

ABSTRACT
Cisplatin is used to treat a variety of malignancies, including testicular germ cell tumours (TGCTs). Although cisplatin‐based chemotherapy yields high response rates, a subset of patients develop cisplatin resistance, limiting treatment options and worsening prognosis. Therefore, there is a high clinical need for new therapeutic strategies targeting cisplatin‐resistant TGCTs. MicroRNA‐371a‐3p (miR‐371), the new serum biomarker for TGCTs, shows significantly increased expression in cisplatin‐resistant TGCT cell lines compared to sensitive parental cell lines. However, the functional impact of miR‐371 on cisplatin sensitivity has not been investigated yet. To evaluate the impact of miR‐371 on cisplatin sensitivity, antagomirs were used to inhibit miR‐371 expression, resulting in a > 98% decrease in miR‐371 expression. Cisplatin sensitivity was significantly increased after miR‐371 inhibition in cisplatin‐resistant and corresponding parental TGCT cell lines, indicating a strongly reduced viability and increased apoptosis after cisplatin treatment in miR‐371‐inhibited cells. Our results suggest that miR‐371 may contribute to the development of cisplatin resistance in TGCTs. Interfering with miR‐371 expression can increase the cisplatin sensitivity of tumour cells, which may represent a promising approach to improve future therapeutic outcomes in patients with TGCTs, especially those with cisplatin‐resistant disease.
Keywords: cisplatin, miR‐371, resistance mechanisms, testicular germ cell tumour
1. Introduction
Testicular germ cell tumours (TGCTs) represent the most prevalent cancer among men aged 15–40 years, with an increasing incidence over the past four decades [1]. Cisplatin‐based chemotherapy is frequently used in treating various malignancies, including head and neck, lung, gastrointestinal tract and genitourinary cancers, for example, urothelial, vulvar, penile squamous cell carcinoma, as well as ovarian and testicular germ cell tumours [2]. Despite the high efficacy of cisplatin‐based regimens in TGCTs, a subset of patients (~10%) develops cisplatin resistance [1, 3], leading to disease recurrence and poorer prognosis [1]. Moreover, even patients who achieve successful outcomes with cisplatin therapy may face acute and lifelong toxicities [1, 4]. Therefore, there is an urgent clinical need to develop new treatment approaches that address cisplatin refractory TGCTs while minimising the cytotoxic impact of cisplatin.
The molecular mechanisms leading to cisplatin resistance in TGCTs are likely multifactorial and can be categorised into four primary mechanisms [5, 6]: (i) decreased cellular import and increased export of cisplatin (pretarget); (ii) decreased accumulation of cisplatin in the cell (on‐target); (iii) reduced induction of apoptosis (posttarget) and (iv) activated compensatory signalling pathways that antagonise the cisplatin resistance mechanism without being a direct target of it (off‐target) [5, 6].
Previous research has highlighted the upregulation of microRNAs (miRs) from the miR‐371‐3 and miR‐302/367 cluster across various cancers, particularly in TGCTs, where they were first proposed as new biomarkers in 2011 [7]. These miRs exhibit superior sensitivity and specificity compared to the established serum tumour markers, including beta‐human chorionic gonadotropin, alpha‐fetoprotein and lactate dehydrogenase [8, 9, 10, 11]. In general, miRs are small noncoding RNAs involved in the epigenetic regulation of gene expression and various processes of tumour progression, including proliferation, angiogenesis, epithelial‐to‐mesenchymal transition, metastasis and DNA repair [12, 13]. Among them, miR‐371‐3p (miR‐371) has emerged as the most sensitive (90.1%) and specific (94.0%) biomarker for TGCT diagnosis, treatment monitoring and detection of residual or recurrent disease [8, 9, 10, 11]. Importantly, miR‐371 is expressed in both seminomas and nonseminomas, except teratomas [8, 9, 10, 11].
This study aimed to investigate the influence of miR‐371 on cisplatin sensitivity in parental TGCT cell lines and their matched cisplatin‐resistant subclones, thereby enhancing our understanding of the molecular basis of cisplatin response and resistance mechanisms.
