. However, installing hydroxyl or amido groups on the ortho position of the pyridinyl substituents afforded efficient tautomerization to the releasing 1,4-dihydropyridazine form and good payload release. This was surprising because contrary to the readily tautomerizing aliphatic 4,5-dihydropyridazines, which can be accelerated by acidic medium or the vicinity of a protic group, bis-pyridinyl-tetrazine-derived 4,5-dihydropyridazines do not tautomerize unassisted, and also do not afford release after addition of acid. Mechanistic and preliminary modeling studies suggest that the tautomerization is induced by hydrogen-bonding (vs. electron donation) with the N1 of the 4,5-tautomer and that the subsequent release, via 1,4-elimination, is possibly enabled by increased electron density caused by electron donation (vs. out-of-plane top ring rotation). The hydroxyl-substituted tetrazines gave much faster tautomerization and release than the amido-substituted ones, but the latter were more reactive, and both motifs afforded almost complete ADC cleavage yields with 10- to 20-fold higher reactivity than the previously used bis-alkyl-tetrazines. The increased reactivity correlated very well with the on-tumor reaction yields between ADC and tetrazine, with higher yields observed for the amido- vs. hydroxyl-substituted activators and for 2- vs. 4-pyridinyl tetrazines. Importantly, we achieved high on-tumor ADC conversion with a 20-fold lower activator dose than what was previously needed with bis-alkyl-tetrazine12. At an order of magnitude lower dose, amido-substituted 1d, selected for its high reactivity (18-fold higher than dimethyltetrazine) and release (96%) afforded the same strong therapeutic benefit in ADC-treated tumor-bearing mice as its less reactive bis-alkyl-tetrazine predecessor.
We expect that the findings presented herein will strongly advance click-to-release utility in vitro as well as in vivo. The near instantaneous and quantitative release afforded by the presented hydroxyl-based activators are ideally suited for time-sensitive in vitro turn-on or turn-off experiments, and hold promise for the design and in vivo application of cell-impermeable analogs with improved pharmacokinetics. The presented amido-substituted activators will enable efficient and well-tolerable in vivo on-target activation as well as off-target deactivation approaches at dosages that are better aligned with clinical translation, as a result of breaking the long-standing inverse correlation between reactivity and release.
Methods
Complete experimental part including Supplementary Methods, synthesis of all compounds, model compound experiments, in vitro and in vivo ADC experiments, DFT calculations, as well as spectra, are given in the Supplementary Information.
DFT calculations
The Amsterdam Density Functional (ADF) program was employed for performing the DFT calculations. The B3LYP exchange-correlation functional in conjunction with a triple-ζ (TZP) Slater-type basis set was used in all calculations. Implicit solvation effects were considered by employing the COSMO model with parameters for water. Please note that these calculations are performed with an implicit solvent and thus should be treated in a qualitative fashion. Internal hydrogen bonding and steric hindrance can be reliably evaluated using implicit solvation as these correspond to intramolecular properties.
MMAE release from ADC in vitro
In a 1.5 mL vial, the following was combined: 650 µL mouse plasma, 600 µL PBS, 20 µL ADC (1.62 µg/µL in PBS) and 7.5 µL d8-MMAE solution (0.167 µg/µL, internal standard). Next, aliquots of 20 µL were prepared to which 5.2 µL of a tetrazine solution (0.25 mM in DMSO:H2O = 1:1) was added and the solution was incubated at 37 °C. At various times, 100 µL of cold MeCN was added to the aliquot and the mixture was vortexed, stored at 4 °C for 30 min. and centrifuged. Supernatant (20 µL) was taken and combined with 100 µL of 1% formic acid (aq) solution. This sample was analyzed with HPLC-SIM-MS to quantify the ratio of free MMAE and d8-MMAE, which is a measure of the release yield.
