

Abstract
The kidney epithelium, with its intricate arrangement of highly specialized cell types, constitutes the organ’s functional core. Loss of kidney epithelium is linked to loss of functional nephrons and a subsequent decline in kidney function. In allogenic kidney transplantation, epithelial injury signatures observed during post-transplant surveillance are strong predictors of adverse kidney allograft outcomes. However, epithelial injury is currently neither monitored clinically nor addressed therapeutically after kidney transplantation. Several factors can contribute to allograft epithelium injury, including rejection, drug toxicity, recurrent infections or postrenal obstruction. Injury mechanisms partially overlap with those in the native kidney during acute kidney injury and chronic kidney disease. Recent studies using advanced transcriptomic analyses of single cells from kidney or urine have identified a role of kidney injury-induced epithelial cell states in exacerbating and sustaining damage after acute kidney injury and in chronic kidney disease. These epithelial cell states and their associated expression signatures are also observed in transplanted kidney allografts. In this review article, we discuss the potential implications of identifying and characterizing transcriptomic epithelial cell states in kidney allografts. We also discuss how injury-associated epithelial cell populations in kidney allografts might be used as diagnostic tools, inform clinical decisions, and constitute molecular targets for therapy.
Introduction
Kidney transplantation stands as the optimal treatment for end-stage kidney disease1. However, the longevity of the transplanted kidney and the organ availability fall short of demand2. Enhancements in diagnostic procedures, clinical decision-making, and discovery of new therapeutic targets could potentially boost the survival rates of kidney transplants and, in turn, improve patient outcomes. Furthermore, the molecular insights gleaned from post-transplant kidneys, as well as in the context of acute kidney injury and chronic kidney disease, could offer reciprocal benefits.
Kidney epithelia are considered the most important target of medical complications of kidney transplantation3–5. Although initial histological and molecular findings are often dominated by immune cell infiltration and signaling, kidney transplant outcomes appear to be mostly determined by the parenchymal injury signature4. Recent studies have observed cell states in kidney transplants in various post-transplant complication settings, which bear similarities to those seen in acute kidney injury or chronic kidney disease6–11. Nonetheless, epithelial damage is neither currently monitored nor directly targeted in treating post-transplant complications.
The objective of this review is to delineate the function and potential of kidney tubular epithelia, their cell states, and related gene expression profiles as tools for therapeutic monitoring and diagnosis. In addition, we explore the potential utility of epithelial transcriptomics in clinical decision-making highlighting innovative therapeutic targets following kidney transplantation. We will first review the emerging knowledge of epithelial gene expression and injury-associated cell states of the native kidney during acute kidney injury (AKI) and chronic kidney disease (CKD). We will then move into the setting of kidney transplantation, where general mechanisms of AKI and CKD concur with injury mechanisms specific to the transplant recipient. We will explore in detail the existing literature on epithelial cell phenotypes after kidney transplantation and discuss the potential clinical applications of diagnostic epithelial cell phenotyping and of targeting specific epithelial cell phenotypes therapeutically.
Cellular epithelial cell states observed in mouse models of AKI and in progression to CKD
AKI refers to severe and diverse clinical conditions characterized by an increase in serum creatinine or a decrease in urinary output12. AKI can manifest in various clinical scenarios, such as sepsis, post-surgery, autoimmune diseases, or drug-induced situations13–15. Even reversible and milder forms of AKI have been linked to a higher long-term mortality rate16–18. The occurrence of AKI significantly heightens the likelihood of subsequent development of CKD, a risk influenced by various molecular processes, some of which will be detailed in the following sections19,20.
The histology of AKI can vary with the underlying pathophysiology but typically includes signs of tubular injury including cell swelling, cytoplasmic vacuolization, tubular dilatation and necrosis, tubular cast formation and infiltration of immune cells into the kidney21,22. AKI induces strong gene expression responses in the kidney following specific patterns over time23,24. Numerous past studies using bulk transcriptomic approaches have revealed orchestrated AKI-associated gene expression responses23–30.
In simplified terms, following AKI, surviving tubular epithelial cells undergo a process of dedifferentiation, replicate to replace adjacent lost tubular epithelial cells and re-differentiate, restoring the healthy kidney epithelium31–34 (successful or adaptive repair). This process might involve subsets of cells bearing stem or progenitor cell characteristics35–39. If such repair processes fail, cells might remain in an aberrant, dysfunctional state, potentially contributing to a decline in kidney function and the development of fibrosis32–34. These processes can maintain ongoing injury and inflammation in the kidney and contribute to chronic kidney disease progression (Fig. 1).
Fig. 1. Induction of cell states in the kidney tubule following AKI.
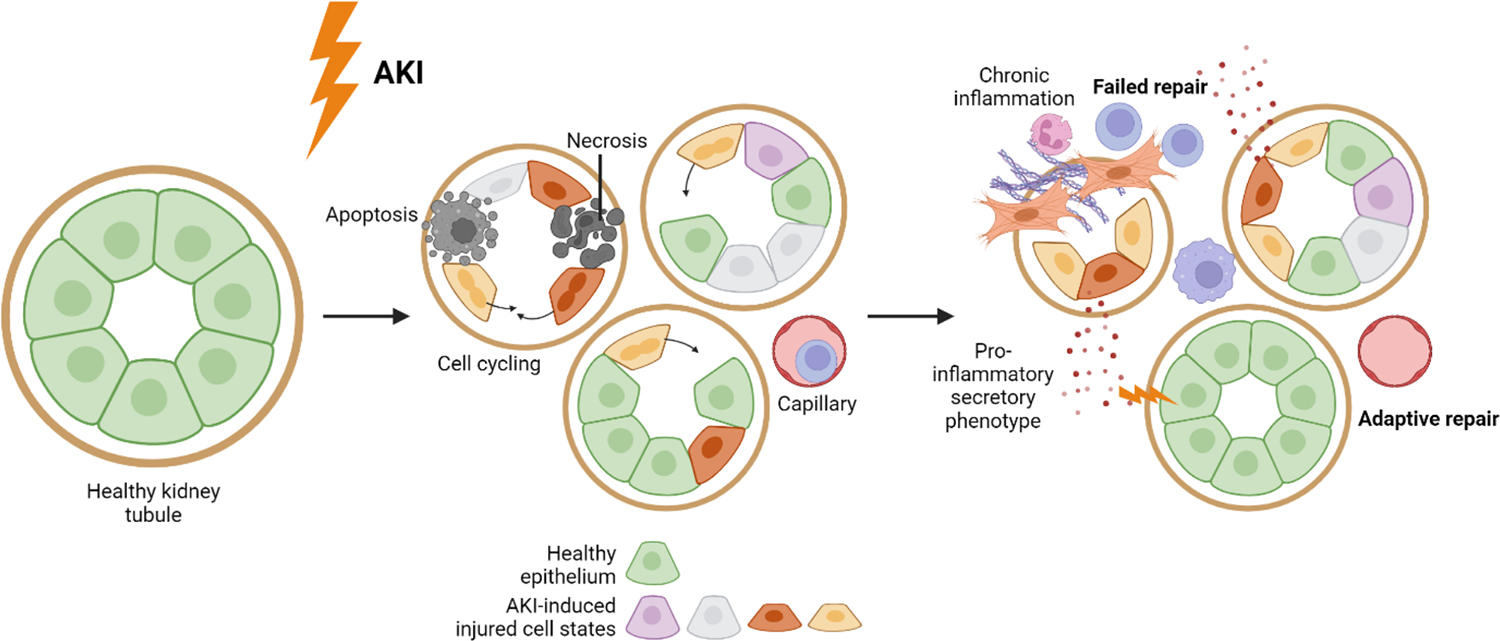
The schematic illustrates a general model of the involvement of tubular epithelial cell states in AKI. This includes the emergence of distinct AKI-induced epithelial cell states, labeled by different colors. An unknown population of injured dedifferentiated epithelial cells undergoes cell cycling and replaces neighboring cells that have been lost. While some injured epithelial cells are able to regenerate and restore normal kidney epithelium through adaptive repair, a subset of dedifferentiated epithelial cells may persist in an aberrant cell state, contributing to chronic injury through a pro-inflammatory secretory phenotype. This further attracts immune cells and potentially causes harm to adjacent healthy kidney epithelium. It is important to note that this figure represents a single time point after AKI, where multiple AKI-induced cell states coexist. It should be noted that the presence and abundance of these cell states may vary between individuals and within any individual depending on the time point after AKI. It is of note that these mechanisms not restricted to the PT.
Our molecular comprehension of AKI is largely based on rodent studies. Various AKI models exist for rodents, with kidney ischemia-reperfusion injury (IRI) being the most frequently utilized approach25–27. In the mouse model of kidney IRI, the kidney’s blood vessels are temporarily clamped for a defined time period to induce AKI, leading to substantial damage, particularly in proximal tubules (PT) and their outer medullary S3 segments, which are located in areas highly sensitive to hypoxia40.
