

Abstract
The design and use of Synthetic Communities, or SynComs, represents one of the most promising strategies for disentangling the complex interactions within microbial communities, and between these communities and their hosts. Compared to natural communities, these simplified consortia provide the opportunity to study ecological interactions at tractable scales, as well as facilitating reproducibility and fostering interdisciplinary science. However, the effective implementation of the SynCom approach requires several important considerations regarding the development and application of these model systems. There are also emerging ethical considerations when both designing and deploying SynComs in clinical, agricultural, or environmental settings. Here, we outline current best practices in developing, implementing and evaluating SynComs across different systems, including a focus on important ethical considerations for SynCom research.
Introduction
Microbial organisms represent the bulk of the diversity present on Earth and as sequencing technologies and computational tools have advanced, we have become increasingly aware of the important role they play across systems. As these organisms live in complex and often highly dynamic communities, there is a clear need to understand when and why microbial taxa coexist, how they interact with one another, and how these interactions translate to function – especially given that these outcomes often cannot be predicted based on knowledge of individual taxa. One of the most promising strategies to disentangle these complex relationships within communities and between communities and their hosts is the design of model consortia, generally referred to as synthetic communities, or SynComs.
SynComs can be defined as “consortia of microorganisms designed to mimic, at some scale, the observed functions and structure of the microbiome in natural conditions”1. This approach was first pioneered in 1965 when Russel Schaedler colonized germ-free mice with defined bacterial isolates2, although the term “Synthetic Community” was first used, to the best of our knowledge, by Kim et al. in 2008 to describe a three species community comprised of soil bacteria3. The approach has since gained popularity in both plant and human systems4–6. Historically, SynComs have been composed of bacterial species and have primarily focused on coexistence, competition, cross-feeding and functions encoded on bacterial genomes or plasmids. Though less common, researchers can also include fungal, protist, archaeal, and viral taxa within these experimental communities7,8. Given their popularity, most of the examples included throughout this piece will focus on bacterial SynComs, though we acknowledge the importance of these multi-kingdom community approaches. It is further important to delineate between the types of synthetic communities discussed in this piece: those composed of naturally sourced organisms meant to model some functions of their originating communities9,10, versus those that represent a group of Synthetic Organisms designed to perform a certain function11,12, usually via genetic engineering, with the latter being more typically used in Synthetic Biology.
Compared to natural communities, SynComs provide several advantages to researchers. Their defined membership enables the reconstitution of identical communities across experiments, allowing reproducibility across time and labs13. Like the development of model systems in biology, this approach allows researchers to integrate knowledge of a given system to accelerate progress and foster interdisciplinary science. While most research on microbiomes relies on destructive sampling, SynComs allow for repeated manipulation of the community to dissect the role of individual species (and their abundances) in its assembly and function.
The SynCom approach can be used to facilitate answers to fundamental research questions, as well as for specific applications, and is equally suitable for host-associated and free-living microbial communities (Figure 1). In both cases, SynComs provide excellent opportunities to model ecological interactions at tractable scales and can offer key insights into community dynamics. As applied systems, non-host associated (environmentally-derived) SynComs can be harnessed for bioremediation, chemical engineering, and biofuel production. Host-associated SynComs can be used to increase crop production in agricultural settings, through both growth promotion and disease protection, as well as to understand and treat microbiome-associated animal diseases. In the context of human health, SynComs can be designed to treat disease and dysbiosis, such as enrichment of opportunistic pathogens, which are associated with infectious (e.g. Clostridioides difficile infection14) or metabolic (e.g. diabetes mellitus15) origins. A better fundamental understanding of how within-microbiome interactions affect the balance of microbial taxa and ability of resident communities to resist pathogen invasion will support the development of targeted microbial interventions to reduce such community disturbances. In all cases, SynComs address the need to understand the host-microbe and microbe-microbe interactions underlying these phenotypes, and act as potential interventions for re-establishing stable microbial communities.
Figure 1. Dual continuums of “question” and “system” for SynCom research.
