

Abstract
Protein post-translational modifications play a vital role in various cellular events essential for maintaining cellular physiology and homeostasis. In cancer cells, aberrant post-translational modifications such as glycosylation, acetylation, and phosphorylation on proteins can result in the generation of antigenic peptide variants presented in complex with MHC molecules. These modified peptides add to the class of tumorspecific antigens and offer promising avenues for targeted anti- cancer therapies. In this review, we focus on the role of phosphorylated peptides (p-peptides) in cancer immunity. We discuss the mechanisms by which the phosphorylated moiety modifies the structural features and binding properties of p-peptides with MHC, compared to their non-phosphorylated counterparts. Additionally, we review recent work on how the HLA-B*07-specific p-peptide, pMLL747–755, interacts with its cognate TCR. Altogether, p-peptides are emerging as a novel class of tumor-specific antigens, expanding the range of targets in cancer immunotherapy.
Keywords: Post-translational modifications, Immunopeptidome, Phosphorylation, Neoantigens, Immune checkpoint blockade therapies
Introduction
Immune checkpoint blockade (ICB) therapies have revolutionized the treatment landscape for various malignancies [1–5]. Clinical benefits from ICB in patients with advanced cancers are directly associated with high tumor mutational burden (TMB) [6,7], as tumors with elevated TMB, such as melanoma, tend to generate highly immunogenic tumor-specific antigens known as neoantigens [6,8–10]. Most neoantigens arise from random passenger mutations, with a significant proportion being unique and not commonly shared across patients.
Aside from mutations, neoantigens can also originate from aberrant post-translational modifications, including glycosylation, O-linked β-N-acetylglucosamine (O-GlcNAc), and phosphorylation [11,12], as well as dysregulated RNA splicing [13–15], proteasome processing, and transporter-associated aberrant antigen processing (TAP) [11,12,16,17]. Some of these post-translational modifications are cancer-specific but not patient-specific, making them promising shared tumor antigens and potential therapeutic targets [18]. For example, Dao et al. and Engelhard et al. highlighted the importance of cancer-specific phosphopeptides (p-peptides), such as those derived from insulin receptor substrate 2 (pIRS21097–1105) and breast cancer antiestrogen resistance 3 (pBCAR3126–134), as viable targets for cancer immunotherapy [19,20]. Additionally, O-GlcNAc-modified peptides associated with MHC-I (HLA-B*07:02) were identified as potential neoantigens in leukemia [21], suggesting that despite a low TMB, leukemias could still be highly immunogenic. Various in vitro-generated, peptide-specific T cell lines have been shown to specifically recognize post-translationally modified peptides, but not their unmodified counterparts, indicating that these modifications can lead to the generation of cancer-specific antigens and TCRs [12,21,22]. Immunogenic peptides resulting from phosphorylation represent an untapped class of neoantigens that could serve as off-the-shelf targets for neoantigen-based cancer immunotherapies. These peptides can also be explored for their potential as “public” tumor antigens, which could be incorporated into shared posttranslationally modified antigen-based treatment regimens across multiple patients. The possible identification and treatment strategy utilizing post translationally modified peptides is illustrated in Figure 1.
Figure 1.
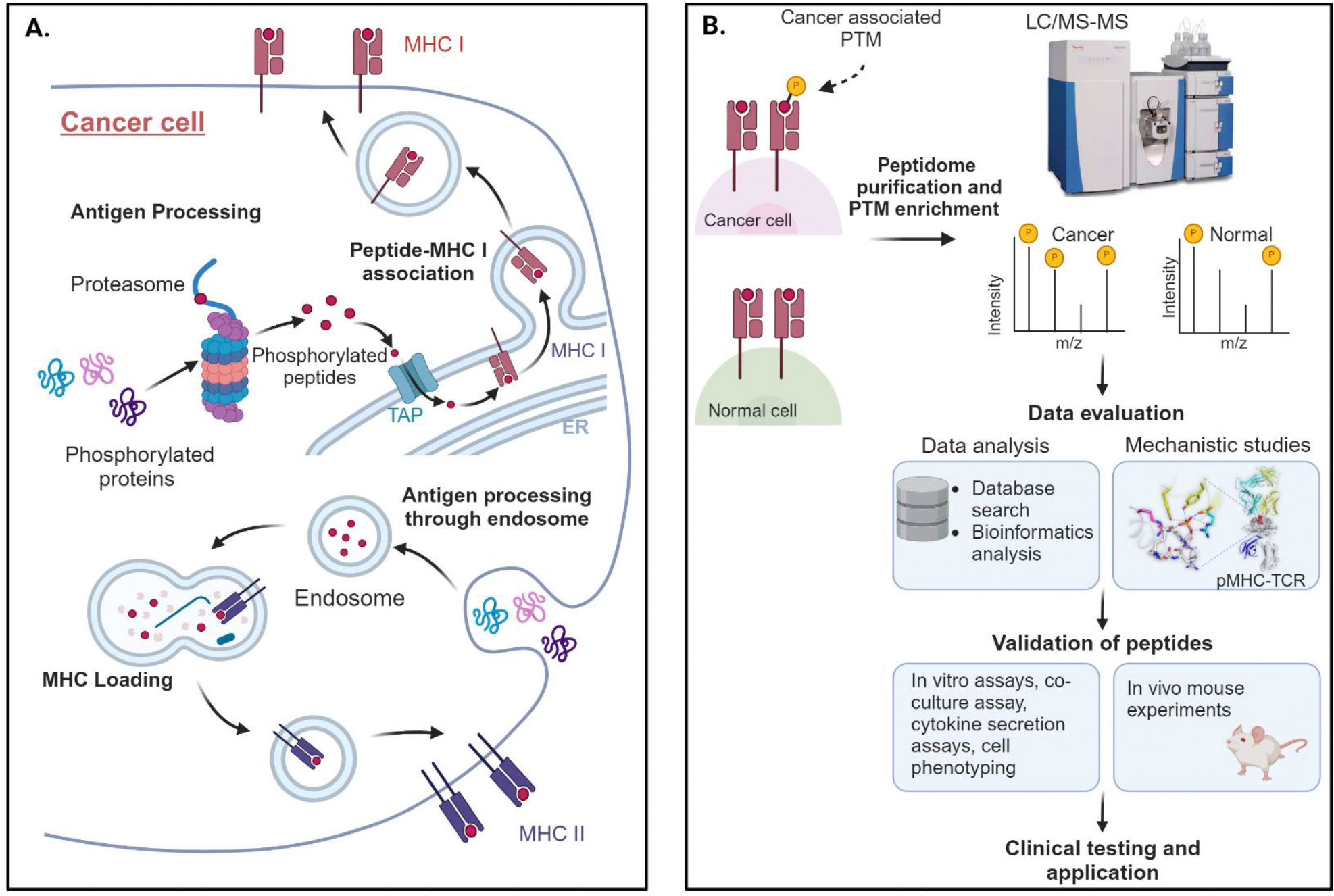
Phosphorylated antigen generation and their utilization for antigen-based treatment workflow in cancers. (A) Phosphorylated proteins are intracellularly processed and presented in complex with MHC I or MHC II molecules on cell surface. (B) The immunopeptidome purification and enrichment from cancer and normal tissues is done and peptide sequences are tested by Liquid Chromatography with tandem mass spectrometry (LC/MS-MS). The phosphorylated peptides from the immunopeptidome are evaluated using in silico tools for data mining and mechanistic studies and validated by both, in vitro and in vivo assays before selection for cancer vaccines. Created with BioRender.com.