Direct assessment of the epithelial gene expression response to AKI using bulk transcriptomic techniques is challenging due to the involvement of many cell types in the kidney during AKI. To facilitate cell type-specific or cell compartment-specific gene expression analysis, previous research has employed methods to tag RNA molecules specific to certain cells or cellular compartments34,41,42. Shen et al. used cre recombinase-driven thiouracil tagging to label nascent RNA molecules of collecting duct cells using either Hoxb7- or Atp6v1b1-driven cre recombinase expression in IRI and volume depletion41. This method allowed them to analyze the newly synthesized RNA specific to these cells at specific time points after the treatment. They found a pro-inflammatory and pro-fibrotic gene expression response in the collecting duct cells 24 hours post-IRI, contrasting with the absence of such response in volume depletion, despite both AKI models causing similar serum creatinine increases41. Similarly, Liu et al. utilized a cre recombinase-dependent approach to investigate cell type-specific RNA expression 24 hours after IRI. They tagged the L10a ribosomal protein subunit with EGFP and then employed translating ribosome affinity purification (TRAP). This technology allows the purification of RNA bound to EGFP-labeled ribosomes by using an anti-EGFP antibody43,44. They studied four different cell compartments, including cells marked by the Six2-cre recombinase (EGFP labeling activated in nephron progenitors and their progeny), Foxd1-cre (interstitial/stromal cells), Cdh5-cre (endothelial cells) and Lyz2-cre (myeloid lineage cells)42. Besides noting an upsurge in pro-inflammatory pathways, the nephron cells (tagged by Six2-cre) exhibited an activation of genes associated with cell cycling, cell junction formation, and cytoskeleton organization, suggestive of ongoing regeneration42. Differential gene expression analysis in other compartments revealed increased signaling in angiogenesis and invasion within stromal cells (tagged by Foxd1-cre), phagocytosis and chemotaxis within myeloid cells (tagged by Lyz2-cre) and immune cell recruitment within endothelial cells (tagged by Cdh5-cre). Interestingly, the authors pointed out that differentially expressed genes between IRI and sham controls showed a large overlap between the different cellular compartments pointing towards shared cellular molecular programs activated by different cell types during AKI. A third study used a Kim1-GFPCreERt2 knockin mouse line to investigate transcriptional changes after AKI34. The transcriptomic analysis specifically in kidney injury molecule Kim1-expressing cells facilitated the selective investigation of expression signatures in acutely injured proximal tubule cells (PTCs) after AKI. The study highlighted that KIM1-expressing PTCs undergo clonal expansion after IRI during kidney tubule repair and that this proliferative state is characterized by an EGFR/FOXM1 signaling axis. The authors could also show that not all injured proximal tubule cells regenerated back to healthy kidney epithelium but rather continued expression of dedifferentiation marker genes such as VIM and SOX934.
While these approaches have provided important insights into the cell type-specific responses to AKI, they are limited to one compartment at a time and depend on the specific activity of cre recombinase within that compartment. This means that they were not suitable to provide an unbiased analysis of cell state and cell type heterogeneity. The advent of single cell transcriptomics enabled such an unbiased investigation of the molecular events in AKI in individual cell types and cell populations and has been successfully applied to mouse and human kidney tissue45–55. In such single-cell transcriptomic analyses, healthy and diseased kidney tissues were harvested and dissociated to thousands of single cells comprising a representation of most if not all cell types within the organ. These individual cells were then subjected to unbiased gene expression analyses using single cell sequencing techniques, such as droplet-based sequencing, providing high-dimensional per cell gene expression information. The initial AKI single-cell studies, by analyzing single cell transcriptomes of dissociated mouse kidney tissue at varying intervals following IRI-induced AKI, consistently reported the appearance of abnormal AKI-associated epithelial cell states within the PTC compartment45,50,51,56. Each study used distinct bioinformatic clustering approaches to analyze the high-dimensional sequencing data, identifying variable numbers of subclusters of AKI-associated cell states within the PT. This included subclusters presumed to be reparative but also presumptive maladaptive cell clusters based on the identified gene expression signatures. Importantly, part of these maladaptive cell states failed to revert back into a healthy, differentiated kidney epithelium45,50,51,56. In the pioneering work by Kirita et al., such maladaptive cells were referred to as “failed repair cells”45. These cells began to appear on day 2 after IRI, made up nearly 30% of PTCs by day 14 after IRI, and continued to exist six weeks after IRI45. The identifying features of failed repair cells included the downregulation of PT marker genes, along with the expression of Vcam1, Dcdc2a, and Sema5a45. Analyses of temporal trajectories indicated that the failed repair cells distinctly diverged from repairing PTCs, thereby reinforcing the concept of an aberrant, persistent cell state within the PT compartment. Gene regulatory network analyses provided evidence of an elevated NF-kappa B signaling pathway within these failed repair cells45. Intriguingly, these cells exhibited evidence of maladaptive signaling to other cell types within the kidney, including pro-inflammatory Ccl2-Ccr2 signaling to immune cells, endothelin signaling to vascular cells, and profibrotic Pdgfrb/Pdgfrd signaling to fibroblasts45. A subsequent study validated and refined these findings by examining the spatial distribution of failed repair cells after IRI-induced AKI56. This study identified Vcam1- and Ccl2-positive, pro-fibrotic, and pro-inflammatory injured PTCs that persisted for weeks post-AKI, without any apparent reversion to a healthy kidney epithelium56. The researchers pointed to NF-kappaB and AP-1 signaling upstream as potential initiators of this failed repair cell state. They demonstrated that Vcam1- and Ccl2-positive cells originated from multiple injury sites, suggesting a propagation of injury from the corticomedullary boundary to the cortex post-AKI56. Maladaptive PTCs were not exclusively observed in injured mouse kidneys but were also detectable at low frequency within apparently healthy kidneys, with their presence increasing during aging56. Additionally, these failed repair cells in the PT demonstrated signs of a senescence-associated secretory phenotype56. Neither study45,56 found evidence of G2/M arrest in failed repair cells, a characteristic previously attributed to maladaptive PT repair57. A more recent study implementing mouse IRI-induced AKI over varied durations identified pyroptosis and ferroptosis as pivotal pathways mediating maladaptive repair following AKI51. Indeed, inhibition of either pyroptosis or ferroptosis resulted in the prevention of maladaptive repair and fibrosis in vivo51. Also, other studies reported ferroptotic stress as a mediator of maladaptive kidney epithelial repair58–61.
To understand the potential contribution of injured kidney epithelial cell states, such as failed repair PTCs, to the development of CKD, it is important to investigate whether these injured cells persist after AKI. This question was addressed in the recent study conducted by Gerhardt et al. who investigated renal epithelial cells up to 6 months post-IRI46. In this study, authors labeled cycling cells (Tamoxifen-inducible Ki67-driven labeling of the nuclear membrane with GFP) after IRI (Tamoxifen applied on days 2 and 3 after IRI) under the assumption that injury induces dedifferentiation of cells and cell cycle entry. The kidney tissue was subsequently examined using single nuclei mRNA sequencing at 4 weeks and 6 months after IRI. As expected, the majority of injured PTCs after IRI was GFP-positive, indicating that these cells indeed entered the cell cycle to potentially replace lost neighboring cells. A fraction of injured GFP-negative PTCs might correspond to either injured PTCs which did not cycle or cycled before application of Tamoxifen. Authors could identify several AKI-induced injured PTC clusters including less severely injured PTCs originating from the S1/S2 and S3 segment of the PT and a Vcam1- and Ccl2-positive cluster corresponding to previously published failed repair cells45. Interestingly, all injured clusters (severely and less severely injured) were also present after 6 months in AKI kidneys. It is yet to be determined whether these findings primarily indicate ongoing injury originating from failed repair PTCs or whether less injured PTCs also persist after AKI. Additionally, the authors observed cells entering the cell cycle after AKI in practically all kidney epithelial and non-epithelial cell types. This expands the perspective on murine AKI and suggests the involvement of the entire kidney tubule and other cell types in mouse IRI-induced AKI.
Given that AKI can arise from numerous clinical situations, it’s plausible that mouse models of IRI-induced AKI may not fully encapsulate all the molecular processes occurring intrarenally in real-world clinical settings. Indeed, recent studies have delved into the molecular alterations in varying experimental mouse models of AKI62,63. In one study, Chen et al. compared different AKI models induced by folic acid, sodium oxalate, cisplatinum, unilateral ureteral obstruction (UUO) and IRI63. Interestingly, the diversification of these models yielded an increase in the complexity of PTC states, with individual cell state compositions among different AKI models. Upon analyzing the ratios of injured PTC states and immune cells, the researchers suggested that molecular similarities existed between AKI induced by folic acid, sodium oxalate and UUO, whereas IRI- and cisplatin-induced AKI appeared molecularly distinct63. Molecular heterogeneity of different AKI states was further explored in a study that compared mouse unilateral IRI-induced AKI with UUO-induced kidney injury (unilateral irreversible ureter ligation) at varying time points (up to 28 days for unilateral IRI and 14 days for UUO)62. While both models triggered the emergence of maladaptive PTCs, the investigators observed differing temporal dynamics of maladaptive cell abundance and unique intermediate PTC states characteristic to each model62. In IRI-induced AKI, maladaptive cells were first detected on day 2, reached their highest presence around day 7, and continued to persist at a reduced percentage on day 28. Conversely, in UUO-induced AKI, the quantity of failed repair cells continuously grew over time, constituting over 50% of all PTC on day 1462. It is yet to be determined whether these substantial variations in injured PTC abundances genuinely signify distinctions between UUO- and IRI-induced AKI or merely reflect different time courses and reversibility of renal injury (transient IRI versus irreversible UUO). Intriguingly, the intermediate injury cell states could be distinguished by distinct metabolic programs. In IRI, an injured PTC population which peaked early (6 hours) after injury exhibited significantly increased fatty acid oxidation. These cells also displayed a considerable potential to return to healthy kidney epithelium. In the UUO model, the intermediate injury state could be characterized by disrupted amino acid metabolism62.
In summary, mouse AKI studies shed light on the emergence of multiple AKI-induced cell states, particularly in the PT. These cell states can vary based on the specific AKI model and the mechanism of kidney injury involved. It is probable that each AKI-induced cell state follows a unique trajectory over time and may have varying abilities to regenerate back to healthy kidney epithelium, ranging from full, partial, to no regeneration at all. Among these injured epithelial cell states, there is a maladaptive cell state and evidence suggests that it is unable to undergo regeneration to restore healthy kidney function. However, also after prolonged time periods after AKI, several other injured PTC states are present in the kidney. It is unclear whether this occurs due to unanticipated incomplete adaptiveness and persistence of several injured cell states, due to cell state transitions or due to continuous re-injury by for instance failed repair epithelia.
Investigation of epithelial cell states associated with human acute and chronic kidney disease
This section aims to examine the occurrence of injured epithelial cell states in human AKI and CKD and explore their relevance in the progression of kidney injury to fibrosis and CKD. Of particular interest for a potential clinical significance of epithelial cell states is the correlation of their presence or abundance with clinical outcomes, their association with relevant pathophysiological processes, and their persistence in AKI and CKD kidney tissue over time, which has been addressed in recent landmark studies48,49,52,55. However, the data available from human AKI and CKD studies is currently considerably less extensive compared to that for mouse AKI models.
