

Abstract
In this study, we constructed a chromosome-scale reference genome assembly for Lixian plateau loach, Triplophysa lixianensis, by integration of MGI short-read, PacBio HiFi long-read and Hi-C sequencing technologies. A 668-Mb haplotypic genome assembly was obtained for a female T. lixianensis, and 98.91% of the assembled sequences were anchored into 25 chromosomes. This assembly owned a moderate repeat content (35.63%) and an annotation of 23,774 protein-coding genes, among them 94.15% were predicted with functions. The assembled genome of T. lixianensis shared a good syntenic relationship with previously published data of its relative T. dalaica. Taken together, our genome data presented here provide a valuable genetic resource for in-depth evolutionary and functional studies, as well as molecular breeding and conservation of this valuable fish species to elevate its ecological and economical values.
Subject terms: Conservation genomics, Ichthyology, DNA sequencing
Background & Summary
The well-known Qinghai-Tibetan Plateau (QTP) is the largest and the highest plateau on earth, and it has been characterized by an extreme environment with low oxygen concentration, rapid fluctuations in temperature, and strong ultraviolet radiation1. Its conditions are strongly affected by the continuing uplift of the plateau, which is considered as one of the most important driving forces for the biological evolution of various organisms on this plateau2. As a consequence, the QTP has become one of the most important biodiversity centers in the world3. Diverse species endemic to the QTP have undergone significant evolutionary genetic changes, and therefore show high adaptability to the harsh environmental conditions by improving their abilities in hypoxia resistance, cold tolerance, and metabolic capacity1,4,5. Thus far, previous studies related to adaptive evolution at a genome level mainly focused on terrestrial animals, such as Tibetans6–8, Tibetan antelope9, Tibetan ground tit10, Tibetan chicken11, Tibetan frog12, and Tibetan sheep13,14. However, only few studies are involved in aquatic animals (especially for teleost) on the QTP. Hence, more investigations are required to reveal potential adaptive mechanisms for the extreme water environments.
Thus far, three endemic lineages of teleost, including Schizothoracinae (family: Cyprinidae), Sisoridae (superfamily: Sisoroidea; order: Siluriformes) and Triplophysa (family: Nemacheilidae; order: Cypriniformes), were reported to inhabit on the QTP15. Among them, Triplophysa contains more species, but these species have smaller body sizes in comparison with those in the other two genera. Meanwhile, a total of 152 Triplophysa species are recorded in the FishBase, and most of them inhabit in adjacent drainage areas from an elevation of 1000 to > 5,200 m16. However, less information is known about genetic basis for adaptation to such hostile environments due to lack of genomic data, especially shortage of high-quality chromosome‐level genome assemblies. In recent years, advancing whole-genome sequencing technology, especially the third-generation sequencing techniques, has presented novel opportunities to explore more genetic bases of environmental adaptations17. Thus far, several chromosome‐level genome assemblies are reported for Triplophysa species, such as T. siluroides18, T. tibetana15, T. bleekeri19, T. dalaica20, and T. yarkandensis21. These genomic resources are valuable for phylogenetic studies of the Triplophysa genus and genomics comparisons to reveal potential mechanisms for residence in such a complex environment. However, environmental adaptations of Triplophysa species to high altitudes are not fully understood due to the severe altitudes and conditions.
Triplophysa lixianensis, a Nemacheiline loach species (Fig. 1a), was identified and named by our research team in 200822. This interesting species is primarily distributed in the upper tributaries of Minjiang River in Sichuan province of China. It can be separated from all other Triplophysa species due to having a unique series of characters, such as posterior chamber of gas bladder being greatly reduced or absent, caudal peduncle columnar with a roughly round cross-section at its beginning, and anterior edge of lower jaw completely exposed or uncovered by the lower lip22. Interestingly, we observed secondary sexual characters in mature male Triplophysa fishes23, although the genetic basis of this phenomenon is still unclear. In our current study, we firstly constructed a chromosome‐level genome assembly for T. lixianensis, and its phylogenetic position was subsequently determined. Our genomics data presented here will be beneficial for in-depth investigations on potential adaptive mechanisms of Triplophysa fishes to high altitudes, and also be useful for exploring the genetic basis for interesting physiological phenomena such as the secondary sexual characters in these valuable fish species.