Here, we focus on the emerging role of phosphorylated tumor antigens in enhancing tumor immunogenicity and developing anti-cancer therapies. Phosphorylated antigens offer a diverse landscape, as variations in phosphorylation sites and patterns give rise to a wide array of potential epitopes. This diversity benefits immunotherapy by providing multiple immune targets, which reduces the likelihood of tumor immune escape. Currently, advanced approaches for identifying the cancer phosphopeptidome, leveraging both sensitive analytical and computational tools, are being used. A list of phosphorylation-associated databases and tools is provided in Table 1.
Table 1.
Protein phosphorylation associated databases and tools.
| Databases and tools | Key Features | Refs | URL |
|---|---|---|---|
| Databases | |||
| PhosphoSitePlus | Online resource for studying experimentally observed PTMs such as phosphorylation, ubiquitination, and acetylation. | [23] | https://www.phosphosite.org |
| PHOSIDA | PTMs such as N-glycosylation, phosphorylation, and acetylation of proteins are listed | [24] | http://www.phosida.com |
| EPSD | Eukaryotic protein phosphorylation sites database | [25] | http://epsd.biocuckoo.cn |
| Phospho3D | Protein 3-D structures and phosphorylation database | [26] | http://www.phospho3d.org |
| LymPHOS 2.0 | Human T-lymphocyte phosphoproteome database | [27] | https://www.lymphos.org |
| RegPhos 2.0 | Open resource to explore protein kinase-substrate phosphorylation networks in mammals. | [28] | http://140.138.144.141/~RegPhos |
| PhosphoNET | Provide information on phosphorylation sites in over 20,000 human proteins. | [29] | http://www.phosphonet.ca |
| BioGRID | Open access PTM database | [30] | https://orcs.thebiogrid.org |
| Analysis tools used in MS-based discovery of PTMs | |||
| MSFragger | Database search tool using fragment ion indexing method to rapidly perform spectra similarity comparisons | [31] | https://msfragger.nesvilab.org/ |
| TagGraph | Tool based on string-based faster search method and probabilistic validation model | [32] | https://sourceforge.net/projects/taggraph/ |
| PTM-Shepherd | Analysis tool of open search results for PTMs and chemical modifications | [33] | https://github.com/Nesvilab/PTM-Shepherd |
Incorporating artificial intelligence (AI) into these predictive tools could significantly enhance the accuracy of phosphorylation site prediction and peptide selection. AI models, trained using large-scale data from comprehensive databases such as UniProt and PhosphoSitePlus, can predict phosphorylation sites with high accuracy. Tools such as NetPhos which utilizes neural networks, facilitate the prediction of potential phosphorylation sites. By integrating AI predictions with experimental validation, these models can be continuously refined through a feedback loop, improving the predictive accuracy of AI-based models over time.
In this review, we summarize the published data on phosphopeptide (p-peptide) neoantigens as possible targets for cancer immunotherapy, describe the mechanisms underlying their immunogenicity, and evaluate their potential clinical applications. We also discuss the likely advantages of p-peptide-based therapies, along with known obstacles and potential solutions.
Phosphopeptide Antigens in Cancer Immunotherapy
A variety of post-translational protein modifications, including glycosylation, acetylation, phosphorylation, and methylation, have been described in cancer-specific and associated peptides. Some post-translationally modified cancer-associated antigens that elicit an anti-cancer response are listed in Table 2. An imbalance between phosphorylation and dephosphorylation, facilitated by kinases and phosphatases, leads to dysregulated signal transduction pathways, often resulting in various malignancies, including cancer [34,35]. Reports indicate the presence of p-peptides in complex with Class I and Class II HLAs on multiple cancer types, including blood cancers, melanoma, breast cancer, and colorectal cancer (Table 2).
Table 2.
Post-translationally modified tumor-associated antigens.
| Modification | Source protein | Associated cancer | Immunogenicity | MHC restriction | Refs |
|---|---|---|---|---|---|
| Glycosylation | MUC1 | Breast cancer | CD8+ T cells | Class I | [30] |
| Myocyte specific enhancer factor 2C | Leukemia | CD8+ T cells | Class I | [21] | |
| RNA binding protein 27 | Leukemia | CD8+ T cells | Class I | ||
| MKL/myocardin-like protein 2 | Leukemia | CD8+ T cells | Class I | ||
| E1A-binding protein p400 | Leukemia | CD8+ T cells | Class I | ||
| Phosphorylation | MART-1 | Melanoma | CD4+ T cells | Class II | [42] |
| IRS2 | Melanoma, Breast | CD8+ T cells | Class I | [19, 20] | |
| BCAR3 | Melanoma | CD8+ T cells | Class I | [20] | |
| β catenin | Melanoma, Ovarian carcinoma | CD8+ T cells | Class I | [20,43] | |
| Cdc25b | Melanoma, ovarian carcinoma | CD8+ T cells | Class I | [43,44] | |
| TRAP-1 | Lung cancer | CD8+ T cells | Class I | [42] | |
| Vimentin | Colorectal cancer | CD4+ T Cells | Class II | [45] | |
| p53 | Head and neck SCC | CD4+ T Cells | Class II | [46] | |
| Acetylation | p53 | Colon, Prostrate | CD4+ T Cells | Class II | [47] |
| Eno1 | Pancreatic ductal carcinoma | CD4+ T Cells | Class II | [48,49] |
One primary source of dysregulated phosphorylation states and increased expression of phosphorylated antigens in cancer and virally infected cells is the suppression of PP2A, a critical phosphatase that regulates various signaling pathways involved in cell apoptosis, transformation, and proliferation [36–38]. High levels of endogenous PP2A negative regulators, such as SET proteins, or inhibitors, such as CIP2A, are associated with cancer progression [39]. Inhibition of PP2A leads to inactivation of retinoblastoma protein (pRb) and the tumor suppressor p53, two frequently mutated proteins in several cancers [40,41]. Unlike normal cells, where phosphorylation is brief and reversible, PP2A suppression in cancer cells extends the lifetime of phosphorylated proteins. This prolonged phosphorylation results in phosphorylated residues remaining even after proteasomal degradation and being presented as pMHC [38]. Decreased activity of another phosphatase, protein phosphatase 1 (PP1), is also associated with increased presentation of p-peptides as pMHC [50].
Although p-peptides have been detected in pMHC complexes isolated from both healthy and cancerous tissues, several epitopes have been found only in cancer cells, most prominently in melanoma and leukemias [43,51–53]. Interestingly, these dysregulated pathways generate the same phosphor-epitopes across multiple cancer types. The immunogenic properties of p-peptides make them attractive targets for cancer immunotherapy, offering a broad and tumorspecific antigen repertoire. Dysregulated phosphorylation of proteins is a hallmark of oncogenic transformation, and p-peptides can add a new layer of antigenicity to other cancer-specific epitopes. Therefore, p-peptides presented by cancer cells could provide an immunological signature of the ‘transformed self’.
Several studies have highlighted the potential of p-peptides as targets in anticancer therapies. Lin et al. [42] demonstrated that immunizing transgenic mice with an HLA-A2-specific p-peptide derived from tumor necrosis factor receptor-associated protein 1 (TRAP1) delayed tumor growth and extended survival. Similarly, HLA-A2-restricted p-peptides derived from IRS2 (pIRS21097–1105 (RVApSPTSGV)), β catenin (pβ catenin30–39 (YLDpSGIHSGA)), and CDC25b (pCDC25b38–46 (GLLGpSPVRA)) were recognized by specific CD8+ T lymphocytes in ovarian carcinoma (COV413), and melanoma cell lines (DM331, SLM2) [43]. Further supporting the clinical relevance, studies involving high risk melanoma patients demonstrated that HLA-A2-specific p-peptides, such as pIRS21097–1105 and pBCAR3126–134 (YLDpSGIHSGA), induced CD8+ T cell responses 42% and 17% of patients, respectively, in a clinical trial (NCT01846143) involving the pBCAR3 phosphopeptide-tetanus vaccine. Importantly, no grade 3–4 adverse effects, dose-limiting toxicity, or death occurred, while on study, underscoring the safety and immunogenicity of p-peptide vaccines [20].