The persistence of injured epithelial cell states can be investigated by analyzing AKI kidney samples at various time points after AKI and comparing them to CKD samples and control kidney tissue. For pathophysiological mechanisms, the available literature on molecular mechanisms for kidney fibrosis and CKD development is extensive and cannot be completely reviewed in detail here64–66. However, through receptor-ligand analysis and spatial transcriptomics, transcriptomic data can be examined for disease-enriched cell-cell interactions, which can then be compared to previously reported causal mechanisms for CKD development. Correlating cell state abundance or signaling with clinical endpoints follows a similar scheme in several studies. For this, gene sets, specific and representative for the investigated cell state can be derived from transcriptomic data (single cell or spatial transcriptomics). These gene sets are then used to calculate scores (e.g. mean expression over all genes) in larger existing bulk transcriptomic cohorts which provides statistical power and enables correlation with associated clinical follow-up data.
In a recent study, Lake et al. analyzed kidney samples from 45 healthy donors and 48 patients suffering from kidney diseases, including AKI and CKD integrating single cell transcriptomics, single cell chromatin accessibility assays, spatial transcriptomics and advanced imaging analyses. They identified altered cell states in AKI and CKD kidneys including altered cell states primarily found within proximal tubules (characterized by VCAM1, DCDC2, HAVCR1 expression) and thick ascending limbs (marked by PROM1, DCDC2)55. The authors reported that altered epithelial cell states showed downregulation of epithelial marker genes and shared common signaling axes including increased epithelial-mesenchymal transition, JAK/STAT, MAPK and TGF beta signaling. By performing trajectory analyses in PT and TAL and associating them with injury signatures at different time points after AKI in mice, the authors aimed to differentiate early, mid and late injury signatures in a temporal manner. This allowed the discovery of an orchestrated timely activation of transcription factors and target genes in human AKI and CKD. Spatial transcriptomics revealed an increased spatial proximity of altered epithelial cells from PT and TAL with stromal cells, lymphocytes and macrophages. These findings support the notion of an association of injured epithelial cell states with a fibrotic niche or microenvironment, which might exacerbate and propagate renal damage66. Receptor ligand analysis showed evidence of an early increased interaction of altered epithelial cell states with immune cells followed by an increased interaction with stromal cells via FGF2, PDGFB and TGFB signaling (Fig. 3). The pivotal role of stromal cells, lymphocytes and macrophages in kidney fibrosis development is undisputed64,67,68. Authors describe, amongst others, a NECTIN2 (injured TAL) - CD226 (T lymphocyte) axis which might help in recruiting and stimulating T cells. The pivotal and necessary roles of T cells for kidney fibrosis development after AKI were previously reported67. FGF2, PDGF and TGFB signaling are among the best-researched pathways in (kidney) fibrosis development and can lead to fibroblast activation and proliferation64–66,69–71. These findings link injured kidney epithelia to the attraction and stimulation of leukocytes and fibroblasts, two hallmarks of kidney fibrosis development. Lake et al. developed a gene expression score to examine evidence of altered cell states in PT and TAL within bulk transcriptomic data. They discovered a significant correlation of this score with a composite kidney endpoint (40% decline in eGFR or the onset of end-stage kidney disease after biopsy) within the Nephrotic Syndrome Study Network cohort. Notably, high scores for altered TAL states were strongly associated with declining kidney function highlighting the potentially important role of TAL injury in the development of fibrosis and progressive CKD.
Fig. 3. Signaling pathways between injured and healthy epithelial cells, immune cells and stromal cells potentially propagating fibrosis.
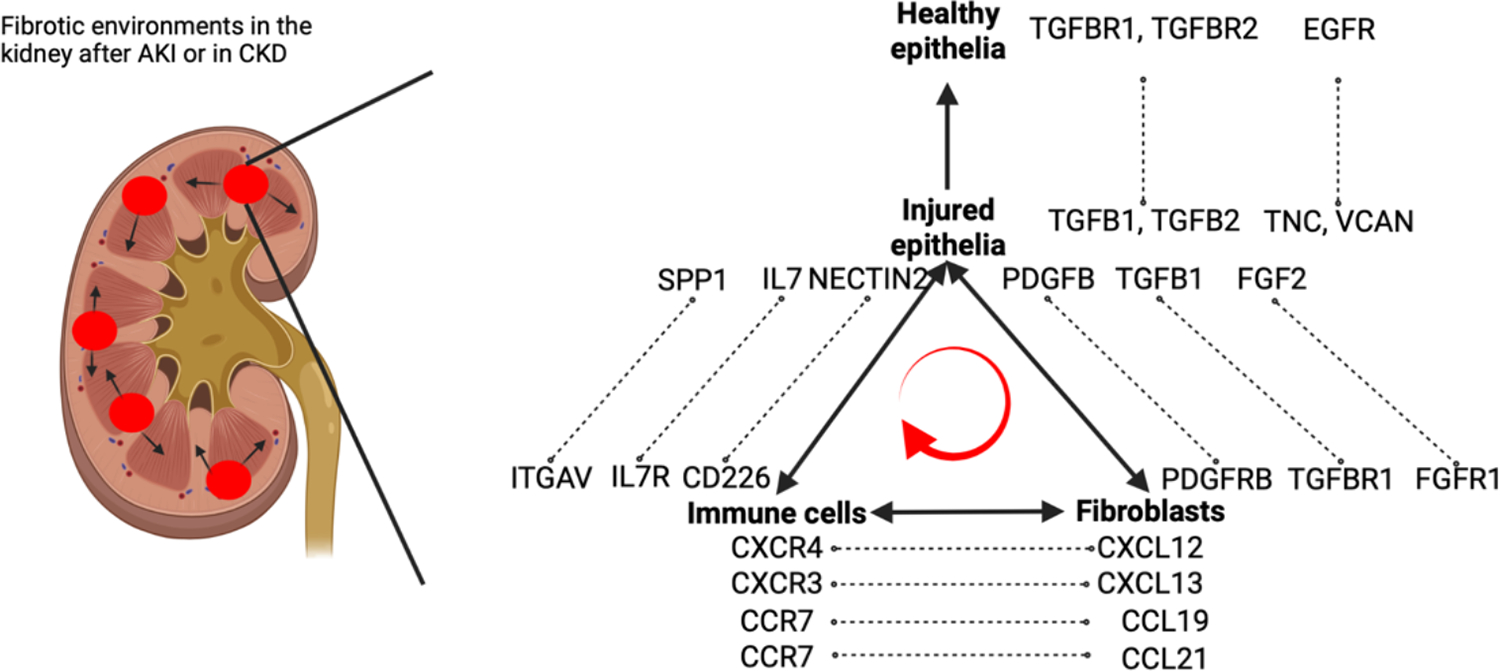
Recent single cell studies have highlighted the spatial proximity of injured epithelial cells, leukocytes, and stromal cells within fibrotic microenvironments. The signaling interactions between these cell types are believed to contribute to persistent inflammation. The pathways, depicted in the figure, are a selection of potential interactions reported in recent human single cell sequencing studies.
Our own group applied single nuclei transcriptomics to post-mortem kidney tissues from critically ill patients with severe respiratory infections and systemic inflammation suffering from severe AKI48. We compared these AKI kidneys with post-mortem non-AKI kidney tissue and with tumor-adjacent normal non-AKI kidney tissues obtained during nephrectomies. Our study identified newly occurring AKI-associated cell states across different cell types of the kidney tubule, including PT, TAL, distal convoluted tubule (DCT) and collecting duct (CD)48. Interestingly, PT, TAL and DCT all exhibited four similar AKI-enriched cell states, which we designated PT-New 1–4, TAL-New 1–4, and DCT-New 1–4. These four cell states showed a marked downregulation of epithelial marker genes and were defined by specific gene expression signatures, which were characterized by expression of genes associated with 1.) oxidative stress, 2.) hypoxia, 3.) inflammation/translation and 4.) epithelial-mesenchymal transition (EMT). The observed marker gene overlap between the four cell states in PT, TAL and DCT suggested a shared response mechanism across the nephron in human AKI48. Interestingly, inter-individual variability between individual patients with AKI was primarily driven by the cell type-specific abundance of these four epithelial cell states, suggesting a potential for personalized assessment and therapy based on epithelial subset abundance (Fig. 2).
Fig. 2. Conserved epithelial injury responses in PT, TAL and DCT cells in human AKI.
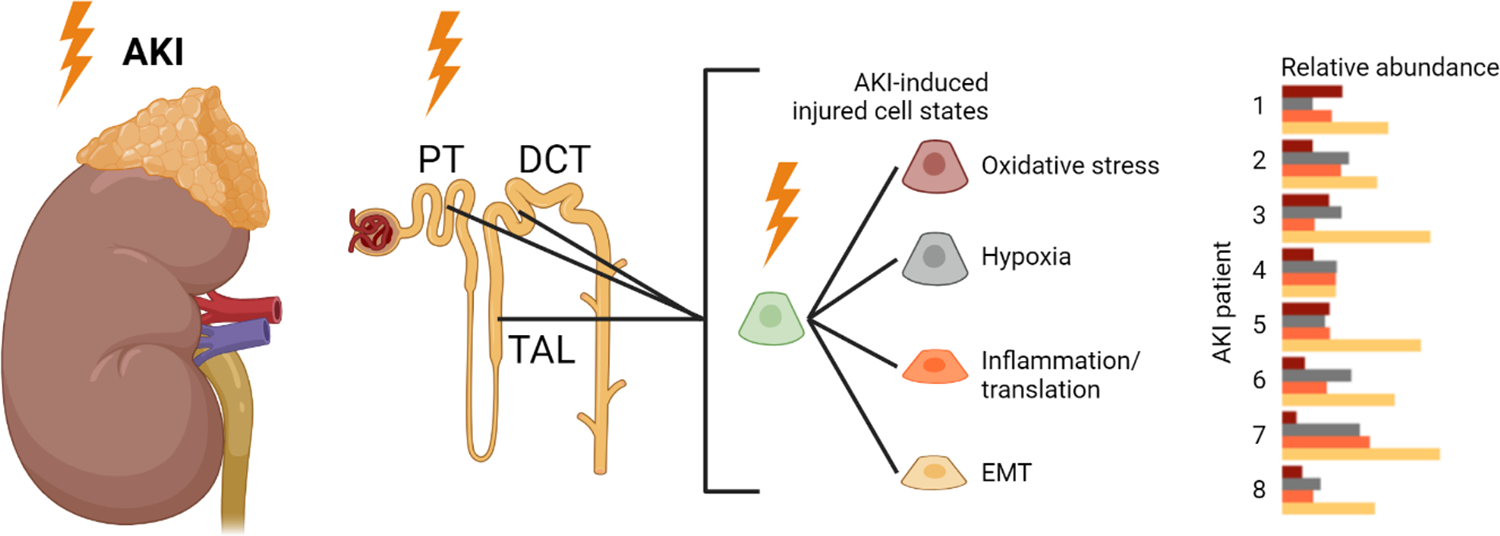
We observed that AKI in the context of severe systemic inflammation induces the emergence of four distinct injured cell states in PT, TAL and DCT associated with oxidative stress, hypoxia, inflammation/translation and EMT. AKI patient heterogeneity can be partly explained by an individual composition of these injured epithelial cell states.
