

Abstract
Background:
Hepcidin, a peptide hormone primarily produced by the liver, regulates iron metabolism by interacting with its receptor, ferroportin. Studies have demonstrated that hepcidin participates in the progression of liver fibrosis by regulating HSC activation, but its regulatory effect on hepatocytes remains largely unknown.
Methods:
A carbon tetrachloride (CCl4)-induced liver fibrosis model was established in C57BL/6 wild-type (WT) and hepcidin knockout (Hamp −/−) mice. Liver injury and inflammation were assessed in WT and Hamp −/− mice at 24 and 48 hours following acute CCl4 exposure. In addition, transcriptomic sequencing of primary hepatocytes was performed to compare gene expression profiles between WT and Hamp −/− mice 24 hours after liver injury. The function of the identified molecule Eif2ak3/PERK (protein kinase R(PKR)-like endoplasmic reticulum kinase), was evaluated both in vitro and in vivo.
Results:
We found that serum hepcidin significantly increased during the progression of liver fibrosis induced by CCl4 and bile duct ligation. In addition, CCl4-treated Hamp −/− mice developed more severe liver injury, liver fibrosis, and hepatocyte apoptosis, with elevated Bax and decreased Bcl-2 expression, compared to the WT mice. Transcriptomic analysis of primary hepatocytes revealed that PERK was upregulated in Hamp −/− mice after CCl4 treatment, promoting apoptosis by regulating Bax and Bcl-2 expression. Subsequently, we demonstrated that hepcidin prevents hepatocyte apoptosis by inhibiting PERK both in vitro and in vivo.
Conclusions:
Hepcidin inhibits hepatocyte apoptosis through suppression of the PERK pathway, highlighting its protective role in liver fibrosis and identifying a potential therapeutic target for the treatment of liver fibrosis.
Keywords: acute liver injury, apoptosis, hepcidin, liver fibrosis, PERK
INTRODUCTION
A multitude of liver diseases, including Metabolic Associated Fatty Liver Disease, alcohol-associated liver disease, and infection–induced liver fibrosis, are often characterized by the accumulation of iron within liver tissue.1,2 An increasing number of studies have focused on the possible role of iron in liver fibrosis. For example, studies have found that iron overload directly induces the activation of and collagen deposition, promoting cirrhosis in mice, while iron-loaded hepatocytes and macrophages release many factors that promote the activation of HSCs and extracellular matrix deposition, accelerating liver fibrosis.3,4,5
Hepcidin, first isolated from human blood by Krause in 2000,6 is a cysteine-rich 25-amino acid antimicrobial peptide that is mainly synthesized by hepatocytes. It controls the delivery of iron to plasma through the internalization and degradation of ferroportin, the only known iron exporter in mammalian cells.7,8 Considerable evidence has shown that hepcidin is an important biochemical parameter in the diagnosis of liver fibrosis as it regulates iron metabolism and is a potential therapeutic target.9 Hepcidin inhibits Smad3 phosphorylation in HSCs through AKT, acting as an endogenous antifibrotic hepatokine.10 Although the activation of HSCs is the central event in fibrosis, the biological status of HSCs is affected by surrounding cells, such as hepatocytes, whose injury and regeneration play an important role in the progression of fibrosis. Thus, whether hepcidin is involved in the progression of chronic liver disease by influencing hepatocytes remains to be clarified.
In this study, we first examined the levels of hepcidin in fibrotic mice and compared the degree of fibrosis in wild-type (WT) and hepcidin knockout (Hamp −/− ) mice while maintaining normal levels of serum iron. Next, we established a carbon tetrachloride (CCl4)-induced acute liver injury model in WT and Hamp −/− mice to elucidate the role of hepcidin in acute liver injury. Finally, to explore the protective mechanism of hepcidin in hepatocytes, primary hepatocytes from WT and Hamp −/− mice were isolated after acute liver injury for an RNA-seq assay to screen regulatory genes involved in hepatocyte apoptosis. This study provides new insights into the protective function of hepcidin in hepatocyte apoptosis following liver injury.
METHODS
Animals model
Male C57BL/6 mice aged 4–8 weeks were purchased from the Vital River Laboratories. After a 3-day adaptation period, mice were housed under specific pathogen-free conditions. Male Hamp −/− mice were provided by Professor Sophie Vaulont from the Center National de la Recherche Scientifique, and Professor Tomas Ganz from the University of California. The mice were raised at Beijing Friendship Hospital, Capital Medical University. Hamp −/− mice were fed a low-iron (4 ppm) diet, and WT mice were fed an ordinary diet ad libitum. All animals were housed at a comfortable temperature and humidity (60 ± 5%) under a 12-hour light-dark cycle. All animal experiments were approved by the Animal Ethics Committee of the Beijing Friendship Hospital, Capital Medical University. CCl4 was dissolved in olive oil (Sigma-Aldrich).
The exposure mode for acute liver injury was i.p. injection. Four-week-old male WT and Hamp −/− mice were randomly divided into the CCl4 and vehicle groups (n = 6). The mice were administered 12.5% CCl4 (CCl4: olive oil = 1:7) or vehicle (olive oil) for 24 and 48 hours. The exposure mode for liver fibrosis was oral gavage. Eight-week-old male WT and Hamp −/− mice were randomly divided into CCl4 and vehicle groups (n = 10) and administered 20% CCl4 (CCl4: olive oil = 1:4) or vehicle (olive oil) for 6 weeks (twice a week). Hamp −/− mice were fed a low-iron (4 ppm) diet.
