

In the title compound, C6H10N2O2, the piperazine-2,3-dione ring adopts a half-chair conformation. In the crystal, the molecules are linked by weak C—H⋯O hydrogen bonds, forming (010) sheets.
Keywords: crystal Structure, half chair, hydrogen bonding
Abstract
In the title compound, C6H10N2O2, the piperazine-2,3-dione ring adopts a half-chair conformation. In the crystal, the molecules are linked by weak C—H⋯O hydrogen bonds, forming (010) sheets.
Structure description
Piperazine and its derivatives are found within biologically active molecules across a diverse range of therapeutic areas, including antifungal, antibacterial, antimalarial, antipsychotic, antidepressant, and antitumor applications targeting colon, prostate, breast, lung, and leukemia cancers (Brockunier et al., 2004 ▸; Bogatcheva et al., 2005 ▸). As part of our studies in this area, we now describe the structure of the title compound, C6H10N2O2.
The asymmetric unit is shown in Fig. 1 ▸. The piperazine-2,3-dione ring adopts a half chair conformation, with C1 and C2 displaced from the other ring atoms by 0.279 (3) and −0.342 (3) Å, respectively. The molecule possesses local C2 symmetry about an axis passing through the midpoints of the C1—C2 and C3—C4 bonds. In the crystal (Fig. 2 ▸), the molecules are connected by weak C2—H2A⋯O1 and C5—H5C⋯O2 hydrogen bonds (Table 1 ▸) to generate (010) layers.
Figure 1.

The asymmetric unit with displacement ellipsoids drawn at the 50% probability level.
Figure 2.

The crystal packing of the title compound.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2A⋯O2i | 0.97 | 2.49 | 3.419 (3) | 161 |
| C5—H5C⋯O2ii | 0.96 | 2.54 | 3.481 (3) | 168 |
Symmetry codes: (i) 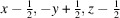 ; (ii)
; (ii)  .
.
A search of the Cambridge Structural Database (CSD; Version 5.43, update November 2022; Groom et al., 2016 ▸) revealed some similar structures to the title compound, including 3,6-dibenzylidene-1,4-dimethylpiperazine-2,5-dione (CSD refcode IQOCEZ; Ge et al., 2019 ▸), 2,5-bis(1-methyl-2-oxoindol-3-ylidene)-1,4-dimethylpiperazine-3,6-dione acetone solvate (PALVUT; Gompper et al., 1992 ▸) and 6-(bromobenzyl)-3-benzylidene-6-erythro-hydroxy-1,4-dimethylpiperazine-2,5-dione (SAWSEO; Sterns et al., 1989 ▸).
Synthesis and crystallization
The title compound was prepared according to the literature method (Haraguchi et al., 2015 ▸). Recrystallization of the solid from dichloromethane solution gave colorless plates, which were suitable for X-ray diffraction.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C6H10N2O2 |
| M r | 142.16 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 293 |
| a, b, c (Å) | 7.3781 (6), 8.0050 (6), 12.1306 (8) |
| β (°) | 99.767 (7) |
| V (Å3) | 706.07 (9) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.10 |
| Crystal size (mm) | 0.37 × 0.32 × 0.29 |
| Data collection | |
| Diffractometer | Agilent Xcalibur, Atlas, Gemini |
| Absorption correction | Analytical (SADABS; Krause et al., 2015 ▸) |
| Tmin, Tmax | 0.507, 0.578 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 2746, 1624, 1194 |
| R int | 0.016 |
| (sin θ/λ)max (Å−1) | 0.681 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.063, 0.181, 1.07 |
| No. of reflections | 1624 |
| No. of parameters | 93 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.45, −0.21 |
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2414314624009362/hb4484sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2414314624009362/hb4484Isup2.hkl
Supporting information file. DOI: 10.1107/S2414314624009362/hb4484Isup3.cml
CCDC reference: 2386002