Fig. 1.
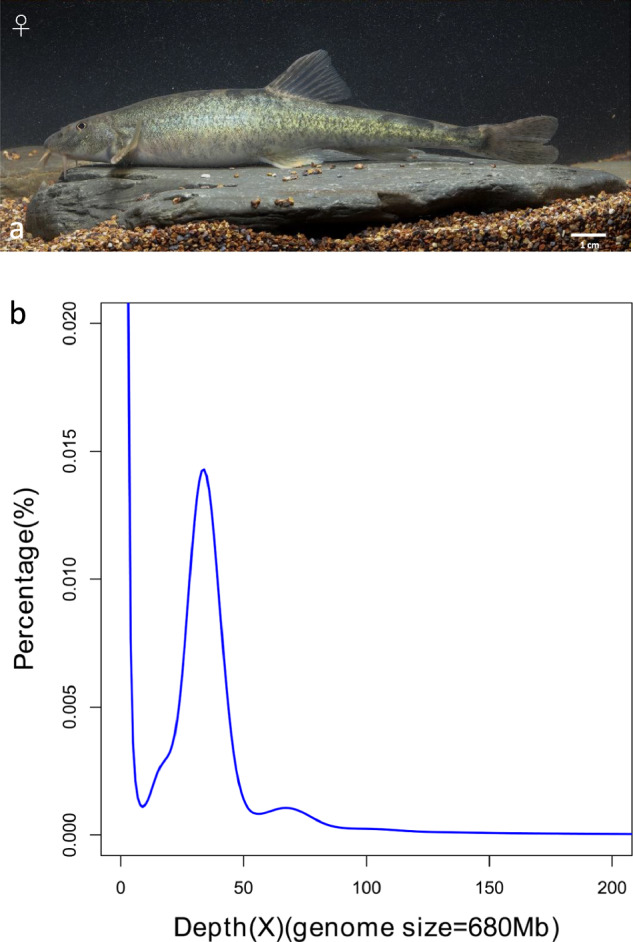
Lixian plateau loach and its Illumina sequencing for a genome survey. (a) Photo of the sequenced loach. (b) K-mer (17-mer) distribution curve for estimation of the genome size of T. lixianensis.
Methods
Sample collection
An adult female T. lixianensis (body length: 14.45 cm, body weight: 17.21 g; Fig. 1a) was collected from Zagunao River (102.9626° E, 31.5059° N), a tributary of Minjiang River of the upper Yangtze River drainage in Sichuan Province of China. Only this female sample was used for genome sequencing since we could not catch any male individual.
Muscle tissues were collected for whole-genome sequencing, including MGI short-read, PacBio HiFi long-read, and Hi-C sequencing (Table 1). Meanwhile, muscle, eye, kidney, intestine, heart, ovary, brain, skin, spleen, stomach, and liver (a total of eleven tissues from the same fish) were collected for transcriptome sequencing (Table 1). These samples were cut into small pieces, immediately frozen in liquid nitrogen, and then stored at −80 °C before use.
Table 1.
Sequencing data of the T. lixianensis genome.
| Sex | Library type | Raw data (Gb) | Clean data (Gb) | Read N50/ length (bp) | Mapping ratio | Coverage (×) | |
|---|---|---|---|---|---|---|---|
| Female | MGI | 45.44 | 42.29 | 150 | 63.3 | ||
| PacBio HiFi | — | 32.50 | 15,400* | 47.79 | |||
| Hi-C | 67 | 66.8 | 150 | 98.52 | |||
| RNA | Brain | 8.04 | 7.39 | 150 | 86.38% | ||
| Eye | 9.09 | 8.36 | 150 | 86.95% | |||
| Muscle | 10.98 | 10.09 | 150 | 81.01% | |||
| Liver | 10.96 | 10.09 | 150 | 73.58% | |||
| Spleen | 11.45 | 10.52 | 150 | 81.35% | |||
| Skin | 7.00 | 6.42 | 150 | 78.07% | |||
| Ovary | 8.57 | 7.88 | 150 | 79.82% | |||
| Intestine | 8.09 | 7.42 | 150 | 80.36% | |||
| Kidney | 7.76 | 7.10 | 150 | 81.27% | |||
| Stomach | 9.52 | 8.74 | 150 | 80.42% | |||
| Heart | 9.35 | 8.60 | 150 | 73.60% | |||
*For the PacBio HiFi sequencing, this number is for read N50; for others, it is for read length.