Additionally, HLA-I-bound p-peptides have been identified in primary colorectal cancer tumors, liver metastases, and colorectal cancer cell lines [54]. CD8+ tumor-infiltrating lymphocytes (TILs) specific to p-peptides pTNS3 (VMIGpSPKKV) and pSELH (RRGpSFEVTL) were detected, and peripheral T cell activation was observed with pIRS2, pTNS3, and pSELH, indicating the potential role for p-peptides in colorectal cancer immunogenicity [54].
Few studies have identified MHC-II p-peptides recognized by CD4+ T cells, which are critical for the generation of effective and long-term anti-tumor immunity [42,55]. Human CD4+ T cells that specifically recognize an HLA-DR1-restricted phosphorylated melanoma-associated MART1 p-peptide (pMART1100–114(APPAYEKLpSAEQ)) were isolated from a cultured melanoma cell line [55]. HLA-DR- associated p-peptides have also been identified on human melanoma and Epstein-Barr virus (EBV)- transformed B lymphoblastoid lines [56]. Interestingly, similar results were reported in a separate study of human MHC class II-restricted p-peptides derived from other melanoma and B lymphoblastoid cell lines [57], suggesting a commonality of p-peptide presentation by MHC class II molecules.
The increased expression of intracellular phosphoproteins in dysregulated signaling pathways supports the malignant characteristics of tumor cells, and p-peptides may provide selective targets for immunotherapy. Studies have documented the differential expression of phosphoproteins in progressive tumors compared to normal and primary tumor sites. Penny et al. identified 120 HLA-I p-peptides from colorectal (CRC) cell lines, primary tumors, and liver metastases, assessing tumor-resident immunity against these p-peptides. Primary tumors displayed 3 times more p-peptides than healthy colon tissues, while liver metastases presented a 1.5-fold increase in p-peptides compared to primary site tumors. Interestingly, similar numbers of p-peptides were found in neighboring healthy liver tissues [54].
A comparative analysis of the phosphoproteome of colon cancer using patient-matched primary (SW480) and metastatic (SW620) cell lines revealed significant phosphorylation changes in critical cancer proteins in the metastatic SW620 cell line [58]. Aikio et al. studied phoshphorylation changes in RNA-binding proteins (RBPs) during prostate cancer development and identified 207 p-peptides originating from 133 RBPs. Phosphorylation patterns were consistent between benign and local prostate cancer. On contrary, there were significant changes between early to metastatic castration-resistant prostate cancer, with a reduction in phosphorylation in nearly one-third of cases and an increase in two-thirds [59]. Another study by Drake et al. identified 18 differentially phosphorylated kinases in clinical metastatic castration-resistant prostate cancer tissues [60]. These findings reveal distinct phosphorylation profiles between primary and metastatic tumors, suggesting that characterizing p-peptides across malignancies and tumor stages may uncover new neoantigens.
Despite these promising results, further research is needed to corroborate these findings, particularly regarding tumor progression, and to provide a comprehensive understanding of p-peptide neoantigens. Many p-peptides are commonly expressed across various cancer types and are recognized by healthy donor T cells, indicating that p-peptides could be safely targeted in a broad range of cancers.
The field of precision immunotherapy is rapidly evolving, with improvements in vaccine delivery methods and the use of combinatory immunotherapy strategies [61]. However, data on the safety and efficacy of post-translational neoantigen-based vaccines remain very limited, although the clinical trials show minimal to no adverse effects [20]. We expect a significant enhancement of antitumor efficacy when anti-cancer vaccines are combined with immune checkpoint inhibitors [62,63]. Further improvements in neoantigen delivery systems, such as self-amplifying mRNA and chemically synthesized minimal mRNA (CmRNA), may further enhance cellular trafficking, target specificity, and immunogenicity compared to in vitro transcribed mRNA (IVT-mRNA) [63,64].
Mechanisms of Phosphopeptide Immunogenicity
Limited mechanistic data have thus far hampered efforts to understand the role of post-translational protein modifications, including phosphorylation, in tumor immunity and to explore their potential applications in cancer immunotherapy. Establishing the molecular basis for p-peptide presentation in complex with MHC and their recognition by cognate TCRs would enable the rational design of immunotherapies targeting p-peptides.
Several studies have indicated that the presence of a phosphate group leads to physicochemical and conformational changes in a p-peptide-MHC complex compared to an unmodified wild-type (WT) peptide-MHC, thereby triggering immune recognition of the former [65–67]. The addition of a phosphate group to any peptide significantly alters its physicochemical properties. For instance, the negatively charged phosphate group (charge -2) is a strong hydrogen bond acceptor, which may support electrostatic interactions between the bound p-peptide and HLA, thereby increasing the stability of the binary complex [66,67].
In most cases, the anionic nature and sheer size of any phosphorylated residue, such as serine, threonine or tyrosine, likely prevents Class I HLAs from binding a p-peptide with phosphorylated anchor residues. These steric limitations, along with those imposed by kinase-specific phosphorylation patterns [68], result in the majority of HLA Class I p-peptides being phosphorylated at serine in position P4 [69] (Figure 2A). Moreover, many HLA-I-specific p-peptides share a common sequence motif with a positively charged basic arginine or lysine residue at P1 and a proline residue at P5 [52,66,70], reflecting the phosphorylation patterns of 1,4-basophilic (e.g., Akt) and proline-directed protein kinases (e.g., MAP kinases and CDKs) [68].
Figure 2.
