

Abstract
Existing evidence indicates that exercise training can enhance neural function by regulating mitochondrial quality control (MQC), which can be impaired by cerebral ischemia, and that sirtuin-3 (SIRT3), a protein localized in mitochondria, is crucial in maintaining mitochondrial functions. However, the relationship among exercise training, SIRT3, and MQC after cerebral ischemia remains obscure. This study attempted to elucidate the relationship among exercise training, SIRT3 and MQC after cerebral ischemia in rats. Male adult SD rats received tMCAO after the transfection of adeno-associated virus encoding either sirtuin-3 (AAV-SIRT3) or SIRT3 knockdown (AAV-sh-SIRT3) into the ipsilateral striata and cortex. Subsequently, the animals were randomly selected for exercise training. The index changes were measured by transmission electron microscopy, Western blot analysis, nuclear magnetic resonance imaging, TUNEL staining, and immunofluorescence staining, etc. The results revealed that after cerebral ischemia, exercise training increased SIRT3 expression, significantly improved neural function, alleviated infarct volume and neuronal apoptosis, maintained the mitochondrial structural integrity, and re-established MQC. The latter promoted mitochondrial biogenesis, balanced mitochondrial fission/fusion, and enhanced mitophagy. These favorable benefits were reversed after SIRT3 interference. In addition, a cellular OGD/R model showed that the increased SIRT3 expression alleviates neuronal apoptosis and re-establishes mitochondrial quality control by activating the β-catenin pathway. These findings suggest that exercise training may optimize mitochondrial quality control by increasing the expression of SIRT3, thereby improving neural functions after cerebral ischemia, which illuminates the mechanism underlying the exercise training-conferred neural benefits and indicates SIRT3 as a therapeutic strategy for brain ischemia.
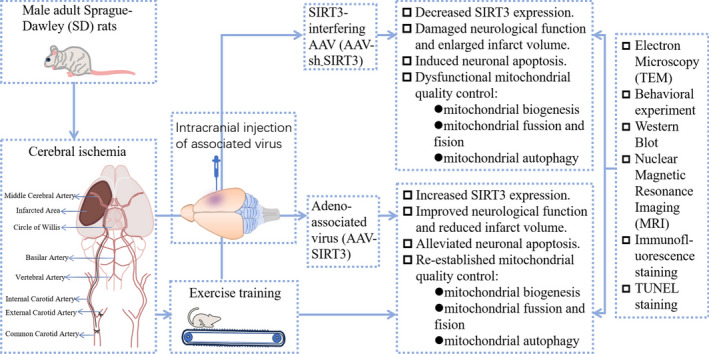
Graphical Abstract
1. The expression of SIRT3 decreases after cerebral ischemia in rats, while exercise training can increase SIRT3 levels in ischemic rats.
2. Exercise training alleviates neuronal apoptosis and re-establishes mitochondrial quality control (MQC) after cerebral ischemia by increasing SIRT3 expression, thereby promoting neurological function recovery.
3. SIRT3 overexpression reduces neuronal apoptosis and re-establishes MQC by activating the β-catenin pathway.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10565-024-09957-3.
Keywords: Cerebral ischemia, Exercise training, SIRT3, Neuronal apoptosis, Mitochondrial Quality Control (MQC)
Introduction
Globally, stroke contributes prominently to mortality and disability (Feigin et al. 2021). As ageing population progresses, the effects of stroke are increasingly prevalent in the general population and the need of rehabilitation exercise is imminent for stroke survivors (Stinear et al. 2020), in that exercise training is an important measure for secondary and tertiary prevention of stroke (Hu et al. 2017b). However, the specific mechanism underlying the benefits of exercise training for stroke treatment remains largely unelucidated.
Of the damages wrought by cerebral ischemia, one hallmark is mitochondrial dysfunction, which can induce the death of neurons (He et al. 2020). As double-membrane organelles, mitochondria engage in various biological processes (Bock and Tait 2020). Given its pivotal role in regulating neuronal death, restoring the functions of mitochondria after ischemic injury may seem to be a powerful alternative for brain ischemia (Yang et al. 2022). Research indicates that mitochondrial quality control (MQC), such as the biogenesis, autophagy, fusion and fission of mitochondria, is critical in preserving both the integrity and functionality of mitochondria (Tang et al. 2021). In MQC, the fusion and fission of mitochondria serve as crucial components in the dynamic management and repair of mitochondria, whereas mitochondrial autophagy and biogenesis facilitate the degradation and rejuvenation of these organelles (Fu et al. 2019). Studies have demonstrated that mitochondria respond to the changing cellular environment via these quality controls [8] and MQC is disrupted after cerebral ischemia (Salmina et al. 2021). Therefore, after cerebral ischemia, reversing the dysregulated MQC is critical for attenuating the adverse effects of mitochondrial damage on neurons (Wu et al. 2021).
Current literature demonstrates that exercise training promotes MQC regulation and that during voluntary exercise, muscle contractions can enhance mitochondrial biogenesis and autophagy, improving the function and structure of mitochondria (Granata et al. 2018). Other studies document that treadmill exercise improves the biogenesis (Rezaee et al. 2019), fusion (Chuang et al. 2017), and autophagy (Hwang et al. 2018) of mitochondria in Parkinson's disease and that sustained short-term exercise can ameliorate cardiac dysfunction and improve MQC in aged mice after myocardial infarction (Zhao et al. 2018). The above findings highlight that regulating MQC via exercise training may act as a potential strategy for disease treatment. However, it remains unsettled whether exercise training can optimize MQC after cerebral ischemia, let alone an exploration into the underlying mechanisms.
