

Abstract
During embryogenesis, vertebral axial patterning is intricately regulated by multiple signaling networks. This study elucidates the role of protogenin (Prtg), an immunoglobulin superfamily member, in vertebral patterning control. Prtg knockout (Prtg−/−) mice manifest anterior homeotic transformations in their vertebral columns and significant alterations in homeobox (Hox) gene expression. Transcriptomic profiling of Prtg−/− mouse embryos highlights Prtg-regulated genes involved in axial development, particularly within the transforming growth factor beta (TGFβ) signaling pathway. Reduced TGFβ signaling in Prtg−/− mouse embryos is evidenced by decreased phosphorylated Smad2 (pSmad2) levels and its downstream target genes in the developing tail. We further show that Prtg interacts with growth differentiation factor 11 (GDF11) to enhance GDF11/pSmad2 signaling activity. Using human-induced pluripotent stem cell-derived presomitic mesoderm-like (hiPSC-PSM) cells, we demonstrate delayed posterior HOX gene expression upon PRTG knockout, which is rescued by GDF11 supplementation. These findings provide compelling evidence that PRTG regulates HOX genes through the GDF11/SMAD2 signaling pathway.
Subject terms: Embryology, Bone development
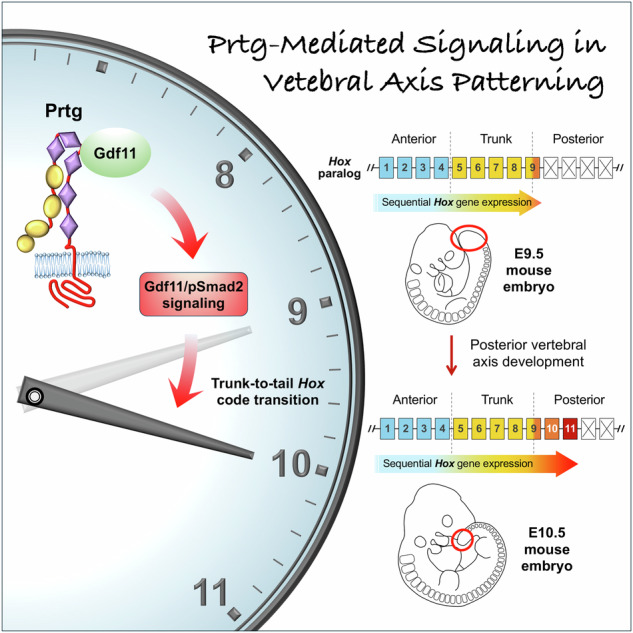
Protogenin regulates vertebral axial patterning in mammalian embryos by interacting with GDF11, enhancing GDF11/pSMAD2 signaling to facilitate trunk-to-tail HOX code transition and ensure proper expression of posterior HOX genes.
Introduction
The vertebral column is one of the most distinctive features of vertebrate animals. It comprises a series of spinal elements with specific morphological characteristics. Based on their morphology, these elements are categorized into cervical, thoracic, lumbar, sacral, and caudal sections1. In humans, the vertebral column consists of 33 bones, including 7 cervical, 12 thoracic, 5 lumbar, and 5 sacral vertebrae, as well as 4 caudal bones that fuse to form the coccyx. Mice possess 7 cervical, 13 thoracic, 6 lumbar, 4 sacral, and more than 10 caudal vertebrae. These bony elements arranged along the anterior-posterior axis constitute the axial patterning of the vertebral column.
Vertebral axial patterning is established during embryogenesis. After the formation of three germ layers, a group of cells located at the caudal end of the embryo, namely, neuromesodermal progenitors (NMPs), continues to generate daughter cells that contribute to both ectodermal and mesodermal lineages2–4. The mesodermal lineage includes presomitic mesodermal (PSM) cells that further proliferate and differentiate to form somites. This process is called somitogenesis5. Since vertebral bones are derived from somites6, the positional identity of each somite, which is determined based on homeobox (Hox) gene expression, contributes to the morphological characteristics of each vertebra7.
Hox proteins belong to a large family of transcriptional factors containing the homeobox DNA binding domain8. Both human and mouse genomes contain 39 Hox genes divided into four clusters: Hoxa, Hoxb, Hoxc, and Hoxd. Within each cluster, Hox genes are further divided into 13 paralogs, designated Hox1 to Hox137. During somitogenesis, Hox genes are activated in a temporally collinear sequence, where low-numbered Hox genes are activated first to specify future anterior vertebrae, while their high-numbered counterparts are activated later to establish future posterior vertebrae. For example, Hox4 and Hox5 define the cervical vertebrae, Hox6 defines the anterior thoracic containing ribs attached to the sternum, and Hox9 defines the posterior thoracic containing ribs not attached to the sternum9–11. The transition from thoracic to lumbar identity is defined by Hox10 expression, while Hox11 expression corresponds to the sacral and caudal vertebrae12.
The expression of Hox genes is regulated by multiple signaling pathways. During gastrulation, activation of the Wnt signaling in NMPs induces the expression of anterior Hox genes Hox1 to Hox513, which define the cervical and anterior thoracic vertebrae. The Wnt signaling further activates its downstream target gene caudal type homeobox 2 (Cdx2), which encodes a homeobox transcription factor inducing the expression of trunk Hox genes (Hox6 to Hox9)14,15 corresponding to the identities of posterior thoracic vertebrae. Thereafter, growth differentiation factor 11 (Gdf11), a member of the transforming growth factor-beta (TGFβ) family, induces the activation of posterior Hox genes (Hox10 to Hox13) to specify lumbar, sacral, and caudal vertebrae identities16–18. This sequential activation of Hox genes establishes the axial patterning of vertebrae.
The TGFβ signaling has been reported to play important roles in embryogenesis and tissue homeostasis. Both human and mouse genomes encode 33 TGF-β family ligands, including GDF11. The TGF-β ligand is first translated as a propolypeptide, which is processed into a mature polypeptide19. Activation of the TGFβ signaling involves the binding of TGFβ ligands to form a heteromeric complex comprising the mature ligand dimer and two pairs of TGFβ type I and type II receptors20. This ligand-receptor complex triggers the phosphorylation of receptor-related Smads, Smad2 and Smad3. Phosphorylated Smad2/3 forms a heterotrimer with the co-Smad, Smad4, and translocates into the nucleus to regulate the expression of target genes. Several studies have demonstrated the role of Gdf11 in the development of posterior vertebrae. In Gdf11 signaling-deficient mice, abnormal tail development and homeotic transformation are associated with impaired activation of caudal Hox genes16,17,21,22. The cis-regulatory elements within the Hox loci contain Smad-binding motifs that respond to Gdf1123,24. In addition, GDF11 administration efficiently activates the expression of HOX10 and more posterior HOX genes in neurons or mesodermal cells derived from human embryonic stem cells or induced pluripotent stem cells (hiPSCs)25,26. Thus, the GDF11/SMAD2 signaling pathway plays a key role in activating posterior HOX genes to induce the trunk-to-tail HOX code transition and promote caudal development. However, regulators mediating the GDF11 signaling activity remain largely unexplored.
Protogenin (Prtg), also known as Igdcc5, encodes a single-pass transmembrane protein belonging to the immunoglobulin superfamily (IgSF). Prtg is the fifth member of the deleted colorectal carcinoma (DCC) subclass containing four immunoglobulin domains and five fibronectin type III domains in its extracellular portion27. During mouse embryogenesis, Prtg is abundantly expressed in the neural tube and mesodermal cells between embryonic days 8 (E8) and 10 (E10) and down-regulated after E10.528,29. Prtg signaling has been shown to suppress premature neuronal differentiation during the early stages of neural development28. Prtg also participates in tooth germ development and differentiation of inner enamel epithelial cells30. In addition, it modulates apoptosis and migration of rostral cephalic neural crest cells31. Recent studies have identified early rhombic lip PRTG-positive stem cells within a human-specific neurovascular niche as critical initiators and maintainers of group 3 medulloblastoma, further highlighting the significance of PRTG in both normal development and pathological conditions32. Despite its prolific expression in the developing mesoderm, the function of Prtg in somitogenesis is yet to be elucidated.
In this study, we demonstrated the roles of Prtg using mice harboring a conventional knockout allele of Prtg (Prtg−/−)31. Prtg−/− mice exhibited a robust phenotype of anterior homeotic transformation in the thoracic vertebrae accompanied by altered expression patterns of thoracic- and lumbar-associated Hox genes. Based on transcriptional profiles and whole-mount staining, we revealed that the TGFβ signaling is significantly down-regulated in the posterior region of E9.5 Prtg−/− embryos. In addition, we demonstrated that Prtg interacts with Gdf11 and enhances the Gdf11/pSmad2 signaling activity. To verify our findings, we developed a novel hiPSC-derived PSM-like (hiPSC-PSM) model that enabled us to accurately reconstitute the delayed expression of posterior Hox genes observed in Prtg−/− mice. Importantly, this delayed expression of posterior HOX genes could be effectively reversed by GDF11 administration, suggesting that PRTG regulates HOX gene expression via the GDF11/SMAD2 signaling.