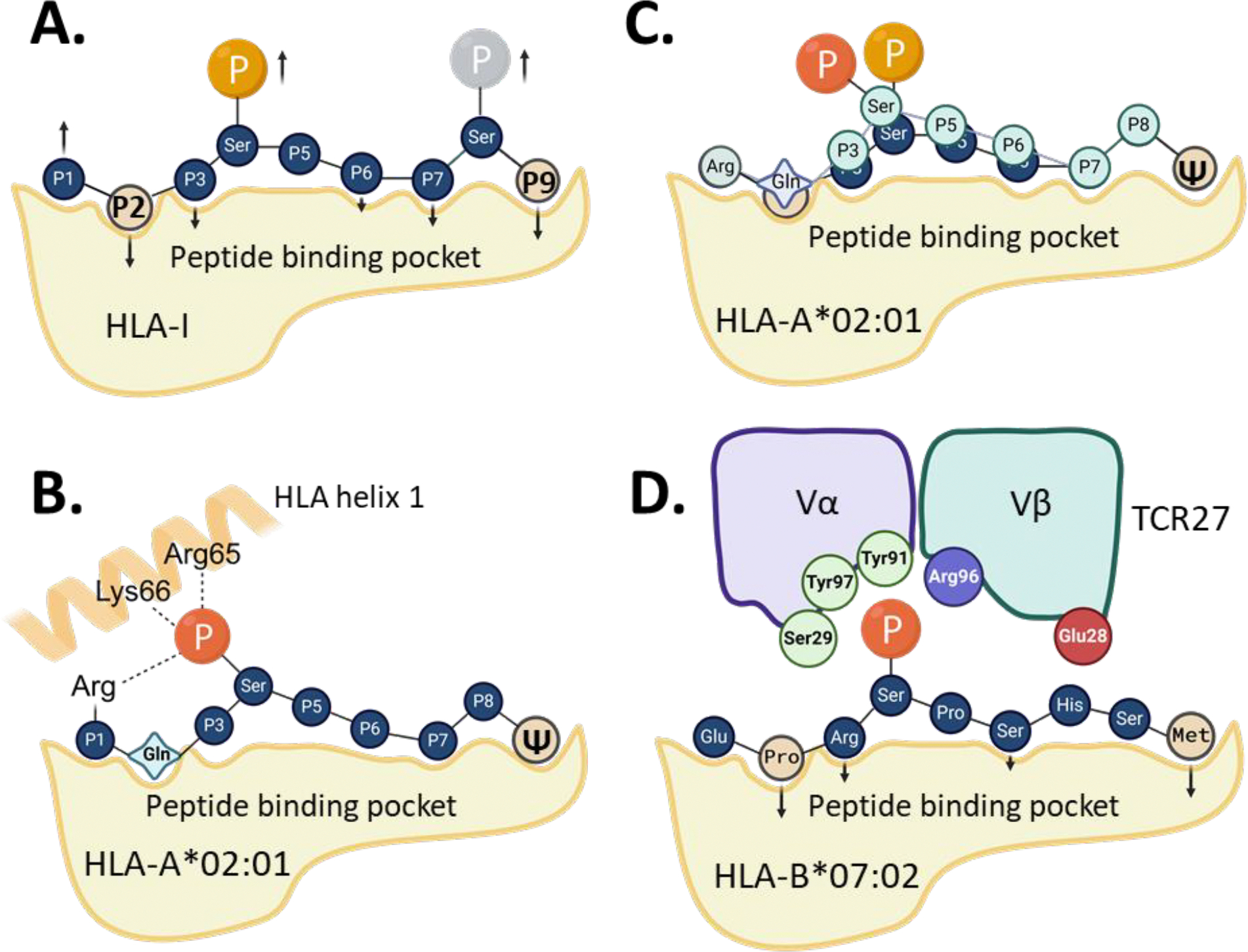
Binding between phosphopeptides and HLA class I molecules. (A) The most typical binding pattern between p-peptide and HLA with phosposerine at position P4 and/or P8 of a 9-mer peptide. The arrows show anchor residues (down) or non-anchor residues (up). P2 and P9 are primary anchor residues, others are usually optional and vary between different peptides. (B) The binding pattern between p-peptides with a consensus sequence of R/KQx(pS)xxxxΨ and HLA-A*02:01. Hydrogen bonds shown as dotted lines. (C) Superimposition between the two typical binding patterns observed for p-peptides in complex with HLA-A*02:01. (D) Schematic representation of the interface between HLA-B*07:02, pMLL p-peptide and TCR27 (variable region only). The TCR residues involved in hydrogen bonding with p-peptide are depicted. Ψ - aliphatic amino acid residues. Created with BioRender.com.
As a consequence of these restrictions, the phosphoserine residue in a typical p-peptide-HLA complex is a non-anchor residue. It remains at least partially exposed to solvent, making it a primary target for TCRs and a major determinant of p-peptide immunogenicity. The only exception to this rule has been reported for HLA-B*40:02-specific p-peptides, whose binding motif includes glutamic or aspartic acid at the anchor position P2, which may be substituted by phosphoserine [71]. However, p-peptides with pSer-P2 are extremely rare, and no crystal structures have been reported so far to confirm this finding.
The mechanistic data available to date, though limited to phosphoserine-containing peptides, demonstrate that the binding affinity between p-peptide and HLA, as well as the conformation of the binary complex, depend on: (1) HLA type and its peptide specificity, (2) the nature of the p-peptide, including its length, sequence, number of phosphate groups, and the positions of phosphorylated residues, and (3) the prevalence of certain protein phosphorylation motifs in cancer cells, which could limit both the number and types of p-peptides presented as pMHC.
In earlier studies by Zarling and co-authors [43,44,53], a substantial number of high-affinity HLA-A*02:01-specific p-peptides were identified with a glutamine residue in position P2. Non- phosphorylated (non-p) peptides with Gln-P2 (or other polar side chains) are usually poor binders to HLA-A*02:01, which typically prefers peptides Leu-P2 [66]. The unexpectedly high affinity and high immunogenicity observed for several Gln-P2-containing p-peptides prompted subsequent structural and mechanistic studies. Mohammed and co-authors determined the crystal structures of HLA-A*02:01 complexed with several p-peptides with various amino acid residues at P2 [66,67], revealing several unusual features of p-peptides recognized by HLA-A*02:01.
For instance, all the p-peptides with the N-terminal consensus sequence of R/KQx(pS) displayed very high binding affinities (with KD values in a low nanomolar range) to HLA-A*02:01. The pMHC crystal structures show that a phosphate group at pSer-P4 maintains electrostatic interactions with basic Arg and Lys side chains (Arg-P1, Arg65 and Lys66), forming an integral part of the p-peptide-HLA interface (Figure 2B) [22,57,67]. By contrast, non-p-peptides with N-terminal sequences of R/KQxS had much lower binding affinities to HLA-A*02:01, with KD values in the micromolar range. Additionally, the conformations of HLA-bound non-p-peptides and the relative orientations of the Arg-P1, Lys65, and Arg66 side chains differed from those observed in a p-peptide-HLA complex [66,67].
An exception to the affinity rule was the pLSP1 (RQA(pS) IELPSMAV) p-peptide, where the binding affinity was similar to that of a WT peptide (RQASIELPSMAV). The main factor was the peptide size (12 amino acid residues), which led to a similar “bulging” ligand conformation observed in both the non-phospho- and p-peptide N termini, including pSer-P4 and Ser-P4 residues, respectively [67]. Notably, the increase in binding affinity following peptide phosphorylation was dependent on the nature of amino acid residue in position P2, as follows: Q ≫ T ≫ V > M. The affinities between non-p-peptides and HLA-A*02:01 followed the reverse pattern: M > V ≫ T > >Q [65, 67].
Studies by Petersen and co-authors [65] further clarified the mechanisms controlling the binding between p-peptides and HLA-A*02:01. The authors solved the crystal structures of HLA-A*02:01 bound to three distinct p-peptide epitopes, alongside their matching non-p-peptides (Table 3), including pβ-catenin30–39 (YLD(pS)GIHSGA), pCDC25b38–46, (GLLG(pS) PVRA), and pIRS21097–1105 (RVA(pS)PTSGV). They also measured the binding affinities between these ligands and HLA and, notably, determined that phosphorylation had no effect on the binding between most peptides and HLA- A*02:01, except for the pIRS2 p-peptide, where the affinity was slightly increased compared to IRS2 peptide.
Table 3.