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
Human parental TGCT cell lines, NCCIT, 2102EP and NT2/D1 (arises from an embryonic carcinoma) and their respective cisplatin‐resistant subclones, NCCIT‐R, 2102EP‐R and NT2/D1‐R (provided with the generous permission of Oing C. et Honecker F. [14, 15]) were grown in a 5% CO2 incubator at 37°C. Cisplatin‐resistant subclones were generated by cultivating the parental cell lines in increasing concentration of cisplatin as published [14, 15, 16]. Monolayer cultures were maintained in RPMI 1640 medium (#31870‐025; Thermo Fisher Scientific, Darmstadt, Germany) supplemented with 10% heat‐inactivated foetal calf serum (#26140‐079; Thermo Fisher Scientific), 0.8% streptomycin–penicillin antibiotics (10,000 units/mL penicillin and 10,000 μg/mL streptomycin) (#15140‐122; Thermo Fisher Scientific) and 1% l‐glutamine (200 mM; #25030‐024; Thermo Fisher Scientific).
2.2. RNA Transfection and RNA Isolation
By using two different antagomiRs, miRCURY LNA miRNA inhibitor HSA‐MIR‐371A‐3P (#267258255; Qiagen, Hilden, Germany, inhibitor 1) and has_miRNA inhibitor (#76209062; IDT, Iowa, USA, inhibitor 2), we established a miR‐371‐inhibition. The miRCURY LNA miRNA inhibitor control (Negative control A [#201802070018‐2; Qiagen]) was used as miR‐inhibitor negative control (CTRL). Tumour cells were transfected with 100 μL transfection mix (10% inhibitor/negative control 10 μM solution, 10% HD‐transfection reagent FuGENE) (#E2311; Promega Corporation, Madison, USA, 90% RPMI) in 1.9 mL culture medium. After incubating the cells for 48 and 72 h, RNA was isolated from cell line pellets using the Total RNA Purification Mini Spin Column Kit (Genaxxon Bioscience GmbH, Ulm, Germany), and RNA quantity and quality were measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
2.3. Quantitative Reverse Transcriptase‐Polymerase Chain Reaction
Complementary DNA (cDNA) was synthesised using the miScript II RT Kit (#218161; Qiagen). Quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR) was performed using 5 ng/mL cDNA and miScript SYBR Green PCR Kit (#218073; Qiagen). The following predesigned Qiagen miScript primer sequences were used: Hs_miR‐371_1 (MS00004060), Hs_miR‐335_1 (MS00003976) and Hs_RNU1A_11 (MS00013986). All samples were run in triplicates, and the relative expression was calculated using the equation RQ = 2−ΔΔCT .
2.4. Measurement of Cell Viability
To measure the cisplatin sensitivity after miR‐371‐inhibition (performed with inhibitor 1), we used a Crystal violet assay. The assay involves exposing the cells (8 × 103 or 1 × 104 cells/well) to different concentrations of cisplatin (0–12 μM) for 24–72 h. Triplicates were made for all conditions. For staining, cells were fixed with 37% paraformaldehyde for 10 min, washed with distilled water and stained with 0.05% crystal violet for 30 min. Cells were washed with distilled water again and dried in room air. To dissolve the dye, 0.1% acetic acid per well was added. The absorbance of the stained cells was measured using an ultraviolet–visible spectrometer (570 nm, Safire Reader [Tecan]). The viability of the cells was calculated based on the absorbance values.
2.5. Flow Cytometry
Flow cytometry was used to analyse the rates of apoptosis (programmed cell death) after miR‐371‐inhibition (performed with inhibitor 1) by using Annexin V/propidium iodide (PI). The staining procedure was carried out according to standard protocols. Early apoptotic cells were defined as Annexin V‐positive/PI‐negative, whereas late apoptotic cells were defined as Annexin V/PI‐positive and necrotic cells were Annexin V‐negative/PI‐positive. Viable cells remained unstained (Annexin V/PI‐negative). A total of 5 × 104 cells was measured for each sample. Single‐cell suspensions of NCCIT and 2102EP and their respective cisplatin‐resistant subclones were stained with 5 μL Annexin V (#640919; BioLegend, San Diego, CA, USA) followed by 20 μg/mL PI (#421301; BioLegend). Data were acquired using a Cytec Aurora flow cytometer (Cytek Biosciences, Fremont, CA, USA) and analysed with the FlowJo software (FlowJo v10.8; BD, https://www.flowjo.com).