In vivo reaction between ADC and activator: tumor blocking study
Groups of tumor bearing mice (n = 4) were injected with 125I-labeled ADC (2 mg/kg, ca. 0.35 MBq/100 µL saline per mouse) followed 48 h later by the activators (dose 1x: 3.3 µmol/kg; dose 5x: 16.7 µmol/kg; 100 µL PBS containing 5% DMSO) and, after 1 h, by [111In]In-DOTA-tetrazine probe (0.33 µmol/kg, ca. 1 MBq/100 µL saline per mouse). One group of mice was injected with the same amount of 125I-labeled ADC followed only by 111In-labeled probe 49 h later. One last group of mice was injected only with the probe (non-specific probe binding). All mice were euthanized 23 h post-probe injection and the 125I and 111In uptakes in tumors were measured by γ-counting (dual-isotope protocol with cross contamination correction), together with standards to determine the % injected dose per gram (%ID/g). The on-tumor reaction yield between ADC and activator was calculated as follows. The 111In uptake in the tumors (%ID/g) was corrected for mean non-specific probe binding and divided by the 125I uptake (%ID/g), to account for different ADC uptake in different sized tumors. The obtained ratios were normalized for the mean value obtained in mice not treated with an activator (mean In-111/I-125 ratio for the “no activator” group). In this latter group, the max probe binding was achieved, signifying 0% reaction yield between ADC and activator.
MMAE measurement in tumors
The concentration of released free MMAE was measured in the tumors of selected groups used for tumor blocking experiments and in those from one more group of mice (n = 4) treated with 4 mg/kg ADC and 33.5 µmol/kg (dose 10x) activator 1d using the same protocol. After γ-counting, tumors samples (0.1–0.2 gr) were transferred into MagNA Lyser green beads tubes (Roche) together with 1 mL methanol and a weighed amount of a d8-MMAE solution as internal standard. The tissues were homogenized (4 × 30 s cycles, 6.5k rpm, with 1 min cooling in between cycles) and the debris was removed by centrifugation (13k rpm, 5 min). The methanol was evaporated under a stream of N2, the residue was reconstituted in PBS (containing BSA digest and 10% MeCN). After overnight standing at −20 °C the solutions were filtered through a 0.22 µm filter. The d0/d8-MMAE metabolites of interest were separated on a C18 RP column (2.1× 100 mm, 3 µm particle size) with a linear 10–80% acetonitrile (containing 0.1% formic acid) gradient (gradient length 2–10 min; flow rate 250 µL/min; injection volume 5 µl). The eluent from 3 to 6 min was introduced into the mass spectrometer. The collision energy for Xevo-TQS micro system (Waters) using nitrogen collision gas was 25 eV and the cell pressure was 0.35 Pa. The source temperature was set at 150 °C and the capillary voltage was maintained at 0.40 kV. The specific m/z transitions of 718.5 to 506.4 and 718.5 to 321.2 for d0-MMAE and 726.6 to 506.3 and 726.6 to 321.2 for d8-MMAE were detected and the d0/d8 peak area ratio was used for quantitation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We thank Bianca Lemmers-van de Weem, Kitty Lemmens-Hermans and Karin de Haas-Cremers (PRIME, Radboud University Medical Center) for technical assistance with animal experiments, and Hans Wessels and Fokje Zijlstra (Radboud University Medical Center) for MMAE measurements in tumor extracts. It is with great sadness that our co-author Henk Janssen passed away during the finalization of this publication. Henk was a very creative and prolific organic chemist, and we dedicate this work to him.
Author contributions
R.V. performed TCO synthesis, model compound studies, and in vitro ADC release studies; F.H. conducted synthesis of tetrazines; I.F. performed DFT calculations; A.v.O. conducted the cytotoxicity studies, R.R. performed in vivo studies, H.J., R.V., R.R., M.R. designed experiments, R.V., R.R., M.R. wrote the paper.
Peer review
Peer review information
Communications Chemistry thanks Haoxing Wu and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer review reports are available.