In non-cerebral ischemia studies, exercise training can improve the expression of Sirtuin-3 (SIRT3) in rats (Muñoz et al. 2018; Nogueira-Ferreira et al. 2019), an NAD + dependent mitochondrial deacetylase that modulates the acetylation of mitochondria-associated proteins (Ansari et al. 2017). As a deacetylase located in mitochondria, SIRT3 positively mitigates mitochondrial dysfunction and attenuates injury by modulating MQC. In sepsis-induced myocardial injury, SIRT3 reduces myocardial damage by activating AMPK-related mitochondrial biogenesis (Xin and Lu 2020); SIRT3 can inhibit excessive mitochondrial fission, mitigating the injury stemmed from cerebral ischemia–reperfusion (Hao et al. 2018). In addition, the SIRT3-promoted autophagy can attenuate ischemic injury by removing damaged nerve cells (Chen et al. 2021). Taken together, these findings demonstrate that SIRT3, like exercise training, is also involved in the MQC regulation. However, the inter-relation among exercise training, SIRT3, and MQC after cerebral ischemia remains blurred.
Available research indicates that SIRT3 can activate the β-catenin pathway and thus mediates the protection conferred by the NLRP3 inflammasomes against osteogenic inhibition induced by titanium particles (Zheng et al. 2021). Evidence documents that the β-catenin pathway engages in various processes, including synaptic remodeling, neuronal genesis, and the maintenance of intracellular homeostasis (Marchetti 2018). The activation of this pathway will recruit β-catenin and facilitate its nuclear entry to modulate the transcription of target genes such as Cyclin D1 and C-myc (Jia et al. 2019). In studies of kidney injury, β-catenin has been shown to alleviate cell death and mitochondrial dysfunction during acute kidney injury, including enhanced mitochondrial biogenesis, an increase in fusion markers (e.g., Mfn2 and OPA1), and a decrease in Drp1, the fission protein, thus restoring the homeostasis of mitochondria (Li et al. 2022). In cases of cerebral ischemia, the activation of the β-catenin pathway can also reduce neuronal apoptosis and maintain the function of mitochondria (Guo et al. 2023). However, in a scenario of cerebral ischemia, the involvement of the β-catenin pathway in the SIRT3-conferred improvement in mitochondrial function remains unexplored.
Therefore, in this study, Sprague–Dawley (SD) rats received transient middle cerebral artery occlusion (tMCAO) and subsequently were subject to treadmill exercise training. SIRT3 was overexpressed in vivo by intracranially injecting the adeno-associated virus (AAV-SIRT3) and SIRT3-interfering AAV (AAV-sh-SIRT3) into rats receiving exercise training to validate the impact of exercise training on cerebral infarction, neural function, neuronal apoptosis, mitochondrial formation, mitochondrial fusion/fission, mitophagy, and mitochondrial integrity after cerebral ischemia. We found that exercise training can re-establish MQC and promote the recovery of neural function by increasing SIRT3 expression. In addition, a cellular OGD/R model showed that SIRT3 overexpression alleviated apoptosis and re-established mitochondrial quality control by activating the β-catenin pathway. The evidence obtained illuminates the underlying mechanism in advocating exercise training for stroke treatment and suggests that SIRT3 is a powerful target in addressing brain ischemia-associated injury.
Materials and methods
Animals
Male adult SD rats (weighed 260–280 g) were obtained from Beijing HFK Bio-Technology. co., Ltd. The animals were raised in a specially designed environment (relative humidity: 50–60%; temperature: 24 ± 2 °C), and accessed food and water freely.
Animal surgery and experimental grouping
SD rats were exposed to tMCAO to initiate cerebral ischemia, following established procedures (Andrabi et al. 2017). Briefly, the SD rats received an intraperitoneal anesthesia with sodium pentobarbital. An incision was created in the midline of the neck to expose and isolate the common carotid artery (CCA) on the right, along with the external carotid artery (ECA) and the internal carotid artery (ICA). After the distal end of the ECA and its branches were ligated, a silicone-coated nylon monofilament was placed into the ICA via the ECA stump and gently pushed to block the middle cerebral artery until encountering slight resistance. Ninety minutes after the tMCAO procedure, the monofilament was gently retracted. The sham group received a similar treatment other than the monofilament insertion. The animals were categorized into: Sham, tMCAO, tMCAO + Exercise (tMCAO + E), tMCAO + AAV-SIRT3, and tMCAO + Exercise + AAV-sh-SIRT3 (tMCAO + E + AAV-sh-SIRT3) groups.
Injection of AAVs
The viruses of AAV-SIRT3 or AAV-sh-SIRT3 were supplied by Hanheng Biotechnology (Shanghai) Co., Ltd. After the dissolution in phosphate buffer saline (PBS), they were administered into the brains of SD rats, targeting the cortex and striatum as outlined (Mouwei et al. 2018). In brief, after anesthesia, the rats were removed to a stereotaxic apparatus and a microlitre syringe (Hamilton, NV) was inserted vertically to a depth of 2.50 mm in the cortex and 4.50 mm in the striatum with the coordinates of 0.20 mm posterior to the Bregma and 2.50 mm lateral to the midline. The virus (1.04 × 1010gc) were delivered slowly (0.1 µl/min). The syringe remained in position for a minimum of 15 min to prevent viral reflux. The efficiency of SIRT3 overexpression in the peri-ischemic region was estimated by GFP fluorescence.
Treadmill training
Exercise training was conducted following a previous treadmill running protocol (Perrino et al. 2011). Before tMCAO surgery, all rodents were positioned on a mobile conveyor belt and taught to jog against the belt’s motion for three days. Those that were able to run regularly were included in the study. Twenty-four hours after the tMCAO surgery, the rats in the tMCAO + E and tMCAO + E + AAV-sh-SIRT3 groups started treadmill exercise training. The intensity of exercise was gradually increased from a speed of 8 m/min at Day 1 to 10 m/min at Day 2 and to a speed of 12 m/min at Day 3, with the latter maintained for 30 min daily until Day 14. In order to motivate the rats to engage in treadmill activity, a gentle electrical shock was administered through a grid situated behind the equipment. For groups that did not receive exercise training, the animals were placed on the treadmill that was not turned on.
Assessment of neurological deficits
Behavioral tests were conducted on experimental rats in a randomized double-blind fashion at Days 1, 7, 14, and 21 after tMCAO. As mentioned (Wang et al. 2021), the neurological functions of rats were examined by the modified Neurological Severity Score (mNSS), including movement, sensation, equilibrium, and responses during abnormal movements. Scores range from 0 to 18, representing the sum of these four indexes, with an elevated score denoting a severer neurological impairment.