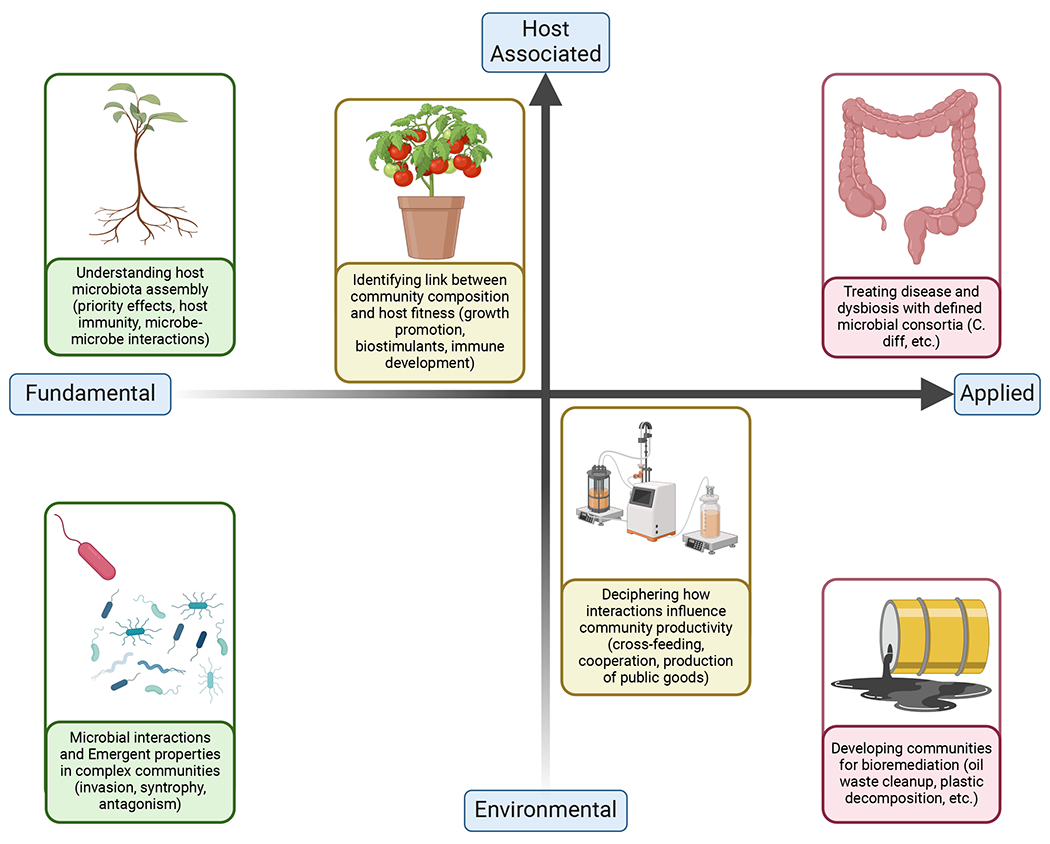
Research questions using SynComs can range from fundamental questions or basic science, that is, trying to understand the rules and functioning underpinning different systems, to applied questions. Here communities are designed to fulfill certain purposes, for example, [AU: please complete this sentence using a brief example from the figure]. Likewise, the system being used can be placed on a continuum from environmental to free living and host-associated microbial communities.
In the following sections we outline current best practices in developing, implementing and evaluating SynComs, including a focus on important ethical considerations for SynCom research and application (Figure 2). This piece is not a comprehensive review of SynCom-associated studies, nor is it the first to outline approaches to developing SynComs1,4,5,11,16,17, but rather serves as a cross-systems primer for those hoping to develop a SynCom for research or application, or for evaluating SynCom-associated work. The information provided herein represents a comprehensive starting point for those unfamiliar with the field, and citations have been chosen carefully to give readers the opportunity to follow up on any points, or explore specific systems, in greater detail.
Figure 2. Flow diagram of approaches used when designing, evaluating and deploying a SynCom.
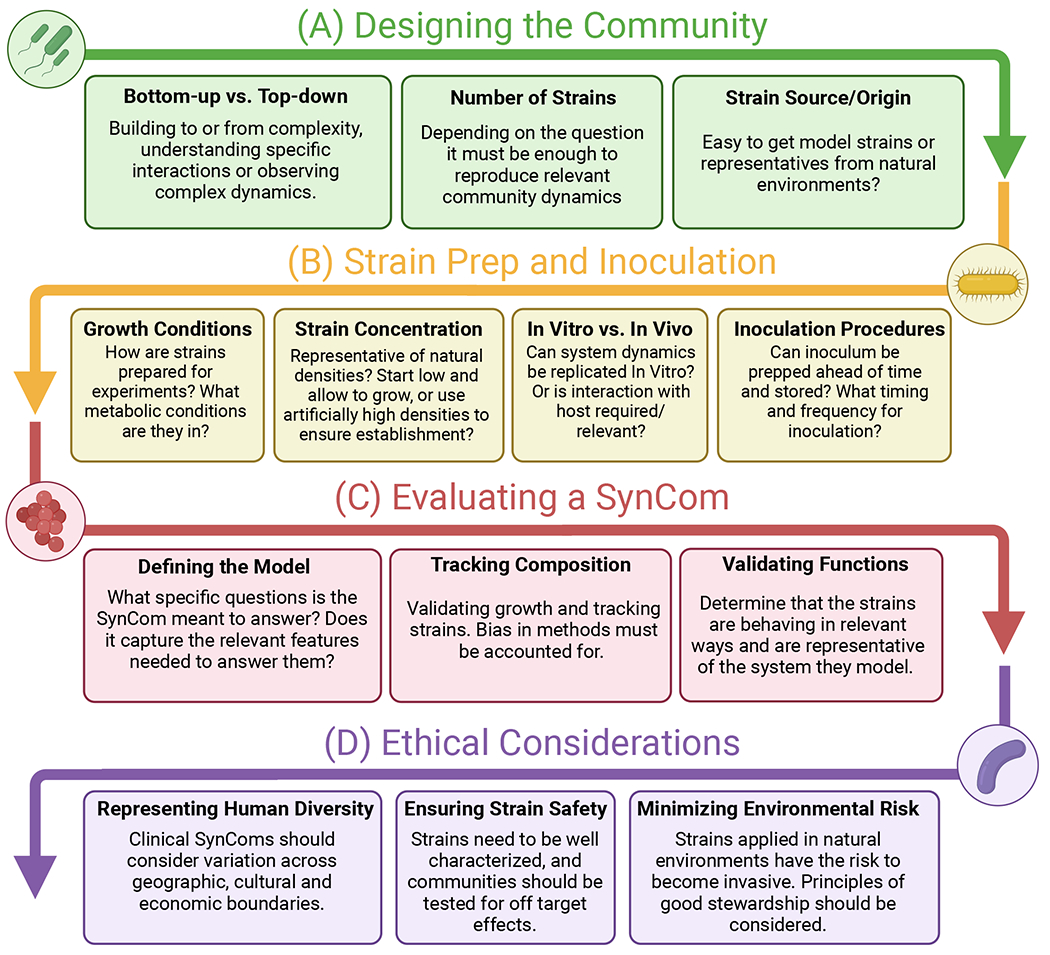
(A) All studies begin by designing the community (green). SynCom design can proceed from either Bottom-up (increase complexity through iterations) or Top-down (reduce complexity through iterations) approaches. When designing communities it is important to consider the number of strains needed to be relevant, as well as the sourcing of those strains. (B) Strains are then prepared and used for inoculation (yellow). Important considerations include the strain growth conditions, applied concentration, experimental system and methods of inoculation. (C) After a SynCom has been implemented, it is critical to evaluate if it provides relevant information about the system being modeled. To do so, the questions must first be well defined, after which the relevant features can be assessed by tracking the composition and functioning of the community. (D) When designing and applying SynComs across both human and environmental systems, there are important ethical considerations to take into account. In human systems, these communities should be representative of diversity seen across geographic, cultural and economic boundaries, and communities applied to patients should be tested for off target effects. When applying a SynCom to a natural system, care must be taken to ensure that these species do not spread and become invasive.