We compared injured PTC states in human AKI to cell states observed during the time course of AKI-to-CKD progression in mouse IRI. Human AKI PTC states exhibiting an oxidative stress signature (i.e. PT-New 1) and a hypoxia signature (PT-New 2) resembled early, less severely injured PT cells of the S1/2 and S3 segments in mice. In contrast, human PTC states characterized by an inflammation/translation signature (called PT-New 3) and an EMT signature (PT-New 4) resembled mouse cell states characterized by advanced injury and/or failed repair. Indeed, the EMT cell state was characterized by positivity of the markers VCAM1 and CCL2 in the PT, similar to what was earlier reported in failed repair PTCs. By reanalyzing the data from Lake et al., we identified similar injury-associated cell states in PT and TAL associating with AKI and CKD tissues55. Early altered TAL and PT cells from Lake et al. resembled PT- and TAL-New 1 and TAL-New 2 cell states, while late injury PT and TAL states from Lake et al. showed a stronger overlap with PT- and TAL-New 4. In contrast, cell populations characterized by inflammation/translation signatures (PT-New 3, TAL-New 3, DCT-New 3) appeared to be uniquely present in critical illness/infection-associated AKI and were not detectable in other forms of AKI or CKD, opening up further potential for their diagnostic usage. In addition to cell-cell interactions of injured epithelia with immune and stromal cells, we also identified new receptor ligand pairs between injured and healthy kidney epithelia, suggesting signaling between adjacent injured and non-injured cells of the nephron. These potential new signaling axes included and EGFR (Fig. 3). Both signaling axes can induce EMT in kidney epithelial cells which constitutes a hallmark of the epithelial response pattern to AKI69,72. This might suggest a model where epithelial injury spreads from injured (potentially maladaptive) kidney epithelia to adjacent uninjured epithelia.
Additional insights were provided by Abedini et al., who amassed a comprehensive dataset comprising single cell and spatial transcriptomic data from 35 healthy control and 38 CKD kidney samples73. The study identified clusters of injured PTCs which showed higher enrichment in CKD samples compared to healthy controls. The authors did not report altered cell states in other kidney cell types. Again, by utilizing spatial transcriptomics, the microenvironment composition around these injured PTC could be analyzed. As expected, injured PTC showed increased presence together with stromal and immune cells in a fibrotic microenvironment which was marked by a heightened production of extracellular matrix73, a hallmark of the fibrogenic niche74. Cell-cell interaction analysis highlighted once more the suggestive role of injured PTC in attracting leukocytes. This included elevated expression of IL7, C3 and SPP1 on injured PTC and their corresponding receptors on leukocyte cell types (Fig. 3). Global knockouts of SPP1 or C3 were previously shown to attenuate leukocyte recruitment, interstitial inflammation and fibrosis in mouse models of kidney fibrosis75,76. Abedini et al. further highlighted chemotactic signaling by stromal cells to immune cells including elevated expression of CXCL12, CXCL13, CCL19 and CCL21 on stromal cells in CKD and their receptors on immune cell subsets, signaling pathways previously reported as regulators of immune cell infiltration in the kidney77 (Fig. 3). To validate the prognostic relevance of such fibrotic microenvironments in CKD, Abedini et al. developed a gene set score specific for their defined fibrotic microenvironment. This enabled the investigation within a larger bulk transcriptomic dataset from kidney biopsies encompassing 298 kidney samples including healthy controls and CKD samples with varying CKD severity73. Indeed, the gene expression signature associated with the fibrotic microenvironment was useful in categorizing kidney samples by disease severity, as quantitated by estimated Glomerular Filtration Rate (eGFR) or the extent of fibrosis. Furthermore, the signature was predictive for a severe decline of eGFR (40% per year) or the onset of end-stage kidney disease. It is of note that the gene set defining the fibrotic microenvironment signature included genes previously associated with failed repair PTC (e.g. VIM, CD44, CLU) and other AKI-induced PTC states (e.g. SERPINA1, KLF6, human leukocyte antigens, AKAP12)48.
Ageing is an important factor heavily impacting wound healing and incidence of AKI and CKD which cannot be fully reviewed in detail here78,79. However, maladaptive or failed repair PTCs or kidney epithelial cells have repeatedly been associated with senescent cells mostly based on correlation of their marker genes with genes involved in the senescence-associated secretory phenotype46,56. Given that cellular senescence is a stress response mechanism, it has to be however noted that many senescence-associated genes are also upregulated in non-senescent states of cellular stress. Moreover, convincing expression of markers such as Cdkn1a or Cdkn2a, markers of cellular senescence, cannot be observed in failed repair PTCs when checking the online resources of mouse and human AKI data45,46,48,55. Hence, to what extent failed repair PTCs fall into the category of truly senescent cells remains unclear.
In summary, the studies discussed underscore the enduring presence of injured or altered kidney epithelial cells after an AKI episode and in CKD. Injured or altered kidney tubular epithelial cells are found within pro-inflammatory and pro-fibrotic microenvironments which additionally involve stromal and immune cells. Concerning their pathophysiological significance, injured epithelial cell states likely play a role in attracting and stimulating leukocytes and fibroblasts, two pivotal components for renal fibrosis development. However, it has to be noted that in many cases epithelial cells are not the only source for the reported chemotactic receptors and ligands. Additionally, pathways such as TGFB signaling can induce a wide range of cellular responses, some of which may also have cytoprotective effects69. While these findings offer promising prospects for further research, it is essential to emphasize the necessity for future studies to validate their significance.
While the findings reported above highlight a potential clinical importance of injured kidney epithelial cell states in kidney disease manifestation and progression, it must be pointed out that future research will be needed to address the question of whether these cell states actively contribute to disease pathophysiology. For instance, it is unclear whether removal of these cells would beneficially affect fibrosis development. Crucially, mouse studies offer data from models with well-defined pathomechanisms and precise time courses of AKI. This can greatly aid in annotating human datasets through cross-species analysis, providing valuable insights into underlying pathomechanisms and time course annotations. The diversity of AKI-induced epithelial cell states identified in the human kidney varied across studies but highlighted the largest transcriptional changes in PT and TAL. As a future perspective, it is however important to increase and establish measures of comparability between results from different single cell studies.
The relevance of epithelial injury after kidney transplantation
Kidney transplantation is the preferred treatment for eligible individuals with advanced chronic or end-stage kidney disease1,80. Unfortunately, the number of patients in need of a kidney transplant drastically exceeds the number of available organs81. Following kidney transplantation, various factors can affect function and survival of kidney allografts, making the clinical situation even more complex than that of the native kidney. Mechanisms of graft injury leading to AKI or progressive CKD include infectious complications, episodes of allograft rejection, drug toxicity, recurrence of the underlying kidney disease, or systemic diseases like diabetes or hypertension82. In clinical reality, it is often difficult to untangle the mechanisms of allograft injury in a given patient, which is in part related to the imperfect diagnostic tools available to diagnose allograft injury and to differentiate between injury mechanisms.83,84. Fine-tuning personalized patient care in kidney transplantation remains a major challenge. Molecular biomarkers beyond creatinine and proteinuria have not lived up to initial expectations and molecular precision medicine is still confined to research settings. Signatures of epithelial injury or the detection of epithelial cell states associated with allograft injury mechanisms and prognosis could address this problem4,85. Single cell transcriptomic analyses of biopsied allografts or of urinary cells, including an analysis of epithelial phenotypes within the injured allograft, offer more refined opportunities for allograft phenotyping and personalized therapy in the future. Hence, applying these methods to monitor allografts and drive specific targeted therapeutic interventions might be a promising strategy to facilitate precision medicine in transplantation.
Detection of epithelial injury signatures after kidney transplantation: evidence from bulk transcriptomics studies
Epithelial damage can manifest in various contexts following kidney transplantation, including the unavoidable IRI resulting from the transplantation procedure itself86, immunological rejection, drug toxicity, ascending urinary tract infections or BK virus nephropathy.3–5,87,88 (Fig. 4). Several studies have utilized bulk transcriptomics on biopsied kidney allograft tissue, providing insights into the molecular changes associated with injury in transplanted kidneys89–91.
Fig. 4.

Potential mechanisms of epithelial injury after kidney transplantation
Cippa et al. investigated 42 kidney allograft biopsies by bulk RNA sequencing, including samples taken before transplantation, shortly after transplantation, and at 3 and 12 months post-transplantation89. The authors identified two distinct temporal transcriptomic trajectories among transplanted kidneys. One trajectory was characterized by increased signaling associated with chronic inflammation, fibrosis, and kidney injury, the other one with kidney recovery and repair89. Transplanted kidneys on the injury/fibrosis trajectory demonstrated a lower GFR at 12 months and exhibited greater degrees of fibrosis based on histopathology evaluation. The authors aimed to identify genes which were determinants of the direction of kidney transplants towards either the recovery or fibrosis trajectory. In the fibrosis trajectory group, the authors observed a significant downregulation of genes involved in mitochondrial homeostasis and an upregulation of genes associated with extracellular matrix organization, including MMP7, which has been reported as a key regulator in kidney fibrosis and is upregulated in injured PT cells in human AKI48,92. Authors also found increased expression of failed repair PTC markers VCAM1 and CCL2 in the fibrosis trajectory in a re-analysis of the same data in a consecutive study56. These findings established an initial connection between genes observed in injured PTC states in AKI and CKD and unfavorable long-term kidney allograft outcomes. It is, however, of note that, due to the bulk transcriptomic approach, the expression of these genes cannot be attributed to a certain cell type or cell state.