Post-stroke motor coordination was examined by the rotarod test. As mentioned (Wang et al. 2021), the rodents were trained on an accelerating rotarod 47,700 (Ugo Basile, Italy) three days before the surgery and tested after the tMCAO. The instrument accelerated from 4 rotations per minute (r/min) to 40 rotations per minute in five minutes. Time of rats on the rod was noted. The test was triplicated at an interval of 15 min. The mean of the dataset was analyzed.
The grip strength test was proceeded with a YLS-13A grip strength tester to assess the neuromuscular function of the animals, as previously outlined (Larcher et al. 2014). Adaptive training commenced 3 days prior to the tMCAO. During the test, the animals were gradually pulled backward until they released the grip plate, with the grip meter automatically recording the maximum muscle force. The measurement was repeated in triplicate with adequate rest in between, with the three grip force readings averaged and recorded.
The cylinder test was employed to evaluate the asymmetry in forelimbs, as reported (van der Kooij et al. 2010). The rodents were placed individually in clear plexiglass drums (20 cm in diameter, 40 cm in height). The tester recorded the initial contact of the front paws (right/left/both) with the wall of the cylinder. Typically, the rats would contact the cylinder wall with both front paws; however, those with cerebral ischemia injury predominantly preferred the use of the ipsilateral (right) forelimb. The proportion of contact by the uninjured (right) front limb was derived from: (right—left) / (left + right + both) × 100%. The test was triplicated to ensure accuracy and reliability.
5-Triphenyltetrazolium (TTC) chloride staining
After the administration of sodium pentobarbital for anesthesia, the rats’ brains were quickly removed, placed in a mold, and then frozen at −20℃ for a duration of 20 min. Next, the brain in the mold was cut into six sections of equal thickness. These sections were then immersed in a light-protected 2% TTC solution (BCBX0337, Sigma, USA) at 37 °C for 20 min, following previously established protocols (Zheng et al. 2022b). Subsequently, images were captured, with the normal brain tissue appearing red and the ischemic region displaying as white. The infarct size was measured with the Image J software. The area of each slice was computed as: (Contralateral area—Ipsilateral non-infarcted tissue area) / (Contralateral area × 2) × 100%.
Magnetic resonance imaging (MRI) analysis
The MRI scans were conducted in vivo at Day 14 after the ischemia on a 7.0 Tesla magnetic resonance scanner equipped with Paravision 6.0 software, with slight adjustments from previous methods (Liang et al. 2017). After the anesthesia with isoflurane, the rats were placed on a specialized bed and their condition was monitored at all times. The volume of infarction was assessed by T2-weighted imaging with a 2D fast-spinecho (TubroRARE) sequence of an echo time of 32 ms and repetition time of 5200 ms. The scan comprised 48 slices, each 0.56 mm thick, covering the entire brain area in a 256 × 256 matrix and 35 × 35 mm field of view (FOV). The percentage of the infarcted volume was quantified from the T2-weighted images with the Image J software.
Primary culture of cortical neurons
The isolation and culture of primary cortical neurons followed a reported protocol (Guo et al. 2023). The pregnant rats were first anesthetized to attain the embryonic rats (age: 16–18 days old), whose brains were obtained. The anterior 1/3 of the brain was collected. After the tissue was fully sliced, papain was added and digested in an incubator at 37 ℃ for 20 min. After the removal of the supernatant, the tissue received DMEM and was gently pipetted to resuspend the cells. Next, the cell suspension was proceeded according to a previous protocol (文献). After the measurement of the cell density, the neurons were seeded at the desired density into poly-D-lysine-coated culture flasks or plates (Ca# E607014; Sangon, China). Finally, the cultures were incubated in cell incubator.
Oxygen–glucose deprivation/reoxygenation (OGD/R)
According to a previous protocol (Chen et al. 2024b), the OGD model was used to simulate in vivo brain ischemia in neurons cultured for 7 to 10 days. Briefly, after the removal of the culture medium, the cell culture flask or plate was washed once with pre-warmed PBS and then received an appropriate amount of glucose-free DMEM (Cat# BL1124A; biosharp, China). The primary neurons were then incubated in a three-gas cell culture incubator for 120 min and further cultured after a medium replacement with a neuronal medium. In the Control group, only the medium was replaced.
Lentivirus transfection and inhibitor
After a 3-day culture, the primary cortical neurons were transfected with lentivirus from Shanghai Hanbio Co., Ltd. (MOI of 3). The lentivirus was diluted in fresh neuronal culture medium and administered to the cells before incubation. After 24 h, the virus-containing medium was substituted with a fresh culture medium. Afterwards, the incubation of the cells continued in cell incubator. XAV-939 (10 µM) was immediately introduced into the SIRT3 medium after the OGD/R.
Isolation of proteins in mitochondria, nuclei, and cytosol
Mitochondria were extracted from freshly harvested brain tissue or primary cortical neurons with a Beyotime kit (Cat# C3606, Beyotime, China). Minced tissues or digested cells were added to Mitochondria Isolation Reagent A on ice, pre-treated with PMSF (Cat# ST506, Beyotime, China) and then underwent centrifugation to increase mitochondria purity. Next, the resulting mitochondria were re-centrifuged. At this point, the precipitate was obtained as the isolated mitochondria and the supernatant was subjected to a 12,000-g centrifugation at 4 °C for 10 min to produce cytosolic proteins devoid of mitochondria.
The Beyotime kit (Cat# P0028, Beyotime, China) was utilized for the isolation of nuclear and cytoplasmic proteins. The minced brain tissues underwent homogenization in a buffer containing 1 mM PMSF, prepared in the appropriate ratios (CPER A: CPER B = 20:1). After a 15-min ice bath, the tissues were centrifuged to isolate cytoplasmic proteins in the supernatant. The remanent pellet underwent lysis in a buffer (CPER A and PMSF). Afterwards, the pellet received CPER B and was further centrifuged to collect the cytoplasmic fraction. Subsequently, the remaining pellet was combined with a buffer to extract nuclear proteins containing PMSF, shaken for 30 min, and finally centrifuged. The resulting supernatant was gathered as the nuclear protein extract.