2.6. Statistical Analysis
Statistical analysis was conducted using GraphPad Prism (Version 9.4.0). The parametric t‐test was used to statistically compare two groups, while the parametric one‐way ANOVA was employed to compare multiple groups. All p‐values were calculated two‐sided, and p < 0.05 was considered statistically significant.
3. Results
3.1. miR‐371 Is Overexpressed in TGCT Cisplatin‐Resistant Cell Lines
In three nonseminomatous TGCT cell lines (NCCIT, 2102EP and NT2/D1) and their matched cisplatin‐resistant subclones (NCCIT‐R, 2102EP‐R and NT2/D1‐R), the expression levels of miR‐371 were determined by qRT‐PCR. The miR‐371 expression was significantly increased in the cisplatin‐resistant TGCT cell lines compared to sensitive parental cell lines (230.6 ± 30.79 [NCCIT vs. NCCIT‐R], 1.997 ± 0.178 [2102EP vs. 2102EP‐R], 218.2 ± 74.84 [NT2/D1 vs. NT2/D1‐R], p < 0.01), as depicted in Figure 1A–C. These data represent an initial indication of the potential involvement of miR‐371 in the development of cisplatin resistance in TGCT cell lines.
FIGURE 1.
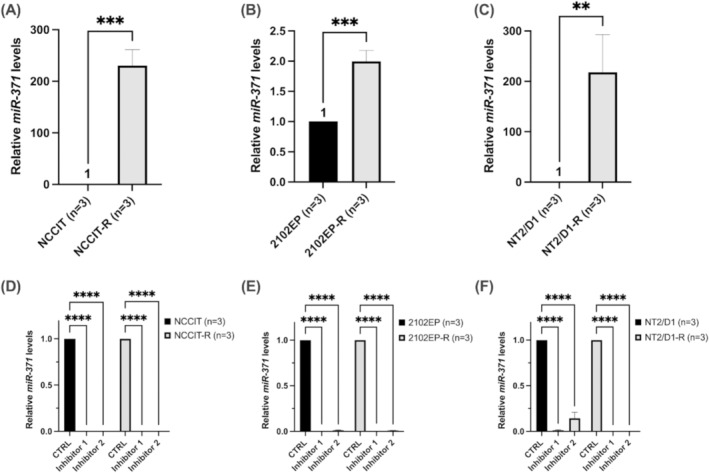
(A–C) Expression analysis of miR‐371 in TGCT cell lines. miR‐371 expression was significantly increased in cisplatin‐resistant testicular germ cell tumour (TGCT) cell lines (NCCIT‐R, 2102EP‐R and NT2/D1‐R) compared to sensitive parental cell lines (NCCIT, 2102EP and NT2/D1). (D–F) Downregulation of miR‐371 levels in TGCT cell lines. By using two different antagomirs (Inhibitors 1 and 2), the inhibition of miR‐371 led to a reduced miR‐371 expression (> 98%) in all parental and corresponding cisplatin‐resistant TGCT cell lines. The relative fold change of miR‐371 expression was determined using the equation RQ = 2−ΔΔCT (unpaired t‐test, **p < 0.01, ***p < 0.001, ****p < 0.0001).
3.2. Enhanced Cisplatin Sensitivity After miR‐371 Downregulation in TGCT Cell Lines
To assess the functional impact of miR‐371 on cisplatin sensitivity in TGCT cell lines, we inhibited miR‐371 by using two different antagomirs (inhibitors 1 and 2), leading to a decreased miR‐371 expression in the cisplatin‐resistant and corresponding parental TGCT cell lines (NCCIT, 2102EP and NT2/D1). A significant miR‐371 downregulation of over 98% was observed in all TGCT cell lines (0.0011 ± 0.0004 [NCCIT], 0.0059 ± 0.0069 [2102EP], 0.0835 ± 0.0780 [NT2/D1], 0.0001 ± 0.0002 [NCCIT‐R], 0.0045 ± 0.0044 [2102EP‐R], 0.0006 ± 0.0006 [NT2/D1‐R], p < 0.0001) with at least one antagomir, as illustrated in Figure 1D–F.