Designing the community
Many strategies exist to design SynComs depending on the objectives and research system of interest. These can be summarized as a continuum from bottom-up to top-down designs (Figure 3). The bottom-up approach relies on the assembly of a specific set of microbial strains of interest, chosen due to their suitability to some criteria of the study (including the feasibility of isolating and culturing them) (Figure 3a). In this case, phenotypically and genomically defined strains are typically combined to characterize microbial interaction dynamics and mechanism, community functions, and emergent properties of known strain assemblages. These simple SynComs have facilitated the discovery of inter-microbial antagonism pathways between microorganisms18, as well as microbial cross-feeding and degradative synergies that are critical for ecosystem functioning19. For example, the OMM mouse community, shows how microbial interactions and cross-feeding can impact their host by shaping their exposure to certain metabolic by-products20. This approach is crucial for identifying the molecular mechanisms driving microbial interactions, but it relies on simplification of the microbial diversity and environmental conditions. Strains are often selected because of the extensive knowledge available on their genetic and phenotypic attributes (i.e. model strains) and not because they co-exist in nature (i.e. from different sources of isolation). Moreover, they are selected because they can grow alone, leading to bias in the types of strains being included. Additionally, recent work has demonstrated that strains coexisting within a stable complex community might fail to coexist in pairwise co-cultures, showing that multi-species coexistence is an emergent phenomenon21.
Figure 3. Examples of bottom-up and top-down design approaches for SynComs.
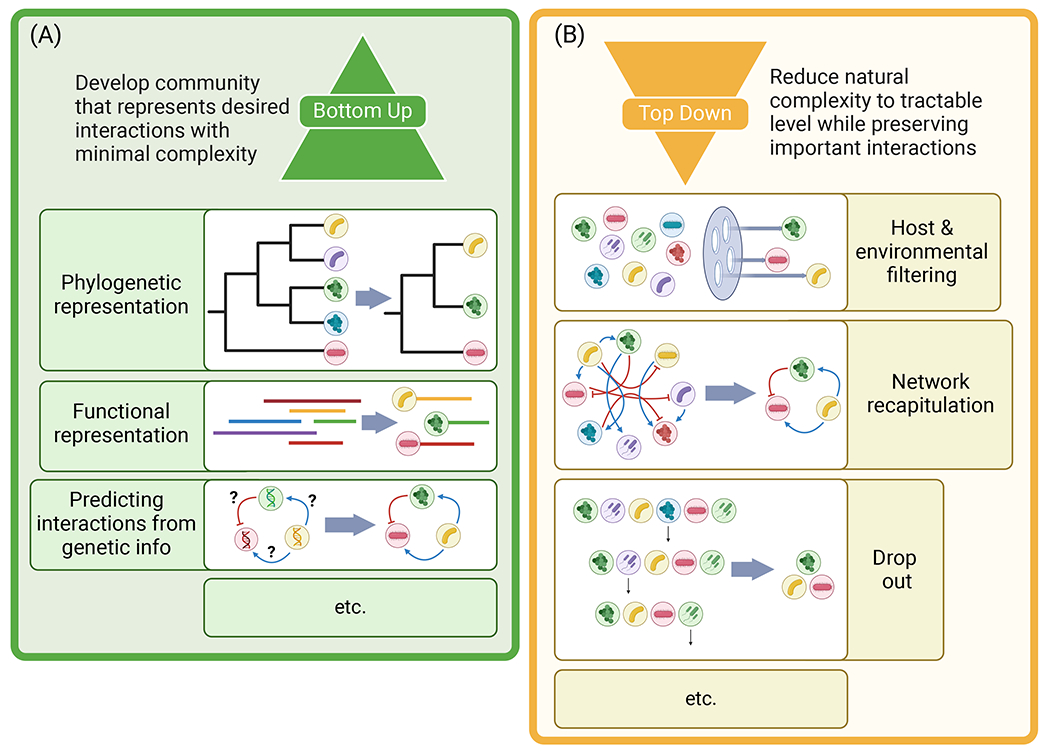
(A) Bottom-up approaches can include selecting strains that represent the phylogenetic diversity of the natural community at some level, identifying strains that perform some functions of interest in the natural community, or through the prediction of key interactions in the community that a researcher might want to model. (B) Top-down designs can employ host or environmental filtering. This is where a larger community is applied into the study environment and only those strains that pass some growth or persistence metrics are included. It can also be achieved through the recapitulation of key features in community interaction networks or through a sequential drop out, where strains are sequentially removed in order to select the minimal complexity required to model the interactions of interest. In practice these approaches are not mutually exclusive, and researchers can choose to employ a combination of bottom-up or top-down strain selection approaches to define their communities.