For the bile duct ligation (BDL) experiment, mice were randomly divided into control and model groups. The normal control group underwent laparotomy with an intact bile duct and portal vein, without undergoing BDL. The model group consisted of 16 mice divided into 2 subgroups: 1-week (BDL-1W) and 2-week (BDL-2W) after ligation, with 8 mice in each subgroup. C57BL/6 mice were fed an ordinary diet after BDL. Male Hamp −/− mice were fed a low-iron diet (4 ppm) after BDL to maintain normal iron levels. The mice were fasted for 12 hours before surgery and weighed. Mice were anesthetized with 1% pentobarbital sodium at a dose of 50 mg/kg, fixed on a fixed plate in the supine position, skin prepared 2 cm under the xiphoid process, and disinfected with iodophor. An incision ~1 cm long was made in the midline of the lower abdomen, below the xiphoid process. The abdominal cavity was opened, the left liver lobe was gently elevated, and the duodenum was carefully lifted with ophthalmic tweezers to expose the portal vein and bile duct. The common bile and portal veins were also isolated. Two 6-0 silk wires were pierced under the portal vein, and ligation was performed at a distance of 0.2–0.5 cm, without cutting the bile duct. The bile duct was then ligated and sutured to close the abdominal cavity.
The 4-week-old C57BL/6 mice and Hamp −/− mice were divided into 4 groups: C57BL/6-CCl4 group, C57-CCl4-PERKi group, Hamp −/−-CCl4 group, and Hamp −/−-CCl4-PERKi group. Mice were administered 12.5% CCl4 (CCl4: olive oil = 1:7) or vehicle (olive oil) for 48 hours. Simultaneously, PERK inhibitor (GSK2656157) was administered at a dose of 20 mg/kg for 48 hours. Half of the right liver lobe was placed in 4% paraformaldehyde for immunohistochemistry (IHC) staining, and the remaining liver tissue was used for protein and RNA extraction.
ELISA
The serum iron detection kits and serum AST and ALT ELISA kits were obtained from Jiancheng Technology Co.,Ltd. The levels of serum IL-6 and TNFα were analyzed using an ELISA Kit (R&D System). All experiments were performed in accordance with the manufacturer’s instructions.
Hematoxylin-eosin staining, IHC, TdT-mediated dUTP nick-end labeling staining, and Prussian blue staining
The right lobe of the liver was fixed with 4% paraformaldehyde. We then performed hematoxylin-eosin staining, Masson staining, TdT-mediated dUTP nick-end labeling (TUNEL) staining, and IHC for Bax, Bcl-2, F4/80, CD45, and Ly6G. Images of 10 random microscope fields of staining in each liver section were recorded for quantitative analysis using the ImageJ software (NIH). Human hepatic iron was detected by Prussian blue staining, and an Enhanced Coloring Agent (Solarbio) was used to enhance the results.
The isolation of hepatocytes, HSCs, KCs, and RNA-seq assay
The liver of WT and Hamp −/− mice were digested by using 2-step collagenase, and then primary hepatocytes and HSCs were isolated, and intrahepatic KCs were sorted as 7AAD−CD45+F4/80+.11,12 Pronase E was purchased from Merck. Type IV collagenase, trypsin inhibitor, HEPES, DMSO, and lipopolysaccharide were purchased from Sigma. RNA-seq data is deposited in GSE276552.
Quantitative real-time RT-PCR
Total RNA from tissue or primary hepatocytes was isolated using TRIzol Reagent (Invitrogen), and first-strand cDNA was synthesized using the ThermoScript RT-PCR System (Thermo Scientific) according to the manufacturer’s instructions. The cDNA was subjected to PCR cycles using SYBR Green quantitative PCR reagent (Promega), and assays were conducted using the CFX96 Real-Time System (Bio-Rad). The cDNA of the primary hepatocytes was detected by RNA sequencing. Primers used are listed in Table 1.
TABLE 1.
PCR primers used in this study
| Gene | Primer sequences |
|---|---|
| Cyclophilin | F-GGAGATGGCACAGGAGGAA |
| R-GCCCGTAGTGCTTCAGCTT | |
| TGFβ1 | F-TGGAGCAACATGTGGAACTCT |
| R-CCTGTATTCCGTCTCCTTGGT | |
| Collagen I | F-TAGGCCATTGTGTATGCAGC |
| R-ACATGTTCAGCTTTGTGGACC | |
| TIMP-1 | F-AGGTGGTCTCGTTGATTTCT |
| R-GTAAGGCCTGTAGCTGTGCC | |
| α-SMA | F-GTTCAGTGGTGCCTCTGTCA |
| R-ACTGGGACGACATGGAAAAG | |
| β-actin | F-ATGGAGGGGAATACAGCCC |
| R-TTCTTTGCAGCTCCTTCGTT | |
| Bcl-2 | F-CTTTCTGCTTTTTATTTCATGAGG |
| R-CAGAAGATCATGCCGTCCTT | |
| Bax | F-TGAAGACAGGGGCCTTTTTG |
| R-AATTCGCCGGAGACACTCG | |
| Fas | F-AGGTGGACTGGATACACAGAC |
| R-TCTCCTGCCCAAACTCTTTGC | |
| Fasl | F-TCCGTGAGTTCACCAACCAAA |
| R-GGGGGTTCCCTGTTAAATGGG | |
| Eif2ak3/PERK | F-GCGTCGGAGACAGTGTTTG |
| R-CGTCCATCTAAAGTGCTGATGAT | |
| Hamp | F-TGCCTGTCTCCTGCTTCT |
| R-TGTCTGCCCTGCTTTCTT | |
| Atf3 | F-GAGGATTTTGCTAACCTGACACC |
| R-TTGACGGTAACTGACTCCAGC | |
| Ddit3 | F-CTGGAAGCCTGGTATGAGGAT |
| R-CAGGGTCAAGAGTAGTGAAGGT |
Cell culture and treatment
The mouse hepatocyte line, NCTC 1469, was purchased from the Basic Medical Cell Center, Chinese Academy of Medical Sciences. The cells were cultured in Dulbecco’s modified Eagle’s medium (Hyclone) supplemented with 10% bovine calf serum (Hyclone), 100 IU/mL penicillin, and 100 mg/mL streptomycin (Hyclone) in a humidified incubator at 37°C with 5% CO2. NCTC 1469 cells were treated with 10 µM PERK inhibitor (Selleck) for 0.5 hours and then treated with 100 ng/mL human hepcidin-25 peptide (Abcam) for 24 hours, and 10 ng/mL TNFα (Peprotech) and 200 ng/mL Actinomycin D (ActD, Solarbio) for 24 hours. NCTC 1469 cells were treated with 100 ng/mL Human hepcidin-25 peptide (Abcam) for 4 hours and then treated with CCl4 for 4 hours. The lentivirus overexpressing the Hamp gene was purchased from Cyagen. Lentiviral particles overexpressing Hamp were used to infect NCTC 1469. After infection, the cells were subjected to puromycin selection to ensure stable Hamp overexpression.