Results
Anterior homeotic transformation of thoracic vertebrae in Prtg-deficient mice
Prtg exhibits unique spatiotemporal changes in expression during early embryonic development in mice28,29. At embryonic day 9.5 (E9.5), Prtg is ubiquitously expressed throughout the body axis. Subsequently, its expression diminishes from head to tail from E10.5 to E11.5. To investigate the potential role underlying the dynamic expression pattern of Prtg, we have previously generated conventional Prtg knockout (Prtg−/−) mice and reported the associated defects in palatine and skull development31. In addition, we identified significant defects in the vertebral patterning of Prtg−/− mice (Fig. 1a). In Prtg−/− neonatal mice, the number of sternum-attached ribs increased from 7 to 10, compared with wild-type (WT) control mice. The number of rib-bearing vertebrae increased from 13 to 15, whereas those of cervical and lumbar vertebrae remained constant. Moreover, the posterior thoracic vertebrae (T8–T15) in Prtg−/− mice adopted shapes resembling those of anterior thoracic vertebrae (Fig. 1b), suggesting an anterior transformation phenotype. This anterior transformation was observed in 96% of Prtg−/− mice, but not in WT or Prtg heterozygous mice (Prtg+/−) (Table 1), and was consistent across different genetic backgrounds (Supplementary Table 1). The number and shape of lumbar vertebrae in Prtg−/− mice were comparable to those in the control mice. To determine whether the additional rib-bearing vertebrae resulted from an increase in somite number, we analyzed somite formation in E11 embryos using in situ hybridization of myogenin (Myog), a marker of differentiated somites. The total number of Myog-positive somites in control and Prtg−/− embryos was consistent, ranging from 29 to 33 somites. However, we observed a posterior displacement of two somites in the anterior boundary of hindlimb buds in Prtg−/− embryos, transitioning from the 24th to the 26th Myog-expression domains (Fig. 1c, d and Supplementary Table 2). These findings highlight the critical role of Prtg in the specification of vertebrae along the anterior-posterior axis.
Fig. 1. Anterior homeotic transformation in the vertebrae of Prtg−/− mice.

a Skeletal and cartilaginous tissue staining of control and Prtg−/− neonatal mice. Two additional thoracic vertebrae (top) and three more sternum-attached rib pairs (bottom) were observed in Prtg−/− mice. b Isolated thoracic and lumbar vertebrae from control and Prtg−/− mice. In Prtg−/− mice, T8 to T10 were transformed into a T7-like shape, while T11 to T13 were transformed into a T8-like shape (n = 32). c Whole-mount in situ hybridization of Myog mRNA in E11.0 control and Prtg−/− embryos. The somite at the anterior edge of the hindlimb bud is indicated by a red arrowhead. The scale bar represents 1 mm; fb forelimb bud, h heart, hb hindlimb bud. d Quantification results of the somite number located at the anterior edge of the hindlimb bud, which has been visualized and manually enumerated as shown in (c), in E11.0 Prtg+/+ (n = 2), Prtg+/− (n = 4), and Prtg−/− (n = 6) embryos from 2 litters. e Whole-mount in situ hybridization of Hoxb6 (n = 10), Hoxb9 (n = 10), Hoxc8 (n = 5), Hoxc9 (n = 8), Hoxd9 (n = 5), Hoxa10 (n = 5), Hoxc10 (n = 4), and Hoxd10 (n = 4) in control and Prtg−/− embryos. In situ hybridization of Hoxa10 was performed on E11.0 and that of others on E10.0. Dashed red lines indicate the regions where Prtg−/− embryos displayed differential expression compared with control embryos. Scale bars represent 1 mm. f Summary of the vertebral phenotypes and Hox expression patterns in control and Prtg−/− mice. Dashed boxes delineate the regions of Hox gene expression in the control group. The intensity of the blue color indicates the level of Hox gene expression observed by in situ hybridization. C cervical vertebra (orange), L lumbar vertebra (light blue), S sacral vertebra (purple), T thoracic vertebra (yellow and green). Red-colored vertebrae indicate additional vertebrae in Prtg−/− mice.
Table 1.
Penetrance of vertebral defects in Prtg mutants
| Transformed ribs | Other transformation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Type of transformation | T1 to C7 | T2 to T1 | T8 to T7 | L1 to T13 | L2 to T13 | L3 to T13 | T8–T10 = T7 | T11–T13 = T8 | S2 to L6 |
| Description | Abnormal T1 rib | Abnormal T2 rib | 8th rib at sternum | 14th rib on L1 (T14) | 15th rib on L2 (T15) | 16th rib on L3 (T16) | T8–T10 vertebrae display a T7-like shape | T11–T13 vertebrae display a T8-like shape | Hindlimb on original S3 |
| Wild type (n = 27) | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| Prtg-/- (n = 32) | 16% (3%)# | 0% (0%)# | 96% (90%)# | 96% (96%)# | 96% (71%)# | 19% (9%)# | 96% | 96% | 96% |
C cervical, L lumbar, S sacral, T thoracic.
#The percentage of animals with the bilateral presence of phenotypes is indicated within parentheses.
Hox gene expression is dysregulated in Prtg−/− embryos
Vertebral morphologies and characteristics are determined by the spatial and temporal expression of Hox genes7. It is possible that Prtg regulates the specification of thoracic vertebrae by modulating Hox gene expression. To explore this hypothesis, we examined Hox gene expression at E10.5, shortly after somite generation occurs in the posterior thoracic-to-lumbar region during development (Fig. 1e). In Prtg−/− embryos, the expression levels of Hox genes associated with cervical and thoracic identity were elevated. Specifically, Hoxc8 expression was increased, while Hoxb6 and Hoxb9 displayed both increased and posteriorly extended-expression patterns. Conversely, Prtg−/− embryos showed a decreased expression of lumbar-associated Hox genes; Hoxa10 was down-regulated, and expression patterns of Hoxc9 and Hoxd9 shifted posteriorly. Hoxc10 and Hoxd10 exhibited both decreased and posteriorly shifted expression patterns in Prtg−/− embryos. Importantly, abnormal expressions of Hox genes in Prtg−/− mice corresponded to the thoracic vertebrae transformation phenotype. These results indicate a potential role of Hox genes in mediating the anterior homeotic transformation phenotypes in Prtg−/− mice (Fig. 1f).
Prtg regulates genes associated with the development of the body axis
Abnormal vertebrae in Prtg−/− mice develop from the 19th to the 27th somite (Fig. 1f), which are formed between E9 and E10. To elucidate the molecular basis underlying the anterior homeotic transformation of vertebrae in Prtg−/− embryos, we conducted mRNA sequencing (mRNA-seq) analysis wherein Prtg+/+ (WT), Prtg+/−, and Prtg−/− embryos were compared with one another. The posterior trunk after the forelimb bud (13th somite) of E9.5 embryos was dissected for mRNA-seq to enrich genes expressed in the thoracic somites (Fig. 2a). The mRNA profiles of WT and Prtg+/− embryos were similar, as only six differentially expressed genes (DEGs) were observed, thus explaining the normal vertebral pattern in Prtg+/− mice (Fig. 2b and Supplementary Fig. 2). To be convenient, Prtg+/+ (WT) and Prtg+/− embryos were combined as the control group for subsequent analyzes. Transcriptomic profiling revealed 529 DEGs, including 267 and 262 up-regulated and down-regulated genes, respectively, in Prtg−/− embryos compared with those in the control group (Fig. 2c and Supplementary Data 1). We further used ingenuity pathway analysis (IPA) to analyze DEGs associated with various signaling pathways, diseases, and physiological functions. The DEGs were significantly associated with axial development and skeleton patterning (Fig. 2d), demonstrating that Prtg plays an essential role in the specification of vertebrae along the anterior–posterior body axis.
Fig. 2. Transcriptome analysis of the posterior trunks of E9.5 Prtg−/− embryos.