Immunological rejection of the allograft following kidney transplantation represents a significant clinical challenge and constitutes a major risk factor of graft loss93. According to the international Banff classification, rejections in kidney transplantation can manifest as T-cell-mediated rejection (TCMR) or antibody-mediated rejection (ABMR), each characterized by distinct histology94,95. Histological hallmarks of ABMR include peritubular capillaritis and glomerulitis, hallmarks of TCMR include tubulitis, arteritis and interstitial inflammation94,95 (Fig. 5). Both, TCMR and ABMR display histopathological features of tubular injury94,95. It should be noted that while TCMR is almost always linked to damage in the kidney parenchyma, ABMR primarily affects the glomeruli and microcirculation, initially exerting less influence on the kidney epithelium but causing more severe effects once glomerular function declines (Fig. 5). TCMR typically occurs within the first year after transplantation, while ABMR is not limited to the early post-transplant period and frequently occurs at later time points93,96,97. The current treatment strategies for rejection aim to target the alloimmune responses98,99. In TCMR, treatment protocols involve corticosteroid pulse therapy and increasing maintenance immunosuppression98. Treatment of ABMR is more challenging and often involves strategies to remove allo-antibodies, achieve immunomodulation, and increase or modify immunosuppressive therapy. Despite all efforts, both, TCMR and ABMR continue to be associated strongly with adverse transplant outcomes.
Fig. 5. Potential models for epithelial injury after TCMR or ABMR.
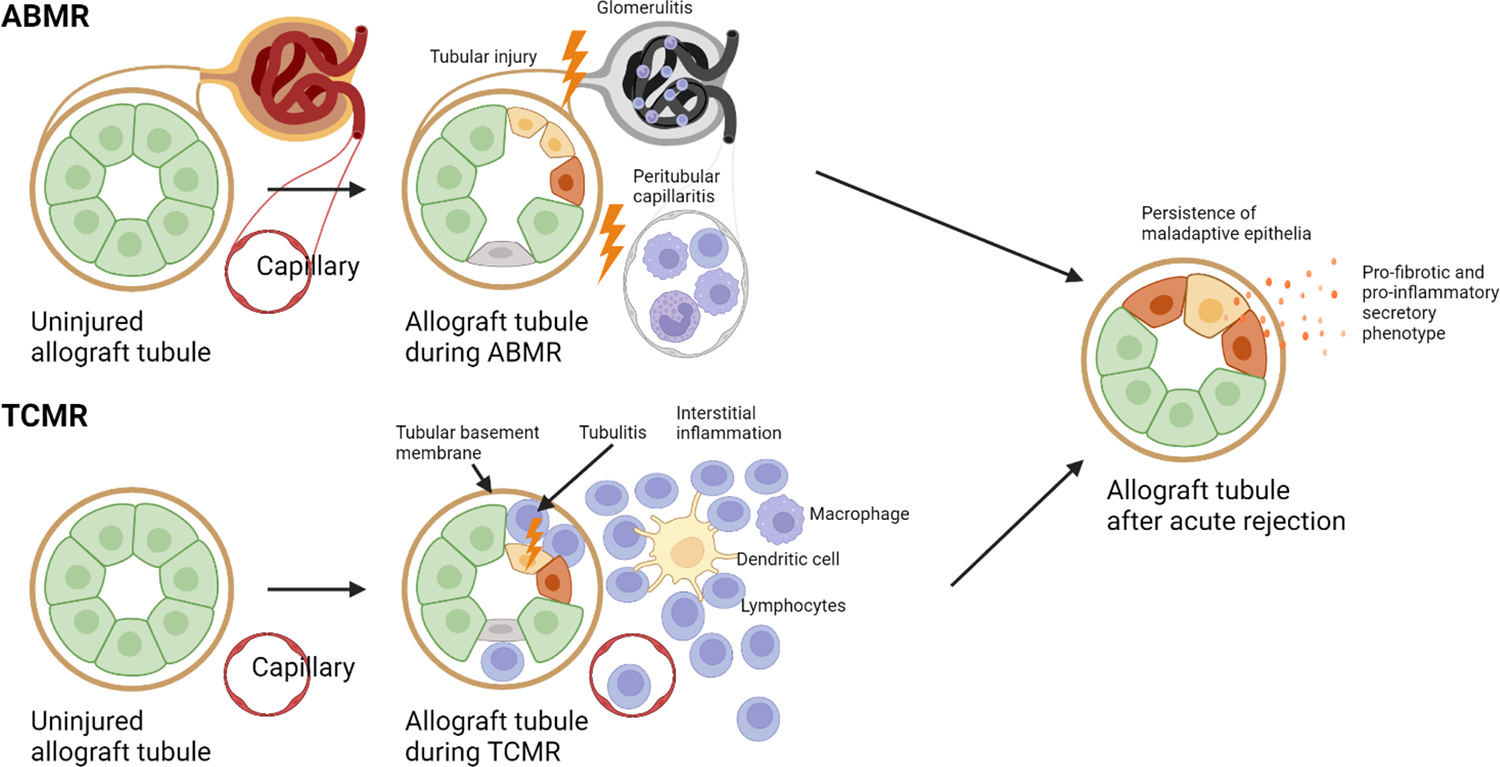
Epithelial injury occurs in, both, TCMR and ABMR. As epithelial injury is the most important predictor of allograft survival after ABMR or TCMR, this might suggest a model of persistence of maladaptive injured epithelial cells propagating further kidney allograft damage. Importantly, whether maladaptive or injured epithelial cell states persisting after ABMR or TCMR are identical or distinct remains unknown.
Large studies employing bulk transcriptomic approaches examined the molecular events associated with TCMR and ABMR, and established correlations of transcriptomic signatures with clinical outcomes4,5,85. In a comprehensive study comprising 1120 patients, including 321 cases of pure ABMR, histopathology and bulk transcriptomic analyses were conducted85. The study included clinical indication biopsies, taken at a median time of 719 days after transplantation, and patients were followed up for a period of 3 years. The authors examined the correlation between various clinical, molecular (using defined gene sets), and histological variables with transplant outcomes at 3 years after the kidney biopsy. Remarkably, in multivariate analysis, the most predictive variables for 3-year graft survival in, both, the full dataset and the ABMR cohort were eGFR and a gene set indicative of parenchymal epithelial injury and AKI85,90. In contrast, neither the presence of donor-specific antibodies nor gene set scores associated with ABMR activity were able to predict allograft outcome. Interestingly, a comparable large study examining gene expression signatures in TCMR yielded similar results4. Similarly, in this study, the most predictive variables for kidney transplant survival at the 3-year mark following biopsy were the previously mentioned parenchymal epithelial injury (or AKI) score, as well as a gene set score derived from kidneys with low GFR4,100. TCMR activity-associated scores showed comparably minimal impact on the 3-year survival outcome4. These findings demonstrate a strong correlation between kidney transplant survival after ABMR and TCMR with gene expression signatures characteristic of epithelial injury.
Two additional studies investigated transcriptional changes in post-transplant biopsies using bulk transcriptomics in a large patient cohort (1526 patients included)100,101. Authors applied unbiased bioinformatics approaches including principal component and archetypal analysis to molecularly characterize transcriptomic responses in post-transplant kidneys. Interestingly, from unbiased analysis, authors observed two archetypes of AKI based on bulk transcriptomics (designated as AKI 1 and 2). While both were associated with poorer transplant outcomes, they differed significantly in their gene expression. Transcriptomic data from AKI 2 showed significantly higher expression of genes associated with inflammation, leukocyte infiltration and response to wounding while these signals were blunted in AKI 1. Less than 1% of biopsies from the AKI 1 group showed TCMR while almost 14% presented with ABMR. Among AKI 2 specimens, 39% presented with TCMR and ca. 18% with ABMR. This sheds new light on the transcriptional heterogeneity in human AKI and epithelial injury post-transplantation.
The significant findings from kidney transplantation studies, as well as bulk and single cell studies in AKI and CKD, raise an important question about the specific cell populations responsible for the observed outcomes and risk factors following TCMR and ABMR. An important question to be addressed in future studies will be whether fibrotic microenvironments containing maladaptive or other injured kidney epithelial cells might be responsible for the reduction in allograft survival after TCMR or ABMR (Fig. 5).
Single cell analyses of kidney transplant injury
Although bulk transcriptomic approaches may not provide a definitive answer to this question, the field is rapidly advancing with a growing body of single cell studies conducted on kidney transplant tissue6–11,102. The number of patients included in these studies remains, however, low and the results have yet to be correlated with clinical outcomes.
In a recent study, two kidney transplant nephrectomies due to multiple rejection episodes (TCMR and ABMR) were compared to six samples of normal kidney tissue from tumor nephrectomies8. The authors were able to show distinct gene expression differences in proximal tubules between rejected and normal kidney tissues. Genes upregulated in proximal tubules in rejected kidneys included injury markers such as SPP1 and NGAL, as well as genes published in human and mouse AKI studies associated with injured PTCs. These genes include markers of several different injured PTC states and include marker genes of less severely injured, potentially early PTC populations such as KLF6 and FTL but also genes from late and severely injured PTC including VIM, ANXA1, TACSTD2, SERPINA1 and genes coding for human leukocyte antigens45,48,56. Again, this suggests the presence of several different injured cell states over time, which are reminiscent of those observed in mouse and human data from AKI and CKD.