In contrast to building SynComs from characterized strains, a top-down design relies on assembling a large diversity of strains (for example, by sampling of a natural source) and reducing their complexity in a stepwise fashion to gain insight into the sub-components of the community (Figure 3b). The objectives for this approach can be wide-ranging, from mimicking the natural phylogenetic diversity, to identifying core taxa or core functions of a microbiome, to understanding the specific role of taxa of interest in complex assemblages. Simplification of the initial community can be achieved through natural or knowledge-driven filtering approaches or bottlenecks. One such straightforward approach is to let the environment or host ‘filter’ or select for strains capable of colonizing and surviving in/on it from the initial strain pool22. Complementary approaches include performing experimental evolution to enrich taxa or functions of interest over multiple cycles of reinoculation23 or applying random filtering such as serial dilution24 to create random subsets of the wider community for subsequent exploration. Alternatively, knowledge-driven filtering can be performed based on existing data that informs add-in/drop-out of taxonomic or functional groups of interest, which can be identified from functional assays or metagenomes25, and/or microbial hubs identified via co-occurrence networks26. These bottom-up and top-down approaches for SynCom development are complementary and necessary to eventually “meet in the middle”, allowing researchers to understand and predict microbiota assembly and functions across scales of complexity.
A crucial aspect in SynCom design is the meticulous sourcing and selection of the strains. Depending on the objectives of the study, strains can be sourced from the study system (e.g. same soil or individual), from across environments, or even from (inter)national strain collections. It is important to acknowledge that the sourcing of strains poses a significant limitation to the SynCom approach, as even complex SynComs may lack certain keystone taxa (i.e. those with outsized impact on community stability or function27) that might be essential for realistic community dynamics4. During the selection process, careful consideration must be given to the number of strains that align with the project’s goals and system complexity28. Factors like ease of cultivation and growth rates play a role in determining the feasibility of incorporating specific strains, but it is critical to appreciate the varying growth capabilities of strains under different conditions (i.e. acknowledging that selection based on one set of criteria likely reduces success or function of the SynCom under different conditions). Mitigating these effects can be achieved by aligning media and culture conditions with the specific requirements of the studied system29. Additionally, it is important to note that the convenience of handling specific strains does not always correspond directly to their significance within the system.
Strain preparation and Inoculation
Various factors must be considered to standardize the use of SynComs across experiments and studies (Box 1). First, the choice between in vitro and in vivo systems is fundamental, necessitating consideration of the ecological relevance and applicability of the chosen system. When studying host-associated communities, an in vitro approach may be most appropriate (at initially for hypothesis generation and when the relevant interactions are solely inter-microbial, but if the interactions of interest are host-microbial, an in vivo approach is necessary, Next, the impact of inoculum concentration must be considered given the density- and frequency-dependent nature of many microbial interactions30. This can be achieved by inoculating at ecologically relevant densities or inoculating at lower densities and allowing the community to establish in situ. There may be compelling reasons to increase the concentration, especially when the SynCom is required to outcompete the resident community (host or environment) within a coalescence framework31. Establishing standardized protocols for SynCom inoculation is essential, including the timing and frequency of inoculation32, growth prior to inoculation (i.e. physiological state of the strains33 and media composition), and subsequent sampling of community dynamics. Additionally, it is necessary to determine if these communities can be stored throughout the duration of the study34, or if they need to be remade each time to ensure that they have the same concentration, evenness, and physiological state.
Box 1. Resources and Best Practices for SynCom Design.
Resources
Computational resources for processing and preparing amplicon sequencing data (short or long reads): dada243, Mothur67, UNOISE368, UCHIME369, Decontam70, LULU71
Resources for tracking and characterizing SynCom/resident microbiota: vegan72, Phyloseq73, MicroViz74, Microbiome R Package75.
Sequencing pipelines for assembling and annotating strain genomes: SPADES76 or Unicycler77 are considered best practices for assembly, while BAKTA78 is the defacto option for annotation.
Methods for identifying keystone species, core or key functional taxa: Abundance Occupancy curves79, LIMITS80, the DKI machine learning framework81, SPIEC-EASI82
Resources for the automated design and predictive effects of synthetic communities: used by Karkaria et al.83 , Toju et al.84 and Paredes et al.85
Best Practices
Methods used to select strains should be documented and published in work referencing the SynCom.
Strains should undergo whole-genome sequencing and these data should be made publicly available.
Strains within SynComs should be made available to other researchers via deposition in a public collection such as ATCC, DSM, CBS, CIRM etc.
Within fields, methods for strain preparation and inoculation should be standardized (i.e. growth conditions prior to experimentation, strain inoculation density, sampling methods during experiment).
Integrity of strain freezer stocks should be maintained, and best practices followed, as it is vital to prevent these strains from becoming lab adapted. Freeze thaw cycles and the number of passages of a strain in non-native conditions should be minimized and documented. Strains should periodically be validated for identity and redundant copies of the library should be stored in separate locations.