DNA extraction and whole-genome sequencing
Genomic DNA (gDNA) was extracted from pooled muscle samples using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Quality and quantity of the isolated DNA were evaluated via agarose gel electrophoresis and an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA).
The gDNA was randomly fragmented to construct a library with an insert size of 350 bp by using MGIEasy universal DNA library prep set (MGI, Shenzhen, China) for subsequent sequencing on a DNBSEQ T7 platform (MGI). A total of 45.44 Gb of paired-end raw reads (150 bp in length) were generated, and then they were filtered by fastp v0.12.624 (parameter: -n 0 -f 5 -F 5 -t 5 -T 5) to remove low-quality reads and adaptor sequences. Finally, approximately 42.29 Gb of clean reads were obtained (Table 1) for estimation of the genome size and further sequence error correction.
For the PacBio HiFi long-read sequencing, about 10 μg of gDNA was used to construct long-read libraries by using a SMRTbell Express Template Prep Kit 2.0 based on PacBio’s standard protocol (Pacific Biosciences, Menlo Park, CA, USA), which were then sequenced on a PacBio Sequel II System. A total of 32.50-Gb HiFi reads with a N50 value of 15,400 bp were obtained (Table 1) using the CCS v6.0.025 (Circular Consensus Sequencing) software with an optimized parameter (-min-passes 3).
For the high-throughput chromosome conformation capture (Hi-C) sequencing, a Hi-C library was constructed by using a GrandOmics Hi-C kit (the applied restriction enzyme is DpnII) according to the manufacturer’s protocol (GrandOmics, Wuhan, China). The Hi-C library was then sequenced on a DNBSEQ T7 platform (MGI) with a paired-end module (PE150). In total, 67 Gb of raw reads were generated. Subsequently, fastp v0.12.624 was applied to filter adaptor sequences and low-quality reads. Finally, high-quality clean data (66.8 Gb; Table 1) were retained for construction of chromosomes.
RNA extraction and transcriptome sequencing (RNA-seq)
Total RNA was extracted from the elven tissues (Table 1) separately by using a standard Trizol protocol (Invitrogen, Frederick, MD, USA), and then purified using a Qiagen RNeasy mini kit (Qiagen, Germantown, MD, USA). RNA concentration and integrity were measured with a NanoDrop 8000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies), respectively. Only those RNA samples with OD260/280 ≥ 1.8 and RNA integrity ≥ 7.0 were selected for transcriptome sequencing. Illumina cDNA libraries were constructed according to the manufacture’s guideline, which were then sequenced on a HiSeq X Ten platform (Illumina, San Diego, CA, USA). Around 9 Gb of transcriptome sequencing data for each tissue (see more details in Table 1) were generated for assistance to gene structure prediction.
Genome-size estimation
To estimate the genome size for T. lixianensis, a putative k-mer analysis was performed by using MGI short clean reads. Through the k-mer counting (KMC) program and genome character estimator (GCE) v1.0.2 software26, a 17-mer frequency was calculated. The genome size was estimated based on the following formula: G = K_num/K_depth, where G is the genome size, K_depth represents the k-mer depth, and K_num stands for the total number of 17-mers. Therefore, the genome size of the female T. lixianensis was estimated to be about 680 Mb (Fig. 1b), which is similar to the reported genome length (692 Mb) of T. rosa, a closely related plateau fish in the same Triplophysa genus27.