Cell death analysis by flow cytometry
NCTC 1469 cells were collected and washed with PBS, and the collected cells were stained with 7-aminoactinomycin D and Annexin V staining for 30 minutes. Finally, 7-aminoactinomycin D and annexin V–positive cells were determined by flow cytometry analysis, according to the manufacturer’s instructions. The PE Annexin V Apoptosis Detection Kit with 7-aminoactinomycin D was obtained from BioLegend.
Western blot
The experiment was performed as described in previous studies.13 Cells were harvested and lysed in RIPA lysis buffer after washing with cold PBS (Solarbio) supplemented with a protease inhibitor cocktail (Roche). Antibodies against GAPDH and β-actin were obtained from Santa Cruz Biotechnology. Antibodies against Bax and Bcl-2 were purchased from Proteintech. Antibodies against P-PERK, PERK, P-EIF2A, and EIF2A were obtained from Cell Signaling Technology. Horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG were obtained from Proteintech. ECL western blot substrate was purchased from Thermo Scientific Pierce.
Statistical analysis
Statistical analyses were performed using the Student t test and 1-way ANOVA. The results are expressed as the mean ± SEM for 3 or 6 independent experiments. Volcano maps were plotted using GraphPad Prism v5.0. All data were analyzed using SPSS 19.0, ImageJ, and significance was assigned at p < 0.05 (*) or p < 0.01 (**).
RESULTS
Hepatic hepcidin is increased in the liver fibrosis mouse model
We established a mouse model of liver fibrosis induced by either 6 weeks of CCl4 treatment or 2 weeks of BDL treatment to investigate hepcidin changes associated with liver fibrosis. Masson’s staining (Figure 1A) revealed a marked increase in collagen deposition in liver fibrosis induced by CCl4 and BDL. In addition, the mRNA expression levels of transforming growth factor-β1 (Tgfb1), Collagen I, α-smooth muscle actin (α-SMA), and Tipm-1 also increased significantly (Figures 1D, E), confirming liver fibrosis induced by CCl4 or BDL. Serum hepcidin levels (Figures 1B, C) also increased in a time-dependent manner. These findings indicated that hepcidin levels are associated with the progression of liver fibrosis.
FIGURE 1.

Hepcidin increased in the liver from mice with CCl4 or BDL-induced liver fibrosis. (A) Changes of Masson staining in livers from CCl4 or BDL-induced liver fibrosis. (B) Levels of serum hepcidin in CCl4-induced liver fibrosis. (C) Levels of serum hepcidin in BDL-induced liver fibrosis. (D) Levels of the mRNA expression of Tgfβ1, Collagen I, α-SMA, and Timp-1 in livers from CCl4-induced liver fibrosis. (E) Levels of the mRNA expression of Tgfβ1, Collagen I, α-SMA, and Timp-1 in livers from BDL-induced liver fibrosis. **p < 0.01, compared with control. Abbreviations: α-SMA, α-smooth muscle actin; BDL, bile duct ligation; CCl4, carbon tetrachloride; Tgfβ1, transforming growth factor-β1.
Hepcidin slows down the progress of liver fibrosis without affecting iron metabolism
Next, we established a liver fibrosis model induced by treatment with CCl4 or BDL in WT and Hamp −/− mice to test whether hepcidin regulates liver fibrosis in an iron-independent manner. As shown in Figures 2A, B, Hamp −/− mice were fed a low-iron diet for 5 weeks to standardize serum iron levels across WT and Hamp −/− mice. Similarly, Prussian blue staining showed no change in hepatic iron levels between WT and Hamp −/− mice (Supplemental Figures S1A and B, http://links.lww.com/HC9/B852). Masson staining (Figures 2C, D) showed that Hamp −/− mice treated with CCl4 or BDL had a more severe deposition of collagen than WT mice. In addition, Hamp −/− mice treated with CCl4 and BDL showed higher mRNA expression levels of α-SMA (Figures 2E, H), Timp-1 (Figures 2F, I), and Collagen I (Figures 2G, J) than WT mice. Collectively, these findings imply that hepcidin slows down the progression of liver fibrosis, whereas iron remains constant.
FIGURE 2.

Hepcidin could delay the progression of liver fibrosis without the regulation of iron metabolism. (A) Changes of serum iron in CCl4-treated Hamp −/− mice for 1–6 weeks. (B) Changes of serum iron in BDL-treated Hamp −/− mice for 1–2 weeks. (C) Changes of Masson staining in CCl4-treated WT and Hamp −/− mice for 1–6 weeks. (D) Changes of Masson staining in BDL-treated WT and Hamp −/− mice for 1–2 weeks. (E–G) Changes of α-SMA, TIMP-1, and Collagen I mRNA expression in CCl4-treated WT and Hamp −/− mice for 0, 4, and 6 weeks. (H–J) Changes of α-SMA, Timp-1, and Collagen I mRNA expression in BDL-treated WT and Hamp−/− mice for 0, 1 and 2 weeks. *p < 0.05, **p < 0.01, compared with control. Abbreviations: α-SMA, α-smooth muscle actin; BDL, bile duct ligation; CCl4, carbon tetrachloride; Hamp −/−, hepcidin knockout; WT, wild type.
