

Abstract
Electrooxidative transformations frequently rely on proton reduction as the terminal electron sink. However, this cathodic counterreaction can be slow in organic solvents and can operate at reducing potentials that are incompatible with catalysts and reagents needed for oxidative reactions. We report aminoborate adducts as redox mediators for proton reduction that operate at mild reducing potentials. This reliable cathodic couple ultimately enables successful oxidative organic transformations, including chlorodeborylation, developed herein, and Cu-catalyzed Chan-Lam coupling, reported previously by our group. Pyridinium borate adducts formed during electrooxidative chlorination of aryl trifluoroborates serve as easily reduced complexes (−1.1 V vs. Fc/Fc+) to catalyze proton reduction. Reactions that promote formation of borate adducts result in high yields, operate at low cell potentials, suppress aryl trifluoroborate decomposition, and mitigate electrode passivation. These studies illustrate the utility of Lewis acid-base complexes in cathodic counterreactions and underscore the importance of developing both anodic and cathodic reactions in electrosynthesis.
Graphical Abstract
Introduction
Electrochemical reactions are most often developed for net-reductive or net-oxidative organic transformations at the respective cathode or anode.1–4 However, these targeted synthetic reactions represent only one half of a complete electrochemical system. The working electrode at which the reaction of interest occurs is inextricably paired with a counter electrode at which the opposite redox process occurs. As a result, the achievable current for a targeted organic reaction at the working electrode cannot exceed that of current generated from the opposite redox reaction at the counter electrode.
While substantial efforts have been dedicated to designing conditions for reactions at the working electrode, the same level of mechanistic insight and design for reactions at the counter electrode is rare. Reductive electrosynthetic reactions are frequently paired with sacrificial metal anodes or amines that undergo oxidative counter reactions as sacrificial electron donors.5–7 In contrast, oxidative electrosynthetic reactions are typically coupled to proton reduction as the counter reaction and terminal electron sink.8 However, direct proton reduction at a heterogeneous electrode can be slow in the absence of high overpotentials, which restricts the overall current. Undesired and irreversible cathodic reactions, like reductive plating of transition metal catalysts designed for oxidative processes, can begin to outcompete proton reduction when electrolysis is performed at synthetic useful currents.9,10 Consequently, the presence of protons does not guarantee a reliable cathodic reaction for a targeted electrooxidation. This is evidenced by reports from our group11 and others12,13 where chemical additives as electron acceptors for reductive half-cell reactions were essential to the success of oxidative synthetic transformations, despite the presence of protic reagents in solution.
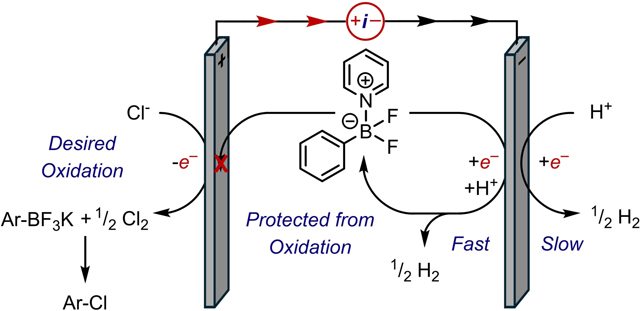
This work highlights the often-overlooked issue of designing cathodic counterreactions for electrooxidative synthesis. We report the electrocatalytic function of aminoborate adducts as redox mediators for proton reduction at mild potentials (−1.1 V vs Fc/Fc+). The development of this cathodic chemistry ultimately enables oxidative organic transformations that otherwise suffer from high overpotentials and electrode passivation (Figure 1.). Mechanistic studies by cyclic voltammetry (CV), X-ray crystallography, and NMR spectroscopy reveal these Lewis acid-base complexes to be the predominant cathodic couple in electrooxidative reactions beyond the case study reported herein.
Figure 1.