Data availability
The data that support the findings reported herein are available in the Supplementary Information, in Supplementary Data 1, or on reasonable request from the corresponding author.
Competing interests
R.R., A.v.O., M.R. are shareholder and/or employee of Tagworks. R.V., R.R., F.H., H.J., M.R. are inventor on corresponding patent family WO2020256544 (Tetrazines for high click release and speed; inventors Marc Stefan Robillard, Freek Johannes Maria Hoeben, Raffaella Rossin, Ronny Mathieu Versteegen, Henricus Marie Janssen). All other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-024-01392-z.
References
- 1.Wang, J., Wang, X., Fan, X. & Chen, P. R. Unleashing the power of bond cleavage chemistry in living systems. ACS Cent. Sci.7, 929–943 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Versteegen, R. M., Rossin, R., ten Hoeve, W., Janssen, H. M. & Robillard, M. S. Click to Release: Instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem. Int. Ed.52, 14112–14116 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Versteegen, R. M. et al. Click-to-Release from trans-cyclooctenes: Mechanistic insights and expansion of scope from established carbamate to remarkable ether cleavage. Angew. Chem. Int. Ed.57, 10494–10499 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Carlson, J. C. T., Mikula, H. & Weissleder, R. Unraveling tetrazine-triggered bioorthogonal elimination enables chemical tools for ultrafast release and universal cleavage. J. Am. Chem. Soc.140, 3603–3612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Onzen, A. H. A. M. et al. Bioorthogonal tetrazine carbamate cleavage by highly reactive trans-cyclooctene. J. Am. Chem. Soc.142, 10955–10963 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Chen, J. et al. Building bioorthogonal click-release capable artificial receptors on cancer cell surface for imaging, drug targeting and delivery. Acta Pharm. Sin. B13, 2736–2746 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mejia-Oneto, J. M., Khan, I., Seebald, L. & Royzen, M. In vivo bioorthogonal chemistry enables local hydrogel and systemic pro-drug to treat soft tissue sarcoma. ACS Cent. Sci.2, 476–482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He, X. et al. An all-in-one tetrazine reagent for cysteine-selective labeling and bioorthogonal activable prodrug construction. Nat. Commun.15, 2831 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li, J., Jia, S. & Chen, P. R. Diels-Alder reaction–triggered bioorthogonal protein decaging in living cells. Nat. Chem. Biol.10, 1003–1005 (2014). [DOI] [PubMed] [Google Scholar]
- 10.McFarland, J. M. et al. Click chemistry selectively activates an auristatin protodrug with either intratumoral or systemic tumor-targeting agents. ACS Cent. Sci.9, 1400–1408 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossin, R. et al. Triggered drug release from an antibody–drug conjugate using fast “Click-to-Release” chemistry in mice. Bioconjug. Chem.27, 1697–1706 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Rossin, R. et al. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun.9, 1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang, G. et al. Bioorthogonal chemical activation of kinases in living systems. ACS Cent. Sci.2, 325–331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao, Q. et al. Synergistic enzymatic and bioorthogonal reactions for selective prodrug activation in living systems. Nat. Commun.9, 5032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vlastara, M. et al. Click-to-Release: Cleavable radioimmunoimaging with [89Zr]Zr-DFO-trans-cyclooctene-trastuzumab increases tumor-to-blood ratio. Theranostics13, 4004–4015 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blackman, M. L., Royzen, M. & Fox, J. M. Tetrazine ligation: Fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc.130, 13518–13519 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauer, J. et al. 1,2,4,5-Tetrazine: Synthesis and reactivity in [4+2] cycloadditions. Eur. J. Org. Chem.1998, 2885–2896 (1998). [Google Scholar]
- 18.Sauer, J., Mielert, A., Lang, D. & Peter, D. Eine studie der Diels-Alder-reaktion, III: Umsetzungen von 1.2.4.5-tetrazinen mit olefinen. Zur struktur von dihydropyridazinen. Chem. Ber.98, 1435–1445 (1965). [Google Scholar]