For drug sensitivity analysis, miR‐371‐inhibited and control cells were exposed to different concentrations of cisplatin (0–12 μM) for 24–72 h. For this purpose, the IC50 was first determined for each parental and corresponding cisplatin‐resistant TGCT cell line. After the addition of different cisplatin concentrations in the range of the IC50, changes in the viability of the cells were measured by crystal violet assays. Cisplatin sensitivity was significantly increased after the miR‐371 inhibition, indicated by a strongly reduced viability after cisplatin addition in miR‐371‐inhibited cells as compared to control cells (p < 0.001) (Figure 2A–D). Representative microscopic images of the different appearances of control and miR‐371‐inhibited TGCT cells (NCCIT, NCCIT‐R, 2010EP, 2102EP‐R) are illustrated in Figure S1. The decrease in cell viability correlated with the duration and concentration of cisplatin treatment, indicating that miR‐371 inhibition may be a promising strategy for enhancing the effectiveness of cisplatin in the treatment of TGCTs.
FIGURE 2.
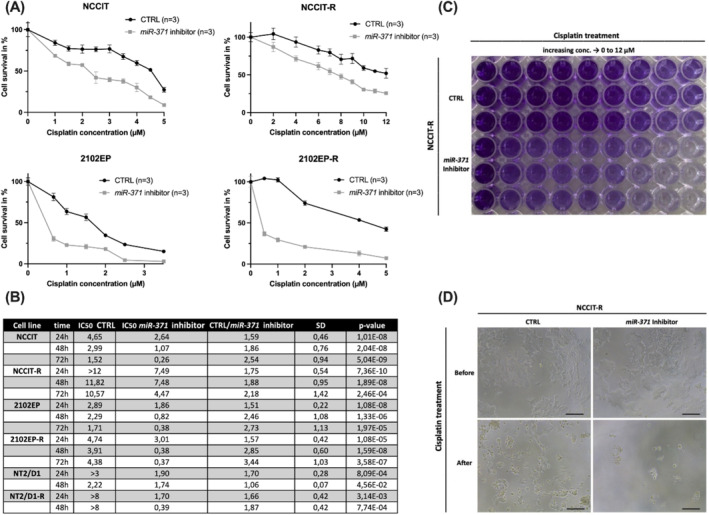
Enhanced cisplatin sensitivity in testicular germ cell tumour (TGCT) cell lines after miR‐371‐downregulation. (A, B) The IC50 was determined for each TGCT cell line (NCCIT, 2102EP, NCCIT‐R, 2102EP‐R). miR‐371‐inhibited and negative control cells were exposed to different concentrations of cisplatin (0–12 μM) for 24–72 h. After miR‐371 inhibition, the cisplatin sensitivity was significantly increased, indicated by a strongly reduced viability after cisplatin addition in miR‐371‐inhibited cells as compared to control cells. (C, D) Representative macroscopic and microscopic images (10× and 40× magnification) of the different appearances of control and miR‐371 inhibited cells (NCCIT‐R). CTRL, miR‐inhibitor negative control.
3.3. Enhanced Apoptosis Rate After miR‐371 Downregulation in TGCT Cell Lines
To assess the apoptotic rates induced by the downregulation of miR‐371, we employed a flow cytometry assay (Annexin V/PI). In the cisplatin‐resistant and corresponding parental TGCT cell lines, cisplatin sensitivity was significantly raised after the miR‐371 inhibition, as evidenced by increased apoptosis following cisplatin treatment in miR371‐inhibited cells compared to control cells (1.669 ± 0.460) (Figure 3A–D; Figure S2). The cisplatin concentration used for each cell line was selected based on their respective IC50 values. Specifically, miR‐371 downregulation led to an increase in the percentage of apoptotic cells in the sample, particularly in the early apoptosis population (1.657 ± 0.631), which was further augmented upon cisplatin addition (2.943 ± 0.863). Our data show that apoptosis is increased after cisplatin treatment in miR‐371‐inhibited cells.