The crystal structures between distinct HLA molecules, p-peptides, phosphomimeticsand WT non-p-peptides.
| PDB ID | HLAtype | Peptide sequence | Protein name | Gene name/Uniprot ID | Main features | Refs |
|---|---|---|---|---|---|---|
| 3L6F | HLA-DRB1*01:01 | APPAYEKL(pS)AEQ | Melanoma antigen recognized by T cells, MART-1 | MLANA/Q16655 | Phosphoserine residue at position P5, exposed | [55] |
| 3BH8 | HLA-A*02:01 | RQA(pS)IELPSM | Lymphocyte specific protein 1 | LSP1/P33241 | High phosphopeptide affinity | [66] |
| 3BH9 | HLA-A*02:01 | RTY(pS)GPMNKV | Protein POF1B | POF1B/Q8WVV4 | High phosphopeptide affinity | |
| 3BHB | HLA-A*02:01 | KMD(pS)FLDMQL | Protein NEDD1 | NEDD1/Q8NHV4 | High phosphopeptide affinity | |
| 3BGM | HLA-A*02:01 | RQA(pS)LSISV | Serine/threonine-protein kinase D2 | PRKD2/Q9BZL6 | High phosphopeptide affinity | |
| 4NNX | HLA-A*02:01 | RQA(pS)LSISV | Serine/threonine-protein kinase D2 | PRKD2/Q9BZL6 | High phosphopeptide affinity | [67] |
| 4NNY | HLA-A*02:01 | RQASLSISV | Serine/threonine-protein kinase D2 | PRKD2/Q9BZL6 | Low WT peptide affinity | |
| 4NO3 | HLA-A*02:01 | RQI(pS)QDVKL | AMP deaminase 2 | Q01433/AMPD2 | High phosphopeptide affinity | |
| 4NO5 | HLA-A*02:01 | RQISQDVKL | AMP deaminase 2 | Q01433/AMPD2 | Low WT peptide affinity | |
| 4NO2 | HLA-A*02:01 | RQA(pS)IELPSMAV | Lymphocyte specific protein 1 | LSP1/P33241 | Phosphopeptide affinity similar to WT | |
| 4NOo | HLA-A*02:01 | RQASIELPSMAV | Lymphocyte specific protein 1 | LSP1/P33241 | Complex with LILRB1, high affinity WT peptide | |
| 3FQN | HLA-A*02:01 | YLDSGIHSGA | Catenin beta-1 | CTNNB1/P35222 | High peptide affinity | [65] |
| 3FQR | HLA-A*02:01 | YLD(pS)GIHSGA | Catenin beta-1 | CTNNB1/P35222 | Phosphopeptide affinity similar to WT | |
| 3FQT | HLA-A*02:01 | GLLGSPVRA | M-phase inducer phosphatase 2 | CDC25B/P30305 | High peptide affinity | |
| 3FQU | HLA-A*02:01 | GLLG(pS)PVRA | M-phase inducer phosphatase 2 | CDC25B/P30305 | Phosphopeptide affinity similar to WT | |
| 3FQW | HLA-A*02:01 | RVASPTSGV | Insulin receptor substrate 2 | IRS2/Q9Y4H2 | High peptide affinity | |
| 3FQX | HLA-A*02:01 | RVA(pS)PTSGV | Insulin receptor substrate 2 | IRS2/Q9Y4H2 | Phosphopeptide affinity higher than WT | |
| 5IEH | HLA-B*40:02 | REF(pS)KEPEL | Inner centromere protein | INCENP/Q9NQS7 | Phosphopeptide affinity similar to WT | [72] |
| 5IEK | HLA-B*40:02 | REFSKEPEL | Inner centromere protein | INCENP/Q9NQS7 | High peptide affinity | |
| 7CIQ | HLA-B*27:05 | RRFSRSPIRR | mRNA splicing cofactor, SON | SON/P18583 | High peptide affinity | [73] |
| 7CIR | HLA-B*27:05 | RRF(pS)RSPIRR | mRNA splicing cofactor, SON | SON/P18583 | Peptide affinity similar to WT | |
| 7DYN | HLA-B*27:05 | RRF(pS)R(pS)PIR R | mRNA splicing cofactor, SON | SON/P18583 | Phosphopeptide affinity much lower than WT | |
| 7CIS | HLA-B*27:05 | RRF(pS)R(pS)PIR | mRNA splicing cofactor, SON | SON/P18583 | Phosphopeptide affinity much lower than WT | |
| 7RZD | HLA-B*07:02 | EPRSPSHSM | Histone-lysine N-methyltransferase 2A | KMT2A/Q03164 | High peptide affinity | [22] |
| 7RZJ | HLA-B*07:02 | EPR(pS)PSHSM | Histone-lysine N-methyltransferase 2A | KMT2A/Q03164 | Phosphopeptide affinity similar to WT | |
| 7S8A | HLA-B*07:02 | EPR(pS)PSHSM | Histone-lysine N-methyltransferase 2A | KMT2A/Q03164 | Phosphopeptide affinity similar to WT | |
| 7S7D | HLA-B*07:02 | EPR(OSE)PSHSM, sulfo- | Histone-lysine N-methyltransfera se 2A | KMT2A/Q03164 | Sulfopeptide affinity similar to WT | |
| 7S79 | HLA-B*07:02 | EPR(E7P)PSHSM, phosphono- | Histone-lysine N-methyltransferase 2A | KMT2A/Q03164 | Phosphonopeptide affinity similar to WT | |
| 7S7F | HLA-B*07:02 | LPA(pS)PAHQL | Histone-lysine N-methyltransferase, H3 lysine-79 specific | DOT1L/Q8TEK3 | Phosphopeptide had slightly lower affinity | |
| 7S7E | HLA-B*07:02 | LPASPAHQL | Histone-lysine N-methyltransferase, H3 lysine-79 specific | DOT1L/Q8TEK3 | High peptide affinity |
These crystallographic studies revealed a potential mechanism behind the observed affinity data. The structures of HLA-A*02:01 bound to pβ-catenin or pCDC25b epitopes were very similar to those crystallized with their non-phosphorylated counterparts. In all three pMHC structures with above p-peptides [65], the phosphate groups were solvent-exposed and displayed signs of increased flexibility, as indicated by the presence of at least two alternating conformations for Ser-P4 in each structure.
The pβ-catenin30–39 peptide has Tyr-P1, whereas Leu-P2 and Ala-P10 serve as its anchor residues. Interestingly, phosphorylation of Ser33 is necessary for ubiquitin-mediated proteasomal degradation of β-catenin [74,75]. Since HLA Class I-restricted peptides are processed via the proteasomal pathway for presentation, there appears to be a link between this specific phosphorylation and p-peptide presentation by HLA-A*02:01.
In contrast, in pCDC25b38–46 (GLLG(pS)PVRA), the pSer residue is not in position P4 but at P5. In the CDC25b peptide-HLA-A*02:01 structure, Ser-P5 acts as an anchor residue. However, when phosphorylated, Ser-P5 becomes a non-anchor residue, resulting in conformational differences between the p-peptide and its WT counterpart. Although this conformational change does affect the binding affinity between HLA and the ligands, the P5-phosphorylated p-peptides could be immunogenic.
In a complex between HLA-A*02:01 and pIRS21097–1105, the predominant alternative conformation of the phosphate moiety is positioned within hydrogen-bonding distance from Lys66 and Arg65, which likely explains the increased stability of this complex compared to the non-p-peptide-MHC complex [65]. In summary, the strong binding between p-peptides and HLA-A*02:01 depends on multiple factors, as described in the examples above. However, in general, the structure of a p-peptide-HLA-A*02:01 complex is always different from that of its non-phosphorylated counterpart. This structural difference could potentially prevent cross-recognition between p-peptides and non-p-peptides by the same TCRs.
Binding between p-peptides and HLA-B molecules share certain features similar to those described above for HLA-A*02:01. However, no p-peptide has been reported yet for HLA-B alleles with a binding affinity significantly greater than that of its corresponding non-p-peptide. For example, in phospho-immunopeptidomics studies of B*07:02, B*27:01, B*39:01, and B*40:02 restricted p-peptides (8– 13mers), phosphorylation was observed at position P4 in more than 60% of all peptide ligands [72]. The majority of p-peptides, except those specific to B*39:01, also had a basic arginine at P1 [72]. The HLA-peptide binding motifs were the same for both p-peptides and non-p-peptides, and the presence or absence of Arg/Lys at P1 or pSer at P4 had no significant effect on the binding affinity between p- peptides and HLA-B molecules [22,72].