Western blot
As reported previously (Chen et al. 2024a), cultured primary neurons and the brain tissues from peri-ischemic cortical were collected and lysed by ultrasound to extract the supernatant as the resulting protein extract. The denatured proteins subsequently underwent separation by electrophoresis. They were next transported to a PVDF membrane and incubated with Bovine serum albumin, primary antibodies, and secondary antibodies (Refer to the supplementary materials for details). Finally, the target protein image was visualized with an ECL detection reagent and the expression was quantified via ImageJ software.
TdT-mediated dUTP nick end labelling (TUNEL) assay
Cell apoptosis in the brain was assessed by TUNEL staining (11,684,817,910; Roche) according to the instructions from the manufacturer (Hu et al. 2017a). Initially, the brain tissues were paraffinized and sliced into sections (thickness: 5 μm), which were deparaffinized, rehydrated, and then treated with 3% H2O2. Subsequently, they were treated with 0.1 M sodium citrate in a microwave for 5 min. Afterwards, the sections were exposed to the TUNEL reaction solution and then underwent DAB staining. The tally of TUNEL-positive cells was acquired from six random fields, as previously described. The proportion of apoptotic cells was expressed in relation to the total cell count.
Transmission electron microscopy (TEM)
As reported (Yang et al. 2018b), the morphology of mitochondria in rat cortical neurons and primary cultured neurons was assessed by TEM. The extracted brain tissue (1 mm3 in size) or digested cells was fixed in Osmium (VIII) oxide and ferrocyanide for 90 min. After the PBS rinsing, the samples underwent dehydration using an ethanol-acetone gradient and were subsequently soaked in an epoxy resin embedding agent. After a complete polymerization, the sample was sectioned into ultra-thin slices (90 to 100 nm) with a Leica UC-7 ultramicrotome. The slices then received a staining with lead citrate and uranium acetate. Finally, the images of samples were captured under a TECNAI transmission electron microscope from FEI Company.
Immunofluorescence staining
Immunofluorescence staining was performed by established protocols (Yingqiong et al. 2017). In brief, the slices underwent a 30-min incubation with glycine (22.5 mg/ml) and then were exposed to a blocking solution (10% goat serum in PBS consisting of 3% BSA and 0.3% Triton) for one hour. Next, they were incubated at 4 °C overnight with primary antibodies (comprising 2.5% goat serum, 0.3% Triton, and 1% BSA) and further with secondary antibodies. They subsequently underwent a staining with the nuclear stain DAPI (see supplementary materials for details). Finally, after another three PBST washes, the slides were imaged by confocal microscopy.
Real-time quantitative PCR (qRT-PCR)
The isolation of total RNA from cortical tissues and primary cultured neurons was performed with RNAfast200 (Cat# 220,010, Fastagen Biotech, China). Next, the resultant RNA was converted into cDNA with a reverse transcription kit from Yesen (Cat# 11141ES60; China). Quantitative RT-PCR was detected by a Prism 7500 thermal cycler and SYBR Green Master Mix. The expression of mRNA was measured by the 2 − ΔΔCT method and normalized against β-actin.
Statistical analysis
Results were processed with GraphPad Prism 8 Software and decribed as mean ± SDM. Data normality was detected by the Shapiro–Wilk test. Variance homogeneity was examined by the Brown-Forsythe test. Data that exhibited a normal distribution and equal variance, inter-group comparisons were conducted by one-way ANOVA and Bonferroni’s post hoc test. The unequal variance was evaluated by one-way ANOVA and Dunnett's T3 post hoc test. The impact of treatment on behavioral performance was examined by two-way repeated-measures ANOVA with Bonferroni post hoc test. The significant difference was designated as: *p < 0.05, ** p < 0.01, ***p < 0.001, as versus the Sham or Control counterparts; #p < 0.05, ##p < 0.01, ###p < 0.001, as versus the tMCAO or OGD/R rats; &p < 0.05, &&p < 0.01, &&&p < 0.001, as versus the tMCAO + E or OGD/R + LV-SIRT3 rats.
Results
Exercise training enhances the neural functional recovery after cerebral ischemia and increases SIRT3 expression in the peripheral ischemic area
To elucidate the potential benefits of exercise training for neurological deficits after cerebral ischemia, the neural functional changes in rats were assessed by mNSS at Days 1, 7, 14, and 21 after tMCAO. The analyses found that in the exercise training group, the mNSS score was dramatically lower than in the non-exercise counterpart at Days 14 and 21 after tMCAO (Fig. 1A). Moreover, western blotting revealed an upregulation of SIRT3 protein expression in the peri-ischemic area of the tMCAO-treated rats after exercise, with the most significant difference appearing at Day 14 (Fig. 1B, C). Similarly, the TTC staining revealed obvious infarct area in tMCAO-treated rats after cerebral ischemia. Compared with the tMCAO group, rats undergoing exercise training reported a markedly-reduced infarction volume, with statistical significance appearing at Days 14 and 21 (Fig. 1D-I). These findings evidence that exercise training can promote the recovery of neural function and increase SIRT3 expression in the peripheral ischemic areas in rats after cerebral ischemia.
Fig. 1.

The neural functional changes and SIRT3 protein expression after exercise training. A mNSS scores at different time points after exercise training (n = 10). B, C Representative immunoblots (B) and quantification of SIRT3 level (C) at different time points (n = 4). D-I TTC staining and quantification of the infarction volume in the experimental groups at different time points (n = 5)
Exercise training reduces cerebellar infarction and promotes the neurological function recovery after cerebral ischemia by increasing SIRT3 expression
To further ascertain the involvement of exercise training and SIRT3 level in cerebral ischemia in rats and the relationship between exercise training and SIRT3 expression, the cortex and striatum of rats respectively received an injection of AAV-SIRT3 and AAV-sh_SIRT3 (Fig. 2B). The results revealed that SIRT3 markedly increased in the rat brains injected with AAV-SIRT3 but decreased in those receiving AAV-sh_SIRT3 (Fig. 2C, D). Afterwards, four neurobehavioral tests were adopted to assess the effects of exercise and SIRT3 overexpression on the motor function of tMCAO rats.