FIGURE 3.
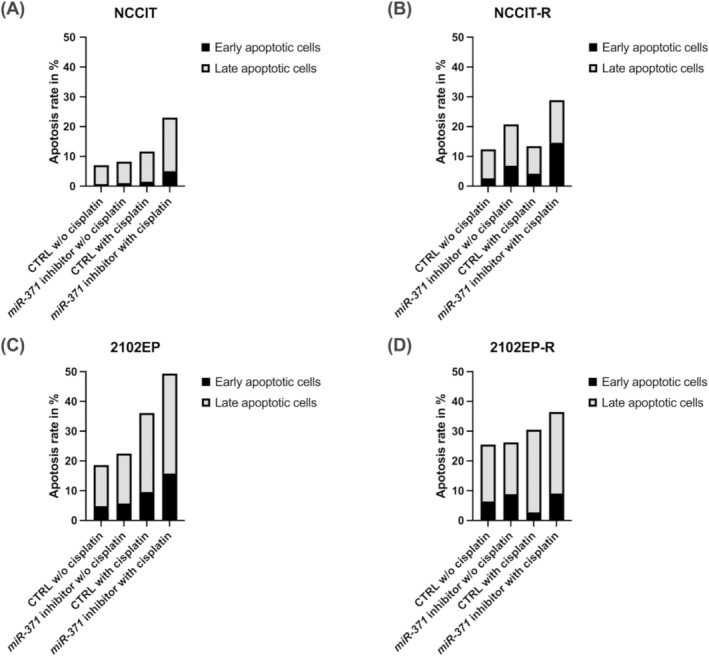
Increased apoptosis rate in testicular germ cell tumour (TGCT) cell lines after miR‐371‐downregulation. (A–D) Flow cytometry assay (Annexin V/PI) was used to measure the apoptotic rates induced by the downregulation of miR‐371. In cisplatin‐resistant, (NCCIT‐R, 2102EP‐R) and sensitive parental (NCCIT, 2102EP) TGCT cell lines, cisplatin sensitivity was significantly enhanced after the miR‐371 inhibition, indicated by increased apoptosis after cisplatin application in miR‐371‐inhibited cells compared to miR‐negative control cells (1.669 ± 0.460). CTRL, miR‐inhibitor negative control.
Analysis of the cell cycle showed no significant differences between miR‐371‐treated and control cells (Figure S3). These findings suggest that miR‐371 downregulation does not significantly impact the cell cycle, but might influence the development of cisplatin resistance.
4. Discussion
Despite advancements in cancer treatments, the mechanisms underlying cisplatin responsiveness and resistance in TGCTs remain poorly understood [4]. Therefore, there is an unmet need for innovative therapeutic approaches to address cisplatin‐resistant TGCTs. Recent studies have reported upregulation of miR‐371‐3 and miR‐302/367 clusters in TGCTs [8, 9, 10, 11], with miR‐371 emerging as the most sensitive and specific biomarker for disease diagnosis, surveillance and recurrence detection [8, 9, 10, 11]. Moreover, increased evidence linked miR‐371 to the epigenetic regulation of gene expression and cisplatin sensitivity [12].
In this report, we demonstrated for the first time that inhibition of miR‐371 significantly enhances cisplatin sensitivity in both cisplatin‐resistant and parental TGCT cell lines. Specifically, miR‐371‐inhibited cells exhibited decreased viability following cisplatin treatment, while cell viability remained unchanged in the absence of cisplatin, regardless of miR‐371 status. Additionally, a significantly higher proportion of cells underwent apoptosis upon cisplatin exposure when miR‐371 was inhibited. These findings suggest that miR‐371 may contribute to the acquisition of cisplatin resistance and serve as a potential therapeutic target to improve cisplatin efficacy in TGCTs.