a Schematic representation of the experimental design for transcriptome analysis. E9.5 embryos were dissected between the 12th and 13th somite, and the posterior trunks were subjected to bulk RNA-seq analysis. b Numbers of differentially expressed genes (DEGs) observed in Prtg+/+ vs Prtg+/−, Prtg+/+ vs Prtg−/−, and Prtg+/− vs Prtg−/− embryos. DEGs were identified using edgeR with FDR < 0.05 as the cutoff. c Heatmap of DEGs in the Prtg+/+, Prtg+/−, and Prtg−/− samples. A total of 529 DEGs were identified between the control (Prtg+/+ and Prtg+/−) and Prtg−/− samples, among which 267 and 262 genes were up-regulated and down-regulated in the Prtg−/− samples, respectively. d Diseases or Functions Annotation generated via Ingenuity Pathway Analysis (IPA) illustrating the significant association of Prtg-regulated genes with body axis development. e Heatmap illustrating the fold change in Hox gene expression observed in Prtg−/− samples relative to control embryos (Prtg+/+ and Prtg+/−), derived from RNA-seq data. f The expression levels of Prtg, Hoxb6, Hoxb7, Hoxb8, Hoxb9, Hoxc9, Hoxd9, Hoxa10, and Hoxc10 in the posterior trunk samples of Prtg+/+, Prtg+/−, and Prtg−/− embryos were quantified using RT-qPCR. Tbp was used as the reference gene. Data are presented as the mean ± SEM. Statistical significance relative to Prtg+/+ is indicated (n = 4 for each bar; *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA).
The anterior homeotic transformation phenotypes of Prtg−/− embryos suggest that Prtg modulates vertebral patterning through Hox genes. Indeed, trunk Hox genes were up-regulated in the Hoxb cluster (Hoxb7, b8, and b9), whereas posterior Hox genes (Hox10–13) in all four clusters were down-regulated (Fig. 2e). To verify our RNA-seq data, we examined Hox gene expression via reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis. Consistently, we noted increased trunk Hox expression (Hoxb6-Hoxb9) and decreased posterior Hox expression (Hoxc9-Hoxc10) in the posterior region of Prtg−/− embryos (Fig. 2f). Taken together, these results suggest that the vertebral transformation in Prtg−/− mice is caused by the down-regulation of posterior Hox genes (paralogous groups 10–13) along with the up-regulation and posterior shift of trunk Hox genes (b6–b9).
The TGFβ signaling is down-regulated in Prtg−/− embryos
To elucidate molecular mechanisms underlying Hox gene regulation by Prtg, DEGs between Prtg−/− and control embryos were subjected to IPA analysis to identify potential upstream regulators. Predicted upstream regulators included signaling pathways related to the TGFβ (TGFB1) and WNT/β-catenin (CTNNB1) signaling pathways, GTPase regulation (HRAS), SOX2 transcription factor, inflammatory responses (NFKBIA and TNF), and apoptosis (TP53 and TP73) (Fig. 3a). Thereafter, we performed gene set enrichment analysis (GSEA) to investigate signaling pathways regulated by Prtg. Among all the pathways examined, the TGFβ pathway exhibited the most significant alteration in Prtg−/− embryos (normalized enrichment score [NES] = 1.82, nominal p-value = 0, false discovery rate [FDR] q-value = 0.014) (Fig. 3b and Supplementary Table 3). Considering the crucial role of the TGFβ signaling in the activation of posterior Hox genes, we hypothesized that Prtg promotes TGFβ signaling activity to regulate Hox gene expression. GSEA results revealed down-regulation of the TGFβ signaling pathway in Prtg−/− embryos, characterized by decreased expression of core components, such as Skil, Ski, Smad7, and Smurf1 (Fig. 3c and Supplementary Table 4). This result was further validated by using RT-qPCR. Expressions of Skil, Smad7, and Smurf1 in the posterior trunks of Prtg−/− embryos were significantly decreased compared with those of the control (Fig. 3d). This result provides evidence for down-regulation of the TGFβ signaling activity in the posterior trunk of Prtg−/− embryos.
Fig. 3. Down-regulation of TGFβ signaling activity in the posterior trunks of E9.5 Prtg−/− embryos.

a Upstream regulator analysis via IPA demonstrated that the DEGs in the Prtg−/− samples were significantly associated with TGFβ1. b GSEA revealed that TGFβ signaling was significantly altered in Prtg−/− embryos. c Heatmap of the expression levels of genes within the TGFβ signaling gene set. d The expression levels of TGFβ signaling target genes in the Prtg+/+, Prtg+/−, and Prtg−/− posterior trunk samples were quantified using qRT-PCR. Tbp was used as the reference gene. Data are presented as the mean ± SEM. Statistical significance relative to Prtg+/+ is indicated (n = 4 for each bar; *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA). e Western blot analysis of the Prtg, phosphorylated Smad2 (pSmad2), total Smad2, and Smad4 levels in Prtg+/+, Prtg+/−, and Prtg−/− posterior trunk samples. Gapdh was used as an internal control. The molecular weight ladders are labeled, and the estimated molecular weight size is indicated by a tilde. f Quantitative results of (e). The level of pSmad2 was significantly decreased in the Prtg−/− samples. Data are presented as the mean ± SEM. Statistical significance relative to Prtg+/+ is indicated (n = 4 for each bar; *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA).
Activation of the TGFβ signaling leads to phosphorylation and activation of downstream signaling transducers, Smad2 and Smad333. Thus, we examined the TGFβ signaling activity by using western blot analysis and quantifying the ratio of phosphorylated Smad2 (pSmad2) to total Smad2. In the posterior trunk of E9.5 Prtg−/− embryos, the pSmad2 ratio was significantly decreased compared with that in the control (Fig. 3e, f). The level of Smad4, a co-Smad involved in both TGFβ and bone morphogenetic protein (BMP) signaling, remained constant. In addition, no obvious change in the expression of those TGFβ ligands, receptors, and Smads was observed in Prtg−/− embryos (Supplementary Fig. 3a and Supplementary Table 5). These results indicate that Prtg deficiency leads to a down-regulation of the TGFβ signaling activity and a reduction in pSmad2 levels.
A decrease of TGFβ signaling activity in the PSM of Prtg−/− embryos
Numerous studies have reported roles of the TGFβ signaling in embryogenesis, including germ layer specification, left–right asymmetry, and axial patterning21,34–36. Since the phenotype of Prtg−/− mice manifest in the vertebral column, we examined whether the TGFβ signaling is down-regulated in the posterior mesoderm of Prtg−/− mice. We performed whole-mount immunostaining of pSmad2 as an indication of TGFβ signaling activity in E9.5 embryos (Fig. 4a and Supplementary Movies 1 and 2). In the control embryos, pSmad2 signaling was detected in the tail region and a portion of the heart. In Prtg−/− embryos, pSmad2 staining was significantly decreased in the tail region, including the tail bud, presomitic mesoderm (PSM), lateral plate mesoderm (LPM), and neural tube, while remaining unchanged in the heart. Conversely, total Smad2/3 staining was uniformly distributed throughout the body and did not display an obvious difference between the control and Prtg−/− embryos (Supplementary Fig. 3b). These results indicate that loss of Prtg leads to down-regulation of TGFβ signaling activity in the tail. Furthermore, we verified the expression of genes downstream of the TGFβ pathway by using whole-mount in situ hybridization. The expression levels of Ski, Skil, and Smurf1 were reduced in the tail bud and PSM of Prtg−/− embryos (Fig. 4c–e and Supplementary Fig. 4b). Interestingly, mesodermal development markers, such as Cyp26a1, T, Tbx6 and Msgn1, were similar between the control and Prtg−/− embryos (Supplementary Fig. 4a), suggesting that down-regulation of TGFβ signaling activity in Prtg−/− embryos do not affect mesodermal development. We also compared Hoxc10 and Hoxd10 expression in E9.5 embryos by using whole-mount staining followed by sectioning (Fig. 4b, f, g and Supplementary Movies 3 and 4). In the paraxial mesoderm, the expression of both Hoxc10 and Hoxd10 was predominantly in the tail bud and PSM (Fig. 4f, g, left panels). In Prtg−/− embryos, Hoxc10 and Hoxd10 expression was posteriorly restricted. In sections of the posterior region, expression levels of Hoxc10 and Hoxd10 were similar in the tail bud region but dramatically decreased in the PSM region of Prtg−/− embryos (Fig. 4f, g, right panels). Collectively, our findings suggest that Prtg regulates TGFβ signaling activity in the developing PSM, which is critical for proper expression of posterior Hox genes.
Fig. 4. Decreased TGFβ signaling activity in the PSM of Prtg−/− embryos.

a Whole-mount immunostaining of Prtg (red), pSmad2 (green), and DAPI (blue) in E9.5 control and Prtg−/− embryos. Digital transverse sections at the indicated levels in the whole-mount embryos are shown at the bottom (n = 8 embryos per group). Scale bars represent 500 µm. b Whole-mount immunostaining of Hoxc10 (green) and DAPI (blue) in E9.5 control and Prtg−/− embryos (n = 4 embryos per group). Digital transverse sections at the indicated levels in the whole-mount embryos are shown at the bottom. Arrowheads indicate the PSM regions. Scale bars represent 500 μm. D dorsal, V ventral. c–e Whole-mount in situ hybridization of Ski (n = 2) (c), Skil (n = 2) (d), and Smurf1 (n = 2) (e) in E9.5 control and Prtg−/− embryos. Dashed lines indicate regions where the expression levels are decreased in Prtg−/− embryos. Scale bars represent 500 µm. f, g Whole-mount in situ hybridization of Hoxc10 (n = 2) (f) and Hoxd10 (n = 2) (g) in E9.5 embryos. Red dashed boxes indicate regions of reduced expression levels of the indicated gene in Prtg−/− embryos. Dashed lines indicate the positions of sections in the right panels. Scale bars represent 500 µm in whole-mount images and 100 µm in sections. fb forelimb bud, h heart, lpm lateral plate mesoderm, nc notochord, np neural plate, nt neural tube, ov otic vesicle, pa1 1st pharyngeal arch, psm presomitic mesoderm, sm somitic mesoderm.