The cathodic counter reaction to synthetically useful oxidative transformations can be improved upon the addition of amino-borate complexes.
These unexpected findings arose from our studies on electrooxidative halodeborylation reactions. Aryl halides generated from such reactions serve as important intermediates in nearly all of organic synthesis,14–17 and can be reliably formed with predetermined regioselectivity based on the position of the boryl group.18,19 Halodeborylation reactions under electrooxidative control can be conducted with simple halide salts20–24 in place of stoichiometric transition metal-halide salts or electrophilic halogenation reagents.25–30 However, such electrochemical reactions remain underdeveloped for halides that are challenging to oxidize, such as chloride or fluoride.31 As an example, the Cai group recently demonstrated electrooxidative iodination and bromination of aryl boronic acids with the corresponding halide salts, but the methodology could not be extended to chlorodeborylation reactions.32 Strategies that address fundamental limitations to electrooxidative halogenation with the full complement of halides would enable new methodologies to prepare high-value haloarenes.
Results and Discussion
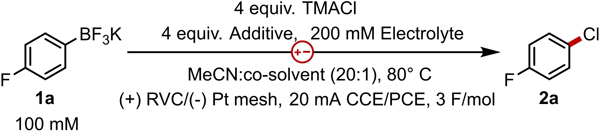
We first evaluated conditions for electrooxidative chlorination on 4-fluorophenyl trifluoroborate (1a) because conversion and yield of this model substrate could be easily monitored by 19F NMR spectroscopy (Table 1). Initial reactions were performed under constant current electrolysis (CCE) in MeCN at a Pt cathode and reticulated vitreous carbon (RVC) anode with tetramethylammonium chloride (TMACl) as the chloride source (entry 1). These initial reactions required high cell potentials (>6 V) to drive the applied current and generated only fluorobenzene as a result of protodeborylation. Additionally, we observed deposits on the cathode that likely cause passivation and force higher operating potentials. We tested reactions with protic co-solvents that could provide protons for reduction to establish a more reliable cathodic couple. However, reactions with added 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) generated product 2a in low yields of 19% yield, and cathodic passivation persisted (entry 2). We observed the first substantial improvement in both product yield (65%) and operating cell potential (Ecell, 2.2 V average) over the initial studies when reactions were conducted with the combination of LiClO4 and pyridine (entry 4). Varying the applied current failed to improve the yield beyond 65% (entry 5), and deposits on the cathode were observed in all reactions where a constant current was maintained. We hypothesized that depolarizing the electrodes may improve the outcome of the reaction by promoting dissolution of deposits or by altering the makeup of reagents within the double layer at the cathode during the rest period.33,34 Electrolysis with pulsed currents in cycles of 60 s on and 30 s off operated at the lowest cell potentials and generated products in near quantitative yields (entry 6). Moreover, the cathode remained pristine following electrolysis. Under these conditions, MeOH can also be utilized as the protic co-solvent with only a minor reduction in yield (entry 7). Finally, control experiments that exclude any component of the standard reaction resulted in low yields and high operating potentials (entries 8–10).
Table 1.