Additional supporting information: crystallographic information; 3D view; checkCIF report
full crystallographic data
1,4-Dimethylpiperazine-2,3-dione. Crystal data
| C6H10N2O2 | F(000) = 304 |
| Mr = 142.16 | Dx = 1.337 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 7.3781 (6) Å | Cell parameters from 9307 reflections |
| b = 8.0050 (6) Å | θ = 3.5–26.4° |
| c = 12.1306 (8) Å | µ = 0.10 mm−1 |
| β = 99.767 (7)° | T = 293 K |
| V = 706.07 (9) Å3 | Plate, colourless |
| Z = 4 | 0.37 × 0.32 × 0.29 mm |
1,4-Dimethylpiperazine-2,3-dione. Data collection
| Agilent Xcalibur, Atlas, Gemini diffractometer | 1194 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.016 |
| ω scans | θmax = 29.0°, θmin = 3.1° |
| Absorption correction: analytical (SADABS; Krause et al., 2015) | h = −10→8 |
| Tmin = 0.507, Tmax = 0.578 | k = −10→5 |
| 2746 measured reflections | l = −6→16 |
| 1624 independent reflections |
1,4-Dimethylpiperazine-2,3-dione. Refinement
| Refinement on F2 | Primary atom site location: dual |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.063 | H-atom parameters constrained |
| wR(F2) = 0.181 | w = 1/[σ2(Fo2) + (0.0839P)2 + 0.2479P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.07 | (Δ/σ)max < 0.001 |
| 1624 reflections | Δρmax = 0.45 e Å−3 |
| 93 parameters | Δρmin = −0.21 e Å−3 |
| 0 restraints |
1,4-Dimethylpiperazine-2,3-dione. Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. All the H atoms were positioned geometrically (C—H = 0.96–0.97 Å) and were refined using a riding model, with Uiso(H) = 1.2Ueq(C) or 1.5Ueq(methyl C). |
1,4-Dimethylpiperazine-2,3-dione. Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O2 | 0.4932 (2) | 0.2262 (2) | 0.62587 (12) | 0.0591 (5) | |
| O1 | 0.8009 (2) | 0.3382 (3) | 0.55770 (14) | 0.0642 (6) | |
| N1 | 0.6498 (2) | 0.3592 (2) | 0.38031 (14) | 0.0427 (5) | |
| N2 | 0.3516 (2) | 0.1982 (2) | 0.44717 (14) | 0.0421 (5) | |
| C4 | 0.6624 (3) | 0.3190 (3) | 0.48783 (16) | 0.0380 (5) | |
| C3 | 0.4923 (3) | 0.2422 (2) | 0.52590 (15) | 0.0364 (5) | |
| C5 | 0.7995 (4) | 0.4489 (4) | 0.3415 (2) | 0.0616 (7) | |
| H5A | 0.899882 | 0.462273 | 0.402473 | 0.092* | |
| H5B | 0.756751 | 0.556833 | 0.313902 | 0.092* | |
| H5C | 0.840112 | 0.386644 | 0.282610 | 0.092* | |
| C1 | 0.4771 (4) | 0.3452 (3) | 0.30300 (18) | 0.0547 (7) | |
| H1A | 0.502520 | 0.333921 | 0.227477 | 0.066* | |
| H1B | 0.406408 | 0.446718 | 0.306177 | 0.066* | |
| C2 | 0.3670 (4) | 0.2011 (3) | 0.32859 (19) | 0.0555 (6) | |
| H2A | 0.245130 | 0.207406 | 0.283752 | 0.067* | |
| H2B | 0.424361 | 0.098582 | 0.309359 | 0.067* | |
| C6 | 0.1915 (3) | 0.1170 (4) | 0.4791 (3) | 0.0649 (8) | |
| H6A | 0.223240 | 0.073267 | 0.553547 | 0.097* | |
| H6B | 0.151882 | 0.027484 | 0.428037 | 0.097* | |
| H6C | 0.093814 | 0.196898 | 0.476737 | 0.097* |
1,4-Dimethylpiperazine-2,3-dione. Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O2 | 0.0637 (11) | 0.0816 (12) | 0.0319 (8) | −0.0121 (9) | 0.0082 (7) | 0.0062 (8) |
| O1 | 0.0445 (9) | 0.0987 (14) | 0.0438 (9) | −0.0153 (9) | −0.0083 (7) | 0.0067 (9) |
| N1 | 0.0455 (10) | 0.0500 (10) | 0.0318 (9) | −0.0055 (8) | 0.0045 (7) | 0.0017 (7) |
| N2 | 0.0376 (9) | 0.0480 (10) | 0.0393 (10) | −0.0067 (8) | 0.0023 (7) | −0.0029 (8) |
| C4 | 0.0358 (10) | 0.0455 (11) | 0.0307 (10) | 0.0003 (9) | −0.0004 (8) | −0.0013 (8) |
| C3 | 0.0388 (10) | 0.0384 (10) | 0.0308 (10) | 0.0031 (8) | 0.0023 (8) | 0.0000 (8) |
| C5 | 0.0650 (15) | 0.0706 (17) | 0.0544 (15) | −0.0116 (13) | 0.0250 (12) | 0.0023 (12) |
| C1 | 0.0653 (15) | 0.0642 (15) | 0.0299 (10) | −0.0058 (12) | −0.0055 (10) | 0.0061 (10) |
| C2 | 0.0572 (14) | 0.0649 (15) | 0.0377 (12) | −0.0054 (12) | −0.0113 (10) | −0.0033 (10) |