To determine a molecular basis for these observations, Alpizar and co-authors solved the crystal structure of HLA-B*40:02 in complex with the p-peptide pINCENP47–55 (pREF(pS)KEPEL) [72]. The structure revealed pSer-P4 as a non-anchor and solvent-accessible residue with a phosphate moiety pointing out of the binding pocket, making it accessible for interaction with the TCR. No significant conformational differences were observed between the HLA-B*40:02 structures in complex with pINCENP or the non-phosphorylated INCENP peptide. Moreover, contrary to what is observed with HLA-A*02:01, the P1 arginine did not interact with pSer-P4 and had no effect on the conformation of pSer-P4 [72].
Recent data reported by Zhao and co-authors [73] described the crystal structures of HLA-B*27:05 in complex with a SON peptide (RRFSRSPIRR) and its mono- or bi-phosphorylated variants (Table 3). Similarly to the previously described HLA-B*40:02 structure, the authors did not observe significant differences between the crystal structures pSON (RRF(pS) RSPIRR) or SON peptide ligands complexed with HLA-B*27:05. There was also no difference in pMHC binding affinities for these ligands. However, the introduction of a second phosphoserine at P6 resulted in a sharp decrease in binding affinity compared to the SON or pSON peptides. Comparison between the crystal structures for mono (phosphorylation at S4) and bi-phosphorylated (phosphorylation at S4 and S6) peptides revealed that phosphorylation at Ser-P6 causes a conformational switch from an anchor (Ser-P6) to a non-anchor (pSer-P6) position, affecting the hydrogen bonding pattern at the ligand-HLA interface and weakening the binding [73]. Other data suggest that the double-phosphorylated epitopes may exist, but predominantly with a phosphoserine located at P4 and in a penultimate position of an epitope, such as P8 of a 9-mer peptide) [69] (Figure 2C).
However, the relationship between phosphate exposure and epitope immunogenicity remains uncertain, as antigen-specific TCRs were not identified in the above studies. Our recent work [22] provided critical insights into molecular mechanisms underlying the immunogenicity of HLA-B*07:02- specific phosphor-neoantigens by describing the crystal structures for pMHC complexes and a p- peptide-specific TCR (TCR27), revealing the mechanism of p-peptide recognition by the TCR [22]. TCR27 is capable of cross-recognizing two p-peptides detected in acute myeloid leukemia (AML) and melanoma [52,70] – pMLL747–755 (EPR(pS)PSHSM) and pDOT1L998–1006 (LPA(pS)PAHQL), respectively. We demonstrated that replacing pSer-P4 in pMLL747–755 with various phosphomimetics reduced or abolished the interaction with TCR27 [22]. To elucidate the mechanism of p-peptide recognition by the TCR, we solved and compared the crystal structures of HLA-B*07:02 in complex with pMLL747–755 (EPR(pS)PSHSM) or pDOT1L998–1006 (LPA(pS)PAHQL), with phosphomimetics (phosphono- and sulfo- derivatives of pMLL), or their non-phosphorylated counterparts. Overall, phosphorylation at Ser-P4 (or its modification by phosphomimetics) had little effect on binding affinities between peptide ligands and HLA-B*07:02, which correlated with the lack of significant structural differences between the corresponding pMHC complexes. Structural similarities were found between these and the previously solved p-peptide-MHC structures with other HLA-B molecules, particularly in terms of pSer-P4 orientation, conformation, and high solvent exposure [72,73].
We also determined the crystal structure of TCR27 and used NMR-guided docking to model the ternary complex between HLA-B*07:02, pMLL747–755, and TCR27. In this complex, the phosphate moiety plays a crucial role by maintaining an extensive hydrogen bond network with surrounding TCR residues [22] (Figure 2D). This finding explains the sharp reduction in binding between pMHC and TCR27 when pSer-P4 replaced with phosphomimetics. Structural and biophysical analysis of TCR27-pMHC complexes revealed that the phosphate group defines the specificity and strength of TCR binding, which is particularly important for generating robust anti-tumor immune responses, as T cells can more effectively distinguish cancer cells from normal cells. Practically, these data present an opportunity for the rational design of TCRs targeting phospho-neoantigens.
To understand the mechanism of p-peptide binding in the context of MHC Class II and its recognition by CD4+ T cells, Li and co-authors [55] determined the crystal structure of pMART1100–114 (APPAYEKL(pS)AEQ) in complex with HLA-DRB*01:01 at a 2.1 Å resolution. MART1 is selectively expressed by melanoma and thus serves as a target for cancer vaccines. The crystal structure of the 15-residue pMART110–114 revealed Tyr104 (P1) and Ala109 (P6) as primary anchor residues, with a fully solvent-exposed phosphorylated Ser108 (P5). The presence of pSer-P5 slightly reduced the binding affinity of pMART1 to HLA compared to the wild-type peptide but resulted in specific recognition by a CD4+ T cell clone, D7-F6. These findings suggest that phosphorylation is a critical determinant of TCR recognition for both MHC I and MHC class II-restricted p-peptides.
Future Prospects
The cancer immunopeptidome is a treasure trove of tumor-specific and associated peptides that can be utilized in various immunotherapy applications, such as cancer vaccines. Immunopeptides may encompass post-translational modifications that are often overlooked by genomic and transcriptomic tools. These modified peptides are an emerging class of potential targets for shared tumor antigens. While this review focuses on phosphorylation, other modifications like glycosylation, acetylation, and citrullination may also result in cancer-specific modified peptides [76–78].
Phosphorylation may enhance epitope specificity, especially if their expression is restricted to cancer cells. These modifications are not processed in the thymus, allowing T cells to pass through central tolerance. Comprehensive studies on T cell responses to p-peptides, particularly in the context of MHC Class I, underscore the substantial potential for use in cancer therapy and vaccines. A key feature of p-peptides that drives the development of p-peptide-targeted agents is the distinct recognition surface presented by phosphorylated epitope sequences compared to their non-phosphorylated counterparts. Studies have clearly demonstrated that TCRs specific to p-peptides can recognize them without cross-reactivity to the wild-type peptides.
Despite challenges with their identification, phosphorylated antigens remain appealing targets for immunotherapeutic treatments due to their shared expression across different cancer types and potential in TCR therapy as shown by Patskovsky et al. [22]. They also hold potential for treating other diseases such as viral infections. Future research could incorporate p-peptides into neoadjuvant settings as cancer vaccines alone or in combination with ICB therapies with a possibility of synergistic effects that could lead to enhanced therapeutic outcomes. Their application as targets for peptide-centric chimeric antigen receptor T cell therapies, as exemplified by recent work by Yamarkovich et al. [79] can also be explored. Another important aspect to explore is the intra-tumor and inter-patient heterogeneity in p-peptide presentation and their potential implications for immunotherapy. A more detailed analysis of heterogeneity, both within tumors and across patients would be a valuable area for future investigation and could potentially lead to identification of potential p-peptides and their application in therapies.
In summary, post-translationally modified peptides, such as p-peptides, represent a novel and highly promising target for cancer therapy. The synergy of p-peptides or other post-translationally modified peptides with other forms of immunotherapy could significantly enhance the efficacy and specificity of anti-cancer treatments in the future.
Acknowledgments
This work was supported by the R01CA243486, R01CA284604, P50CA225450 (to M.K.) and the New York University Center for Blood Cancer (CBC) Developmental Grant supported by an NIH P30CA016087 grant (to M.K. and Y.P.).
Conflict of Interest
MK previously received support from Agenus for published work on phosphoantigens described here. MK serves as an advisor for Merck Sharp and Dohme. MK currently receives research support from Merck Sharp and Dohme, Genentech, Biogen and Novartis which is not related to this work.