Reactions conducted on a 0.30 mmol scale. Yields were determined by 19F-NMR analysis using 1,3,5-trifluoro-benzene as the internal standard.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Electrolyte | Co-Solv. | Additive | Electrolysis Conditions | Yield 2a(%) | Measured Ecell |
| 1 | KPF6 | – | – | CCE | 0 | 6.4 V |
| 2 | KPF6 | HFIP | – | CCE | 19 | 4.0 V |
| 3 | KPF6 | HFIP | Pyridine | CCE | 37 | 3.4 V |
| 4 | LiCIO4 | HFIP | Pyridine | CCE | 65 | 2.2 V |
| 5 | LiCIO4 | HFIP | Pyridine | CCE (10 mA) | 6 | 2.2 V |
| 6 | LiCIO 4 | HFIP | Pyridine | 60s on 30s off | 95 | 1.8 V |
| 7 | LiCIO4 | MeOH | Pyridine | 60s on 30s off | 80 | 2.0 V |
| 8 | LiCIO4 | HFIP | Pyridine | – | 0 | – |
| 9 | LiCIO4 | HFIP | – | 60s on 30s off | 11 | 4.0 V |
| 10 | LiCIO4 | – | Pyridine | 60s on 30s off | 5 | 3.7 V |
We were surprised that the successful development of this electrooxidative reaction relied almost exclusively on improving the cathodic chemistry. This underscores the importance of establishing both anodic and cathodic reactions in undivided electrosynthetic reactions. As such, we investigated the roles of additives on both half-cell reactions by cyclic voltammetry (CV). Illustrated in Figure 2a, CVs of the standard reaction mixture (black trace) at oxidizing potentials reveal an irreversible oxidation with an onset (Eonset) of 0.9 V vs Fc/Fc+. Referenced electrolysis experiments during synthetic reactions reveal that the anode operates at 1.4 V (dashed vertical line). Independent CVs of the individual reactants reveal that both Cl- and organoborate 1a can undergo oxidation at this potential, with respective Eonset values of 0.9 V (blue trace) and 1.3 V (gray trace). These CVs suggest that competitive oxidation of the arylborate would be possible at the operating potentials of the anode during the electrolysis. However, we discovered that addition of pyridine to 1a completely suppressed any oxidative processes (red trace). This key additive likely serves to protect the substrate from oxidative degradation (vide infra).35
Figure 2.

(a) CVs of standard reaction conditions (black trace), TMACl (blue trace), 1a (grey trace), 1a and pyridine but without TMACl (red trace). (b) CVs of standard reaction conditions (black trace), HFIP and pyridine (blue trace), 1a (grey trace), 1a and pyridine (red trace). CVs are run in acetonitrile with LiClO4 as supporting electrolyte and are reported in IUPAC convention. Standard reaction conditions comprise analytes 1a, pyridine, TMACl, and HFIP.
CVs of the standard reaction mixture at reducing potentials (Figure 2b, black trace) reveal a cathodic current with an Eonset of just −1.0 V vs Fc/Fc+. This redox event is consistent with the operating potential of the cathode during bulk reactions (−1.2 V vs Fc/Fc+, vertical dashed line). However, none of the individual reaction components undergo redox at this mild potential (gray and blue traces). CVs of protic co-solvent and substrate alone require potentials of −1.8 V before similar cathodic currents are achieved to those of the standard reaction mixture at −1.2 V. The cathodic response measured in standard reactions was only achieved from a combination of 1a and pyridine in a supporting electrolyte of Li+ (red trace).
We sought to characterize the species generated from the combination of 1a, pyridine, and LiClO4 because of the dramatic effect it had on the redox reactions at both electrodes and, ultimately, on the success of the oxidative transformation. Analysis of the combination of reagents by 19F NMR spectroscopy revealed two distinct fluorine resonances with a 1:2 integral ratio instead of the expected 1:3 ratio that would result from the Ar-F and Ar-BF3 of 1a (see the SI, Figure S10). X-ray diffraction of single crystals grown of a para-phenyl analog revealed that trifluoroborate 1b was converted into the Lewis acid-base adduct 3b in the presence of pyridine and Li+ salts (Figure 3a). The solid-state structure of 3b is consistent with the solution state structures of 3a determined by 1H, 19F, and 11B NMR spectroscopy. Formation of the aryl difluoro(pyridinium)borate is likely promoted by Li-mediated abstraction of fluoride from the trifluoroborate, followed by trapping of the Lewis acidic ArBF2 by pyridine.
Figure 3.

(a) Reaction scheme for the formation of the aryl-BF2-pyridine Lewis acid-base adduct with X-ray crystal structure of isolated complex. (b) CVs of 1c (black trace), 1c and pyridine (red trace), 1c and pyridine and HFIP (increasing concentrations)(blue traces). CVs are run in acetonitrile with LiClO4 and TBAClO4 as supporting electrolyte and are reported in IUPAC convention. (c) Proposed mechanism of complex reduction and HER.