Hepcidin inhibits liver injury and inflammation in acute liver injury
Liver cell injury begins with a sequence of events that is independent of the cause of inflammation and provides signals for the development of liver fibrosis.14 Next, we established CCl4-induced acute liver injury models in WT and Hamp −/− mice to explore the role of hepcidin in liver injury. As shown in Supplemental Figures S2A, B, http://links.lww.com/HC9/B852, the serum ALT and AST levels in Hamp −/− mice treated with CCl4 were higher than those in WT mice, indicating that hepcidin has a protective effect against liver injury. After 24 hours of treatment with CCl4, the Hamp −/− mice were slightly lower than that of WT mice; however, after 48 hours, the liver weights of the 2 groups did not differ significantly (Supplemental Figure S2C, http://links.lww.com/HC9/B852). In addition, the liver/body weight ratio (Supplemental Figure S2D, http://links.lww.com/HC9/B852) was comparable between WT and Hamp −/− mice at 24 and 48 hours after acute liver injury. There was no significant difference in the serum iron levels (Supplemental Figure S2E, http://links.lww.com/HC9/B852) before and after treatment with CCl4. In addition, hematoxylin-eosin staining showed that hepatic cells were irregularly arranged in the lobules of the liver after treatment with CCl4, and there was more regional necrosis in the CCl4 group of Hamp −/− mice than in the WT mice (Supplemental Figure S3A, http://links.lww.com/HC9/B852), implying that hepcidin protected against liver injury by protecting hepatic cells. In addition, the expression levels of F4/80 (Figures 3A, D), Ly6G (Figures 3B, E), and CD45 (Figures 3C, F) were higher in CCl4-treated Hamp −/− mice than in WT mice. The levels of TNFα and IL-6 quantified in sera (Supplemental Figures S3B, C, http://links.lww.com/HC9/B852) and livers (Supplemental Figures S3D, E, http://links.lww.com/HC9/B852) were also higher in CCl4-treated Hamp −/− mice than in CCl4-treated WT mice. Taken together, these results indicate that hepcidin can partially alleviate liver injury and inflammation.
FIGURE 3.

Hamp −/− mice treated with CCl4 had higher expression of F4/80, Ly6G, and CD45 than WT mice treated with CCl4. (A) IHC of F4/80, (B) IHC of Ly6G, (C) IHC of CD45 in livers of WT and Hamp −/− mice with CCl4-induced acute liver injury for 24 and 48 hours. (D) Positive F4/80 cells, (E) positive Ly6G cells, (F) positive CD45 cells in livers of WT and Hamp −/− mice with CCl4-induced acute liver injury for 24 and 48 hours. Images of 10 random microscope fields of staining in each liver section were recorded for quantitative analysis using ImageJ software. *p < 0.05, **p < 0.01, compared with control. Abbreviations: CCl4, carbon tetrachloride; Hamp −/−, hepcidin knockout; IHC, immunohistochemistry; WT, wild type.
Hepcidin suppresses the apoptosis of hepatocytes by regulating the expression of Bax and Bcl-2 in acute liver injury
To explore whether hepcidin regulates hepatic apoptosis, TUNEL staining was performed to detect hepatic cell apoptosis following acute liver injury. As shown in Figures 4A, D, Hamp −/− mice exposed to CCl4 showed more aberrant apoptosis than WT mice, indicating that hepcidin inhibited hepatic cell apoptosis. As detected by IHC (Figures 4B, C, E, F) and qPCR assays (Supplemental Figure S4A, B, http://links.lww.com/HC9/B852), after acute liver injury, the expression of Bax was increased while the expression of Bcl-2 was decreased. However, there was no significant difference in Bcl-2 expression in mice exposed to CCl4 at 48 hours, possibly because of the large difference between the groups. The mRNA expression of Fas increased within 24 hours but remained unchanged at 48 hours (Supplemental Figure S4C, http://links.lww.com/HC9/B852). Taken together, these data revealed that hepcidin inhibited the apoptosis of hepatic cells by regulating the expression of apoptosis-related genes.
FIGURE 4.

Hepcidin suppresses hepatic cell apoptosis in WT and Hamp −/− mice with CCl4-induced acute liver injury for 24 and 48 hours. (A) TUNEL staining, (B) IHC of BAX, (C) IHC of BCL-2 in livers of WT and Hamp −/− mice with CCl4-induced acute liver injury for 24 and 48 hours. (D) Positive TUNEL cells, (E) positive BAX cells, (F) positive BCL-2 cells in livers of WT and Hamp −/− mice with CCl4-induced acute liver injury for 24 and 48 hours. Images of 10 random microscope fields of staining in each liver section were recorded for quantitative analysis using ImageJ software. *p < 0.05, **p < 0.01, compared with control. Abbreviations: CCl4, carbon tetrachloride; Hamp −/−, hepcidin knockout; IHC, immunohistochemistry; TUNEL, TdT-mediated dUTP nick-end labeling; WT, wild type.