Prtg interacts with Gdf11 and modulates its signaling activity
We observed a reduction in pSmad2 levels in the tails of Prtg−/− embryos (Fig. 4), which corresponds with the expression pattern of Gdf1137. In contrast, the pSmad2 levels in the heart of Prtg−/− embryos remained unchanged, where the TGFβ signaling is mediated by Tgfb1 and Tgfb238. Moreover, it is well established that Gdf11 regulates posterior Hox genes, which were similarly affected in Prtg−/− embryos16,39. Our RNA-seq results also revealed a decrease in Isl1 expression and a slight increase in Lin28 expression (Supplementary Data 1), indicating a down-regulation of Gdf11 signaling activity. Based on these findings, we hypothesized that Prtg regulates Gdf11 signaling activity to modulate pSmad2 levels. To test this hypothesis, we co-expressed Prtg and Gdf11 in P19 embryonic carcinoma cells and accessed TGFβ signaling activity by using a CAGA-driven luciferase reporter. Our results showed that Prtg enhances Gdf11-mediated TGFβ signaling activity, whereas knockdown of Prtg (shPrtg) led to reduced TGFβ signaling activity (Fig. 5a). In contrast, TGFβ signaling activity mediated by Inhba (activin A) or Tgfb1 remained unaffected by Prtg. Furthermore, pSmad2 levels in cells expressing Gdf11 showed a significant correlation with expression levels of Prtg (Fig. 5b, c), indicating that Prtg specifically facilitates Gdf11-mediated TGFβ signaling.
Fig. 5. Prtg interacts with Gdf11 and modulates its signaling activity.

a (CAGA)12-MLP-Luc reporter activities in P19 cells transfected with Gdf11, Inhba, Tgfb1, or a control, along with either Prtg overexpression or knockdown (shPrtg) vectors. Control and Prtg overexpression vectors contain a shRNA with a scrambled shPrtg sequence (shCtrl). Data are presented as the mean ± SEM. Statistical significance is indicated (n = 3 for each bar; *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA). b Protein levels of Prtg, pSmad2, and Smad2&3 were measured by western blot. Gapdh serves as an internal control. c Quantification of pSmad2 levels in (b). Data are presented as the mean ± SEM. Statistical significance is indicated (n = 3 for each bar; *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA). d Co-immunoprecipitation of P19 cells transfected with HA-tagged Prtg and flag-tagged TGFβ ligands. IP immunoprecipitation, WB western blot. e Schematic illustration of Prtg variants: full-length Prtg (Full), Prtg lacking the intracellular domain (ET), and Prtg lacking the extracellular domain (TC). f Co-immunoprecipitation of Prtg Full, Prtg ET, and Prtg TC with Gdf11 in P19 cells. Arrowheads indicate Prtg variants in the immunoblot images. g (CAGA)12-MLP-Luc reporter activities in P19 cells expressing Gdf11 with Prtg Full, Prtg ET, or Prtg TC. Data are presented as the mean ± SEM. Statistical significance is indicated (n = 3 for each bar; *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA). h Levels of pSmad2 in P19 cells expressing Gdf11 with Prtg variants were analyzed by western blot. i Quantification of pSmad2 levels (h). Data are presented as the mean ± SEM. Statistical significance is indicated (n = 3 for each bar; *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA).
Given that the expression of Gdf11 was unchanged in Prtg−/− embryos (Supplementary Fig. 3a and Supplementary Table 5), it is possible that Prtg regulates Gdf11 signaling activity through a post-translational mechanism. Indeed, we demonstrated that Prtg interacts with Gdf11 in P19 cells by using co-immunoprecipitation, while no interaction was observed between Prtg and activin A (Inhba) or Tgfb1 (Fig. 5d). Furthermore, we showed that the extracellular domain of Prtg is required for its interaction with Gdf11 (Fig. 5e, f). Using a TGFβ activity reporter assay, we found that the extracellular domain of Prtg (Prtg ET) alone was capable of facilitating Gdf11-mediated TGFβ signaling activity, whereas truncation of the extracellular domain (Prtg TC) completely abolished this ability (Fig. 5g). The levels of pSmad2 detected by western blot exhibited similar patterns as those observed with the TGFβ activity reporter assay (Fig. 5h, i). These results suggest that the extracellular domain of Prtg, but not the intracellular domain, is crucial for its interaction with and regulation of Gdf11 signaling activity. Taken together, our findings indicate that Prtg promotes Gdf11 signaling activity through protein–protein interaction.
Recapitulation of Prtg−/− phenotypes in the hiPSC-derived PSM model
Our results from studying Prtg−/− mice suggest that the vertebral transformation defect is caused by down-regulation of the Gdf11/Smad2 signaling activity and subsequent reduction of posterior Hox genes. To determine whether Prtg regulates posterior Hox gene expression via the Gdf11/Smad2 signaling, we employed an in vitro model of axial and somite development derived from hiPSCs. Specifically, we adapted protocols from Lippmann, et al. and Matsuda, et al. to direct differentiation of hiPSCs into PSM through stepwise addition of activin A (ACT A), fibroblast growth factor 2 (FGF2), and CHIR99021 (a small-molecule agonist of the WNT/β-catenin pathway) (Fig. 6a)26,40. Over the course of 7 days, we observed a decrease in the expression of a stem cell marker POU5F1/OCT4 based on RT-qPCR and western blot analyses (Fig. 6b, c, h). Expressions of primitive streak (PS) markers TBXT and MIXL1 were increased from day 1, followed by a decrease after day 2. Expressions of PSM markers TBX6 and MSGN1 were detectable from day 1 to day 7. The expression profile of these marker genes indicated a process of paraxial mesodermal differentiation: hiPSCs initially differentiate into PS cells on day 1 and subsequently progress into PSM-like lineage. To evaluate the capability of our in vitro-induced PSM-like cells to further differentiate into somitic mesoderm, we exposed them to a combination of SB431542 (a TGFβ signaling inhibitor), LDN193189 (a BMP signaling inhibitor), PD173074 (an FGF signaling inhibitor), and XAV939 (a Wnt signaling inhibitor) for 2 days (Supplementary Fig. 5a). These cells differentiated into somitic mesoderm based on the expression of MEOX1, a somite marker (Supplementary Fig. 5b–f). Taken together, at each stage of our induction and differentiation protocol, expected markers according to studies in animal models were appropriately expressed41, confirming that our stepwise protocol recapitulated the developmental trajectory of the somitic mesoderm.
Fig. 6. Recapitulation of PRTG knockout (PRTGKO) phenotypes observed in embryos using an in vitro hiPSC-derived PSM model.

a Schematic illustration of the differentiation protocol for generating induced pluripotent stem cell-derived presomitic mesoderm-like (iPSC-PSM) cells. PS, primitive streak; PSM, presomitic mesoderm. b The protein levels of PRTG, pSMAD2, SMAD2/3, HOXC10, and OCT4 at different time points in differentiated iPSC-PSM cells from control clones were measured using a western blot. The molecular weight of nearby ladders is labeled. The estimated molecular weight size is labeled using a tilde. c Quantification results of Fig. 5b. Data are presented as the mean ± SEM (n = 4). d Schema illustrating the generation of PRTGKO (A5, A7, E8, and F4) and non-edited (G2 and N2) hiPSC clones via CRISPR/Cas9 gene editing. e PRTG expression in each hiPSC clone was validated using a western blot on day 4 of the iPSC-PSM model. f The protein levels of pSMAD2, SMAD2/3, and HOXC10 on day 7 of the hiPSC-PSM model were measured using a western blot. g Quantification results of Fig. 5f. Data are presented as the mean ± SEM (n = 4 for control group and n = 8 for PRTGKO group) (**p < 0.01; ***p < 0.001; by student’s t-test). h The expression levels of PRTG, stem cell marker (POU5F1, also known as OCT4), PS markers (TBXT and MIXL1), PSM markers (TBX6 and MSGN1), HOXB1, HOXB6, HOXC9, HOXD9, HOXC10, HOXD10, and HOXC11 from day 0 to day 7 of the differentiated hiPSC-PSM model were measured using RT-qPCR. RPL13A was used as the reference gene. Data are presented as the mean ± SEM (n = 4 for control group and n = 8 for the PRTGKO group) (*p < 0.05; **p < 0.01; ***p < 0.001; by two-way ANOVA).