CV scans of pyridinium borate 3c reveal the same irreversible reductive event observed from CV analyses of the standard reaction mixture (Figure 3b, red trace). While these data implicate 3c as a key component of the cathodic reaction, the arylborate cannot be irreversibly consumed at the cathode as the terminal electron sink because it is also the substrate for oxidative reactions that were shown to generate products in near-quantitative yields. This quandary suggested that the pyridinium borate is not destroyed upon reduction as the irreversible CV would suggest, but rather that it serves as a redox mediator for reduction of the terminal electron acceptor. Specifically, we detected solvated H2 by 1H-NMR spectroscopy in reaction mixtures following electrolysis (see the SI, Figure S11). These results suggest that reduced 3c mediates proton reduction to H2 at mild potentials in place of heterogeneous proton reduction at more negative potentials where electrode passivation can occur.
Although the precise mechanism of the hydrogen evolution reaction (HER) remains unclear, we propose that reduction of 3c generates an unstable intermediate that fragments to release fluoride and a boryl radical anion 4c, as indicated by the irreversible CV of 3c.36–38 Boryl radical anions are known to mediate H-atom abstraction,39,40 which can occur from acetonitrile solvent. This hypothesis is supported by H/D exchange studies of MeCN under the reaction conditions (see the SI, Figure S12). Finally, the hydridic borohydride 5c can react with protic reagents like HFIP to generate H2 and regenerate 3c upon recapture by pyridine. This final step is supported by CVs of 3c that reveal increased cathodic currents with added HFIP (Figure 3b, blue traces) and is indicative of the electrocatalytic process illustrated in Figure 3c. This reductive couple enables the anodic half-cell reaction, which we propose constitutes oxidation of Cl– followed by electrophilic aromatic substitution at the ipso carbon of the aryl borate complex 3c.29,32
We similarly observed improved yields, lower cell potentials, and mitigated passivation for electrooxidative Chan-Lam coupling reactions under conditions that formed aminoborate adducts. (Figure 4).41 CVs of solutions containing the boronic acid substrate and an amine base (Figure 4, red trace) result in a positive shift of the peak cathodic current by 450 mV relative to the boronic acid alone (black trace). NMR spectroscopic studies implicate the formation of an ammonium-borate adduct that is analogous to those generated during halodeborylation (see the SI, Figure S13). Additionally, the peak cathodic current of the ammonium-borate complex is nearly double that of any other reducible species present under the reaction conditions, despite the milder reduction potential. These studies suggest that ammonium-borate adducts could potentially serve as general cathodic chemistries for electrooxidative transformations involving boron-based reagents.
Figure 4.

Similar reduction improvement observed upon the complexation of aryl boronic acid and alkyl amines. CV’s of phenylboronic acid (black trace), phenylboronic acid, aniline, and triethylamine (red trace).
With the successful development of the cathodic reaction, electrooxidative chlorodeborylation reactions can be performed for a variety of aryl, heteroaryl, and vinyl trifluoroborate salts (Chart 1). Reactions of electron rich aryl substrates generated chloroarenes in excellent yields (2c-2f, 2h). Reactions with electron-poor arenes that are traditionally unreactive in electrophilic aromatic substitution reactions were isolated in good to moderate yields (2b, 2i-2o). Substrates that are exceedingly electron rich (2g) or exceedingly electron deficient (2p) undergo competitive oxidation and reduction, respectively, and are incompatible under the developed conditions. Finally, ortho-substituted trifluoroborates (2d, 2h, 2k, 2t, 2u) as well as vinyl (2q) and heteroaryl substrates (2r-2u) undergo chlorination in good yields. Milder conditions that employ lithium chloride instead of TMACl and that operate at room temperature instead of 80 °C could also be performed with a slight reduction in yield (see SI Chart S1).