To delve deeper into the source of hepcidin secretion, we examined hepcidin RNA expression levels in hepatocytes, HSCs, and KCs. The findings revealed that hepatocytes were the primary cells responsible for hepcidin secretion (Figure 5A). To confirm the protective effects of hepcidin on hepatocytes, NCTC 1469 cells were cultured with or without hepcidin, before stimulation with TNFα and Dactinomycin (ActD) in vitro. As shown in Figures 5B, C, the ratio of cell apoptosis was reduced by 28.75% in the hepcidin treatment group compared with the TNFα + ActD group (14.56 ± 1.61 vs. 10.37 ± 2.94). Western blot results (Figures 5D, E) showed that the expression of BAX was reduced after hepcidin treatment in the TNFα + ActD group. In addition, cells transfected with a virus-overexpressing Hamp exhibited reduced apoptosis compared to the control group (Figures 5F–H). Furthermore, hepcidin decreased the expression of Bax and increased that of Bcl-2 (Figures 5I, J). In summary, these findings demonstrate that hepcidin suppresses hepatocyte apoptosis by modulating Bax and Bcl-2 expression.
FIGURE 5.

Hepcidin inhibits hepatocyte line NCTC 1469 apoptosis through regulating Bax and Bcl-2. (A) Hamp expression in hepatocytes, HSCs, and KCs. (B, C) The results of flow cytometry 3 times in NCTC 1469 after treatment with TNFα, ActD, and hepcidin peptide. (D, E) Western blot showed the protein expression of apoptotic-related gene BAX and BCL-2 in NCTC 1469 after treatment with TNFα, ActD, and hepcidin peptide. (F–H) The results of flow cytometry 3 times after treatment with TNFα, ActD in cells transfected with Hamp virus. (I, J) RT-qPCR showed the expression of Bax, Bcl-2 after treatment with TNFα, and ActD in cells transfected with the Hamp virus. *p < 0.05, **p < 0.01, compared with control. Abbreviation: ActD, Dactinomycin.
Hepcidin inhibits the apoptosis of hepatocytes through PERK
To clarify the mechanism by which hepcidin suppresses hepatocyte apoptosis, we isolated primary hepatocytes from WT and Hamp −/− mice 24 hours after CCl4 i.p. injection and analyzed the differentially expressed genes using an RNA-seq assay. Fold change value (FC value) >2 and FC value <0.5 were set as significantly different values. As shown in Figure 6A, there were 1493 upregulated genes (red) and 1371 downregulated genes (green). The top 12 genes with significant differences in apoptosis were selected for further investigation (Figure 6B). These genes are listed in Table 2. Among them, eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3)/ protein kinase R (PKR)-like endoreticulum kinase (PERK) was increased 30-fold in the CCl4-treated group than the control group in Hamp −/− mice (Figure 6C), consistent with the qRT-PCR results (Figure 6D). PERK is one of the 3 receptors that initiates the unfolded protein response to endoplasmic reticulum stress.15 We speculate that the antiapoptotic effect of hepcidin on hepatocytes may be mediated by the endoplasmic reticulum stress-PERK pathway. Western blot analysis revealed that, in the CCl4-induced acute liver injury model, phosphorylated PERK (P-PERK) level was significantly elevated in the livers of Hamp −/− mice compared to WT mice (Figure 6E). Similarly, PERK expression in the livers of Hamp −/− mice showed a time-dependent increase following CCl4 treatment (Figure 6F). Furthermore, in liver injury models induced by CCl4 and BDL, P-PERK levels were higher in the livers of Hamp −/− mice relative to WT mice (Supplemental Figures S5A, B, http://links.lww.com/HC9/B852). The activation of PERK leads to the phosphorylation of eIF2 (eukaryotic translation initiation factor 2 alpha), which triggers significant changes in the expression of various genes, including those associated with the unfolded protein responses, such as Atf3 (Activating transcription factor 3) and Ddit3 (DNA damage-inducible transcript 3).15 As anticipated, RNA-seq analysis revealed that the expression levels of Atf3 and Ddit3 were upregulated in the livers of CCl4-treated Hamp −/− mice compared to control mice (Supplemental Figures S5C, D, http://links.lww.com/HC9/B852), consistent with the RT-qPCR results (Supplemental Figures S5E, F, http://links.lww.com/HC9/B852). Altogether, hepcidin inhibits the apoptosis of hepatocytes through the PERK pathway.
FIGURE 6.

Hepcidin regulates the expression of Bcl-2 and Bax through PERK. (A) Volcano plot (p < 0.05 and FC >2 or FC <0.5 were differentially expressed. The red expressed upregulated genes, and the green expressed downregulated genes). (B) The expression of DEGs, (C, D) RNA-seq and RT-qPCR showed the expression of Eif2ak3/PERK in primary hepatocytes from WT and Hamp −/− mice treated with CCl4. (E) The protein expression levels of P-PERK and PERK in WT and Hamp −/− mice with CCl4-induced acute liver injury for 48 hours. (F) The mRNA expression of PERK in WT and Hamp −/− mice with CCl4-induced liver fibrosis for 6 weeks. (G) The mRNA expression of Bax, (H) the mRNA expression of Bcl-2 after treated with TNFα, ActD, hepcidin peptide, PERK inhibitor in NCTC 1469. (I) The protein expression of BAX and BCL-2 after being treated with TNFα, ActD, hepcidin peptide, and PERK inhibitor in NCTC 1469. *p < 0.05, **p < 0.01, compared with control. Abbreviations: ActD, Dactinomycin; CCl4, carbon tetrachloride; Hamp −/−, hepcidin knockout; WT, wild type.
TABLE 2.