Existing literature highlights DNA repair mechanisms and impaired apoptosis as key contributors to cisplatin resistance in TGCT patients [4, 17]. Unlike many other tumours, TGCTs rarely harbour TP53 mutations, though such alterations may occur in mediastinal TGCTs, which may also involve mutations in the RAS pathway [18, 19]. While extragonadal pure seminomas demonstrate similar overall survival rates regardless of primary site, retroperitoneal nonseminomas exhibit higher 5‐year overall survival rates compared to their mediastinal counterparts, emphasising the impact of tumour microenvironment on cisplatin sensitivity [20]. Importantly, TP53 alterations have predominately been observed in cisplatin‐resistant TGCT patients [3]. Increased expression or amplification of MDM2 and MDM4, negative regulators of p53, has been associated with more aggressive and therapy‐resistant TGCT phenotypes [21]. Therefore, treatment with the MDM inhibitor Nutlin‐3 has been shown to significantly elevate TP53 expression, thereby activating p53‐dependent proapoptotic pathways [21]. In contrast, nonfunctional TP53 is present in approximately 95% of ovarian carcinoma cases, contributing substantially to cisplatin resistance due to defective apoptosis induction [22]. In renal cell carcinoma cell lines, CDKN1A knockdown increased p53 protein levels and sensitised cells to cisplatin‐induced apoptosis via p53 [23]. Furthermore, Voorhoeve et al. [24] demonstrated that the miR‐371‐3 cluster neutralises p53 function in TGCTs by targeting the tumour suppressor LATS2. Thus, miR‐371 inhibition may enhance cisplatin sensitivity through the restoration of p53‐mediated apoptotic pathways.
In the context of precision medicine, identifying tumour‐specific aberrations that dive into oncogenic growth and resistance through activation of oncogenes or suppression of tumour suppressor genes, including epigenetic modifications, has facilitated the evaluation of targeted therapies. The combination of nonselective chemotherapy with agents targeting resistance‐mediating pathways may improve therapeutic efficacy and reduce adverse effects [25]. Modulating miR‐371 expression in combination with targeted cancer therapies may offer a promising approach to delaying or overcoming acquired cisplatin resistance. Given the overall high cisplatin sensitivity of TGCTs, integrating platinum‐sensitising agents with platinum‐based regimens may overcome resistance in challenging clinical cases, while potentially reducing chemotherapeutic dosages in responsive patients to minimise acute and long‐term toxicities [25]. Importantly, miR‐371 inhibition not only increases cisplatin sensitivity in resistant tumours but may also enhance sensitivity in cisplatin‐responsive tumours, offering an avenue to reduce chemotherapy doses and associated toxicities [16].
Limitations of this study include the challenge of translating in vitro findings into clinical practice, particularly the exclusive use of embryonal carcinoma cell lines, which limits the generalisation of results across the broader spectrum of TGCTs, including yolk sac tumours and choriocarcinoma. Furthermore, the study focused solely on miR‐371‐3p, without evaluating the potential contributions of other miRNAs within the miR‐371‐3 and miR‐302/367 clusters. Our methodology emphasised miRNA inhibition but did not include the use of miRNA mimics, which could have provided insights into the opposing effects of miR‐371 on cisplatin sensitivity. A significant challenge to the clinical application of miRNA inhibitors (‘antimiRs’) remains the risk of severe off‐target toxicities. Consequently, further investigations are required, including in vitro studies utilising a more diverse array of TGCT cell lines, as well as comprehensive in vivo and clinical studies, to thoroughly evaluate the safety and therapeutic potential of miRNA inhibitors.
5. Conclusion
In conclusion, our results suggest that miR‐371 may contribute to the development of cisplatin resistance in TGCTs, and especially downregulation of miR‐371 expression may increase the cisplatin sensitivity of tumour cells. This may represent a promising approach to improve therapeutic outcomes in patients with TGCTs, especially those with cisplatin‐resistant disease.