We further examined PRTG expression in our hiPSC-PSM model. PRTG was initially detected on day 2, reaching its peak on days 3 and 4, followed by a decline on day 5, as observed at levels of both transcript and protein (Fig. 6b, c, h). To validate the axial patterning using our hiPSC-PSM model, we generated PRTG knockout (PRTGKO) hiPSC lines using the CRISPR/Cas9 method (Fig. 6d, e and Supplementary Figs. 6 and 7). All PRTGKO hiPSC clones (A5, A7, E8, and F4) exhibited similar characteristics as WT cells (N2 and G2), displaying normal stem cell morphology, high alkaline phosphatase activity, and the pluripotent cell marker OCT4 (Supplementary Fig. 8). Upon differentiation into PSM, the expression profiles of markers for different stages, including POU5F1, TBXT, TBX6, MSGN1, and MEOX1, were comparable between the control and PRTGKO lines (Fig. 6h). In addition, flow cytometry and immunocytochemistry analyses on day 5 of hiPSC-PSM differentiation revealed that appropriately 85% of cells were TBX6-positive, with no significant difference in TBX6-positive ratio between clones (Supplementary Fig. 9). These results demonstrate that PRTGKO hiPSCs undergo normal differentiation into PSM-like cells.
We assessed HOX gene expression in PRTGKO cells using our iPSC-PSM differentiation protocol. In the hiPSC-PSM model, HOX gene expression followed a collinear pattern similar to that in embryonic development (Fig. 6h and Supplementary Fig. 10). Specifically, HOXB1 (an anterior HOX gene) and HOXB6 (a trunk HOX gene) exhibited increased expression on post-induction days 1 and 2, while the posterior HOX genes HOXC9 and HOXC10 displayed up-regulation after day 3. The more posterior HOX gene HOXC11 remained silent until day 7. In PRTGKO cells, we did observe a delay in posterior HOX gene expression. In WT clones, HOXC9 and HOXC10 were expressed on day 3, whereas in PRTGKO cells, their expression became noticeable only after day 4. Importantly, we observed a significant decrease in the level of pSMAD2 on day 7 of differentiation in PRTGKO hiPSC-PSM cells (Fig. 6f, g). These findings demonstrate that, akin to mouse embryonic development, PRTG deficiency in a human cell model leads to diminished TGFβ signaling activity and delayed and/or reduced expression of posterior HOX genes.
GDF11/SMAD2 signaling acts downstream of PRTG in regulating the expression of posterior HOX genes
To determine whether enhancing TGFβ signaling activity could rescue the expression of posterior HOX genes in PRTGKO hiPSC-PSM cells, we administrated recombinant human GDF11 to the culture medium from day 3 to day 5 to activate the TGFβ signaling in these cells (Fig. 7a). GDF11 supplementation increased the level of pSMAD2 in PRTGKO cells (Fig. 7b, c), suggesting an elevation of TGFβ signaling activity. Notably, the expression of posterior HOX genes, including HOXC10, HOXD10, HOXA11, and HOXC11, was significantly up-regulated in PRTGKO hiPSC-PSM cells upon GDF11 administration on day 5 (Fig. 7d). Additionally, inhibition of SMAD2 phosphorylation by SB431542 suppressed the expression of HOXC10, whereas the induction of SMAD2 phosphorylation by other TGFβ ligands, such as activin A and TGFB1, enhanced its expression (Supplementary Fig. 11). These results suggest that the induction of posterior HOX genes is mediated by the TGFβ/SMAD2 pathway, and that restoring TGFβ/SMAD2 signaling activity with GDF11 is sufficient to rescue the delayed expression of posterior HOX genes in PRTG-deficient iPSC-PSM cells. Together with the phenotypic analysis of Prtg−/− embryos and data from P19 cells, we demonstrate that the GDF11/SMAD2 signaling acts downstream of PRTG in regulating posterior HOX gene expression, therefore modulating vertebral axial patterning.
Fig. 7. GDF11 administration rescues the delayed expression of posterior HOX genes in PRTG-deficient hiPSC-PSM cells.

a Schematic illustration of the experimental design. b Protein levels of pSMAD2, total SMAD2/3, and HOXC10 on day 5 of the PRTGKO hiPSC-PSM model, with or without GDF11 treatment, were measured using western blot. c Quantification results of (b). Data are presented as the mean ± SEM (n = 8 in the control group and n = 16 in the PRTGKO group; *p < 0.05; ***p < 0.001; by one-way ANOVA). d The expression levels of HOXB1, HOXB4, HOXB6, HOXB9, HOXC9, HOXD9, HOXA10, HOXC10, HOXD10, HOXA11, HOXC11, HOXD11, and HOXB13 on day 5 of the PRTGKO and GDF11-treated PRTGKO iPSC-PSM cells were measured using RT-qPCR. RPL13A was used as the reference gene. Expression levels were normalized to the levels of control iPSC-PSM cells (N2 and G2 clones). Data are presented as the mean ± SEM (n = 8 in each group; *p < 0.05; **p < 0.01; ***p < 0.001; by student’s t-test).
Discussion
The spatial and temporal expression of Hox genes during early embryonic development subdivides the vertebral column into distinct regions. Several signaling pathways, including the Wnt and TGFβ, are known to define the anterior, trunk, and posterior/tail Hox gene expression. In addition, other molecules such as miR-196, Nr6a1, and retinoic acid have been shown to influence Hox expression22,42,43. However, many factors involved in the regulation of the Hox gene remain to be uncovered. Here, we highlight the role of Prtg in governing the trunk-to-tail transition of Hox gene expression (Fig. 8). In Prtg-deficient mice, we observed a reduction in the level of pSmad2 in the tail region of E9.5 embryos, leading to the delayed activation of Hox10 and Hox11. Consequently, this delay resulted in the anterior homeotic transformation of the posterior thoracic vertebrae. Furthermore, we showed that Prtg interacts with Gdf11 and facilitates Gdf11-mediated TGFβ signaling activity. To further investigate these effects, we established an in vitro hiPSC-derived PSM-like model and successfully recapitulated the reduction of pSMAD2 and delayed expression of HOX10 and HOX11 in Prtg−/− mouse embryos by using PRTGKO cells. Our results demonstrate that GDF11 addition effectively elevated the level of pSMAD2 and rescued the expression of posterior HOX genes in PRTGKO hiPSC-PSM cells. These findings underscore the critical role of PRTG in the GDF11/SMAD2-mediated activation of posterior HOX genes.
Fig. 8. Delayed transition of thoracic-to-lumbar Hox code in Prtg knockout mice.

A schematic representation illustrating the process of thoracic-to-lumbar Hox code transition in wild-type and Prtg knockout embryos. Arrows above the Hox genes denote the direction of Hox code translation. In wild-type mice, Prtg interacts with Gdf11 to enhance TGFβ signaling, thereby facilitating the transition of the Hox code from thoracic to lumbar identity, which occurs in the tail region of E9.5 embryos. In Prtg knockout mice, decreased TGFβ signaling results in a delayed transition of the Hox code.
The abundant expression of Prtg during early embryogenesis highlights its role in regulating developmental processes. Our study indicates that Prtg regulates Gdf11 signaling to control axial patterning of the somitic mesoderm. Interestingly, Gdf11-mutant mice display severe defects in the developmental process, including down-regulation of T, Tbx6, and Msgn1, which are genes associated with the mesodermal fate21. In contrast, in Prtg−/− embryos, the expression of T, Tbx6, and Msgn1 remains mostly unchanged, and no obvious morphological abnormalities on E9.5 to E11.0 were observed (Supplementary Fig. 4a and Supplementary Data 1). These results suggest that, despite of down-regulation of the Gdf11 signaling in Prtg−/− embryos, it remains sufficient to support most developmental processes. However, the timing of trunk-to-tail Hox code transition, which may be sensitive to Gdf11 signaling activity, appears to be delayed.
The expression of Prtg shows a gradual decline in the tail region overall at E9.5 (Fig. 4a), although it remains highly expressed in the PSM of the tail. Notably, Prtg has also been found to be enriched in early NMPs compared with late NMPs44, indicating that the level of Prtg is crucial during the trunk-to-tail transition. Gdf11 and miR-196, both expressed in the tail region of the E9.5 embryo, synergistically suppress the trunk Hox code in vitro22. Interestingly, embryos deficient for either Gdf11 or miR-196 show increased expression of Prtg21,43, suggesting that Prtg is down-regulated by some posterior regulatory factors. Therefore, Prtg might initially function as an early stimulator of the Gdf11 signaling to promote posterior fate, but is suppressed later following the activation of those posterior regulators.