Differentially expressed genes (DEGs)
| Gene | Description |
|---|---|
| Cidec | Cell death–inducing DFFA-like effector c |
| Dusp10 | Dual specificity phosphatase 10 |
| Tinagl1 | Tubulointerstitial nephritis antigen-like 1 |
| Fos | FBJ osteosarcoma oncogene |
| Lats2 | Large tumor suppressor 2 |
| Ctss | Cathepsin S |
| Eif2ak3/ PERK | Eukaryotic translation initiation factor 2 alpha kinase 3/RNA activated protein kinase (PKR)-like endoplasmic reticulum kinase |
| Nfkb1 | Nuclear factor of kappa light polypeptide gene enhancer in B cells 1, p105 |
| Bid | BH3 interacting domain death agonist |
| Jun | Jun proto-oncogene |
| Mfn2 | Mitofusin 2 |
The inhibition of PERK alleviates liver injury
To further verify the effect of hepcidin on the suppression of hepatocyte apoptosis through PERK, PERK inhibitor GSK2656157 or hepcidin peptide was used to observe hepatocyte apoptosis in vitro. As presented in Figures 6G–I, either hepcidin or the PERK inhibitor reduced the expression level of Bax and increased the Bcl-2 level in hepatocytes after TNFα and ActD treatment, suggesting that hepcidin inhibits hepatocyte apoptosis through the PERK pathway. To determine whether PERK inhibition could mitigate liver injury, we assessed the effects of the PERK inhibitor on CCl4-induced liver damage in both Hamp −/− mice and WT mice. The PERK inhibitor significantly reduced serum AST and ALT levels (Figures 7A, B). Hematoxylin-eosin staining demonstrated that the inhibitor markedly alleviated liver injury (Figure 7C). TUNEL staining revealed that the PERK inhibitor significantly suppressed hepatic apoptosis (Figures 7D, E). Moreover, the expression of P-PERK, P-EIF2A, and BAX was downregulated in CCl4-induced liver injury, while BCL-2 expression was upregulated (Figure 7F). Collectively, these results indicate that PERK inhibition can effectively attenuate liver injury.
FIGURE 7.

The inhibition of PERK alleviates liver injury. (A, B) Levels of serum AST (A) and ALT (B) in WT and Hamp −/− mice with CCl4-induced liver injury after treatment with PERK inhibitor. (C) H&E staining of liver tissues from WT and Hamp −/− mice after treatment with PERK inhibitor in the CCl4-induced liver injury model. (D, E) TUNEL staining and quantification of apoptotic cells in liver tissues of WT and Hamp −/− mice after treatment with PERK inhibitor in the CCl4-induced liver injury model. (F) Protein expression levels of P-PERK, PERK, P-EIF2A, EIF2A, BAX, and BCL-2 in WT and Hamp −/− mice with CCl4-induced acute liver injury for 48 hours after treatment with PERK inhibitor. PERKi, PERK inhibitor. **p < 0.01, compared with control. Abbreviations: CCl4, carbon tetrachloride; H&E, hematoxylin-eosin; Hamp −/−, hepcidin knockout; TUNEL, TdT-mediated dUTP nick-end labeling; WT, wild type.
DISCUSSION
The role of hepcidin in liver fibrosis has been increasingly investigated in recent years. However, previous studies have mostly focused on the effect of changes in iron metabolism caused by hepcidin on fibrosis. Our current study showed that Hamp −/− mice maintained on a low-iron diet to control for iron-related effects developed more severe liver fibrosis compared to WT mice. This finding indicated that hepcidin can alleviate fibrosis through pathways that are not exclusively linked to iron metabolism. We discovered a novel mechanism by which hepcidin influences liver injury and fibrosis, specifically through the regulation of hepatocyte apoptosis.
Liver injury could be driven by the apoptosis of hepatocytes and the formation of apoptotic corpuscles, which further cause inflammatory cells (macrophages, lymphocytes, and monocytes) to infiltrate the liver and accelerate the production of soluble media and oxidative stress, thereby promoting liver fibrosis.16,17,18,19 In our study, we observed that in Hamp −/− mice, the serum ALT and AST levels, as well as the expression levels of F4/80, Ly6G, and CD45, were all significantly increased compared to those in WT mice after CCl4 treatment, indicating that hepcidin had a protective effect on hepatocytes and inflammation.
Inactivation of HSCs, molecular dysregulation in activated HSCs, induction of senescence, and apoptosis of HSCs are strategies proposed to utilize HSCs as a target in the reversal of liver fibrosis.20 There is growing evidence that inhibiting hepatic apoptosis can reduce the activation of HSCs and the progression of liver fibrosis.16,21,22 Therefore, hepatocyte apoptosis plays a vital role in the progression of liver fibrosis, and blocking hepatocyte apoptosis is of great significance for suppressing the progression of liver fibrosis.16 In this study, we found a greater percentage of apoptosis in hepcidin-depleted cells. The expression level of the apoptosis-related gene Bax increased, while that of the apoptosis suppressor Bcl-2 decreased in CCl4-treated Hamp −/− mice. An in vitro study confirmed that hepcidin inhibited hepatocyte apoptosis by regulating the expression of Bax and Bcl-2. These data suggest that hepcidin suppresses hepatocyte apoptosis by regulating apoptosis-related genes and mitigating subsequent inflammation.
PERK is one of the 3 receptors that initiate the unfolded protein response to endoplasmic reticulum stress.23 When unfolded and misfolded proteins accumulate in the endoplasmic reticulum, PERK is activated to initiate the unfolded protein response, which interferes with the accumulation of unfolded and misfolded proteins in the endoplasmic reticulum and enables the endoplasmic reticulum to function normally.23 However, prolonged unfolded protein response activation can induce apoptosis.15,24,25,26,27 PERK promotes cell death by activating DNA damage-inducible transcript 3 (CHOP), a proapoptotic transcription factor.23,28 We found that in Hamp −/− mice, PERK in the CCl4-treated group was up to 30-fold higher than that in the control group. Our data further revealed that the expression level of PERK was significantly increased in Hamp −/− mice during liver fibrogenesis, indicating that hepcidin could decrease the level of PERK to protect hepatic cells against injury and become a key factor in protecting hepatic cells from damage.