Author Contributions
Richard Weiten: conceptualization (equal), data curation (equal), formal analysis (equal), methodology (equal), project administration (equal), software (equal), supervision (equal), visualization (equal), writing – original draft (equal). Theadora Engler: data curation (equal), formal analysis (equal), investigation (equal), methodology (equal), writing – original draft (equal). Hubert Schorle: resources (equal), writing – review and editing (equal). Jörg Ellinger: resources (equal), supervision (equal), writing – review and editing (equal). Miriam Saponaro: investigation (supporting), methodology (supporting), writing – review and editing (equal). Abdullah Alajati: resources (supporting), writing – review and editing (equal). Daniel Nettersheim: conceptualization (equal), methodology (equal), project administration (equal), supervision (equal), writing – review and editing (lead). Isabella Syring‐Schmandke: conceptualization (equal), project administration (equal), resources (equal), supervision (equal), writing – review and editing (lead).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figures S1–S3.
Acknowledgements
T. Engler is supported by the BONFOR Program of the medical faculty of the University of Bonn, grant ID 2020‐4‐09. This work was supported by the Open Access Publication Fund of the University Hospital Bonn.
Funding: The authors received no specific funding for this work.
Richard Weiten, Theadora Engler, Daniel Nettersheim and Isabella Syring‐Schmandke contributed equally.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Patrikidou A., Cazzaniga W., Berney D., et al., “European Association of Urology Guidelines on Testicular Cancer: 2023 Update,” European Urology 84, no. 3 (2023): 289–301. [DOI] [PubMed] [Google Scholar]
- 2. Országhová Z., Kalavska K., Mego M., and Chovanec M., “Overcoming Chemotherapy Resistance in Germ Cell Tumors,” Biomedicine 10, no. 5 (2022): 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loveday C., Litchfield K., Proszek P. Z., et al., “Genomic Landscape of Platinum Resistant and Sensitive Testicular Cancers,” Nature Communications 11, no. 1 (2020): 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Singh R., Fazal Z., Freemantle S. J., and Spinella M. J., “Mechanisms of Cisplatin Sensitivity and Resistance in Testicular Germ Cell Tumors,” Cancer Drug Resist 2, no. 3 (2019): 580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Galluzzi L., Senovilla L., Vitale I., et al., “Molecular Mechanisms of Cisplatin Resistance,” Oncogene 31, no. 15 (2012): 1869–1883. [DOI] [PubMed] [Google Scholar]
- 6. Galluzzi L., Vitale I., Michels J., et al., “Systems Biology of Cisplatin Resistance: Past, Present and Future,” Cell Death & Disease 5, no. 5 (2014): e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murray M. J., Halsall D. J., Hook C. E., Williams D. M., Nicholson J. C., and Coleman N., “Identification of microRNAs From the miR‐371~373 and miR‐302 Clusters as Potential Serum Biomarkers of Malignant Germ Cell Tumors,” American Journal of Clinical Pathology 135, no. 1 (2011): 119–125. [DOI] [PubMed] [Google Scholar]
- 8. Dieckmann K. P., Spiekermann M., Balks T., et al., “MicroRNAs miR‐371‐3 in Serum as Diagnostic Tools in the Management of Testicular Germ Cell Tumours,” British Journal of Cancer 107, no. 10 (2012): 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Syring I., Bartels J., Holdenrieder S., Kristiansen G., Müller S. C., and Ellinger J., “Circulating Serum miRNA (miR‐367‐3p, miR‐371a‐3p, miR‐372‐3p and miR‐373‐3p) as Biomarkers in Patients With Testicular Germ Cell Cancer,” Journal of Urology 193, no. 1 (2015): 331–337. [DOI] [PubMed] [Google Scholar]
- 10. Leão R., Albersen M., Looijenga L. H. J., et al., “Circulating MicroRNAs, the Next‐Generation Serum Biomarkers in Testicular Germ Cell Tumours: A Systematic Review,” European Urology 80, no. 4 (2021): 456–466. [DOI] [PubMed] [Google Scholar]