Evaluating a SynCom
When designing a SynCom it is important to remember the oft cited quote by George Box, “all models are wrong, some are useful”. SynComs, after all, are meant to be tractable models of natural systems. The question is not whether they represent those systems perfectly, but rather if they represent the features of the system that the researchers aim to study. It is therefore of critical importance that the SynCom is designed with specific questions and context in mind, that these questions are well articulated, and that the features selected to be represented in that model are relevant to the system in its natural or applied environment. For fundamental questions, employing a simplified SynCom can prove highly advantageous in demonstrating the feasibility or existence of specific functions or interactions (Fig. 1). For example, Yang et al.35 employed a community consisting of 6 species from the same genus, and although highly simplified compared to natural communities, this SynCom allowed them to test the role of community diversity in robustness against invasion, though of course a more complex community might reveal additional contributing factors. More complex questions might require more complex communities. To identify a conserved set of host (Arabidopsis thaliana) genes that are upregulated in response to colonization by bacteria, Maier et al.36 constructed a SynCom consisting of 38 strains representing the breadth of phyla naturally associated with the plant. Establishing that this was a general plant response required a SynCom that captured more of the natural diversity that is found to associate with their host. While these two communities are quite different, each is sufficient to represent models of the interactions of interest.
When evaluating the effectiveness of a SynCom, it is important to focus on the system being modeled rather than the composition or complexity compared to other established communities (Fig. 3). For every SynCom study, there will be a tradeoff between tractability and relevance. Simple communities are easier to work with, but less representative of real systems. They run the risk of missing emergent properties and context-specific outcomes, such as higher order competitive interactions, priority effects, or the impact of rare keystone members, making them potentially less generalizable. In contrast, while more complex models might capture these effects, they can be harder to implement, show lower reproducibility, and offer significant challenges when it comes to data interpretation.
Researchers must think critically about their questions and ensure that their community is sufficiently designed to answer them, while also transparently communicating the limitations of their model. To ensure your SynCom aligns with the questions being asked, methods can be implemented to validate community performance. This includes understanding what features of your community you need to validate (sequencing depth, growth, survival, interactions, productivity, host phenotypes, ecosystem function etc.) and identifying methods to do so (see Box 1 for suggested computational resources). In light of new methods for barcoding/labeling strains37 and multi-omics approaches, both validating composition (or change in composition) of the SynCom and evaluating the functions of the SynCom can be done simultaneously, but depending on if the study is focusing on ecological (validate composition) or functional (validate function) properties of the community both approaches might not be needed.
The classic approach to understanding bacterial community composition is through 16S rRNA gene sequencing (though other targets such as gyrB and rpoB can be used). However, this method only resolves relative abundance of the bacteria present, and can run into issues with copy number variation, primer bias, as well as difficulties delineating at the species level and the inability to distinguish between living and dead bacteria. When employing a well-defined SynCom, many of these issues can be addressed or avoided by employing alternative/additional methods. Plating, if community members can be morphologically distinguished, allows for a relatively cheap and effective determination of living bacterial numbers and diversity. For more complex communities, absolute abundance can also be approximated through qPCR38,39or ddPCR40 using general or species-specific primers. Further corrections for copy number can be employed if the SynCom member genomes have been sequenced41, and primer bias can be addressed through comparisons to known mock communities42, for example, using the Zymo community standards. When addressing strain resolution, methods like DADA243 can distinguish between strains if they differ by at least one base pair in the sequenced region (though this is not the case for all taxonomic groups). Other approaches such as long read sequencing44 or metagenomic barcoding can be used to distinguish more closely related strains. Finally, live/dead PCR using PMA has been employed to remove relic DNA from sequencing samples, limiting quantification to cells that are still intact (and therefore likely alive) at the time of sequencing45.
Perhaps more complicated is determining that a SynCom is performing the functions of interest. When evaluating these functions, more complex “Omics” enabled methods can be employed. Metagenomics can be used to quantify the genetic and functional composition of the community, including both gene presence and relative abundance46, and could be used to determine if these are representative of the natural system. Further approaches could be applied to approximate a community metagenome by normalizing genome assemblies (for gene content) against 16S rRNA gene sequencing data. RNAseq can be applied to determine whether the host is responding to the SynCom under the conditions of interest, as well as evaluating the microbial responses through community-wide RNAseq47. Additionally, both host and community can be evaluated simultaneously using dual RNA-seq48. The extent to which host and community responses are reflective of the actual system can be further quantified by comparing gene expression in the natural and model systems49.
Ethical considerations in SynCom development and application
When developing SynComs to model or treat human disease it is important that these systems are representative of diverse human groups (i.e. those living across rural and urban settings, from different geographic areas, and across the socio-economic continuum) and focus on both well-studied and typically neglected diseases50. Their design should consider that microbiome composition can vary across geographic and economic boundaries51,52 , either by including these groups when defining important strains or by designing communities to specifically represent them. When a SynCom is intended for clinical application it must be composed of known, culturable, and reproducible communities that are verified to be free from harmful pathogens or virulence factors that could pose risks to human health. This is now possible because the genomes and features of the community (e.g. metatranscriptome, metaproteome) can be more easily characterized. Even after this, however, rigorous multi-center, longitudinal cohort studies are required to identify SynCom off-target effects, such as unintentional transfer of pathobionts from donor to recipient53, inadvertent propagation of genes (e.g., antimicrobial resistance genes), unintended impacts on the endogenous microbiota such as competitive enrichment of other pathogens54, or unanticipated effects on host metabolism or susceptibility to disease. Finally, it is important to consider the long-term effects on the resident community, as well as the potential for transmission beyond the original recipient, including horizontal transmission within the household, as well as vertical transmission from parent to child55.