De novo genome assembly and chromosome construction
For the initial genome assembly, 32.5 Gb of HiFi long reads (Table 1) were de novo assembled into contigs through Hifiasm v0.16.028 with default parameters. The primary genome assembly was 668 Mb in length, which is consistent with the estimated genome sizes (Fig. 1b). Nextpolish v1.2.429 was employed to correct the genome assembly using the MGI clean data with default parameters, and then a polished genome assembly was obtained.
Based on this polished genome, the Hi-C sequencing reads were employed to construct haplotypic chromosomes. First, the Hi-C clean reads were mapped onto the assembled contigs using bowtie2 v2.2.530 (–very-sensitive -L 20–score-min L, −0.6, −0.2–end-to-end). Subsequently, the HiC-Pro v2.8.131 pipeline was applied to detect valid ligation products, and only those valid contact paired reads were retained for further analysis. With these valid reads, the assembled contigs were oriented, ordered, and clustered onto chromosomes using LACHESIS32 with optimized parameters (CLUSTER_MIN_RE_SITES = 100, CLUSTER NONINFORMATIVE RATIO = 1.4, CLUSTER_MAX_LINK_DENSITY = 2.5, ORDER MIN N RES IN SHREDS = 60, ORDER MIN N RES IN TRUNK = 60). Juicebox v1.11.0833 was applied to visualize before manually adjusting candidate assemblies. We hence obtained the final genome assembly with a total size of 668.27 Mb, of which 98.91% are anchored into 25 chromosomes (Fig. 2). The scaffold and contig N50 values of this chromosome-scale genome assembly are up to 25.35 Mb and 12.41 Mb, respectively.
Fig. 2.
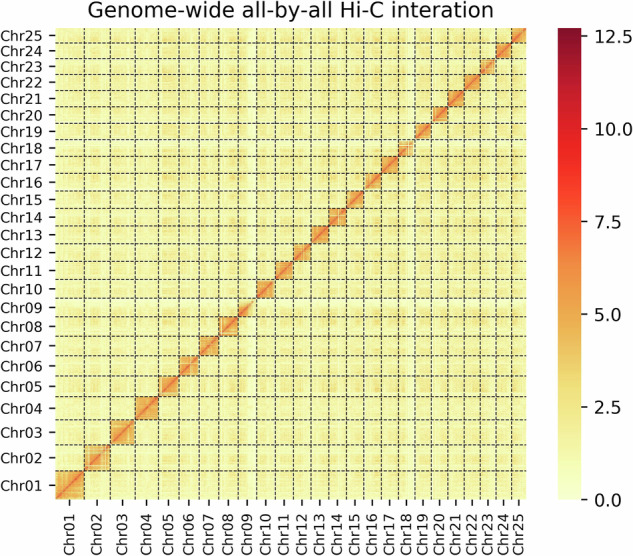
Genome-wide analysis of chromatin interactions at a 500-kb resolution in the assembled T. lixianensis genome. Color blocks represent the interactions, with various strength from yellow (low) to red (high).
We then employed two routine methods to evaluate genome completeness. First, the conserved genes (248 genes) existing in representative eukaryotes were selected to construct a core gene library for CEGMA34 evaluation. Our results revealed that the majority of core eukaryotic genes (97.98%) were successfully identified. Second, BUSCO v5.030 (Benchmarking Universal Single-Copy Orthologs) was employed to search against the actinopterygii_odb10 database. It was also validated that the assembled genome contained 96.6% [S:95.3%, D:1.3%, F:0.8%, M:2.6%] of the total of 3,640 conserved genes. Both good results prove that the final genome assembly has considerable integrity, continuity, and accuracy as a high-quality reference.
Annotation of repeat elements
For prediction of repetitive elements (REs), we first annotated tandem repeats by using GMATA35 and Tandem Repeats Finder (TRF)36, where GMATA identified the simple repeats sequences (SSRs) and TRF recognized all tandem repeat elements in the whole genome. Transposable elements (TEs) in the genome were predicted by combination of homology-based and de novo methods. For the homology approach, TEs were identified using RepeatMasker v4.0.6 and RepeatProteinMask v4.0.637. For the de novo approach, RepeatModeler v1.0.838 and LTR_FINDER v1.0.639 were employed to generate a de novo repeat library, and then RepeatMasker was applied to annotate REs against this repeat library.