Our study suggests that Prtg plays a crucial role in promoting the transition of the trunk-to-tail Hox code by facilitating the Gdf11/Smad2 signaling pathway. Although Prtg has not been previously reported to directly control the Gdf11/Smad2 signaling in mammals, several studies have implicated the potential involvement of IgSF DCC subclass members in regulating the TGFβ pathway. For instance, in Drosophila, Plum, a distant homolog of Prtg and Igdcc4 (also known as Nope), promotes the signaling activity of myoglianin, the Drosophila homolog of Mstn and Gdf1145. This regulation has been linked to axon pruning in the developing nervous system and synaptic function in the neuromuscular junction46. Additionally, WAP, follistatin/kazal, immunoglobulin, kunitz, and netrin domain containing 2 (WFIKKN2), a negative regulator of Gdf11, has been reported as a ligand for Igdcc3, Igdcc4, and Prtg in a recent study47,48. These findings raise the possibility that regulation of the Gdf11/Smad2 signaling by Prtg may involve additional molecules and have roles in other developmental processes, providing ways for future study.
We have demonstrated that Prtg regulates Gdf11 through direct interaction, as validated by co-immunoprecipitation experiments showing that Prtg forms a protein complex with Gdf11. This interaction modulates Gdf11-mediated TGFβ signaling, consistent with our findings that Prtg enhances the GDF11/pSMAD2 signaling in both embryos and the hiPSC-PSM cell model. Although Prtg interacts with Gdf11, it does not seem to function as a Gdf11 receptor, as the intracellular domain of Prtg does not have any known kinase activity and is not required for enhancing the Gdf11/pSmad2 signaling (Fig. 5). We propose that Prtg may function as a co-receptor within the Gdf11 signaling complex. Being present on the cell surface, Prtg could facilitate Gdf11 binding to its receptors, in a manner similar to how betaglycan enhances the binding of TGF-β2 to TGF-β receptors, or how Cripto enables Nodal binding to activin receptors and promotes Nodal signaling19. In addition, like other TGFβ ligands, Gdf11 is produced as a pro-peptide and requires proteolytic cleavage for activation19. Our results indicate that Prtg interacts with the Gdf11 pro-peptide, suggesting that Prtg may play a role in the proteolytic processing of Gdf11. Nevertheless, further investigation is needed to determine whether Prtg binds to Gdf11 receptors or participates in the proteolytic processing of Gdf11 to facilitate its signaling.
Another ligand of Prtg, Dnajb11 (also known as ERdj3), has been demonstrated to interact with Prtg to regulate neuronal differentiation28. Our previous study also shows that interaction between Prtg and Radil affects integrin activation, which contributes to the regulation of migration and apoptosis in rostral cephalic neural crest cells31. Moreover, a recent study heightened PRTG expression in gastric cancer and Helicobacter pylori-infected tissues, with PRTG activation linked to cGMP/PKG axis stimulation, promoting the proliferation, metastasis, and chemoresistance of gastric cancer cells49. Despite that Prtg is involved in various signaling pathways, whether these pathways participate in the regulation of Prtg on Hox gene expression or axial vertebral patterning has not been reported. Further investigation is required to determine whether these Prtg-modulated signaling pathways regulate Hox gene expression and axial vertebrate patterning during development.
The advantage of hiPSCs in reconstituting development in vivo has recently been evaluated. When cultured under defined conditions, hiPSCs differentiate into cell types such as NMPs, neurons, and somitic mesodermal cells26,40. Their differentiation potential provides a relatively homogeneous sample for studying molecular mechanisms, thus simplifying the complicated condition in vivo. The hiPSC-derived organoids that form segment somite-like structures have recently raised the potential for modeling somitogenesis50–52. However, in previous studies, these somite-like organoids did not fully activate the collinearity of HOX genes. Moreover, the expression of HOXD10 and more posterior HOX genes was not detected, and the organoid failed to accomplish fully human somitogenesis. The defective development of the caudal body in these organoids closely resembles the phenotype in Gdf11 mutant mice21. Interestingly, in our hiPSC-PSM model, HOXD10 and even more posterior Hox genes were induced. Considering that the activation of posterior HOX genes requires GDF11/SMAD2 signaling activity, the gradual elevation of the pSMAD2 level in our hiPSC-PSM model after day 4 might have facilitated the expression of more posterior HOX genes. Indeed, GDF11 treatment has been used to trigger caudal HOX gene expression in studies that generated hiPSC-derived cells with regional identities26. As the mechanism that activates endogenous GDF11 expression during embryogenesis remains undiscovered, our hiPSC-PSM model potentially offers valuable insights into achieving complete somitogenesis.
In conclusion, our study provides evidence that Prtg interacts with Gdf11 and modulates Gdf11-mediated Smad2 activation in the developing paraxial mesoderm, thereby promoting posterior Hox gene expression and facilitating the trunk-to-tail transition during vertebral patterning. Our in vitro hiPSC-PSM model enables us to further investigate the molecular mechanism by which PRTG regulates GDF11 signaling activity. The roles of PRTG-mediated GDF11 signaling in other developmental processes, as well as the mechanism by which this interaction facilitates GDF11/SMAD2 signaling activity, are key areas of interest for future research.
Methods
Animals and genotyping
The conventional Prtg knockout allele was generated as described previously31. All procedures involving mice were conducted in strict accordance with university guidelines. Ethical approval for animal experiments was obtained from the Institutional Animal Care and Use Committee of National Yang Ming Chiao Tung University (IACUC no. 1110111nr). For the genotyping of postnatal mice, tissue samples (a piece of the tail or foot finger) were lysed by incubating them with 300 µL of 50 mM NaOH at 95 °C for 30 min, followed by neutralization with 50 µL of 1 M Tris (pH 7.9). For embryo genotyping, the yolk sac was lysed in 30 µL of 50 mM NaOH, heated at 95 °C for 30 min, and subsequently neutralized with 5 µL of 1 M Tris (pH 7.9). Extracted DNA was subjected to PCR using Taq DNA polymerase (Geneaid) with the primer sequences listed in Supplementary Table 6.
Skeletal and cartilaginous tissue staining
Neonatal mice were euthanized, skinned, and eviscerated before staining. Cartilage was stained with Alcian Blue 8GX (Sigma-Aldrich) and skeletal tissue with Alizarin Red S (Sigma-Aldrich). Specimens were stored in 20% glycerol for photography. See Supplementary Methods for details.
Digoxigenin-labeled riboprobe synthesis
DNA templates were generated using high-fidelity PCR and subcloned into a pCRII-TOPO Vector (Invitrogen) or directly used for probe synthesis. Digoxigenin-labeled riboprobes were synthesized using T7 RNA polymerase (Roche) or SP6 RNA polymerase (Thermo Fisher Scientific) and purified using a Blood/Cell RNA Mini Kit (Geneaid). See supplementary methods for details. The primers used for probe preparation are listed in Supplementary Table 6.
Whole-mount in situ hybridization
Embryos were fixed in 4% paraformaldehyde (PFA)/phosphate-buffered saline (PBS) with 0.1% Tween 20 (PBST) overnight, dehydrated with methanol, rehydrated with PBST, bleached with 6% H2O2, and permeabilized with proteinase K (Roche). Digoxigenin-labeled riboprobes were used to probe target genes, and alkaline phosphatase-conjugated anti-digoxigenin Fab (Roche) was used to label the riboprobes. Color staining was performed using nitro blue tetrazolium (NBT, Sigma-Aldrich) and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP, Sigma-Aldrich). See Supplementary Methods for details.
Total mRNA-seq and transcriptome analyses
Total RNA from E9.5 posterior tissues was purified using a Tissue Total RNA Mini Kit (Geneaid), and mRNA-seq was performed by GENEWIZ using an Illumina HiSeq sequencer. DEGs were generated using edgeR and analyzed via Qiagen IPA and GSEA. See Supplementary Methods for details.
RT-qPCR
Total RNA was extracted using the Tissue Total RNA Mini Kit (Geneaid) or Blood/Cell RNA Mini Kit (Geneaid), and genomic DNA was removed via on-column DNase digestion. SuperScript IV Reverse Transcriptase (Invitrogen) and anchored oligo dT(20)VN (Integrated DNA Technologies) were used to generate complementary DNA. RT-qPCR was performed using an ABI StepOnePlus instrument. TaqMan assays were performed using the PrimeTime Gene Expression Master Mix (Integrated DNA Technologies), and SYBR Green assays were performed using the PowerTrack SYBR Green Master Mix (Applied Biosystems). See Supplementary Methods for details. The sequences of the primers and probes are listed in Supplementary Table 6.
Western blot
Protein extracted from tissues or cells was cleaned by centrifugation, separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred onto a polyvinylidene fluoride membrane (Millipore). The membrane was blocked with skimmed milk, probed using a primary antibody, and subsequently probed with horseradish peroxidase (HRP)-conjugated secondary antibody. A chemiluminescent HRP substrate was used for protein detection. See Supplementary Methods for details. The antibodies used for western blot are listed in Supplementary Table 7.