| C6 | 0.0440 (13) | 0.0738 (17) | 0.0774 (19) | −0.0156 (12) | 0.0118 (12) | −0.0092 (14) |
1,4-Dimethylpiperazine-2,3-dione. Geometric parameters (Å, º)
| O2—C3 | 1.218 (2) | C5—H5B | 0.9600 |
| O1—C4 | 1.222 (2) | C5—H5C | 0.9600 |
| N1—C4 | 1.331 (3) | C1—C2 | 1.474 (3) |
| N1—C1 | 1.452 (3) | C1—H1A | 0.9700 |
| N1—C5 | 1.461 (3) | C1—H1B | 0.9700 |
| N2—C3 | 1.333 (3) | C2—H2A | 0.9700 |
| N2—C6 | 1.457 (3) | C2—H2B | 0.9700 |
| N2—C2 | 1.462 (3) | C6—H6A | 0.9600 |
| C4—C3 | 1.537 (3) | C6—H6B | 0.9600 |
| C5—H5A | 0.9600 | C6—H6C | 0.9600 |
| C4—N1—C1 | 121.47 (18) | N1—C1—C2 | 112.27 (18) |
| C4—N1—C5 | 120.23 (19) | N1—C1—H1A | 109.2 |
| C1—N1—C5 | 117.27 (18) | C2—C1—H1A | 109.2 |
| C3—N2—C6 | 119.7 (2) | N1—C1—H1B | 109.2 |
| C3—N2—C2 | 121.30 (18) | C2—C1—H1B | 109.2 |
| C6—N2—C2 | 118.05 (19) | H1A—C1—H1B | 107.9 |
| O1—C4—N1 | 124.1 (2) | N2—C2—C1 | 110.93 (19) |
| O1—C4—C3 | 118.12 (18) | N2—C2—H2A | 109.5 |
| N1—C4—C3 | 117.77 (17) | C1—C2—H2A | 109.5 |
| O2—C3—N2 | 123.9 (2) | N2—C2—H2B | 109.5 |
| O2—C3—C4 | 118.32 (18) | C1—C2—H2B | 109.5 |
| N2—C3—C4 | 117.82 (17) | H2A—C2—H2B | 108.0 |
| N1—C5—H5A | 109.5 | N2—C6—H6A | 109.5 |
| N1—C5—H5B | 109.5 | N2—C6—H6B | 109.5 |
| H5A—C5—H5B | 109.5 | H6A—C6—H6B | 109.5 |
| N1—C5—H5C | 109.5 | N2—C6—H6C | 109.5 |
| H5A—C5—H5C | 109.5 | H6A—C6—H6C | 109.5 |
| H5B—C5—H5C | 109.5 | H6B—C6—H6C | 109.5 |
| C1—N1—C4—O1 | −175.3 (2) | N1—C4—C3—O2 | −170.0 (2) |
| C5—N1—C4—O1 | −7.2 (3) | O1—C4—C3—N2 | −170.1 (2) |
| C1—N1—C4—C3 | 5.3 (3) | N1—C4—C3—N2 | 9.4 (3) |
| C5—N1—C4—C3 | 173.39 (19) | C4—N1—C1—C2 | −35.3 (3) |
| C6—N2—C3—O2 | −3.8 (3) | C5—N1—C1—C2 | 156.3 (2) |
| C2—N2—C3—O2 | −172.4 (2) | C3—N2—C2—C1 | −37.7 (3) |
| C6—N2—C3—C4 | 176.84 (19) | C6—N2—C2—C1 | 153.5 (2) |
| C2—N2—C3—C4 | 8.3 (3) | N1—C1—C2—N2 | 49.3 (3) |
| O1—C4—C3—O2 | 10.5 (3) |
1,4-Dimethylpiperazine-2,3-dione. Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2A···O2i | 0.97 | 2.49 | 3.419 (3) | 161 |
| C5—H5C···O2ii | 0.96 | 2.54 | 3.481 (3) | 168 |
Symmetry codes: (i) x−1/2, −y+1/2, z−1/2; (ii) x+1/2, −y+1/2, z−1/2.
References
- Agilent (2012). CrysAlis PRO and CrysAlis RED. Agilent Technologies Ltd, Yarnton, England.
- Bogatcheva, E., Hanrahan, C., Nikonenko, B., Samala, R., Chen, P., Gearhart, J., Barbosa, F., Einck, L., Nacy, C. A. & Protopopova, M. (2005). J. Med. Chem.49, 3045–3048. [DOI] [PMC free article] [PubMed]
- Brockunier, L. L., He, J., Colwell, L. F. Jr, Habulihaz, B., He, H., Leiting, B., Lyons, K. A., Marsilio, F., Patel, R. A., Teffera, Y., Wu, J. K., Thornberry, N. A., Weber, A. E. & Parmee, E. R. (2004). Bioorg. Med. Chem. Lett.14, 4763–4766. [DOI] [PubMed]
- Ge, Y., Han, Z., Wang, Z. & Ding, K. (2019). J. Am. Chem. Soc.141, 8981–8988. [DOI] [PubMed]
- Gompper, R., Kellner, R. & Polborn, K. (1992). Angew. Chem. Int. Ed. Engl.31, 1202–1205.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Haraguchi, R., Takada, Y. & Matsubara, S. (2015). Org. Biomol. Chem.13, 241–247. [DOI] [PubMed]
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst.48, 3–10. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Spek, A. L. (2020). Acta Cryst. E76, 1–11. [DOI] [PMC free article] [PubMed]
- Sterns, M., Patrick, J. M., Patrick, V. A. & White, A. H. (1989). Aust. J. Chem.42, 349.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2414314624009362/hb4484sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2414314624009362/hb4484Isup2.hkl
Supporting information file. DOI: 10.1107/S2414314624009362/hb4484Isup3.cml
CCDC reference: 2386002
Additional supporting information: crystallographic information; 3D view; checkCIF report