Fig. 2.

The effects of exercise training on functional recovery and infarct volume after tMCAO. A The timeline of experiment. B Representative immunofluorescence image of SIRT3 overexpression in the brain cortex using a GFP-containing AAV (magnification 100 ×). C, D Representative immunoblots (C) and quantification of SIRT3 expression (D) of experimental groups; n = 4. E–H The the mNSS scoring (E), cylinder test (F), grip strength test (G), and rotarod test (H) were conducted to assess motor function across various groups; n = 10. I Representative T2-weighted MRI images in different experimental groups. J, K Representative immunoblots (J) and quantification of SIRT3 (K) of experimental groups; n = 4
The mNSS score, assessed at Days 1, 7, 14 and 21 after tMCAO, revealed a reduction in neurological deficit in the rats receiving exercise training and the AAV-SIRT3 injection at Day 14 when compared with the ischemic rats. However, the beneficial effects of exercise training were compromised by AAV-sh_SIRT3 treatment (Fig. 2E). Likewise, in the cylinder test, the exercise training group displayed reduced ipisateral forepaw preference at Day 14 after tMCAO, while the AAV-SIRT3-treated rats reported a decreased asymmetry at Day 7 after tMCAO as compared with the tMCAO rats. The improvement in the exercise training group was inhibited by AAV-sh_SIRT3 treatment (Fig. 2F). The grip strength was improved in both the exercise training and AAV-SIRT3-treated groups at Day 7 after tMCAO, which was partially reversed by inhibiting SIRT3 expression (Fig. 2G). Similarly, both the exercise training and AAV-SIRT3-treated groups spent significantly longer duration on the rotarod at Day 14 after tMCAO than the tMCAO counterparts. In contrast, the inhibition of SIRT3 expression after exercise training notably reduced the duration rats spent on the rotarod (Fig. 2H).
Besides, compared with the tMCAO rats, the exercise training and the AAV-SIRT3-treated groups reported a marked decrease in the volume of brain infarction detected at Day 14 after tMCAO, which was increased the tMCAO + E + AAV-sh_SIRT3 counterparts compared with the exercise training group (Fig. 2I). The expression level of SIRT3 in different experimental groups was assessed by WB technology after 14 days of exercise. Consistent with previous research, the SIRT3 level was decreased after tMCAO; both exercise training and SIRT3 overexpression led to an increase in SIRT3 levels; however, the interference with SIRT3 expression after exercise resulted in a reduction of SIRT3 levels (Fig. 2J, K). The results evidence that exercise training and AAV-SIRT3 treatment exert beneficial effects on the neural recovery in rats after cerebral ischemia and that exercise training promotes the recovery from cerebral ischemia by increasing SIRT3 expression.
Exercise training reduces neuronal apoptosis after ischemia in rats by promoting SIRT3 expression
Subsequently, the study delved into the impacts of exercise training and the overexpression of SIRT3 on cell apoptosis. The results revealed a notable increase in Bax and cleaved caspase-3 protein and a significant decline in Bcl-2 protein in tMCAO rats at Day 14 after tMCAO. Both exercise training and SIRT3 overexpression downregulated the protein expression of cleaved caspase-3 and Bax and markedly upregulated that of Bcl-2 when compared with tMCAO group, which were partially offset in the exercise training group injected with AAV-sh-SIRT3. No marked difference in the pro-Caspase-3 level was observed among the groups (Fig. 3A-E).
Fig. 3.

The effects of exercise training on neuronal apoptosis after tMCAO. A-E Representative immunoblots (A) and quantification of cleaved caspase-3 (B), Caspase-3 (C), Bcl-2 (D) and Bax (E) of experimental groups; n = 4. F, G Representative images of TUNEL staining (F) and quantification of TUNEL-positive cells (G) of experimental groups, with black arrows indicating TUNEL-positive cells; n = 3, scale bar: 100 um
Consistently, at Day 14 after the surgery, the tMCAO group demonstrated an elevation of TUNEL-positive cells in the ischemic peri-infarct cortex. However, compared with the tMCAO counterpart, the percentage of positive cells significantly decreased after exercise training and SIRT3 injection, and increased in the AAV-sh_SIRT3-injected group after exercise training (Fig. 3F, G). The results suggest that exercise training can diminish the expressions of mitochondrial pro-apoptotic proteins, boost those of anti-apoptotic proteins, and decrease TUNEL-positive cells by upregulating SIRT3 expression.
Exercise training maintains mitochondrial integrity after ischemia in rats by increasing SIRT3 expression
As mitochondrial integrity plays a critical role in preventing apoptosis, we respectively examined the protein expression of Cyt c in cytoplasm and mitochondria to confirm the impact of exercise training on mitochondrial integrity. Cyt c, primarily situated in mitochondria as a pro-apoptotic factor, entered the cytoplasm from the mitochondria upon the disruption of mitochondrial integrity. The results reported that the level of Cyt c in cytoplasm progressively increased while that in mitochondria gradually declined in the cortex of the tMCAO group at Day 14. In contrast, in relation to the tMCAO group, the exercise training and SIRT3 overexpression markedly reduced the level of cytoplasmic Cyt c but dramatically increased that of mitochondrial Cyt c. After the transfection of SIRT3-interfering virus during exercise training, Cyt c was upregulated in the cytoplasm and downregulated in mitochondrial. The protein level of AIF in nucleus remarkably increased in tMCAO group and decreased in the exercise training and SIRT3 overexpression groups. Compared with the tMCAO + E counterpart, the AIF protein expression also increased in the tMCAO + E + AAV-sh_SIRT3 group (Fig. 4A-D). Moreover, fluorescence staining of the peripheral cerebral ischemic area of rats showed an increased nuclear Cyt c diffusion in the tMCAO treatment group, which was reduced in the rats receiving exercise training group and the tMCAO + AAV-SIRT3 treatment. This effect was largely reversed in the tMCAO + E + AAV-sh_SIRT3 rats, with an increased intracellular diffusion of Cyt c (Fig. 4E, F).