Another recent single cell transcriptomics study of kidney samples of transplant patients with chronic allograft dysfunction provided further evidence for a role of epithelial gene expression in the development of fibrosis and allograft loss10. This study included eight kidney transplant biopsies, five from patients with chronic allograft dysfunction defined by interstitial fibrosis and tubular atrophy, and three from stable allografts. The authors identified subpopulations of dedifferentiated proximal tubular cells actively expressing extracellular matrix components, thereby potentially propagating inflammation and attracting leukocytes. Once again, these dedifferentiated injured proximal tubular cells expressed markers highly reminiscent of the maladaptive epithelial injury response from the AKI studies described above.
In summary, large studies using bulk transcriptomic approaches show that kidney epithelial injury is the most important predictor of transplant function after rejection4,85. They derive these results by correlating a score based on a gene set derived from human AKI90 with clinical end points. Many of these genes are indeed marker genes of injured epithelial cell populations including failed repair PTC in single cell studies from AKI and CKD and kidney post-transplantation specimens. Additionally, results from clustering analyses of single cell data after kidney transplantation find injured clusters highly reminiscent of injured epithelial cell populations induced by AKI and CKD. It is currently unclear whether any of the injured epithelial cell populations after kidney transplantation are responsible for the reduced allograft survival after rejection.
The potential clinical applications of monitoring or targeting epithelial cell states and epithelial signaling after kidney transplantation
Although the data from single cell resolution gene expression studies in kidney transplantation is still limited, epithelial cell states and signaling in the context of kidney transplantation could potentially have a wide range of clinical applications. Direct access to kidney epithelial cells post-transplantation can be achieved by examining kidney tissue through protocol or indication biopsies, or by analyzing kidney epithelial cells shed into the urine (Fig. 6A). In fact, single-cell sequencing has been successfully executed from the urine of patients with various kidney diseases49,103–106. This non-invasive technique additionally allows for frequent sampling. Klocke et al. have recently shown that epithelial cell populations observed in human AKI can be recovered from single-cell transcriptomes of kidney cells shed into the urine of AKI patients, including failed repair cells49. However, it’s worth noting that urinary cell populations have thus far been correlated with their tissue counterparts based on marker gene comparison, which may be compromised by abnormal gene expression in urinary cells due to harsh urinary conditions. To our knowledge, there are no published studies examining urinary kidney epithelial cells from post-transplant patients using single-cell transcriptomics. Yet, flow cytometry analysis of urinary cells has proven informative in distinguishing different causes of transplant dysfunction107,108. For instance, Goerlich et al. demonstrated that counting T cells and tubular epithelial cells through flow cytometry could differentiate kidney transplant recipients with rejection from patients without rejection, whereas measuring T cell abundance alone could not107.
Fig. 6. Potential applications using injured epithelial cells and epithelial cell signaling after kidney transplantation.
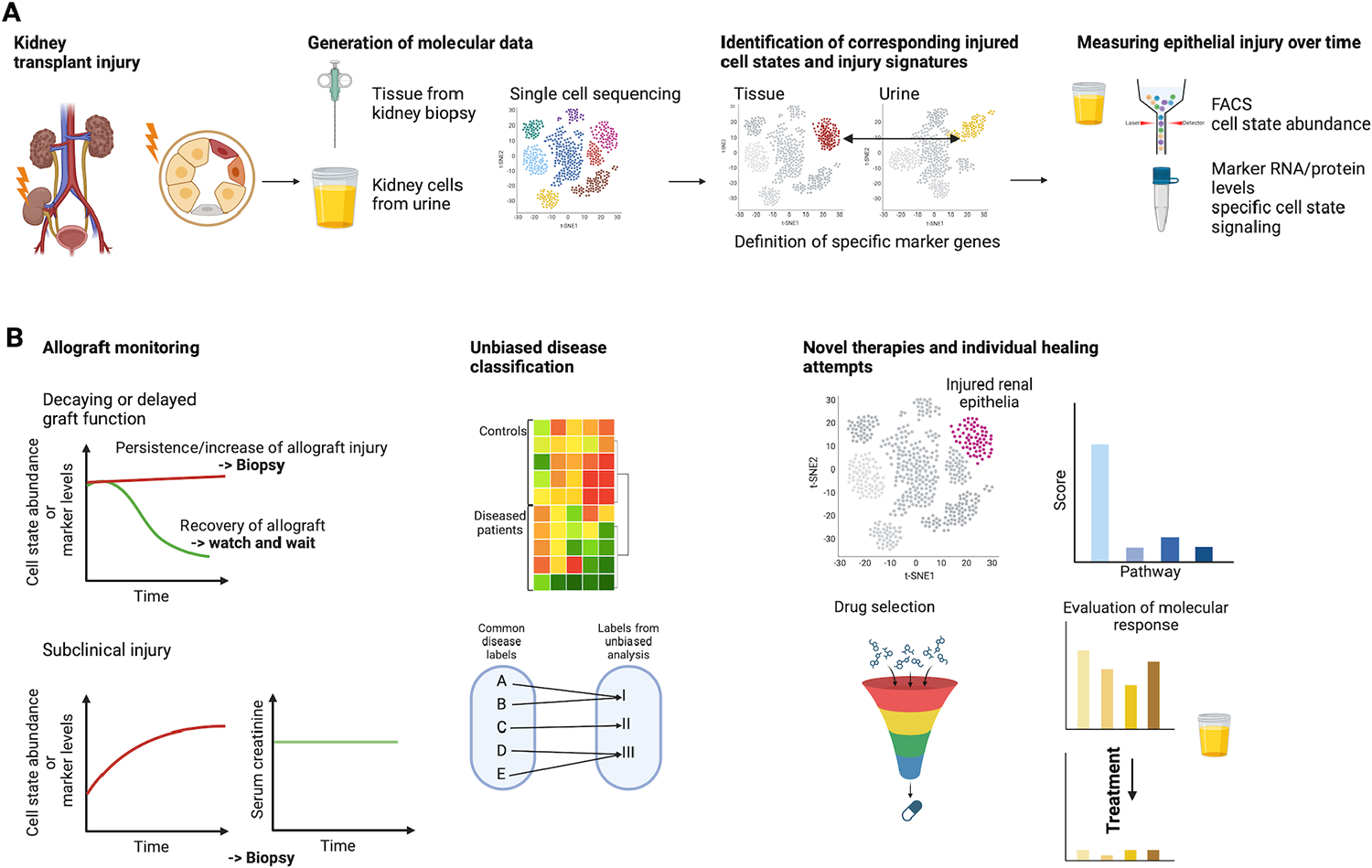
A. Epithelial cell information can be obtained from kidney tissue from biopsies or kidney cells shed into the urine. Having, both, tissue and urine single cell data can help in associating kidney tissue injury signatures with the corresponding cell population in the urine. The so-identified injured cell populations in patient urine can be non-invasively measured over time using either single cell sequencing or other faster and more cost-effective techniques such as FACS or measuring RNA or protein levels of specific targets derived from single cell sequencing data. B. Applications of data from injured epithelial cell states as mentioned in the text including allograft monitoring (decaying/delayed graft function), unbiased disease classification and novel therapies.
It has to be noted that carrying out single-cell sequencing analyses is currently costly and time-consuming, limiting current options for everyday clinical application. However, this might quickly change as single cell technologies and bioinformatic expertise will become more affordable and accessible. Additionally, findings derived from single cell transcriptomics can be combined with other quicker and more cost-effective techniques such as FACS or determining RNA or protein levels of selected markers (Fig. 6A).
While it still remains hypothetical, it interesting to conjecture how diagnostic information obtained from single cell transcriptomic analyses of epithelial cell states in kidney tissue or urine could be used to improve the management of kidney transplant recipients.
We envision several scenarios where such diagnostic information might be useful (Fig. 6B):
Diagnosing mechanisms of delayed or decaying allograft function: Monitoring the identity and abundance of abnormal epithelial cell states in transplanted allografts or urine samples might allow for a more personalized fine tuning of post-transplant care. A frequent question arising in post-transplant patients is to diagnostically distinguish immunological rejection, toxicity of calcineurin inhibitors (drugs that are frequently applied in kidney transplantation), and AKI from other causes. Differentiating such pathomechanisms based on epithelial cell states (yet to be defined) and assessing therapeutic response to changes in management would potentially address important clinical needs.
Diagnosing “subclinical” injury: Current markers of allograft injury (serial measurements of serum creatinine and proteinuria) lack sensitivity to detect continuous, mild forms of injury underlying progression of chronic allograft dysfunction. Quantitating abnormal epithelial cell states at baseline (following transplantation) and over time might help in detecting ongoing immunological injury or drug toxicity. This might allow timely detection and therapy of such injury.
Unbiased disease classification and prognostic assessment: While current classification of allograft injury is undertaken based on clinical parameters and histopathology, a molecular classification has the potential to remove ambivalence and introduce objectiveness and reproducibility. Utilizing gene expression data and categorization algorithms, such as hierarchical clustering or advanced machine learning techniques additionally has the potential to uncover novel disease entities and uncover more specifically the molecular mechanisms driving disease progression and response to treatments.
Guiding novel therapies: In the future, abnormal epithelial cell states might be therapeutically targetable (see next section). Therefore, detecting and monitoring epithelial states would rationalize such therapies and allow therapeutic surveillance. In addition, high resolution molecular phenotyping may uncover epithelial cell signaling pathways for which targeted treatments already exist and enable individual healing attempts (e. g., anti-IL6 or anti-TNF therapies).
Potential avenues for therapeutic interventions using information from epithelial cell signaling after kidney transplantation
While there have been promising and convincing results linking epithelial cell states and signaling to various clinical contexts, including AKI, CKD, and post-kidney transplantation, it is essential to acknowledge that most of the evidence linking epithelial injury states to clinical endpoints is largely based on associations. Nevertheless, earlier studies have provided initial evidence that abnormal epithelial cell states and active signaling from these cells participate in kidney disease progression and could potentially be used as therapeutic targets after kidney transplantation (Table 1)
Table 1.