A total of 238.1 Mb (35.63%) repetitive sequences were annotated in the assembled genome (Table 2), in which DNA transposons made up the greatest proportion (15.12%), followed by long interspersed nuclear elements (LINE; 6.21%) and long terminal repeats (LTR; 5.68%). Compared with the genome of T. dalaica (REs account for 35.01%), T. lixianensis displayed a similar RE percentage. Subsequently, the repetitive regions of the assembled genome of T. lixianensis were masked prior to subsequent gene structure prediction.
Table 2.
Statistics of the assembled T. lixianensis genome.
| Category | Data |
|---|---|
| Genome survey (Mb) | 680 |
| Genome length (bp) | 668,279,432 |
| Longest scaffold (bp) | 40,579,028 |
| Number of scaffolds | 41 |
| Contig N50 (bp) | 12419652 |
| Scaffold N50 (bp) | 25358741 |
| GC content | 39.0% |
| Short reads mapping rate | 99.37% |
| CEGMA | 97.98% |
| BUSCO | 96.6% |
| Anchor ratio | 98.91% |
| Number of chromosomes | 25 |
| Chromosome length (bp) | 660,999,891 |
| Repetitive sequence | 35.63% |
Gene annotation and functional assignment
Prediction of protein-coding genes was conducted with three methods, including homology, de novo and tanscriptome-based annotations. First of all, AUGUSTUS v3.2.140 was employed to fulfil the ab inito gene predictions. Subsequently, GeMoMa v1.6.441 was applied for the homology-based prediction. We aligned homology proteins from seven representative fish species, including fathead minnow (Pimephales promelas), golden-line barbels (Sinocyclocheilus rhinocerous and S. anshuiensis), high-plateau loach (Triplophysa bleekeri), largescale shoveljaw fish (Onychostoma macrolepis), Rohu (Labeo rohita), and tiger barb (Puntigrus tetrazona) (downloaded from the NCBI database). Finally, the transcriptome (RNA-seq) data from eleven tissues of T. lixianensis were assembled into unigenes using Trinity v2.5.142 with mapping ratio ranging from 73.58% to 86.95% (Table 1), and then gene structures were predicted using PASA v2.3.343. Finally, gene sets were integrated by the Evidence Modeler (EVM) pipeline v1.043.
A total of 23,774 protein-coding genes were annotated in the female T. lixianensis genome. Moreover, BLASTP was conducted to annotate gene functions by comparing the predicted protein sequences with five public databases, including SwissProt, Gene Ontology (GO), Non-Redundant Protein Sequence (NR), Kyoto Encyclopedia of Genes and Genomes (KEGG) and EuKaryotic Orthologous Groups (KOG), with an E-value cutoff of <1e−5. In total, 22,383 (94.15%) genes were predicted with successful hit(s) in at least one database. The BUSCO completeness value was calculated to be 93.8% of the total predicted protein-coding genes (Table 3).
Table 3.
Functional annotation and BUSCO evaluation of the total protein-coding genes.
| Category | Female | |
|---|---|---|
| Number | Percentage (%) | |
| Total | 23,774 | 100 |
| NR | 22,311 | 93.85 |
| Swissprot | 19,896 | 83.69 |
| KEGG | 15,657 | 65.86 |
| GO | 15,117 | 63.59 |
| KOG | 14,612 | 61.46 |
| Overall | 22,383 | 94.15 |
| BUSCO | 3,415 | 93.8 |
Overall represents the number of annotated genes with at least one hit from the five public databases.
Based on the annotated protein-coding sequences and gene structures, JCVI v19021344 was applied to perform a chromosomal collinearity analysis between T. lixianensis and T. dalaica. It seems that both genomes have a good collinearity relationship, and their chromosomes present a good match with one-to-one correspondence (Fig. 3), indicating that our assembled genome of T. lixianensis is truly complete and high-quality.
Fig. 3.

Genome synteny between T. lixianensis and its relative T. dalaica20.