Fig. 4.

The impact of exercise training on the integrity of mitochondrial membrane structure after brain ischemia in rats. A-D Representative immunoblots (A) and quantification of mitochondrial Cyt c (B), cytosolic Cyt c (C), nuclear AIF (D) in the cortex of experimental rats; n = 4. E, F Representative immunofluorescence image (E) and quantification of Cyt c (F) of experimental groups; n = 3, scale bar: 20 um. G Representative TEM images of mitochondrial cristae in the cortex of experimental groups; scale bar: 200 nm
TEM was performed to analyze the mitochondrial ultrastructure in the ischemic periphery of rats. In relation to the Sham counterpart, the tMCAO group displayed a disorganized mitochondrial structure and significantly-reduced mitochondrial crista density (Fig. 4G). Exercise training and SIRT3 overexpression improved mitochondrial structure and partially restored the mitochondrial crista density. These effects were partially reversed in the tMCAO + E + AAV-sh_SIRT3 group, resulting in a decrease in mitochondrial crista density. The results evidence that exercise training can maintain mitochondrial integrity by increasing SIRT3 expression.
Exercise training promotes mitochondrial biogenesis and regulates mitochondrial fission/fusion balance in rats after cerebral ischemia by increasing SIRT3 expression
To detect the effect of exercise training on mitochondrial quality, we further investigated whether mitochondrial quality control was impaired in tMCAO rats and restored by exercise training. The expression of mitochondria-generating proteins was detected. The findings indicated a decline in the expression of TFAM after tMCAO. Additionally, compared with the tMCAO rats, exercise training and SIRT3 overexpression increased the production of all mitochondria-associated proteins, such as NRF-1, PGC-1α, and TFAM proteins, which was suppressed by the administration of SIRT3 interfering virus (Fig. 5A-D).
Fig. 5.

The impact of exercise training on mitochondrial biogenesis and dynamics after cerebral ischemia in rats. A-D Representative immunoblots (A) and quantification of PGC-1α (B), NRF-1 (C) and TFAM (D) of experimental groups; n = 4. E-J Representative immunoblots (E) and quantification of OPA1 (F), Mfn 1 (G), Mfn 2 (H), Drp1 (I), and Fis (J) in the different groups; n = 4
Subsequently, we quantified the markers of mitochondrial fission–fusion signaling. After tMCAO, the expression of Drp1 and Fis (fission proteins) was upregulated while that of OPA1 and Mfn2 (fusion proteins) markedly declined; after exercise training and SIRT3 overexpression, that of Drp1 and Fis decreased while that of OPA1 and Mfn2 notably increased. These regulatory benefits of exercise training were partially reversed by SIRT3 interfering virus, decreasing the expression of OPA1 and Mfn2, and increasing that of Drp1 and Fis. Nevertheless, the level of Mfn1 did not markedly differ across the groups (Fig. 5E-J). These results demonstrate that exercise training can promote mitochondrial generation and regulate the mitochondrial fission/fusion balance by increasing SIRT3 expression.
Exercise training promotes mitochondrial autophagy in rats after cerebral ischemia by increasing SIRT3 expression
To detect the role of exercise training in the regulation of mitochondrial autophagy after brain ischemia, the level of mitophagy-related markers was quantified. After tMCAO, Parkin expression decreased while LC3 II expression increased. Notably, exercise training and SIRT3 overexpression increased the proteins expression of Parkin and PINK1, which was decreased by the interference with SIRT3 expression after exercise training (Fig. 6A-D).
Fig. 6.

The impact of exercise training on mitochondrial autophagy in rats after cerebral ischemia. A-D Representative immunoblots (A) and quantification of PINK1 (B), Parkin (C) and LC3 II (D) of experimental groups; n = 4. E, F Representative confocal images (E) and quantification (F) of mitochondrial autophagy in experimental groups (magnification 600 ×); n= 3
To visualize mitochondrial autophagy, we conducted immunofluorescence staining for PINK1, a reliable marker for monitoring mitophagy, along with mitochondria co-labeled with Tomm20. The analysis revealed that after cerebral ischemia, the exercise training and the SIRT3 overexpression groups reported a higher number of PINK1-positive mitochondria than the tMCAO group (Fig. 6E, F). Consistent with the Western-blot analysis, the injection of SIRT3 interfering virus also partially mitigated the impact of exercise training, leading to a decrease in PINK1-positive mitochondria. These data illuminate the significance of exercise training in modulating mitochondrial autophagy and maintaining mitochondrial quality through the upregulation of SIRT3 expression.
SIRT3 overexpression inhibits the apoptosis of primary neurons after OGD/R by activating β-catenin pathway
To verify whether SIRT3 activates the β-catenin pathway, we used the XAV-939 to selectively inhibit the β-catenin-mediated transcription. The analysis reported that, unlike the control group, the expression of p-β-catenin was elevated while that of c-Myc, β-catenin, and Cyclin D1 proteins was decreased. In contrast, in relation to that of the OGD/R group, the treatment with LV-SIRT3 reduced the level of p-β-catenin and increased that of β-catenin, c-Myc, and Cyclin D1 proteins. Additionally, XAV-939 reversed the LV-SIRT3-induced changes in these proteins (Fig. 7A-E). The data suggest that SIRT3 overexpression can activate the β-catenin pathway.
Fig. 7.

The effect of SIRT3 overexpression on apoptosis of primary cortical neurons after OGD/R after XAV-939 administration. A-H Representative immunoblots (A, F) and quantification of p-β-catenin (B), β-catenin (C), C-Myc (D), CyclinD1 (E), Bcl-2 (G) and Bax (H) in the different groups; n = 3. I Fluorescent-stained images of neurons
Additionally, the neuronal morphology and apoptosis were assessed. The results revealed significant damage to neuronal morphology after OGD/R treatment, along with an increase in Bax levels and a decrease in Bcl-2. After treatment with LV-SIRT3, neuronal morphology was restored, and the level of Bcl-2 increased, while that of Bax decreased. Conversely, the pathway inhibitor XAV-939 reversed the effects of SIRT3 overexpression, upregulating the level of Bax and inhibiting that of Bcl-2 (Fig. 7F-I). These results suggest that SIRT3 inhibits the apoptosis of primary cortical neurons by activating the β-catenin pathway.