Chart 1.

Isolated yields of products generated via electrochemical chloro-deborylation on a 0.30 mmol scale (GC yields are in parentheses for low-boiling products that cannot be readily isolated). (a) (20:1) MeCN:TFE, 2.5 V PPE. (b) (20:1:1) MeCN:TFE:AcOH, 100 mM pyridine. (c) 1.5 F/mol instead of 3.0 F/mol. (d) 1.0 mmol scale.
In summary, we report an electrochemical chlorodeborylation methodology that circumvents common oxidative electrosynthetic limitations. Counterintuitively, the success of this electrooxidative transformation relied on the study and development of the reductive counterreaction. The formation of a pyridinium-borate adduct of the substrate serves to catalyze proton reduction at the cathode, allowing for higher currents at lower overpotentials. The adduct also suppresses aryl trifluoroborate oxidation at the anode leading to higher yields. Finally, these mechanistic studies illustrate the utility of Lewis acid-base complexes as cathodic mediators and underscore the importance of developing both anodic and cathodic reactions in electrosynthesis.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (NIH R35 GM138373) and the Ohio State University Sustainability Institute for a seed grant in support of this work. We would like to thank Dr. Curtis Moore for collection and analysis of X-ray diffraction data.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, cell design, characterization of compounds, spectroscopic data, X-ray data, electro-chemical data, and additional experiments are supplied (PDF)
The authors declare no competing financial interest.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- (1).Lodh J; Paul S; Sun H; Song L; Schöfberger W; Roy S. Electrochemical Organic Reactions: A Tutorial Review. Front. Chem. 2023, 10 (January), 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Frontana-Uribe BA; Little RD; Ibanez JG; Palma A; Vasquez-Medrano R. Organic Electrosynthesis: A Promising Green Methodology in Organic Chemistry. Green Chem. 2010, 12 (12), 2099–2119. [Google Scholar]
- (3).Novaes LFT; Liu J; Shen Y; Lu L; Meinhardt JM; Lin S. Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 2021, 50 (14), 7941–8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hilt G. Basic Strategies and Types of Applications in Organic Electrochemistry. ChemElectroChem 2020, 7 (2), 395–405. [Google Scholar]
- (5).Nédélec J-Y; Périchon J; Troupel M. Organic Electroreductive Coupling Reactions Using Transition Metal Complexes as Catalysts. In Electrochemistry IV Electroorganic Synthesis: Bond Formation at Anode and Cathode; 1997; pp 141–173. [Google Scholar]
- (6).Li J; He L; Liu X; Cheng X; Li G. Electrochemical Hydrogenation with Gaseous Ammonia. Angew. Chemie 2019, 131 (6), 1773–1777. [DOI] [PubMed] [Google Scholar]
- (7).Zhang X; Jiang R; Cheng X. Electrochemical Tandem Olefination and Hydrogenation Reaction with Ammonia. J. Org. Chem. 2021, 86 (22), 16016–16025. [DOI] [PubMed] [Google Scholar]
- (8).Yuan Y; Lei A. Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions. Acc. Chem. Res. 2019, 52 (12), 3309–3324. [DOI] [PubMed] [Google Scholar]
- (9).Jiao KJ; Xing YK; Yang QL; Qiu H; Mei TS Site-Selective C-H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis. Acc. Chem. Res. 2020, 53 (2), 300–310. [DOI] [PubMed] [Google Scholar]