Data Records
Files of the MGI, PacBio, Hi-C and transcriptome sequencing, along with serially assembled genomes for the female Lixian plateau loach were deposited at NCBI under the accession number PRJNA1119268. Raw reads are available in the Sequence Reads Archive (SRA) with the accession number SRP51272645. The final genome assembly was deposited at NCBI GenBank with the accession number GCA_041430785.146. Annotation files of the assembled T. lixianensis genome are available in Figshare47.
Technical Validation
The quality scores across all bases of the MGI raw sequencing data were inspected using FastQC v0.11.9 (https://github.com/s-andrews/FastQC). We conducted a 17-mer distribution analysis to estimate the target genome size based on the MGI clean data. The integrity of assembled genome and protein-coding genes was evaluated using BUSCO with the actinopterygii_odb10 database as the reference. More than 96% of complete BUSCOs were identified in assembled genome. The comparisons of 25 chromosomes between T. lixianensis and T. dalaica proved high conservation of synteny between this pair of relatives, indicating that our genome assembly and annotation for T. lixianensis are indeed complete and of high quality.
Acknowledgements
This work was financially supported by the Natural Science Fund of Sichuan Province of China (no. 2023NSFSC1221), the Project of Sichuan Provincial Department of Science and Technology (no. ZYZFSC22004), the Research Fund from Key Laboratory of Sichuan Province for Fishes Conservation and Utilization in the Upper Reaches of Yangtze River (no. NJTCSC23-3). Meanwhile, we thank Shengtao Guo, Ji Liang, Wenchu Yan, and Hanxi Chen for their helps in sample collection and species identification.
Author contributions
Z.S., Z.W. and Q.S. conceived and designed the study. Z.W. and C.H. collected the samples. X.Z., Z.W. and C.H. performed data analysis. Z.W. and C.H. conducted experiments for species identification. X.Z., Z.W. and C.H. wrote the manuscript. Z.S., Q.S. and Z.W. revised the manuscript. All authors read and approved the final manuscript for publication.
Code availability
The versions and parameters of bioinformatics tools applied in this study have been described in the Methods section. If no parameter is provided, the default is set. No custom code was used.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chunlin He, Xinhui Zhang.
Contributor Information
Qiong Shi, Email: shiqiong@szu.edu.cn, Email: shiqiong@genomics.cn.
Zhaobin Song, Email: zbsong@scu.edu.cn.
References
- 1.Qiao, Q. et al. The genome and transcriptome of Trichormus sp. NMC-1: insights into adaptation to extreme environments on the Qinghai-Tibet Plateau. Sci Rep.6, 29404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao, Z. & Li, S. Extinction vs. rapid radiation: The Juxtaposed evolutionary histories of Coelotine spiders support the Eocene-Oligocene orogenesis of the Tibetan plateau. Syst Biol.66, 988–1006 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Myers, N., Mittermeier, R. A., Mittermeier, C. G., da Fonseca, G. A. & Kent, J. Biodiversity hotspots for conservation priorities. Nature.403, 853–858 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Beall, C. M. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc Natl Acad Sci USA.104(Suppl 1), 8655–8660 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beall, C. Adaptation to high altitude: Phenotypes and genotypes. Annual Review of Anthropology.43, 251–272 (2014). [Google Scholar]
- 6.Simonson, T. S., McClain, D. A., Jorde, L. B. & Prchal, J. T. Genetic determinants of Tibetan high-altitude adaptation. Hum Genet.131, 527–533 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Hu, H. et al. Evolutionary history of Tibetans inferred from whole-genome sequencing. PLoS Genet.13, e1006675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He, Y. et al. De novo assembly of a Tibetan genome and identification of novel structural variants associated with high-altitude adaptation. Natl Sci Rev.7, 391–402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge, R. L. et al. Draft genome sequence of the Tibetan antelope. Nat Commun.4, 1858 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu, Y. et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nat Commun.4, 2071 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Wang, M. S. et al. Genomic analyses reveal potential independent adaptation to high altitude in Tibetan chickens. Mol Biol Evol.32, 1880–1889 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Sun, Y. B. et al. Whole-genome sequence of the Tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes. Proc Natl Acad Sci USA.112, E1257–1262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei, C. et al. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci Rep.6, 26770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu, X. J. et al. The Genome landscape of Tibetan sheep reveals adaptive introgression from Argali and the history of early human settlements on the Qinghai-Tibetan Plateau. Mol Biol Evol.36, 283–303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang, X. et al. Chromosome-level genome assembly of Triplophysa tibetana, a fish adapted to the harsh high-altitude environment of the Tibetan Plateau. Mol Ecol Resour.19, 1027–1036 (2019). [DOI] [PubMed] [Google Scholar]