Whole-mount immunofluorescent staining
Embryos were collected using ice-cold PBS and fixed with 4% PFA/PBS for 4 h at 4 °C. Following fixation, the embryos were permeabilized using Tris-buffered saline (TBS) containing 1% Triton X-100, blocked with blocking buffer, and incubated with a primary antibody at 4 °C. The embryos were subsequently washed with blocking buffer and incubated with a secondary antibody overnight at 4 °C. Thereafter, they were cleaned with RapiClear (SunJin Lab) and imaged using a confocal microscope. See Supplementary Methods for details. The antibodies used for immunofluorescent staining are listed in Supplementary Table 7.
Vector construction
The mouse open reading frames of Prtg, Gdf11, Inhba, Tgfb1, and Pcsk5 were amplified by PCR from the cDNA of E9.5-E10.5 mouse embryos and subsequently cloned into an expression vector derived from UI4-puro-SIBR. The DNA fragments containing either the Prtg knockdown sequence or its scramble control were cloned into the UI4-puro-SIBR, resulting in shPrtg and shCtrl, respectively. All plasmids were verified by restriction enzyme mapping and Sanger sequencing. Primers and oligonucleotides used for cloning are listed in Supplementary Table 6. The constructed sequences with corresponding restriction enzyme sites are provided in Supplementary Data 2.
P19 cell culture and transfection
P19 embryonic carcinoma cells, purchased from Bioresource Collection and Research Center (BCRC Taiwan), were cultured in α-MEM (Gibco) supplemented with 10% FBS (Gibco) and 1× PSG (Gibco). Cells were passaged every two or three days when reached 80–90% confluence. During the passage, cells were rinsed with PBS, detached using TrypLE Express (Gibco), and reseeded on a new tissue culture dish. Transfection was performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s manual. See Supplementary Methods for details.
TGFβ luciferase reporter assay
TGFβ signaling activities were measured by a CAGA-driven luciferase reporter vector53. Two days after transfection, cells were lysed in a passive lysis buffer (Promega), and luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega). See Supplementary Methods for details.
Co-immunoprecipitation (Co-IP)
Two days after transfection, P19 cells were lysed in an IP lysis buffer. The lysates were clarified by centrifugation and incubated overnight with anti-HA antibody-conjugated resin. The resin was then washed three times with IP lysis buffer, and the protein complexes were extracted by heating in SDS sample buffer. See Supplementary Methods for details.
hiPSC culture
The hiPSC line NTUH-iPSC-02-02 (abbreviated as N2) was purchased from the Bioresource Collection and Research Center of the Food Industry Research and Development Institute, Taiwan. The use of these hiPSC lines followed the Policy Instructions of the Ethics of Human Embryo and Embryonic Stem Cell Research guidelines, in Taiwan. In addition, this study was approved by the institutional review boards of National Yang Ming Chiao Tung University (#YM110194W). Human iPSCs were routinely maintained in StemFlex medium (Gibco) on vitronectin (VTN-N, Gibco) coated dishes at 37 °C in a 5% CO2 incubator and passaged with ethylenediaminetetraacetic acid/Dulbecco’s PBS according to the manufacturer’s instructions. See supplementary methods for details.
Generation of PRTGKO hiPSC lines using the CRISPR/Cas9 technique
DNA oligonucleotides for guide RNA (gRNA) targeting were designed using the TrueGuide CRISPR gRNA Design Tool (Thermo Fisher Scientific). The gRNA was complexed with the Cas9 protein (Invitrogen) and electroporated into hiPSCs. After recovery, individual colonies were expanded to establish isogenic cell lines. PRTG knockout in these cell lines was verified using western blot and confirmed via Sanger sequencing. See Supplementary Methods for details.
Induction of presomitic and somitic mesodermal cells
One day after cells were attached to a vitronectin-coated 24-well tissue culture plate, the medium was replaced with advanced Dulbecco’s Modified Eagle Medium (DMEM)/F-12 (Gibco) containing 1× N-2 supplement (Gibco), 1x GlutaMAX supplement (Gibco), 0.5× non-essential amino acid (NEAA) supplement (Gibco), 50 ng/mL Activin A (PeproTech), 5 µM CHIR99021 (Tocris Bioscience), and 20 ng/mL FGF2 (PeproTech) to induce PS differentiation. For further PSM differentiation, the medium was exchanged with advanced DMEM/F-12 containing 1× N-2 supplement, 1× GlutaMAX, 0.5× NEAA, 5 µM CHIR99021, and 20 ng/mL FGF2. SM differentiation was then achieved by replacing the medium with advanced DMEM/F-12 containing 1× N-2 supplement, 1× GlutaMAX, 0.5× NEAA, 10 µM SB431542 (Tocris Bioscience), and 250 nM LDN193189 (Tocris Bioscience) for 1 day, followed by a final medium change to advanced DMEM/F-12 containing 1× N-2 supplement, 1× GlutaMAX, 0.5× NEAA, 1 µM XAV939 (Tocris Bioscience), and 100 nM PD173074 (Tocris Bioscience) for an additional day. See Supplementary Methods for details.
Immunofluorescence
hiPSCs were seeded on vitronectin-coated coverslips and subjected to iPSC-PSM differentiation. On day 5 of differentiation, cells were fixed with 4% PFA in PBS, permeabilized with methanol, blocked with 5% BSA in TBST, and incubated with primary antibodies overnight. The following day, coverslips were rinsed with TBST and incubated with secondary antibodies and DAPI. After staining, the coverslips were rinsed three times with TBST and mounted on slides with Fluoromount-G for imaging. See Supplementary Methods for details.
Flow cytometry
Day 5 hiPSC-PSM cells were detached with TrypLE and fixed with 4% PFA in PBS. Fixed cells were then permeabilized with methanol, blocked with 10% horse serum in TBST, and stained with primary and secondary antibodies. The stained cells were subsequently analyzed using a Beckman CytoFLEX S flow cytometer. See Supplementary Methods for details.
Statistics and reproducibility
Data are presented as means ± SEM. Statistical significance was assessed using a t-test, one-way ANOVA with Tukey testing, or two-way ANOVA with the original FDR method of Benjamini and Hochberg as a post hoc test. The t-test was conducted by Microsoft Excel. One-way ANOVA and two-way ANOVA were performed using GraphPad Prism software. Statistical significance is indicated in the figures as *p < 0.05, **p < 0.01, and ***p < 0.001. Each data point represents a single measurement or the mean of technical replicates from an embryo or sample collected from independently treated cells. The number of replicates is specified in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
The authors acknowledge Dr. David Turner for providing the vectors US2-puro, UI4-puro-SIBR, UI4-GFP-SIBR, and pRL-US2, as well as the National Genomics Center for Clinical and Biotechnological Applications of the National Core Facility for Biopharmaceuticals (NCFB), National Science and Technology Council (NSTC, Taiwan) for sequencing services. We also thank the Human Disease iPSC Service Consortium funded by the NSTC (112-2740-B-001-004) for iPSC generation and technical support. This work was financially supported by the Brain Research Center of National Yang Ming Chiao Tung University from the featured areas research center program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan, and by NSTC grants to J.-Y.Y. (NSTC 112-2311-B-A49-003) and to Y.-H.W. (NSTC 111-2320-B-A49-027 and NSTC 113-2628-B-A49-015-MY3).
Author contributions
Conceptualization: Y.-S.H. and Y.-H.W. Experimentation and data analysis: Y.-S.H., W.-M.L., Y.-C.W., W.-C.K., and Y.-Y.C. Research supervision: M.-J.F., J.-Y.Y., and Y.-H.W. Manuscript preparation: Y.-S.H., J.-Y.Y., and Y.-H.W. Funding acquisition: M.-J.F., J.-Y.Y., and Y.-H.W. All authors discussed the results, provided feedback, and approved the final manuscript.
Peer review
Peer review information
Communications Biology thanks Arnon Dias Jurberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Edwina McGlinn and Dario Ummarino.
Data availability
RNA-sequencing data are deposited in the NCBI database under accession number GSE256393. Uncropped blot images are provided in Supplementary Fig. 12. Constructed sequences generated in this study are listed in Supplementary Data 2. Source data underlying the graphs and charts in the main figures are included in Supplementary Data 3. All detailed experimental procedures are available in the Supplemental Information.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Wei-Mi Lin, Yu-Chiuan Wang, Wei-Chih Kuo.
Contributor Information
Jenn-Yah Yu, Email: jyyu@nycu.edu.tw.
Yu-Hui Wong, Email: yuhui.wong@nycu.edu.tw.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-024-07342-8.