The PERK/CHOP signaling pathway can directly reduce the expression level of Bcl-2 and upregulate the expression level of Bax.29,30 Our in vitro and in vivo results showed that either hepcidin or a PERK inhibitor suppressed Bax expression and upregulated Bcl-2 expression in apoptotic hepatocytes. Taken together, our data showed that hepcidin inhibits hepatocyte apoptosis mainly through PERK by regulating the expression of Bax and Bcl-2.
Hepcidin, a small-molecule peptide secreted by hepatocytes, has no immunogenicity or hepatotoxicity. Therefore, hepcidin can exert protective effects against liver fibrosis by suppressing hepatocyte apoptosis. In addition, our findings identify a new molecular mechanism: hepcidin inhibits the apoptosis of hepatocytes by regulating apoptosis-related genes through PERK (Figure 8, by Figdraw), which provides an experimental basis for the further clinical application of hepcidin in the treatment of liver fibrosis.
FIGURE 8.
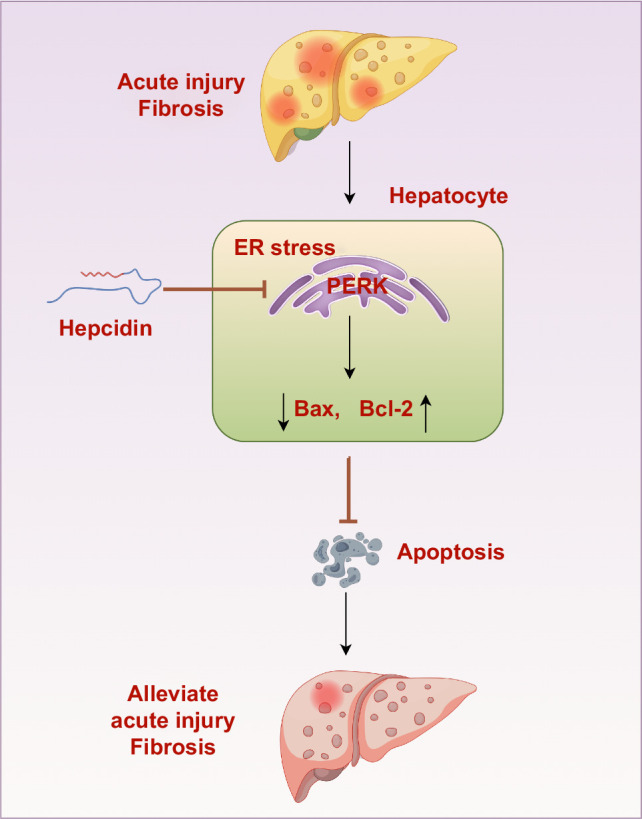
The mechanisms by which hepcidin inhibits liver fibrosis. Hepcidin could effectively inhibit the apoptosis of hepatocytes by regulating the expression of apoptosis-related genes through PERK, therefore possibly exerting protective effects on acute liver injury and liver fibrosis.
Supplementary Material
AUTHOR CONTRIBUTIONS
All the listed authors participated in the study and have seen and approved the submission of this manuscript. Changying Li, Guojin Pang, and Weihua Zhao participated in the research, analyzed the data, and initiated the original draft of the article. Yingying Liu, Xiaoli Huang, Wei Chen, Xinyan Zhao, Tianhui Liu, Ping Wang, Xu Fan, Ming Gao performed the experiments and collected the data. Min Cong established the hypotheses, supervised the study, analyzed the data, and co-wrote the manuscript.
ACKNOWLEDGMENTS
The authors thank Professor Sophie Vaulont of the National Center for Scientific Research of France, Professor Tomas Ganz of the University of California, USA, and Professor Sijin Liu for providing Hamp −/− mice. They also thank the AiMi Academic Services (www.aimieditor.com) for English language editing and review services.
FUNDING INFORMATION
This work was supported by grants from the National Natural Science Foundation of China (81570542 and 82170614), Beijing Natural Science Foundation (7142043), and Wang Baoen Liver Fibrosis Foundation (CFHPC2021042).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: ActD, Actinomycin D; Atf3, activating transcription factor 3; BDL, bile duct ligation; CCl4, carbon tetrachloride; Ddit3, DNA damage-inducible transcript 3; eIF2α, eukaryotic translation initiation factor 2 alpha; Eifak3/Perk, eukaryotic translation initiation factor 2 alpha kinase 3/ protein kinase R(PKR)-like endoplasmic reticulum kinase; Hamp −/− , hepcidin knockout; IHC, immunohistochemistry; T gf-β1, transforming growth factor-β1; TUNEL, TdT-mediated dUTP nick-end labeling; WT, wild type; α-SMA, α-smooth muscle actin.
Changying Li, Guojin Pang, and Weihua Zhao participated in the research, analyzed the data, and initiated the original draft of the article. They contributed equally to this work.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Changying Li, Email: changyingli1006@163.com.
Guojin Pang, Email: 13263292199@163.com.
Weihua Zhao, Email: sunnyzhaoweihua@163.com.
Yingying Liu, Email: 13027730353@163.com.
Xiaoli Huang, Email: hxlguaiguai@126.com.
Wei Chen, Email: cw_2011@126.com.
Xinyan Zhao, Email: zhao_xinyan@ccmu.edu.cn.
Tianhui Liu, Email: liu_tianhui@163.com.
Ping Wang, Email: wp1998@163.com.
Xu Fan, Email: xufan@ccmu.edu.cn.
Ming Gao, Email: minggao@rcees.ac.cn.
Min Cong, Email: maomao0623@sina.com.