SIRT3 overexpression repairs mitochondrial structure and re-establishes mitochondrial quality control (MQC) after OGD/R by activating the β-catenin pathway
To validate that SIRT3 exerts neuroprotective effects via β-catenin, electron microscopy was used to observe the mitochondrial structure. The results revealed that the mitochondrial structure was damaged after OGD/R treatment but was repaired after LV-SIRT3 treatment; the repair process was inhibited by XAV-939 treatment (Fig. 8A). The results of RT-PCR showed that the mRNA levels of PGC-1α, NRF-1, and TFAM increased after treatment with LV-SIRT3, while they were reversed by XAV-939 intervention (Fig. 8B-D). We subsequently measured the levels of Mfn2 and Drp1 in the cells. After OGD/R treatment, the expression of Mfn2 decreased, while Drp1 expression increased. Treatment with LV-SIRT3 decreased the level of Drp1 and increased that of Mfn2. Conversely, the pathway inhibitor XAV-939 reversed the effects of SIRT3 overexpression, inhibiting the level of Mfn2 and upregulating that of Drp1 (Fig. 8E-G). Additionally, the assessment of mitophagy revealed an upregulation in the fluorescence expression of PINK1. In contrast, the XAV-939 treatment group showed a decrease in the fluorescence expression of PINK1 (Fig. 8H). These findings illuminate that SIRT3 repairs mitochondrial structure and re-establishes MQC after OGD/R by activating the β-catenin pathway.
Fig. 8.

The impact of SIRT3 overexpression on the structure of mitochondria and MQC in cultured neurons after OGD/R via the β-catenin pathway. A Representative transmission electron microscope image of mitochondria of primary cortical neurons; scale bar: 300 nm. B-D qPCR analysis of the relative mRNA levels of experimental groups; n = 3. E–G Representative immunoblots (E) and quantification of Mfn2 (F) and Drp1 (G) of primary neurons of experimental groups; n = 3. H Representative confocal images of mitochondrial autophagy of experimental groups; scale bar: 200 um
Discussion
This study reported that exercise training promoted the recovery of neurological function, alleviated brain ischemic injury, reduced neuronal apoptosis, and re-established mitochondrial quality control by promoting SIRT3 expression. The study elucidates that SIRT3 can act as a therapeutic alternative for brain ischemia.
As well-established in evidence-based medicine, exercise training is an effective method to reduce disability in stroke patients and plays a pivotal part in promoting the post-ischemic recovery of neural function (Brazzelli et al. 2011; Zhang et al. 2020a). In this study, exercise training continuously improved the neurological function in cerebral ischemic rats, which was reflected in the steady decline in mNSS score, which was significant at Days 14 and 21 after exercise training. Available evidence has found that exercise training can diminish the volume of cerebral infarction (Cheng et al. 2020). Consistently, in our study, the cerebral infarction diminished and was statistically significant after either 14 or 21 consecutive days of exercise training. Existent studies demonstrate that after cerebral ischemic injury, SIRT3 expression in the brain can be markedly down-regulated (Yang et al. 2018a), which can be boosted in rodents' brain by exercise training (Pan et al. 2021; Wang et al. 2020). Therefore, we detected SIRT3 expression after exercise training and found a higher SIRT3 protein expression in rats receiving exercise training than in rats treated with tMCAO alone, which was statistically significant at Day 14.
SIRT3, an NAD-dependent protein deacetylase, regulates apoptosis and mitochondrial function (Chen et al. 2024b), including the control of mitochondrial quality in mitochondrial biogenesis, dynamics and mitophagy. To verify this mechanism, we introduced AAV-SIRT3 and AAV-sh_SIRT3 to investigate the neurological function of rats by assessing their performance in the behavioral paradigms. Consistent with our hypothesis, SIRT3 overexpression and exercise training both mitigated neurological deficits and the cerebral infarction, and improved the muscle strength and motor coordination, which were reversed by interference with SIRT3 virus after exercise training. These findings highlight that after cerebral ischemia, SIRT3 is indeed involved in the exercise training-improved neural function.
In addition, we examined the apoptosis of neural cells in ischemic rats. Apoptosis, characterized by DNA fragmentation, is deemed a crucial factor in exacerbating tissue damage and cellular demise following cerebral ischemia (Sp et al. 2020). In the mechanism of cellular apoptosis, Bax serves as a pro-apoptotic regulator, while Bcl-2 controls the regulatory proteins of apoptosis, like caspase-3, an effector protease crucial in apoptotic proteolysis (Yang et al. 2019). Existent studies suggest that exercise training can attenuate neural apoptosis by regulating the expressions of apoptosis-related proteins (Terashi et al. 2019). To elucidate the anti-apoptotic impact of exercise training, we examined essentially apoptotic TUNEL-positive cells (Zhang T 2021) in tMCAO rats, which showed that both exercise training and SIRT3 overexpression reduced TUNEL-positive cells and the expression of Bax and cleaved caspase-3, and upregulated that of Bcl-2 in the peri-ischemic region. Nevertheless, these effects were reversed in the tMCAO + E + AAV-sh_SIRT3 group. These findings indicate that exercise training might attenuate apoptosis after cerebral ischemia by stimulating SIRT3 expression.
Cerebral ischemic injury can disrupt the integrity of mitochondria, triggering subsequent apoptosis (Guo et al. 2023). After cerebral ischemia, mitochondrial permeability increases due to abnormal mitochondrial swelling, inducing the flux of Cyt c and AIF from mitochondria to cytoplasm. The former binds to Apaf-1 and Caspase 9, forming the "apoptosome", which then activates Caspase 3, ultimately leading to apoptosis (Shahid et al. 2020). The latter enters the nucleus from the cytoplasm, bringing about DNA degradation and cell death (Zhong et al. 2018). SIRT3 can inhibit Cyt c release (Feng et al. 2018) to maintain mitochondrial integrity. Consistently, in the current study, after cerebral ischemia, the influx of mitochondrial Cyt c into the cytoplasm increased the Cyt c expression in the cytoplasm and the nuclear transfer increased the expression of nuclear AIF; both exercise training and SIRT3 overexpression upregulated the expression of mitochondrial Cyt c and downregulated that of cytoplasmic Cyt c and nuclear AIF, indicating a partial restoration of mitochondrial integrity; and the transfection of SIRT3-interfering virus upregulated the expression of cytoplasmic Cyt c and nuclear AIF. Collectively, the findings evidence the critical role of SIRT3 in modulating the mitochondrial integrity.