- 19.Albu, S. A. et al. 125I-Tetrazines and inverse-electron-demand Diels–Alder chemistry: A convenient radioiodination strategy for biomolecule labeling, screening, and biodistribution studies. Bioconjug. Chem.27, 207–216 (2016). [DOI] [PubMed] [Google Scholar]
- 20.García-Vázquez, R. et al. Development of 18F-labeled bispyridyl tetrazines for in vivo pretargeted PET imaging. Pharmaceuticals15, 245 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossin, R. et al. In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem. Int. Ed.,49, 3375–3378 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Rossin, R., van Duijnhoven, S. M. J., Läppchen, T., van den Bosch, S. M. & Robillard, M. S. Trans-cyclooctene tag with improved properties for tumor pretargeting with the Diels–Alder reaction. Mol. Pharm.11, 3090–3096 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Kuba, W. et al. Oxidative desymmetrization enables the concise synthesis of a trans-cyclooctene linker for bioorthogonal bond cleavage. Chem. Eur. J.29, e202203069 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, B. et al. A concise synthetic approach to highly reactive Click-to-Release trans-cyclooctene linkers. Chem. Eur. J.29, e202300755 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Sondag, D. et al. Readily accessible strained difunctionalized trans-cyclooctenes with fast click and release capabilities. Chem. Eur. J.29, e202203375 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossin, R. et al. Highly reactive trans-cyclooctene tags with improved stability for Diels-Alder chemistry in living systems. Bioconjug. Chem.24, 1210–1217 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Taylor, M. T., Blackman, M. L., Dmitrenko, O. & Fox, J. M. Design and synthesis of highly reactive dienophiles for the tetrazine-trans-cyclooctene ligation. J. Am. Chem. Soc.133, 9646–9649 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarris, A. J. C. et al. Fast and pH-independent elimination of trans-cyclooctene by using aminoethyl-functionalized tetrazines. Chem. Eur. J.24, 18075–18081 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Devaraj, N. K., Thurber, G. M., Keliher, E. J., Marinelli, B. & Weisslede, R. Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc. Natl Acad. Sci.109, 4762–4767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houszka, N., Mikula, H. & Svatunek, D. Substituent effects in bioorthogonal Diels–Alder reactions of 1,2,4,5-tetrazines. Chem. Eur. J.29, e202300345 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svatunek, D., Wilkovitsch, M., Hartmann, L., Houk, K. N. & Mikula, H. Uncovering the key role of distortion in bioorthogonal tetrazine tools that defy the reactivity/stability trade-off. J. Am. Chem. Soc.144, 8171–8177 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang, H. et al. Enabling Universal Access to Rapid and Stable Tetrazine Bioorthogonal Probes through Triazolyl-Tetrazine Formation. JACS AU4, 2853–2861 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karver, M. R., Weissleder, R. & Hilderbrand, S. A. Synthesis and evaluation of a series of 1,2,4,5-tetrazines for bioorthogonal conjugation. Bioconjug. Chem.22, 2263–2270 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alouane, A., Labruère, R., Le Saux, T., Schmidt, F. & Jullien, L. Self-immolative spacers: Kinetic aspects, structure–property relationships, and applications. Angew. Chem. Int. Ed.54, 7492–7509 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Hay, M. P., Sykes, B. M., Denny, W. A. & O’Connor, C. J. Substituent effects on the kinetics of reductively-initiated fragmentation of nitrobenzyl carbamates designed as triggers for bioreductive prodrugs. J. Chem. Soc. Perkin Trans.1, 2759–2770 (1999). [Google Scholar]
- 36.Parit, S. et al. Antibody-drug conjugates: A promising breakthrough in cancer therapy. Int. J. Pharm.659, 124211 (2024). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data that support the findings reported herein are available in the Supplementary Information, in Supplementary Data 1, or on reasonable request from the corresponding author.