References
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010. Aug 19;363(8):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015. Jul 2;373(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015. Jun 25;372(26):2521–32. [DOI] [PubMed] [Google Scholar]
- 4.Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019. Aug 1;30(8):1321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang C, Xia BR, Zhang ZC, Zhang YJ, Lou G, Jin WL. Immunotherapy for Ovarian Cancer: Adjuvant, Combination, and Neoadjuvant. Front Immunol. 2020. Oct 6;11:577869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aggarwal C, Ben-Shachar R, Gao Y, Hyun SW, Rivers Z, Epstein C, et al. Assessment of Tumor Mutational Burden and Outcomes in Patients With Diverse Advanced Cancers Treated With Immunotherapy. JAMA Netw Open. 2023. May 1;6(5):e2311181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hotz MJ, O’Halloran EA, Hill MV, Hayden K, Zaladonis AG, Deng M, et al. Tumor mutational burden and somatic mutation status to predict disease recurrence in advanced melanoma. Melanoma Res. 2022. Apr 1;32(2):112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. 2023. Jan 6;8(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng M Tumor mutation burden for predicting immune checkpoint blockade response: the more, the better. J Immunother Cancer. 2022. Jan;10(1):e003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang AC, Zappasodi R. A decade of checkpoint blockade immunotherapy in melanoma: understanding the molecular basis for immune sensitivity and resistance. Nat Immunol. 2022. May;23(5):660–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vlad AM, Muller S, Cudic M, Paulsen H, Otvos L Jr, Hanisch FG, et al. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J Exp Med. 2002. Dec 2;196(11):1435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apostolopoulos V, Yuriev E, Ramsland PA, Halton J, Osinski C, Li W, et al. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc Natl Acad Sci U S A. 2003. Dec 9;100(25):15029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang E, Aifantis I. RNA Splicing and Cancer. Trends Cancer. 2020. Aug;6(8):631–44. [DOI] [PubMed] [Google Scholar]
- 14.Hoyos LE, Abdel-Wahab O. Cancer-Specific Splicing Changes and the Potential for Splicing-Derived Neoantigens. Cancer Cell. 2018. Aug 13;34(2):181–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahles A, Lehmann KV, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell. 2018. Aug 13;34(2):211–24.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marijt KA, Doorduijn EM, van Hall T. TEIPP antigens for T-cell based immunotherapy of immune-edited HLA class Ilow cancers. Mol Immunol. 2019. Sep;113:43–9. [DOI] [PubMed] [Google Scholar]
- 17.van Hall T, Wolpert EZ, van Veelen P, Laban S, van der Veer M, Roseboom M, et al. Selective cytotoxic T-lymphocyte targeting of tumor immune escape variants. Nat Med. 2006. Apr;12(4):417–24. [DOI] [PubMed] [Google Scholar]
- 18.RodrÍguez E, Schetters STT, van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol. 2018. Mar;18(3):204–11. [DOI] [PubMed] [Google Scholar]
- 19.Dao T, Mun SS, Molvi Z, Korontsvit T, Klatt MG, Khan AG, et al. A TCR mimic monoclonal antibody reactive with the “public” phospho-neoantigen pIRS2/HLA-A*02:01 complex. JCI Insight. 2022. Mar 8;7(5):e151624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelhard VH, Obeng RC, Cummings KL, Petroni GR, Ambakhutwala AL, Chianese-Bullock KA, et al. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J Immunother Cancer. 2020. May;8(1):e000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malaker SA, Penny SA, Steadman LG, Myers PT, Loke JC, Raghavan M, et al. Identification of Glycopeptides as Posttranslationally Modified Neoantigens in Leukemia. Cancer Immunol Res. 2017. May;5(5):376–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patskovsky Y, Natarajan A, Patskovska L, Nyovanie S, Joshi B, Morin B, et al. Molecular mechanism of phosphopeptide neoantigen immunogenicity. Nat Commun. 2023. Jun 23;14(1):3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015. Jan;43(Database issue):D512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gnad F, Gunawardena J, Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011. Jan;39(Database issue):D253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin S, Wang C, Zhou J, Shi Y, Ruan C, Tu Y, et al. EPSD: a well-annotated data resource of protein phosphorylation sites in eukaryotes. Brief Bioinform. 2021. Jan 18;22(1):298–307. [DOI] [PubMed] [Google Scholar]
- 26.Zanzoni A, Ausiello G, Via A, Gherardini PF, Helmer-Citterich M. Phospho3D: a database of three-dimensional structures of protein phosphorylation sites. Nucleic Acids Res. 2007. Jan;35(Database issue):D229–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen TD, Vidal-Cortes O, Gallardo O, Abian J, Carrascal M. LymPHOS 2.0: an update of a phosphosite database of primary human T cells. Database (Oxford). 2015. Dec 26;2015:bav115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang KY, Wu HY, Chen YJ, Lu CT, Su MG, Hsieh YC, et al. RegPhos 2.0: an updated resource to explore protein kinase-substrate phosphorylation networks in mammals. Database (Oxford). 2014. Apr 25;2014(0):bau034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safaei J, Maňuch J, Gupta A, Stacho L, Pelech S. Prediction of 492 human protein kinase substrate specificities. Proteome Sci. 2011. Oct 14;9 Suppl 1(Suppl 1):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oughtred R, Stark C, Breitkreutz BJ, Rust J, Boucher L, Chang C, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019. Jan 8;47(D1):D529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods. 2017. May;14(5):513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devabhaktuni A, Lin S, Zhang L, Swaminathan K, Gonzalez CG, Olsson N, et al. TagGraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nat Biotechnol. 2019. Apr;37(4):469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geiszler DJ, Kong AT, Avtonomov DM, Yu F, Leprevost FDV, Nesvizhskii AI. PTM-Shepherd: Analysis and Summarization of Post-Translational and Chemical Modifications From Open Search Results. Mol Cell Proteomics. 2021;20:100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turdo A, D’Accardo C, Glaviano A, Porcelli G, Colarossi C, Colarossi L, et al. Targeting Phosphatases and Kinases: How to Checkmate Cancer. Front Cell Dev Biol. 2021. Oct 28;9:690306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vainonen JP, Momeny M, Westermarck J. Druggable cancer phosphatases. Sci Transl Med. 2021. Apr 7;13(588):eabe2967. [DOI] [PubMed] [Google Scholar]
- 36.Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J. 2016. Mar;283(6):1004–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruvolo PP. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016. Aug 3;6:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahoney KE, Shabanowitz J, Hunt DF. MHC Phosphopeptides: Promising Targets for Immunotherapy of Cancer and Other Chronic Diseases. Mol Cell Proteomics. 2021;20:100112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lucas CM, Harris RJ, Holcroft AK, Scott LJ, Carmell N, McDonald E, et al. Second generation tyrosine kinase inhibitors prevent disease progression in high-risk (high CIP2A) chronic myeloid leukaemia patients. Leukemia. 2015. Jul;29(7):1514–23. [DOI] [PubMed] [Google Scholar]