- 16.He, C., Song, Z. & Zhang, E. Triplophysa fishes in China and the status of its taxonomic studies. Sichuan Journal of Zoology.30, 150–155 (2011). [Google Scholar]
- 17.Hu, T., Chitnis, N., Monos, D. & Dinh, A. Next-generation sequencing technologies: An overview. Hum Immunol.82, 801–811 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Yang, L. et al. A chromosome-scale reference assembly of a Tibetan loach, Triplophysa siluroides. Front Genet.10, 991 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan, D. et al. Chromosomal genome of Triplophysa bleekeri provides insights into its evolution and environmental adaptation. Gigascience.9, giaa132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou, C. et al. The Chromosome-level genome of Triplophysa dalaica (Cypriniformes: Cobitidae) provides insights into its survival in extremely alkaline environment. Genome Biol Evol.13, evab153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.She, J., Chen, S., Liu, X. & Huo, B. Chromosome-level assembly of Triplophysa yarkandensis genome based on the single molecule real-time sequencing. Sci Data.11, 39 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He, C., Song, Z. & Zhang, E. Triplophysa lixianensis, a new nemacheiline loach species (Pisces: Balitoridae) from the upper Yangtze River drainage in Sichuan Province, South China. Zootaxa.1739, 41–52 (2008). 41–52. [Google Scholar]
- 23.Hou, F. X., He, C. L., Zhang, X. F. & Song, Z. B. Secondary sexual characters in males of Triplophysa fishes. Acta Zootaxonomica Sinica.35, 101–107 (2010). [Google Scholar]
- 24.Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics.34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics.13, 278–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv:1308.2012 (2013).
- 27.Zhao, Q., Shao, F., Li, Y., Yi, S. V. & Peng, Z. Novel genome sequence of Chinese cavefish (Triplophysa rosa) reveals pervasive relaxation of natural selection in cavefish genomes. Mol Ecol.31, 5831–5845 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods.18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics.36, 2253–2255 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods.9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol.16, 259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol.31, 1119–1125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst.3, 99–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics.23, 1061–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Wang, X. & Wang, L. GMATA: An integrated software package for genome-scale SSR mining, marker development and viewing. Front Plant Sci.7, 1350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res.27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics.Chapter 4, 4 10 11–14 10 14 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA.117, 9451–9457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res.35, W265–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res.34, W435–439 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol.1962, 161–177 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc.8, 1494–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol.9, R7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang, H. et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics.15, 312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.NCBI Sequence Read Archive.https://identifiers.org/ncbi/insdc.sra:SRP512726 (2024).
- 46.NCBI Assembly.https://identifiers.org/ncbi/insdc.gca:GCA_041430785.1 (2024).
- 47.Zhang, X. Annotation file of T. lixianensis. Figshare.10.6084/m9.figshare.26326063.v1 (2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- NCBI Sequence Read Archive.https://identifiers.org/ncbi/insdc.sra:SRP512726 (2024).
- NCBI Assembly.https://identifiers.org/ncbi/insdc.gca:GCA_041430785.1 (2024).
- Zhang, X. Annotation file of T. lixianensis. Figshare.10.6084/m9.figshare.26326063.v1 (2024).
Data Availability Statement
The versions and parameters of bioinformatics tools applied in this study have been described in the Methods section. If no parameter is provided, the default is set. No custom code was used.