- (10).Cheng X; Lei A; Mei TS; Xu HC; Xu K; Zeng C. Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis. CCS Chem. 2022, 4 (4), 1120–1152. [Google Scholar]
- (11).Hintz H; Bower J; Tang J; Lalama M; Hintz H; Bower J; Tang J; Lalama M; Sevov C. Copper-Catalyzed Electrochemical C – H Fluorination. Chem Catal. 2023, 3 (1), 100491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xiang J; Shang M; Kawamata Y; Lundberg H; Reisberg SH; Chen M; Mykhailiuk P; Beutner G; Collins MR; Davies A; Del Bel M; Gallego GM; Spangler JE; Starr J; Yang S; Blackmond DG; Baran PS Hindered Dialkyl Ether Synthesis with Electrogenerated Carbocations. Nature 2019, 573 (7774), 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shrestha A; Lee M; Dunn AL; Sanford MS Palladium-Catalyzed C-H Bond Acetoxylation via Electrochemical Oxidation. Org. Lett. 2018, 20 (1), 204–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dombrowski AW; Gesmundo NJ; Aguirre AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y. Expanding the Medicinal Chemist Toolbox: Comparing Seven C(Sp2)-C(Sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett. 2020, 11 (4), 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Spargo PL Organic Halides. Contemp. Org. Synth. 1994, 1 (2),113–124. [Google Scholar]
- (16).Fu GC The Development of Versatile Methods for Palladium-Catalyzed Coupling Reactions of Aryl Electrophiles through the Use of P(t-Bu)3 and PCy3 as Ligands. Acc. Chem. Res. 2008, 41 (11), 1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Alberico D; Scott ME; Lautens M. Aryl-Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107 (1), 174–238. [DOI] [PubMed] [Google Scholar]
- (18).Zhu C; Falck JR Transition Metal-Free Ipso-Functionalization of Arylboronic Acids and Derivatives. Adv. Synth. Catal. 2014, 356 (11–12), 2395–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Szumigala RH; Devine PN; Gauthier DR; Volante RP Facile Synthesis of 2-Bromo-3-Fluorobenzonitrile: An Application and Study of the Halodeboronation of Aryl Boronic Acids. J. Org. Chem. 2004, 69 (2), 566–569. [DOI] [PubMed] [Google Scholar]
- (20).Jagatheesan R; Christopher C; Ramesh P; Sambathkumar, & S. Exclusively Explored Electrochemical Halogenation of Aryl Compounds; Periodical Updates: Since. Synth. Commun. 2020, 50, 2391–2412. [Google Scholar]
- (21).Scheide MR; Nicoleti CR; Martins GM; Braga AL Electrohalogenation of Organic Compounds. Org. Biomol. Chem. 2021, 19 (12), 2578–2602. [DOI] [PubMed] [Google Scholar]
- (22).Liang Y; Lin E; Adeli Y; Jin R; Jiao N. Efficient Electrocatalysis for the Preparation of (Hetero)Aryl Chlorides and Vinyl Chloride with 1,2-Dichloroethane. Angew. Chem Int. Ed. 2019, 58, 4566–4570. [DOI] [PubMed] [Google Scholar]
- (23).Duan Y; Luo S. Phase-Transfer Catalysis for Electrochemical Chlorination and Nitration of Arenes.Pdf. Angew. Chem Int. Ed. 2024, 63, e202319206. [DOI] [PubMed] [Google Scholar]
- (24).Yuan Y; Yao A; Zheng Y; Gao M; Zhou Z; Qiao J; Hu J; Ye B; Zhao J; Wen H; Lei A. Electrochemical Oxidative Clean Halogenation Using HX/NaX with Hydrogen Evolution. iScience 2019, 12, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schimler SD; Sanford MS Copper-Mediated Functionalization of Aryl Trifluoroborates. Synlett 2016, 27 (15), 2279–2284. [Google Scholar]