References
- 1.Mallo, M., Vinagre, T., & Carapuco, M. The road to the vertebral formula. Int J. Dev. Biol.53, 1469–1481 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Martin, B. L. Mesoderm induction and patterning: insights from neuromesodermal progenitors. Semin Cell Dev. Biol.127, 37–45 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tzouanacou, E., Wegener, A., Wymeersch, F. J., Wilson, V. & Nicolas, J. F. Redefining the progression of lineage segregations during mammalian embryogenesis by clonal analysis. Dev. Cell17, 365–376 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Henrique, D., Abranches, E., Verrier, L. & Storey, K. G. Neuromesodermal progenitors and the making of the spinal cord. Development142, 2864–2875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubaud, A. & Pourquie, O. Signalling dynamics in vertebrate segmentation. Nat. Rev. Mol. Cell Biol.15, 709–721 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Scaal, M. Early development of the vertebral column. Semin Cell Dev. Biol.49, 83–91 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Wellik, D. M. Hox patterning of the vertebrate axial skeleton. Dev. Dyn.236, 2454–2463 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Holland, P. W. & Hogan, B. L. Expression of homeo box genes during mouse development: a review. Genes Dev.2, 773–782 (1988). [DOI] [PubMed] [Google Scholar]
- 9.McIntyre, D. C. et al. Hox patterning of the vertebrate rib cage. Development134, 2981–2989 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Horan, G. S., Wu, K., Wolgemuth, D. J. & Behringer, R. R. Homeotic transformation of cervical vertebrae in Hoxa-4 mutant mice. Proc. Natl. Acad. Sci. USA91, 12644–12648 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horan, G. S. et al. Compound mutants for the paralogous hoxa-4, hoxb-4, and hoxd-4 genes show more complete homeotic transformations and a dose-dependent increase in the number of vertebrae transformed. Genes Dev.9, 1667–1677 (1995). [DOI] [PubMed] [Google Scholar]
- 12.Wellik, D. M. & Capecchi, M. R. Hox10 and Hox11 genes are required to globally pattern the mammalian skeleton. Science301, 363–367 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Neijts, R. et al. Polarized regulatory landscape and Wnt responsiveness underlie Hox activation in embryos. Genes Dev.30, 1937–1942 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazzoni, E. O. et al. Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signals. Nat. Neurosci.16, 1191–1198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neijts, R., Amin, S., van Rooijen, C. & Deschamps, J. Cdx is crucial for the timing mechanism driving colinear Hox activation and defines a trunk segment in the Hox cluster topology. Dev. Biol.422, 146–154 (2017). [DOI] [PubMed] [Google Scholar]
- 16.McPherron, A. C., Lawler, A. M. & Lee, S. J. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat. Genet.22, 260–264 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Suh, J. et al. Growth differentiation factor 11 locally controls anterior–posterior patterning of the axial skeleton. J. Cell Physiol.234, 23360–23368 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wymeersch, F. J., Wilson, V., & Tsakiridis, A. Understanding axial progenitor biology in vivo and in vitro. Development148, dev180612 (2021). [DOI] [PubMed]
- 19.Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal.12, eaav5183 (2019). [DOI] [PMC free article] [PubMed]
- 20.Tzavlaki, K. & Moustakas, A. TGF-beta signaling. Biomolecules10, 487 (2020). [DOI] [PMC free article] [PubMed]
- 21.Aires, R. et al. Tail bud progenitor activity relies on a network comprising Gdf11, Lin28, and Hox13 genes. Dev. Cell48, 383–395 e388 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Hauswirth, G. M. et al. Breaking constraint of mammalian axial formulae. Nat. Commun.13, 243 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaunt, S. J., George, M. & Paul, Y. L. Direct activation of a mouse Hoxd11 axial expression enhancer by Gdf11/Smad signalling. Dev. Biol.383, 52–60 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Gaunt, S. J. Gdf11/Smad signalling and Cdx proteins cooperate to activate the Hoxc8 early enhancer in HepG2 cells. Int J. Dev. Biol.61, 427–432 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Mouilleau, V. et al. Dynamic extrinsic pacing of the HOX clock in human axial progenitors controls motor neuron subtype specification. Development148, dev194514 (2021). [DOI] [PMC free article] [PubMed]
- 26.Lippmann, E. S. et al. Deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem Cell Rep.4, 632–644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toyoda, R., Nakamura, H. & Watanabe, Y. Identification of protogenin, a novel immunoglobulin superfamily gene expressed during early chick embryogenesis. Gene Expr. Patterns5, 778–785 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Wong, Y. H. et al. Protogenin defines a transition stage during embryonic neurogenesis and prevents precocious neuronal differentiation. J. Neurosci.30, 4428–4439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vesque, C., Anselme, I., Couve, E., Charnay, P. & Schneider-Maunoury, S. Cloning of vertebrate protogenin (Prtg) and comparative expression analysis during axis elongation. Dev. Dyn.235, 2836–2844 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Takahashi, K. F. et al. Protogenin, a new member of the immunoglobulin superfamily, is implicated in the development of the mouse lower first molar. BMC Dev. Biol.10, 115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang, Y. C. et al. Protogenin prevents premature apoptosis of rostral cephalic neural crest cells by activating the alpha5beta1-integrin. Cell Death Dis.4, e651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Visvanathan, A. et al. Early rhombic lip Protogenin(+ve) stem cells in a human-specific neurovascular niche initiate and maintain group 3 medulloblastoma. Cell187, 4733–4750.e4726 (2024). [DOI] [PubMed] [Google Scholar]
- 33.Nakao, A. et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J.16, 5353–5362 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitman, M. Nodal signaling in early vertebrate embryos: themes and variations. Dev. Cell1, 605–617 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Lee, Y. J. et al. Growth differentiation factor 11 signaling controls retinoic acid activity for axial vertebral development. Dev. Biol.347, 195–203 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh, S. P. et al. Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes Dev.16, 2749–2754 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakashima, M., Toyono, T., Akamine, A. & Joyner, A. Expression of growth/differentiation factor 11, a new member of the BMP/TGFbeta superfamily during mouse embryogenesis. Mech. Dev.80, 185–189 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Azhar, M. et al. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev.14, 391–407 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurberg, A. D., Aires, R., Varela-Lasheras, I., Novoa, A. & Mallo, M. Switching axial progenitors from producing trunk to tail tissues in vertebrate embryos. Dev. Cell25, 451–462 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Matsuda, M. et al. Recapitulating the human segmentation clock with pluripotent stem cells. Nature580, 124–129 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Tani, S., Chung, U. I., Ohba, S. & Hojo, H. Understanding paraxial mesoderm development and sclerotome specification for skeletal repair. Exp. Mol. Med.52, 1166–1177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang, Y. C. et al. Nr6a1 controls Hox expression dynamics and is a master regulator of vertebrate trunk development. Nat. Commun.13, 7766 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong, S. F. et al. Independent regulation of vertebral number and vertebral identity by microRNA-196 paralogs. Proc. Natl. Acad. Sci. USA112, E4884–E4893 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gouti, M. et al. A gene regulatory network balances neural and mesoderm specification during vertebrate trunk development. Dev. Cell41, 243–261.e247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu, X. M. et al. Plum, an immunoglobulin superfamily protein, regulates axon pruning by facilitating TGF-beta signaling. Neuron78, 456–468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sahota, V. K. et al. Plum modulates myoglianin and regulates synaptic function in D. melanogaster. Open Biol.13, 230171 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondas, K., Szlama, G., Nagy, A., Trexler, M. & Patthy, L. Biological functions of the WAP domain-containing multidomain proteins WFIKKN1 and WFIKKN2. Biochem Soc. Trans.39, 1416–1420 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Nickerson, K. R. et al. WFIKKN2 is a bifunctional axon guidance cue that signals through divergent DCC family receptors. Preprint at bioRxiv10.1101/2023.06.15.544950 (2023).
- 49.Xiang, T. et al. The novel ZEB1-upregulated protein PRTG induced by Helicobacter pylori infection promotes gastric carcinogenesis through the cGMP/PKG signaling pathway. Cell Death Dis.12, 150 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanaki-Matsumiya, M. et al. Periodic formation of epithelial somites from human pluripotent stem cells. Nat. Commun.13, 2325 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miao, Y. et al. Reconstruction and deconstruction of human somitogenesis in vitro. Nature614, 500–508 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamanaka, Y. et al. Reconstituting human somitogenesis in vitro. Nature614, 509–520 (2023). [DOI] [PubMed] [Google Scholar]
- 53.Itoh, S. et al. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J.17, 3091–3100 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
RNA-sequencing data are deposited in the NCBI database under accession number GSE256393. Uncropped blot images are provided in Supplementary Fig. 12. Constructed sequences generated in this study are listed in Supplementary Data 2. Source data underlying the graphs and charts in the main figures are included in Supplementary Data 3. All detailed experimental procedures are available in the Supplemental Information.