- 11. Dieckmann K. P., Radtke A., Geczi L., et al., “Serum Levels of MicroRNA‐371a‐3p (M371 Test) as a New Biomarker of Testicular Germ Cell Tumors: Results of a Prospective Multicentric Study,” Journal of Clinical Oncology 37, no. 16 (2019): 1412–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arif K. M. T., Elliott E. K., Haupt L. M., and Griffiths L. R., “Regulatory Mechanisms of Epigenetic miRNA Relationships in Human Cancer and Potential as Therapeutic Targets,” Cancers (Basel) 12, no. 10 (2020): 2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah J. A., Khattak S., Rauf M. A., Cai Y., and Jin J., “Potential Biomarkers of miR‐371‐373 Gene Cluster in Tumorigenesis,” Life (Basel) 11, no. 9 (2021): 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fenske A. E., Glaesener S., Bokemeyer C., et al., “Cisplatin Resistance Induced in Germ Cell Tumour Cells Is due to Reduced Susceptibility Towards Cell Death but Not to Altered DNA Damage Induction or Repair,” Cancer Letters 324, no. 2 (2012): 171–178. [DOI] [PubMed] [Google Scholar]
- 15. Oechsle K., Honecker F., Cheng T., et al., “Preclinical and Clinical Activity of Sunitinib in Patients With Cisplatin‐Refractory or Multiply Relapsed Germ Cell Tumors: A Canadian Urologic Oncology Group/German Testicular Cancer Study Group Cooperative Study,” Annals of Oncology 22, no. 12 (2011): 2654–2660. [DOI] [PubMed] [Google Scholar]
- 16. Skowron M. A., Hoffmann M. J., Watolla M. M., and Nettersheim D., “Evaluation of Chemotherapeutic Drugs for Treatment of (Cisplatin‐Resistant) Germ Cell Cancer Cell Lines,” Methods in Molecular Biology 2195 (2021): 99–111. [DOI] [PubMed] [Google Scholar]
- 17. Skowron M. A., Oing C., Bremmer F., et al., “The Developmental Origin of Cancers Defines Basic Principles of Cisplatin Resistance,” Cancer Letters 519 (2021): 199–210. [DOI] [PubMed] [Google Scholar]
- 18. Bagrodia A., Lee B. H., Lee W., et al., “Genetic Determinants of Cisplatin Resistance in Patients With Advanced Germ Cell Tumors,” Journal of Clinical Oncology 34, no. 33 (2016): 4000–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor J., Donoghue M. T., Ho C., et al., “Germ Cell Tumors and Associated Hematologic Malignancies Evolve From a Common Shared Precursor,” Journal of Clinical Investigation 130, no. 12 (2020): 6668–6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bokemeyer C., Nichols C. R., Droz J. P., et al., “Extragonadal Germ Cell Tumors of the Mediastinum and Retroperitoneum: Results From an International Analysis,” Journal of Clinical Oncology 20, no. 7 (2002): 1864–1873. [DOI] [PubMed] [Google Scholar]
- 21. Koster R., Timmer‐Bosscha H., Bischoff R., Gietema J. A., and de Jong S., “Disruption of the MDM2‐p53 Interaction Strongly Potentiates p53‐Dependent Apoptosis in Cisplatin‐Resistant Human Testicular Carcinoma Cells via the Fas/FasL Pathway,” Cell Death & Disease 2, no. 4 (2011): e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fajac A., Da Silva J., Ahomadegbe J. C., et al., “Cisplatin‐Induced Apoptosis and p53 Gene Status in a Cisplatin‐Resistant Human Ovarian Carcinoma Cell Line,” International Journal of Cancer 68, no. 1 (1996): 67–74. [DOI] [PubMed] [Google Scholar]
- 23. Park S. H., Park J. Y., and Weiss R. H., “Antisense Attenuation of p21 Sensitizes Kidney Cancer to Apoptosis in Response to Conventional DNA Damaging Chemotherapy Associated With Enhancement of Phospho‐p53,” Journal of Urology 180, no. 1 (2008): 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voorhoeve P. M., le Sage C., Schrier M., et al., “A Genetic Screen Implicates miRNA‐372 and miRNA‐373 as Oncogenes in Testicular Germ Cell Tumors,” Cell 124, no. 6 (2006): 1169–1181. [DOI] [PubMed] [Google Scholar]
- 25. Oing C., Skowron M. A., Bokemeyer C., and Nettersheim D., “Epigenetic Treatment Combinations to Effectively Target Cisplatin‐Resistant Germ Cell Tumors: Past, Present, and Future Considerations,” Andrology 7, no. 4 (2019): 487–497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.