- 40.Vélez-Cruz R, Johnson DG. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int J Mol Sci. 2017. Aug 16;18(8):1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017. Sep 7;170(6):1062–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin MH, Shen KY, Liu BS, Chen IH, Sher YP, Tseng GC, et al. Immunological evaluation of a novel HLA-A2 restricted phosphopeptide of tumor associated Antigen, TRAP1, on cancer therapy. Vaccine X. 2019. Mar 11;1:100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zarling AL, Polefrone JM, Evans AM, Mikesh LM, Shabanowitz J, Lewis ST, et al. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc Natl Acad Sci U S A. 2006. Oct 3;103(40):14889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zarling AL, Obeng RC, Desch AN, Pinczewski J, Cummings KL, Deacon DH, et al. MHC-restricted phosphopeptides from insulin receptor substrate-2 and CDC25b offer broad-based immunotherapeutic agents for cancer. Cancer Res. 2014. Dec 1;74(23):6784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohara M, Ohara K, Kumai T, Ohkuri T, Nagato T, Hirata-Nozaki Y, et al. Phosphorylated vimentin as an immunotherapeutic target against metastatic colorectal cancer. Cancer Immunol Immunother. 2020. Jun;69(6):989–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohara K, Ohkuri T, Kumai T, Nagato T, Nozaki Y, Ishibashi K, et al. Targeting phosphorylated p53 to elicit tumor-reactive T helper responses against head and neck squamous cell carcinoma. Oncoimmunology. 2018. Aug 1;7(9):e1466771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumai T, Ishibashi K, Oikawa K, Matsuda Y, Aoki N, Kimura S, et al. Induction of tumor-reactive T helper responses by a posttranslational modified epitope from tumor protein p53. Cancer Immunol Immunother. 2014. May;63(5):469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou W, Capello M, Fredolini C, Piemonti L, Liotta LA, Novelli F, et al. Mass spectrometry analysis of the post-translational modifications of alpha-enolase from pancreatic ductal adenocarcinoma cells. J Proteome Res. 2010. Jun 4;9(6):2929–36. [DOI] [PubMed] [Google Scholar]
- 49.Capello M, Caorsi C, Bogantes Hernandez PJ, Dametto E, Bertinetto FE, Magistroni P, et al. Phosphorylated alpha-enolase induces autoantibodies in HLA-DR8 pancreatic cancer patients and triggers HLA-DR8 restricted T-cell activation. Immunol Lett. 2015. Sep;167(1):11–6. [DOI] [PubMed] [Google Scholar]
- 50.Wu JQ, Guo JY, Tang W, Yang CS, Freel CD, Chen C, et al. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat Cell Biol. 2009. May;11(5):644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol. 2017. Nov 1;199(9):3360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. 2016. Nov 21;7:13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zarling AL, Ficarro SB, White FM, Shabanowitz J, Hunt DF, Engelhard VH. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J Exp Med. 2000. Dec 18;192(12):1755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Penny SA, Abelin JG, Malaker SA, Myers PT, Saeed AZ, Steadman LG, et al. Tumor Infiltrating Lymphocytes Target HLA-I Phosphopeptides Derived From Cancer Signaling in Colorectal Cancer. Front Immunol. 2021. Aug 24;12:723566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Depontieu FR, Sidney J, Salay TM, Engelhard VH, Hunt DF, et al. Structural basis for the presentation of tumor-associated MHC class II-restricted phosphopeptides to CD4+ T cells. J Mol Biol. 2010. Jun 18;399(4):596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Depontieu FR, Qian J, Zarling AL, McMiller TL, Salay TM, Norris A, et al. Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc Natl Acad Sci U S A. 2009. Jul 21;106(29):12073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyer VS, Drews O, Günder M, Hennenlotter J, Rammensee HG, Stevanovic S. Identification of natural MHC class II presented phosphopeptides and tumor-derived MHC class I phospholigands. J Proteome Res. 2009. Jul;8(7):3666–74. [DOI] [PubMed] [Google Scholar]
- 58.Schunter AJ, Yue X, Hummon AB. Phosphoproteomics of colon cancer metastasis: comparative mass spectrometric analysis of the isogenic primary and metastatic cell lines SW480 and SW620. Anal Bioanal Chem. 2017. Mar;409(7):1749–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aikio E, Koivukoski S, Kallio E, Sadeesh N, Niskanen EA, Latonen L. Complementary analysis of proteome-wide proteomics reveals changes in RNA binding protein-profiles during prostate cancer progression. Cancer Rep (Hoboken). 2023. Oct;6(10):e1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell. 2016. Aug 11;166(4):1041–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin X, Tang S, Guo Y, Tang R, Li Z, Pan X, et al. Personalized neoantigen vaccine enhances the therapeutic efficacy of bevacizumab and anti-PD-1 antibody in advanced non-small cell lung cancer. Cancer Immunol Immunother. 2024. Jan 27;73(2):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imani S, Tagit O, Pichon C. Neoantigen vaccine nanoformulations based on Chemically synthesized minimal mRNA (CmRNA): small molecules, big impact. NPJ Vaccines. 2024. Jan 18;9(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Xiao Z, Wang D, Jia L, Nie S, Zeng X, et al. The screening, identification, design and clinical application of tumor-specific neoantigens for TCR-T cells. Mol Cancer. 2023. Aug 30;22(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palmer CD, Rappaport AR, Davis MJ, Hart MG, Scallan CD, Hong SJ, et al. Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Nat Med. 2022. Aug;28(8):1619–29. [DOI] [PubMed] [Google Scholar]
- 65.Petersen J, Wurzbacher SJ, Williamson NA, Ramarathinam SH, Reid HH, Nair AK, et al. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc Natl Acad Sci U S A. 2009. Feb 24;106(8):2776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohammed F, Cobbold M, Zarling AL, Salim M, Barrett-Wilt GA, Shabanowitz J, et al. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nat Immunol. 2008. Nov;9(11):1236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mohammed F, Stones DH, Zarling AL, Willcox CR, Shabanowitz J, Cummings KL, et al. The antigenic identity of human class I MHC phosphopeptides is critically dependent upon phosphorylation status. Oncotarget. 2017. Apr 8;8(33):54160–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A. A curated compendium of phosphorylation motifs. Nat Biotechnol. 2007. Mar;25(3):285–6. [DOI] [PubMed] [Google Scholar]
- 69.Solleder M, Guillaume P, Racle J, Michaux J, Pak HS, Müller M, et al. Mass Spectrometry Based Immunopeptidomics Leads to Robust Predictions of Phosphorylated HLA Class I Ligands. Mol Cell Proteomics. 2020. Feb;19(2):390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cobbold M, De La Peña H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med. 2013. Sep 18;5(203):203ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marcilla M, Alpízar A, Lombardía M, Ramos-Fernandez A, Ramos M, Albar JP. Increased diversity of the HLA-B40 ligandome by the presentation of peptides phosphorylated at their main anchor residue. Mol Cell Proteomics. 2014. Feb;13(2):462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alpízar A, Marino F, Ramos-Fernández A, Lombardía M, Jeko A, Pazos F, et al. A Molecular Basis for the Presentation of Phosphorylated Peptides by HLA-B Antigens. Mol Cell Proteomics. 2017. Feb;16(2):181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Y, Sun M, Zhang N, Liu X, Yue C, Feng L, et al. Phosphosite-dependent presentation of dual phosphorylated peptides by MHC class I molecules. iScience. 2022. Mar 1;25(4):104013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002. Mar 22;108(6):837–47. [DOI] [PubMed] [Google Scholar]
- 75.Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem. 1997. Oct 3;272(40):24735–8. [DOI] [PubMed] [Google Scholar]
- 76.Katayama H, Kobayashi M, Irajizad E, Sevillarno A, Patel N, Mao X, et al. Protein citrullination as a source of cancer neoantigens. J Immunother Cancer. 2021. Jun;9(6):e002549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015. Sep;15(9):540–55. [DOI] [PubMed] [Google Scholar]
- 78.Zhong Q, Xiao X, Qiu Y, Xu Z, Chen C, Chong B, et al. Protein posttranslational modifications in health and diseases: Functions, regulatory mechanisms, and therapeutic implications. MedComm (2020). 2023. May 2;4(3):e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yarmarkovich M, Marshall QF, Warrington JM, Premaratne R, Farrel A, Groff D, et al. Targeting of intracellular oncoproteins with peptide-centric CARs. Nature. 2023. Nov;623(7988):820–27. [DOI] [PMC free article] [PubMed] [Google Scholar]