Likewise, there are several critical factors that must be considered when SynComs are to be used in natural or agricultural settings. We are optimistic that the field can learn from, and avoid, past mistakes made with novel biological technologies (i.e. antibiotics or species introduction biocontrol) as SynComs become more widely applied. Given the high densities at which microbial amendments are typically introduced, these impacts could be more disruptive than ‘natural’ microbial dispersal56. Moreover, since many members of SynComs have been specifically selected to grow well (often across diverse habitats), they should be considered to pose a risk for invasion, possibly leading to loss of natural microbial diversity. As such, the development and deployment of SynComs outside of the laboratory should adhere to the four principles of ethics (do good, don’t harm, respect, and act justly) and the eleven guiding principles for microbiome research57. Practically, studies should be undertaken to assess the associated risks under the conditions that these SynComs might be applied. Further, while biocontainment has been a long-standing focus for engineered microbial organisms58, it has received far less attention for Syncoms and probiotics more generally. As use of Syncoms in medical, agricultural, and environmental settings becomes more prominent, this will need more thorough assessment.
Beyond SynCom release, there are also ethical considerations around their use in research. These include embracing FAIR data principles to ensure that all data underlying published findings are findable, accessible, interoperable and reusable59. There are numerous data and strain repositories that can be used to achieve this goal, but true reusability requires useful metadata59–61 (Box 1). The National Microbiome Data Collaborative (https://microbiomedata.org/) offers many examples of how this can be done and is itself an exemplary effort of how to bring these issues to the attention of the research community.
Future Perspectives
In summary, while SynComs represent an important resource for increasing our fundamental knowledge of microbial systems, as well as a valuable applied tool, there are critical considerations when designing and implementing them. While this piece provides a high-level overview of those considerations, more system-specific reading will certainly be useful as the reader begins to construct or evaluate a SynCom. We suggest the following reviews as excellent next steps depending on the specific system in question; plant5,17 (including the review by Northen et al., in this issue62 ) animal63, agriculture1,64, human4,16,65.
Despite the progress made in developing the SynCom approach, the field is still in its infancy and researchers must continue to collaboratively establish and share best practices. As research becomes more collaborative and more standardized, the field may move towards “model” SynComs for use across research groups. However, it is essential to first identify the most effective systems and communities, and in doing so we will likely need to expand our efforts to include less culturable organisms, as well as increasing diversity across kingdoms and trophic levels. Additionally, as more research groups begin working with SynComs, it is imperative to explore methods for integrating findings across different models to uncover common principles and patterns, including standardizing the reporting of metadata associated with these studies. Likewise, the field should develop best practices for calibrating and testing the effectiveness of communities as models for specific research questions. Looking ahead, the potential role of artificial intelligence66 in advancing the development and study of SynComs should also be considered, as tools to accomplish this are beginning to be implemented.
Acknowledgements
This manuscript was inspired by an ASM Microbe panel discussion involving many of the authors and organized by the ASM EEB track in 2023, we would like to specifically thank Benjamin Callahan for early discussions on this topic. EM would like to acknowledge funding from the NSF EAGER award # 1838299 as well as support from the NSF Postdoctoral Research Fellowships in Biology award # 2209151. BK is a is a Chan Zuckerberg San Francisco Biohub investigator. BJ and KP would like to acknowledge funding from the National Institutes of Health award # U19 AI157981. GA and MS were supported by the 3rd Programme for Future Investments (France 2030) and operated by the SUCSEED project (ANR- 20- PCPA-0009) funded by the ‘Growing and Protecting crops Differently’ French Priority Research Program (PPR-CPA), part of the national investment plan operated by the French National Research Agency (ANR). LPM would like to acknowledge funding from Conahcyt (Consejo Nacional de Humanidades, Ciencias y Tecnologías) with award #’s A1-S-9889 and CBF2023-2024-2642.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.de Souza RSC, Armanhi JSL & Arruda P. From Microbiome to Traits: Designing Synthetic Microbial Communities for Improved Crop Resiliency. Frontiers in Plant Science 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaedler RW, Dubs R & Costello R ASSOCIATION OF GERMFREE MICE WITH BACTERIA ISOLATED FROM NORMAL MICE. J Exp Med 122, 77–82 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim HJ, Boedicker JQ, Choi JW & Ismagilov RF Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proceedings of the National Academy of Sciences 105, 18188–18193 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Leeuwen PT, Brul S, Zhang J & Wortel MT Synthetic microbial communities (SynComs) of the human gut: design, assembly, and applications. FEMS Microbiol Rev 47, fuad012 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vorholt JA, Vogel C, Carlström CI & Müller DB Establishing Causality: Opportunities of Synthetic Communities for Plant Microbiome Research. Cell Host & Microbe 22, 142–155 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Lebeis SL et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349, 860–864 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Thonar C, Frossard E, Šmilauer P. & Jansa J Competition and facilitation in synthetic communities of arbuscular mycorrhizal fungi. Molecular Ecology 23, 733–746 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Durán P et al. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell 175, 973–983.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehlferber EC et al. Phyllosphere microbial associations improve plant reproductive success. Front Plant Sci 14, 1273330 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flores-Núñez VM et al. Synthetic Communities Increase Microbial Diversity and Productivity of Agave tequilana Plants in the Field. Phytobiomes Journal 7, 435–448 (2023). [Google Scholar]