REFERENCES
- 1. Hino K, Yanatori I, Hara Y, Nishina S. Iron and liver cancer: An inseparable connection. FEBS J. 2022;289:7810–7829. [DOI] [PubMed] [Google Scholar]
- 2. Marmur J, Beshara S, Eggertsen G, Onelöv L, Albiin N, Danielsson O, et al. Hepcidin levels correlate to liver iron content, but not steatohepatitis, in non-alcoholic fatty liver disease. BMC Gastroenterol. 2018;18:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li SW, Liu CM, Guo J, Marcondes A, Deeg J, Li X, et al. Iron overload induced by ferric ammonium citrate triggers reactive oxygen species-mediated apoptosis via both extrinsic and intrinsic pathways in human hepatic cells. Hum Exp Toxicol. 2016;35:598–607. [DOI] [PubMed] [Google Scholar]
- 4. Li X, Li S, Lu M, Yang G, Shen Y, Zhou X. Proteomic profiling of iron overload-induced human hepatic cells reveals activation of TLR2-mediated inflammatory response. Molecules. 2016;21:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mehta KJ, Farnaud SJ, Sharp PA. Iron and liver fibrosis: Mechanistic and clinical aspects. World J Gastroenterol. 2019;25:521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krause A, Neitz S, Mägert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480:147–150. [DOI] [PubMed] [Google Scholar]
- 7. Liu J, Sun B, Yin H, Liu S. Hepcidin: A promising therapeutic target for iron disorders: A systematic review. Medicine (Baltimore). 2016;95:e3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ganz T, Nemeth E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematology Am Soc Hematol Educ Program. 2011;2011:538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vela D. Low hepcidin in liver fibrosis and cirrhosis; a tale of progressive disorder and a case for a new biochemical marker. Mol Med. 2018;24:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han CY, Koo JH, Kim SH, Gardenghi S, Rivella S, Strnad P, et al. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt. Nat Commun. 2016;7:13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Charni-Natan M, Goldstein I. Protocol for primary mouse hepatocyte isolation. STAR Protoc. 2020;1:100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun G, Wang Y, Yang L, Zhang Z, Zhao Y, Shen Z, et al. Rebalancing liver-infiltrating CCR3(+) and CD206(+) monocytes improves diet-induced NAFLD. Cell Rep. 2023;42:112753. [DOI] [PubMed] [Google Scholar]
- 13. Gao M, Li C, Xu M, Liu Y, Cong M, Liu S. LncRNA MT1DP aggravates cadmium-induced oxidative stress by repressing the function of Nrf2 and is dependent on interaction with miR-365. Adv Sci (Weinh). 2018;5:1800087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaeschke H. Inflammation in response to hepatocellular apoptosis. Hepatology. 2002;35:964–966. [DOI] [PubMed] [Google Scholar]
- 15. Hu H, Tian M, Ding C, Yu S. The C/EBP Homologous Protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. 2018;9:3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanda T, Matsuoka S, Yamazaki M, Shibata T, Nirei K, Takahashi H, et al. Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol. 2018;24:2661–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Asadizade S, Hatami M, Salehipour Bavarsad S, Kabizade B, Shakerian E, Rashidi M. Curcumin modulates NOX gene expression and ROS production via P-Smad3C in TGF-beta-activated hepatic stellate cells. Iran Biomed J. 2024;28:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang JX, Mikami K, Venugopal S, Li Y, Török NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sugimoto K, Takei Y. Pathogenesis of alcoholic liver disease. Hepatol Res. 2017;47:70–79. [DOI] [PubMed] [Google Scholar]
- 20. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang C, Bai Y, Li T, Liu J, Wang Y, Ju S, et al. Ginkgetin exhibits antifibrotic effects by inducing hepatic stellate cell apoptosis via STAT1 activation. Phytother Res. 2024;38:1367–1380. [DOI] [PubMed] [Google Scholar]
- 22. New-Aaron M, Koganti SS, Ganesan M, Kanika S, Kumar V, Wang W, et al. Hepatocyte-specific triggering of hepatic stellate cell profibrotic activation by apoptotic bodies: The role of hepatoma-derived growth factor, HIV, and ethanol. Int J Mol Sci. 2023;24:5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsu SK, Chiu CC, Dahms HU, Chou CK, Cheng CM, Chang WT, et al. Unfolded protein response (UPR) in survival, dormancy, immunosuppression, metastasis, and treatments of cancer cells. Int J Mol Sci. 2019;20:2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mehrbod P, Ande SR, Alizadeh J, Rahimizadeh S, Shariati A, Malek H, et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence. 2019;10:376–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun P, Jin J, Wang L, Wang J, Zhou H, Zhang Q, et al. Porcine epidemic diarrhea virus infections induce autophagy in Vero cells via ROS-dependent endoplasmic reticulum stress through PERK and IRE1 pathways. Vet Microbiol. 2021;253:108959. [DOI] [PubMed] [Google Scholar]
- 26. Li X, Zheng J, Chen S, Meng F, Ning J, Sun S. Oleandrin, a cardiac glycoside, induces immunogenic cell death via the PERK/elF2alpha/ATF4/CHOP pathway in breast cancer. Cell Death Dis. 2021;12:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai). 2014;46:629–640. [DOI] [PubMed] [Google Scholar]
- 29. You C, Zhang Z, Ying H, Yang Z, Ma Y, Hong J, et al. Blockage of calcium-sensing receptor improves chronic intermittent hypoxia-induced cognitive impairment by PERK-ATF4-CHOP pathway. Exp Neurol. 2023;368:114500. [DOI] [PubMed] [Google Scholar]
- 30. Ning B, Zhang Q, Wang N, Deng M, Fang Y. beta-Asarone regulates ER stress and autophagy via inhibition of the PERK/CHOP/Bcl-2/Beclin-1 pathway in 6-OHDA-induced Parkinsonian rats. Neurochem Res. 2019;44:1159–1166. [DOI] [PubMed] [Google Scholar]