- (26).Wu H; Hynes J. Copper-Catalyzed Chlorination of Functionalized Arylboronic Acids. Org. Lett. 2010, 12 (6),1192–1195. [DOI] [PubMed] [Google Scholar]
- (27).Molloy JJ; O’rourke KM; Frias CP; Sloan NL; West MJ; Pimlott SL; Sutherland A; Watson AJB Mechanism of Cu-Catalyzed Aryl Boronic Acid Halodeboronation Using Electrophilic Halogen: Development of a Base-Catalyzed Iododeboronation for Radiolabeling Applications. Org. Lett. 2019, 21 (7), 2488–2492. [DOI] [PubMed] [Google Scholar]
- (28).Yao ML; Reddy MS; Li Y; Walfish I; Blevins DW; Kabalka GW Chemoselective Bromodeboronation of Organotrifluoroborates Using Tetrabutylammonium Tribromide: Application in (Z)-Dibromoalkene Syntheses. Org. Lett. 2010, 12 (4), 700–703. [DOI] [PubMed] [Google Scholar]
- (29).Molander GA; Cavalcanti LN Metal-Free Chlorodeboronation of Organotrifluoroborates. J. Org. Chem. 2011, 76 (17), 7195–7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kabalka GW; Mereddy AR A Facile Synthesis of Aryl Iodides via Potassium Aryltrifluoroborates. Tetrahedron Lett. 2004, 45 (2), 343–345. [Google Scholar]
- (31).Rein J; Annand JR; Wismer MK; Fu J; Siu JC; Klapars A; Strotman NA; Kalyani D; Lehnherr D; Lin S. Unlocking the Potential of High-Throughput Experimentation for Electrochemistry with a Standardized Microscale Reactor. ACS Cent. Sci. 2021, 7 (8), 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Fu Z; Hao G; Fu Y; He D; Tuo X; Guo S; Cai H. Transition Metal-Free Electrocatalytic Halodeborylation of Arylboronic Acids with Metal Halides MX (X = I, Br) to Synthesize Aryl Halides. Org. Chem. Front. 2020, 7 (3), 590–595. [Google Scholar]
- (33).Atkins AP; Chaturvedi AK; Tate JA; Lennox AJJ Pulsed Electrolysis: Enhancing Primary Benzylic C(Sp3)-H Nucleophilic Fluorination. Org. Chem. Front. 2023, 11 (3),802–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Atkins AP; Lennox AJ J. Application of Pulsed Electrolysis in Organic Electrosynthesis. Curr. Opin. Electrochem. 2024, 44, 101441. [Google Scholar]
- (35).Morris JH; Gysling HJ; Reed D. Electrochemistry of Boron Compounds. Chem. Rev. 1985, 85 (1), 51–76. [Google Scholar]
- (36).Rossler SL; Jelier BJ; Magnier, Emmanuel, Dagousset, G.; Carreira, E. M.; Togni, A. Pyridinium Salts as Redox-Active Functional Group Transfer Reagents. Angew. Chem Int. Ed. 2020, 59, 9264–9280. [DOI] [PubMed] [Google Scholar]
- (37).Baban JA; Marti VPJ; Roberts BP Ligated Boryl Radicals. Part 2. Electron Spin Resonance Studies of Trialkylamine-Boryl Radicals. J. Chem. Soc. Perkin Trans. 2 1985, No. 11,1723–1733. [Google Scholar]
- (38).Baban JA; Roberts BP An e.s.r. Study of Amine-Boryl Radicals (R3N–BH2˙) in Solution. J. Chem. Soc., Chem. Chommun. 1983, No. 21, 1224–1226. [Google Scholar]
- (39).Sheeller B; Ingold KU Absolute Rate Constants for Some Reactions of the Triethylamine-Boryl Radical and the Borane Radical Anion. J. Chem. Soc. Perkin Trans. 2 2001, No. 4, 480–486. [Google Scholar]
- (40).Paul V; Roberts BP Polarity Reversal Catalysis of Hydrogen Atom Abstraction Reactions. J. Chem. Soc. Chem. Commun. 1987, No. 17, 1322–1324. [Google Scholar]
- (41).Walker BR; Manabe S; Brusoe AT; Sevov CS Mediator-Enabled Electrocatalysis with Ligandless Copper for Anaerobic Chan-Lam Coupling Reactions. J. Am. Chem. Soc 2021, 143, 6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.