- 11.Johns NI, Blazejewski T, Gomes AL & Wang HH Principles for designing synthetic microbial communities. Current Opinion in Microbiology 31, 146–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarty NS & Ledesma-Amaro R Synthetic Biology Tools to Engineer Microbial Communities for Biotechnology. Trends Biotechnol 37, 181–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasse J et al. Multilab EcoFAB study shows highly reproducible physiology and depletion of soil metabolites by a model grass. New Phytologist 222, 1149–1160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Theriot CM & Young VB Interactions Between the Gastrointestinal Microbiome and Clostridium difficile. Annual Review of Microbiology 69, 445–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singer-Englar T, Barlow G & Mathur R Obesity, diabetes, and the gut microbiome: an updated review. Expert Review of Gastroenterology & Hepatology 13, 3–15 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Vázquez-Castellanos JF, Biclot A, Vrancken G, Huys GR & Raes J Design of synthetic microbial consortia for gut microbiota modulation. Current Opinion in Pharmacology 49, 52–59 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Marín O, González B & Poupin MJ From Microbial Dynamics to Functionality in the Rhizosphere: A Systematic Review of the Opportunities With Synthetic Microbial Communities. Front. Plant Sci 12, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson SB, Bertolli SK & Mougous JD Interbacterial antagonism: at the center of bacterial life. Curr Biol 30, R1203–R1214 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Souza G et al. Ecology and evolution of metabolic cross-feeding interactions in bacteria. Natural Product Reports 35, 455–488 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Pérez Escriva P, Fuhrer T & Sauer U Distinct N and C Cross-Feeding Networks in a Synthetic Mouse Gut Consortium. mSystems 7, e01484–21 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang C-Y, Bajić D, Vila JCC, Estrela S & Sanchez A Emergent coexistence in multispecies microbial communities. Science 381, 343–348 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Finkel OM et al. A single bacterial genus maintains root growth in a complex microbiome. Nature 587, 103–108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anand G, Goel V, Dubey S & Sharma S Tailoring the rhizospheric microbiome of Vigna radiata by adaptation to salt stress. Plant Growth Regul 93, 79–88 (2021). [Google Scholar]
- 24.Auchtung JM, Preisner EC, Collins J, Lerma AI & Britton RA Identification of Simplified Microbial Communities That Inhibit Clostridioides difficile Infection through Dilution/Extinction. mSphere 5, e00387–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar N, Hitch TCA, Haller D, Lagkouvardos I & Clavel T MiMiC: a bioinformatic approach for generation of synthetic communities from metagenomes. Microbial Biotechnology 14, 1757–1770 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caballero-Flores G, Pickard JM & Núñez G Microbiota-mediated colonization resistance: mechanisms and regulation. Nat Rev Microbiol 21, 347–360 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee S, Schlaeppi K & van der Heijden MGA Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol 16, 567–576 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Emmenegger B et al. Identifying microbiota community patterns important for plant protection using synthetic communities and machine learning. Nat Commun 14, 7983 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerna D, Clara D, Allwardt D, Mitter B & Roach T Tailored Media Are Key to Unlocking the Diversity of Endophytic Bacteria in Distinct Compartments of Germinating Seeds. Microbiology Spectrum 10, e00172–22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abisado RG, Benomar S, Klaus JR, Dandekar AA & Chandler JR Bacterial Quorum Sensing and Microbial Community Interactions. mBio 9, 10.1128/mbio.02331-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocca JD, Muscarella ME, Peralta AL, Izabel-Shen D & Simonin M Guided by Microbes: Applying Community Coalescence Principles for Predictive Microbiome Engineering. mSystems 6, 10.1128/msystems.00538-21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Debray R et al. Priority effects in microbiome assembly. Nat Rev Microbiol 20, 109–121 (2022). [DOI] [PubMed] [Google Scholar]
- 33.Mutlu A, Kaspar C, Becker N & Bischofs IB A spore quality-quantity tradeoff favors diverse sporulation strategies in Bacillus subtilis. ISME J 14, 2703–2714 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parnell JJ, Vintila S, Tang C, Wagner MR & Kleiner M Evaluation of ready-to-use freezer stocks of a synthetic microbial community for maize root colonization. Microbiology Spectrum 12, e02401–23 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang T et al. Resource availability modulates biodiversity-invasion relationships by altering competitive interactions: Resource availability modulates biodiversity. Environmental Microbiology 19, 2984–2991 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Maier BA et al. A general non-self response as part of plant immunity. Nat. Plants 7, 696–705 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ordon J et al. Chromosomal barcodes for simultaneous tracking of near-isogenic bacterial strains in plant microbiota. Nat Microbiol 9, 1117–1129 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jian C, Luukkonen P, Yki-Järvinen H, Salonen A & Korpela K Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLOS ONE 15, e0227285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zemb O et al. Absolute quantitation of microbes using 16S rRNA gene metabarcoding: A rapid normalization of relative abundances by quantitative PCR targeting a 16S rRNA gene spike-in standard. Microbiologyopen 9, e977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morella NM, Yang SC, Hernandez CA & Koskella B Rapid quantification of bacteriophages and their bacterial hosts in vitro and in vivo using droplet digital PCR. Journal of Virological Methods 259, 18–24 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Kembel SW, Wu M, Eisen JA & Green JL Incorporating 16S Gene Copy Number Information Improves Estimates of Microbial Diversity and Abundance. PLOS Computational Biology 8, e1002743 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brooks JP et al. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiology 15, 66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Callahan BJ et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Callahan BJ et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Research 47, e103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carini P et al. Effects of Spatial Variability and Relic DNA Removal on the Detection of Temporal Dynamics in Soil Microbial Communities . mBio 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Souza RSC, Armanhi JSL, Damasceno N de B, Imperial J. & Arruda P. Genome Sequences of a Plant Beneficial Synthetic Bacterial Community Reveal Genetic Features for Successful Plant Colonization. Frontiers in Microbiology 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giannoukos G et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol 13, r23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westermann AJ, Gorski SA & Vogel J Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10, 618–630 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Lewin GR et al. Application of a quantitative framework to improve the accuracy of a bacterial infection model. Proceedings of the National Academy of Sciences 120, e2221542120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohajeri MH et al. The role of the microbiome for human health: from basic science to clinical applications. Eur J Nutr 57, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mallott EK et al. Human microbiome variation associated with race and ethnicity emerges as early as 3 months of age. PLoS Biol 21, e3002230 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupta VK, Paul S & Dutta C Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeFilipp Z et al. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N Engl J Med 381, 2043–2050 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Varga JJ et al. Antibiotics Drive Expansion of Rare Pathogens in a Chronic Infection Microbiome Model. mSphere 7, e00318–22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bogaert D et al. Mother-to-infant microbiota transmission and infant microbiota development across multiple body sites. Cell Host Microbe 31, 447–460.e6 (2023). [DOI] [PubMed] [Google Scholar]
- 56.Ladau J et al. Microbial invasions and inoculants: a call to action. (2023). [Google Scholar]
- 57.Lange L et al. Microbiome ethics, guiding principles for microbiome research, use and knowledge management. Environmental Microbiome 17, 50 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pantoja Angles A, Valle-Pérez AU, Hauser C & Mahfouz MM Microbial Biocontainment Systems for Clinical, Agricultural, and Industrial Applications. Frontiers in Bioengineering and Biotechnology 10, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huttenhower C, Finn RD & McHardy AC Challenges and opportunities in sharing microbiome data and analyses. Nat Microbiol 8, 1960–1970 (2023). [DOI] [PubMed] [Google Scholar]
- 60.Yilmaz P et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol 29, 415–420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mirzayi C et al. Reporting guidelines for human microbiome research: the STORMS checklist. Nat Med 27, 1885–1892 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Northen T PLACEHOLDER. [Google Scholar]
- 63.Jennings SAV & Clavel T Synthetic Communities of Gut Microbes for Basic Research and Translational Approaches in Animal Health and Nutrition. Annual Review of Animal Biosciences 12, 283–300 (2024). [DOI] [PubMed] [Google Scholar]
- 64.Shayanthan A, Ordoñez PAC & Oresnik IJ The Role of Synthetic Microbial Communities (SynCom) in Sustainable Agriculture. Front. Agron 4, (2022). [Google Scholar]
- 65.Cheng AG et al. Design, construction, and in vivo augmentation of a complex gut microbiome. Cell 185, 3617–3636.e19 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emmenegger B et al. Identifying microbiota community patterns important for plant protection using synthetic communities and machine learning. Nat Commun 14, 7983 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schloss PD et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Applied and Environmental Microbiology 75, 7537–7541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edgar RC UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. Preprint at 10.1101/081257 (2016). [DOI] [Google Scholar]
- 69.Edgar RC, Haas BJ, Clemente JC, Quince C & Knight R UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davis NM, Proctor DM, Holmes SP, Reiman DA & Callahan BJ Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frøslev TG et al. Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nat Commun 8, 1188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dixon P VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 14, 927–930 (2003). [Google Scholar]
- 73.McMurdie PJ & Holmes S phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barnett D. J. m, Arts I. C. w & Penders J. microViz: an R package for microbiome data visualization and statistics. Journal of Open Source Software 6, 3201 (2021). [Google Scholar]
- 75.Lahti L & Shetty S microbiome R package. (2017) doi: 10.18129/B9.bioc.microbiome. [DOI] [Google Scholar]
- 76.Bankevich A et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wick RR, Judd LM, Gorrie CL & Holt KE Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Computational Biology 13, e1005595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwengers O et al. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom 7, 000685 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shade A & Stopnisek N Abundance-occupancy distributions to prioritize plant core microbiome membership. Current Opinion in Microbiology 49, 50–58 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Fisher CK & Mehta P Identifying Keystone Species in the Human Gut Microbiome from Metagenomic Timeseries Using Sparse Linear Regression. PLoS One 9, e102451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X-W et al. Identifying keystone species in microbial communities using deep learning. Nat Ecol Evol 8, 22–31 (2024). [DOI] [PubMed] [Google Scholar]
- 82.Kurtz ZD et al. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLOS Computational Biology 11, e1004226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karkaria BD, Fedorec AJH & Barnes CP Automated design of synthetic microbial communities. Nat Commun 12, 672 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Toju H et al. Scoring Species for Synthetic Community Design: Network Analyses of Functional Core Microbiomes. Front. Microbiol 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Paredes SH et al. Design of synthetic bacterial communities for predictable plant phenotypes. PLOS Biology 16, e2003962 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]