Examples of potential therapeutic approaches targeting maladaptive epithelial cell signaling after kidney transplantation
| Therapeutic approach | Mechanism of action |
|---|---|
| Interleukin 6 blockage109 | Inhibition of T-cell invasion by targeting interleukin 6 production from basophils attracted by injured epithelial cells |
| Basophil depletion109 | Inhibition of basophils attracted by injured epithelial cells, leading to reduced production of interleukin 6 and decreased attraction of T cells |
| Pyroptosis/ferroptosis inhibition51,58,59,61 | By inhibiting pyroptosis/ferroptosis in maladaptive epithelial cells, they can be redirected towards adaptive repair |
| CAR T cell therapy110 | Targeted destruction of maladaptive epithelial cells |
| Senolytic treatment111,112 | Removal of potentially senescent maladaptive kidney cells |
| Epithelial cell differentiation and metabolism113–116 | Promotion of differentiation and signaling towards baseline metabolism, inhibition of dedifferentiation |
Based on these studies, multiple therapeutic avenues might exist, including promotion of adaptive repair (e. g. by inhibiting pyroptosis/ferroptosis, or activating signaling axes that facilitate differentiation and physiological metabolism), restraining dedifferentiation, inhibiting detrimental signaling originating from maladaptive kidney epithelia (interleukin 6 blockage, basophil depletion) or implementing therapies that aim to eliminate these dysfunctional kidney cells (e.g. via CAR T cell-based treatments, or senolytic therapy). All these approaches could hypothetically be considered to prevent IRI-induced AKI and its sequelae shortly after kidney transplantation, to prevent long-term injury after alloimmune complications (TCMR, ABMR) or to address the unresolved yet frequent issue of chronic allograft dysfunction.
It is evident that establishing whether a proposed therapeutic approach achieves significant clinical improvements requires the inclusion of a large number of patients and long-term follow-up clinical data. However, modern molecular data, such as those obtained from single cell sequencing, may provide a valuable surrogate to assess molecular efficacy. Once the therapeutic approach and target patient population are defined, single cell sequencing analyses, particularly of injured epithelial cell states before and after treatment, could potentially be used as a phenotypic readout offering insights into the molecular response and the success or failure of the proposed treatment strategy. Thereby, single cell transcriptomics-based analysis of allografts might aid in identifying eligible patients and in delivering tailored study treatment strategies in small, refined patient cohorts.
Conclusions and future perspective
An emerging body of studies highlights the importance of disease-associated kidney epithelial cell states that 1.) arise in the setting of AKI, CKD, and in transplanted allografts, 2.) display distinct molecular phenotypes, and 3.) potentially promote disease pathogenesis, e. g. the formation of fibrogenic niches by recruiting interstitial cells and leukocytes. It is clear that there is phenotypical overlap between these cell states in different disease settings, including in AKI and CKD of the native kidney and in conditions affecting the transplanted kidney allograft. Nevertheless, the field is still emerging and future studies will need to refine the precise epithelial cellular states, their disease association and their disease-relevant diagnostic information, both in experimental model systems and in patient cohorts. In addition, it will be important to experimentally address the functional roles of each cellular subtype and its molecular signaling pathways. The clinical setting of kidney transplantation is particularly useful to explore the utility of assessing epithelial cell states, because of the strong medical need to optimally tailor post-transplant care by fine-grained phenotyping.
Acknowledgments
All figures were created using BioRender.com. This work was supported by grants to Kai M. Schmidt-Ott from the Deutsche Forschungsgemeinschaft (DFG; CRC 1365, GRK 2318, RU 2841), ERA PerMed (OnAKI-ICI) and Ministry for Science and Culture of Lower Saxony (MWK), to Jonathan M. Barasch from NIDDK NIH HHS/United States (R01 DK124667/DK, T32 DK108741/DK, UG3 DK114926/DK, U54 DK104309/DK) and to Christian Hinze from MWK as project of the “Center for Organ Regeneration and Replacement (CORE)”, Transplant Center, Hannover Medical School.
References
- 1.Abecassis M, et al. Kidney transplantation as primary therapy for end-stage renal disease: a National Kidney Foundation/Kidney Disease Outcomes Quality Initiative (NKF/KDOQITM) conference. Clin J Am Soc Nephrol 3, 471–480 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lentine KL, et al. OPTN/SRTR 2021 Annual Data Report: Kidney. Am J Transplant 23, S21–S120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halloran PF, Famulski KS & Reeve J Molecular assessment of disease states in kidney transplant biopsy samples. Nat Rev Nephrol 12, 534–548 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Madill-Thomsen KS, et al. Relating Molecular T Cell- mediated Rejection Activity in Kidney Transplant Biopsies to Time and to Histologic Tubulitis and Atrophy-fibrosis. Transplantation (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halloran PF, et al. Review: The transcripts associated with organ allograft rejection. Am J Transplant 18, 785–795 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, et al. Single-cell analysis reveals immune landscape in kidneys of patients with chronic transplant rejection. Theranostics 10, 8851–8862 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malone AF, et al. Harnessing Expressed Single Nucleotide Variation and Single Cell RNA Sequencing To Define Immune Cell Chimerism in the Rejecting Kidney Transplant. J Am Soc Nephrol 31, 1977–1986 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rashmi P, et al. Multiplexed droplet single-cell sequencing (Mux-Seq) of normal and transplant kidney. Am J Transplant 22, 876–885 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu H, et al. Single-Cell Transcriptomics of a Human Kidney Allograft Biopsy Specimen Defines a Diverse Inflammatory Response. J Am Soc Nephrol 29, 2069–2080 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDaniels JM, et al. Single nuclei transcriptomics delineates complex immune and kidney cell interactions contributing to kidney allograft fibrosis. Kidney Int (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suryawanshi H, et al. Detection of infiltrating fibroblasts by single-cell transcriptomics in human kidney allografts. PLoS One 17, e0267704 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellum JA, Lameire N & Group KAGW Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit Care 17, 204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoste EAJ, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol 14, 607–625 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Vijayan A Tackling AKI: prevention, timing of dialysis and follow-up. Nat Rev Nephrol 17, 87–88 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ronco C Acute kidney injury: from clinical to molecular diagnosis. Crit Care 20, 201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kane-Gill SL, et al. Risk factors for acute kidney injury in older adults with critical illness: a retrospective cohort study. Am J Kidney Dis 65, 860–869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coca SG, Yusuf B, Shlipak MG, Garg AX & Parikh CR Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53, 961–973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newsome BB, et al. Long-term risk of mortality and end-stage renal disease among the elderly after small increases in serum creatinine level during hospitalization for acute myocardial infarction. Arch Intern Med 168, 609–616 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Abdala PM, Swanson EA & Hutchens MP Meta-analysis of AKI to CKD transition in perioperative patients. Perioper Med (Lond) 10, 24 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guzzi F, Cirillo L, Roperto RM, Romagnani P & Lazzeri E Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. Int J Mol Sci 20(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fogo AB, Lusco MA, Najafian B & Alpers CE AJKD Atlas of Renal Pathology: Ischemic Acute Tubular Injury. Am J Kidney Dis 67, e25 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Fogo AB, Lusco MA, Najafian B & Alpers CE AJKD Atlas of Renal Pathology: Toxic Acute Tubular Injury. Am J Kidney Dis 67, e31–32 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Liu J, et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cippa PE & McMahon AP Proximal tubule responses to injury: interrogation by single-cell transcriptomics. Curr Opin Nephrol Hypertens (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu K, et al. Unique Transcriptional Programs Identify Subtypes of AKI. J Am Soc Nephrol 28, 1729–1740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vigolo E, et al. Canonical BMP signaling in tubular cells mediates recovery after acute kidney injury. Kidney Int 95, 108–122 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Marko L, et al. Tubular Epithelial NF-kappaB Activity Regulates Ischemic AKI. J Am Soc Nephrol 27, 2658–2669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Supavekin S, et al. Differential gene expression following early renal ischemia/reperfusion. Kidney Int 63, 1714–1724 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Yuen PS, Jo SK, Holly MK, Hu X & Star RA Ischemic and nephrotoxic acute renal failure are distinguished by their broad transcriptomic responses. Physiol Genomics 25, 375–386 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshida T, et al. Monitoring changes in gene expression in renal ischemia-reperfusion in the rat. Kidney Int 61, 1646–1654 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Humphreys BD, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2, 284–291 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Gerhardt LMS & McMahon AP Identifying Common Molecular Mechanisms in Experimental and Human Acute Kidney Injury. Semin Nephrol 42, 151286 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang-Panesso M & Humphreys BD Cellular plasticity in kidney injury and repair. Nat Rev Nephrol 13, 39–46 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Chang-Panesso M, et al. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J Clin Invest 129, 5501–5517 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humphreys BD Kidney injury, stem cells and regeneration. Curr Opin Nephrol Hypertens 23, 25–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindgren D, et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am J Pathol 178, 828–837 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angelotti ML, et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 30, 1714–1725 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Loverre A, et al. Increase of proliferating renal progenitor cells in acute tubular necrosis underlying delayed graft function. Transplantation 85, 1112–1119 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Peired AJ, et al. Acute kidney injury promotes development of papillary renal cell adenoma and carcinoma from renal progenitor cells. Sci Transl Med 12(2020). [DOI] [PubMed] [Google Scholar]
- 40.Scholz H, et al. Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat Rev Nephrol 17, 335–349 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Shen TH, et al. Snapshots of nascent RNA reveal cell- and stimulus-specific responses to acute kidney injury. JCI Insight 7(2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, et al. Cell-specific translational profiling in acute kidney injury. J Clin Invest 124, 1242–1254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doyle JP, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirita Y, Wu H, Uchimura K, Wilson PC & Humphreys BD Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A 117, 15874–15883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerhardt LMS, et al. Lineage Tracing and Single-Nucleus Multiomics Reveal Novel Features of Adaptive and Maladaptive Repair after Acute Kidney Injury. J Am Soc Nephrol 34, 554–571 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dixon EE, Wu H, Muto Y, Wilson PC & Humphreys BD Spatially Resolved Transcriptomic Analysis of Acute Kidney Injury in a Female Murine Model. J Am Soc Nephrol 33, 279–289 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinze C, et al. Single-cell transcriptomics reveals common epithelial response patterns in human acute kidney injury. Genome Med 14, 103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klocke J, et al. Urinary single-cell sequencing captures kidney injury and repair processes in human acute kidney injury. Kidney Int 102, 1359–1370 (2022). [DOI] [PubMed] [Google Scholar]
- 50.Rudman-Melnick V, et al. Single-Cell Profiling of AKI in a Murine Model Reveals Novel Transcriptional Signatures, Profibrotic Phenotype, and Epithelial-to-Stromal Crosstalk. J Am Soc Nephrol 31, 2793–2814 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balzer MS, et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat Commun 13, 4018 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Legouis D, et al. Single cell profiling in COVID-19 associated acute kidney injury reveals patterns of tubule injury and repair in human. bioRxiv, 2021.2010.2005.463150 (2021). [Google Scholar]
- 53.Jansen J, et al. SARS-CoV-2 infects the human kidney and drives fibrosis in kidney organoids. Cell Stem Cell 29, 217–231 e218 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuppe C, et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 589, 281–286 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lake BB, et al. An atlas of healthy and injured cell states and niches in the human kidney. Nature 619, 585–594 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gerhardt LMS, Liu J, Koppitch K, Cippa PE & McMahon AP Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc Natl Acad Sci U S A 118(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L, Besschetnova TY, Brooks CR, Shah JV & Bonventre JV Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16, 535–543, 531p following 143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedmann Angeli JP, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ide S, et al. Ferroptotic stress promotes the accumulation of pro-inflammatory proximal tubular cells in maladaptive renal repair. Elife 10(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linkermann A, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111, 16836–16841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Z, et al. XJB-5–131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis 11, 629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Dixon EE, Wu H & Humphreys BD Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab 34, 1977–1998 e1979 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Z, et al. Single-cell sequencing reveals homogeneity and heterogeneity of the cytopathological mechanisms in different etiology-induced AKI. Cell Death Dis 14, 318 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7, 684–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Humphreys BD Mechanisms of Renal Fibrosis. Annu Rev Physiol 80, 309–326 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Li L, Fu H & Liu Y The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol 18, 545–557 (2022). [DOI] [PubMed] [Google Scholar]
- 67.Tapmeier TT, et al. Pivotal role of CD4+ T cells in renal fibrosis following ureteric obstruction. Kidney Int 78, 351–362 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Anders HJ, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest 109, 251–259 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gewin L The many talents of transforming growth factor-beta in the kidney. Curr Opin Nephrol Hypertens 28, 203–210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Floege J, Eitner F & Alpers CE A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol 19, 12–23 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Livingston MJ, et al. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 19, 256–277 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu H, et al. Tenascin-C promotes acute kidney injury to chronic kidney disease progression by impairing tubular integrity via alphavbeta6 integrin signaling. Kidney Int 97, 1017–1031 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abedini A, et al. Spatially resolved human kidney multi-omics single cell atlas highlights the key role of the fibrotic microenvironment in kidney disease progression. bioRxiv, 2022.2010.2024.513598 (2022). [Google Scholar]
- 74.Fu H, et al. Tenascin-C Is a Major Component of the Fibrogenic Niche in Kidney Fibrosis. J Am Soc Nephrol 28, 785–801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou X, et al. Complement 3 activates the renal renin-angiotensin system by induction of epithelial-to-mesenchymal transition of the nephrotubulus in mice. Am J Physiol Renal Physiol 305, F957–967 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Nagao T, et al. Osteopontin plays a critical role in interstitial fibrosis but not glomerular sclerosis in diabetic nephropathy. Nephron Extra 2, 87–103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sato Y & Yanagita M Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm Regen 37, 17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coca SG Acute kidney injury in elderly persons. Am J Kidney Dis 56, 122–131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stevens LA, Viswanathan G & Weiner DE Chronic kidney disease and end-stage renal disease in the elderly population: current prevalence, future projections, and clinical significance. Adv Chronic Kidney Dis 17, 293–301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tonelli M, et al. Systematic review: kidney transplantation compared with dialysis in clinically relevant outcomes. Am J Transplant 11, 2093–2109 (2011). [DOI] [PubMed] [Google Scholar]
- 81.Bastani B The present and future of transplant organ shortage: some potential remedies. J Nephrol 33, 277–288 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Ponticelli C, Villa M, Cesana B, Montagnino G & Tarantino A Risk factors for late kidney allograft failure. Kidney Int 62, 1848–1854 (2002). [DOI] [PubMed] [Google Scholar]
- 83.Kwon H, et al. Pure T-cell mediated rejection following kidney transplant according to response to treatment. PLoS One 16, e0256898 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kant S, Dasgupta A, Bagnasco S & Brennan DC BK Virus Nephropathy in Kidney Transplantation: A State-of-the-Art Review. Viruses 14(2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Einecke G, et al. Factors associated with kidney graft survival in pure antibody-mediated rejection at the time of indication biopsy: Importance of parenchymal injury but not disease activity. Am J Transplant 21, 1391–1401 (2021). [DOI] [PubMed] [Google Scholar]
- 86.Requiao-Moura LR, Durao Mde S. Junior, Matos AC & Pacheco-Silva A Ischemia and reperfusion injury in renal transplantation: hemodynamic and immunological paradigms. Einstein (Sao Paulo) 13, 129–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mbianda C, El-Meanawy A & Sorokin A Mechanisms of BK virus infection of renal cells and therapeutic implications. J Clin Virol 71, 59–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Halloran PF, et al. Molecular diagnosis of ABMR with or without donor-specific antibody in kidney transplant biopsies: Differences in timing and intensity but similar mechanisms and outcomes. Am J Transplant 22, 1976–1991 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cippa PE, et al. Transcriptional trajectories of human kidney injury progression. JCI Insight 3(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Famulski KS, et al. Molecular phenotypes of acute kidney injury in kidney transplants. J Am Soc Nephrol 23, 948–958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.O’Connell PJ, et al. Biopsy transcriptome expression profiling to identify kidney transplants at risk of chronic injury: a multicentre, prospective study. Lancet 388, 983–993 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ke B, Fan C, Yang L & Fang X Matrix Metalloproteinases-7 and Kidney Fibrosis. Front Physiol 8, 21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nankivell BJ, et al. The natural history of chronic allograft nephropathy. N Engl J Med 349, 2326–2333 (2003). [DOI] [PubMed] [Google Scholar]
- 94.Loupy A, et al. The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am J Transplant 20, 2318–2331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jeong HJ Diagnosis of renal transplant rejection: Banff classification and beyond. Kidney Res Clin Pract 39, 17–31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chandran S & Mannon RB T cell-mediated rejection in kidney transplant recipients: The end(point) is also the beginning. Am J Transplant 22, 683–684 (2022). [DOI] [PubMed] [Google Scholar]
- 97.Bohmig GA, Eskandary F, Doberer K & Halloran PF The therapeutic challenge of late antibody-mediated kidney allograft rejection. Transpl Int 32, 775–788 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ho J, et al. Effectiveness of T cell-mediated rejection therapy: A systematic review and meta-analysis. Am J Transplant 22, 772–785 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Townamchai N & Avihingsanon Y Updated management for antibody-mediated rejection: opportunity to prolong kidney allograft survival. Curr Opin Nephrol Hypertens 32, 13–19 (2023). [DOI] [PubMed] [Google Scholar]
- 100.Halloran PF, et al. Discovering novel injury features in kidney transplant biopsies associated with TCMR and donor aging. Am J Transplant 21, 1725–1739 (2021). [DOI] [PubMed] [Google Scholar]
- 101.Halloran PF, et al. Archetypal Analysis of Injury in Kidney Transplant Biopsies Identifies Two Classes of Early AKI. Front Med (Lausanne) 9, 817324 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lamarthee B, et al. Transcriptional and spatial profiling of the kidney allograft unravels a central role for FcyRIII+ innate immune cells in rejection. Nat Commun 14, 4359 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Y, et al. Single-cell RNA-Seq analysis identified kidney progenitor cells from human urine. Protein Cell 12, 305–312 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Abedini A, et al. Urinary Single-Cell Profiling Captures the Cellular Diversity of the Kidney. J Am Soc Nephrol 32, 614–627 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Latt KZ, et al. Urine Single-Cell RNA Sequencing in Focal Segmental Glomerulosclerosis Reveals Inflammatory Signatures. Kidney Int Rep 7, 289–304 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cheung MD, et al. Single-Cell RNA Sequencing of Urinary Cells Reveals Distinct Cellular Diversity in COVID-19-Associated AKI. Kidney360 3, 28–36 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goerlich N, et al. Kidney transplant monitoring by urinary flow cytometry: Biomarker combination of T cells, renal tubular epithelial cells, and podocalyxin-positive cells detects rejection. Sci Rep 10, 796 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Galante NZ, et al. Noninvasive immune monitoring assessed by flow cytometry and real time RT-PCR in urine of renal transplantation recipients. Transpl Immunol 16, 73–80 (2006). [DOI] [PubMed] [Google Scholar]
- 109.Doke T, et al. Single-cell analysis identifies the interaction of altered renal tubules with basophils orchestrating kidney fibrosis. Nat Immunol 23, 947–959 (2022). [DOI] [PubMed] [Google Scholar]
- 110.Amor C, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Docherty MH, Baird DP, Hughes J & Ferenbach DA Cellular Senescence and Senotherapies in the Kidney: Current Evidence and Future Directions. Front Pharmacol 11, 755 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mylonas KJ, et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci Transl Med 13(2021). [DOI] [PubMed] [Google Scholar]
- 113.Dhillon P, et al. The Nuclear Receptor ESRRA Protects from Kidney Disease by Coupling Metabolism and Differentiation. Cell Metab 33, 379–394 e378 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao J, et al. Genomic integration of ERRgamma-HNF1beta regulates renal bioenergetics and prevents chronic kidney disease. Proc Natl Acad Sci U S A 115, E4910–E4919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taguchi K, et al. Cyclin G1 induces maladaptive proximal tubule cell dedifferentiation and renal fibrosis through CDK5 activation. J Clin Invest 132(2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lovisa S, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21, 998–1009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]