Furthermore, cerebral ischemia can seriously compromise the mitochondrial structure, resulting in severe neurological dysfunction (Guo et al. 2023). In this study, electron microscopy revealed an impaired mitochondrial morphology in ischemic rats, with the mitochondrial membrane structure blurred and the number of mitochondrial cristae reduced. Exercise training and SIRT3 overexpression restored mitochondrial membrane structure and increased mitochondrial cristae after cerebral ischemia, which was respectively offset by SIRT3 interference. These findings illuminate that exercise training can maintain mitochondrial integrity and alleviate apoptosis after cerebral ischemia by increasing SIRT3 expression.
Apart from the damage to mitochondrial structure, cerebral ischemia can also impair mitochondrial function. The maintenance of mitochondrial function depends on MQC, which mainly involves the autophagy, fission, fusion, and biogenesis of mitochondria (Zheng et al. 2022a). The latter is the process of replacing damaged mitochondria with new ones and restoring mitochondrial function (Yuan et al. 2023), in which NRF-1, PGC-1α, and TFAM are the pivotal protein factors (Jornayvaz and Shulman 2010). Studies have shown that mitochondrial biogenesis can be rapidly triggered 24 h after cerebral ischemia in rats, resulting in increased expression of mitochondria-related proteins PGC-1, NRF, and TFAM (Zhou et al. 2022). However, other studies show that the levels of TFAM and PGC-1 decrease 3 days after cerebral ischemia (Chang et al. 2023). These inconsistencies may be attributed to the different timings of brain ischemia.
This study showed that the expression of mitochondrial production-associated protein TFAM decreased at Day 14 after cerebral ischemia. However, exercise training and SIRT3 overexpression boosted the protein level of PGC-1, NRF and TFAM, while interfering SIRT3 expression after exercise training inhibited mitochondrial generation, manifested as a decrease in the level of NRF, PGC-1, and TFAM. The available literature has long documented that exercise can confer favorable effects on mitochondrial function, partly by initiating the biogenesis of mitochondria (Park et al. 2021), as validated in the cerebral ischemia model of the current study. These results indicate that exercise training may promote mitochondrial biogenesis after cerebral ischemia by upregulating SIRT3 expression.
In the pathology of cerebral ischemic injury, the disruption of the mitochondrial fusion and fission is a fundamental event (Campos et al. 2023), as the fusion and fission of mitochondria are indispensable for preserving mitochondrial function (Zhang et al. 2019). Mitochondrial fusion involves the outer and inner membrane fusion (OMM, IMM, respectively) (Zheng et al. 2022a). The former is regulated by Mfn1 and Mfn2 and the latter mainly depends on OPA1 (Zheng et al. 2022a). The pathological fission of mitochondria mainly counts on dynamin-related protein 1 (Drp1) and mitochondrial fission protein 1 (Fis1) (Song et al. 2022). Other research shows insufficient fusion and excessive mitochondrial fission during ischemia (An et al. 2021). However, exercise training can stabilize mitochondrial function by regulating mitochondrial dynamics (Campos et al. 2023). Accordingly, we examined the expression of proteins linked to the fission and fusion of mitochondria. The results revealed that the fission-promoting proteins increased and the fusion-promoting proteins decreased after cerebral ischemia; that exercise training and SIRT3 overexpression promoted fusion and inhibited fission in the mitochondria; and that the administration of SIRT3-interfering virus increased mitochondrial fission and decreased fusion. The findings demonstrate that exercise training can modulate the mitochondrial dynamics by promoting the expression of SIRT3.
When dysfunctional mitochondria within a cell fail to undergo self-repair via fission and fusion, they are selectively eliminated through mitochondrial autophagy (Zhang et al. 2020b), which is primarily regulated by the PINK1 pathway (Anzell et al. 2018). In this process, upon the occurrence of mitochondrial damage, PINK1 stabilizes on the outer mitochondrial membrane, stimulating the activation and recruitment of Parkin, thus setting mitophagy in motion (Matsuda et al. 2010). Studies indicate that an increase in mitochondrial autophagy may produce neuroprotection from brain ischemia (Cai et al. 2021; Shen et al. 2017) and that exercise training can modulate signaling biomarkers of mitochondrial autophagy and activate the PINK1 signaling pathway (Drake et al. 2019). In this study, after cerebral ischemia, mitochondrial autophagy was suppressed and LC3 II expression increased and Parkin expression declined. The accumulation of LC3 II protein may be due to the inhibition of LC3 II turnover (Zhao et al. 2018). However, some research suggests that mitochondrial autophagy is activated after cerebral ischemia (He et al. 2022), which may be explained by the notion that mitochondrial autophagy is a spontaneous neuroprotective response that is briefly triggered after brain ischemia (Yang et al. 2022). Meanwhile, this study confirmed that the protein expressions of PINK1 and Parkin and the percentage of PINK1-positive mitochondria increased in both exercise training and SIRT3 overexpression groups, which were reversed by the transfection of SIRT3-interfering virus. These results highlight that exercise training may modulate the autophagy of mitochondria by increasing SIRT3 expression after cerebral ischemia.
In non-cerebral ischemia, SIRT3 exerts a protective effect by activating β-catenin. The nuclear transfer of β-catenin from the cytoplasm is crucial for the biological function of the β-catenin pathway. This notion is validated with the findings of the in vitro model of cerebral ischemia (OGD/R) with primary cultured neurons, which evidence that SIRT3 can indeed regulate the level of key molecules in the β-catenin signaling pathway and downstream genes. Furthermore, the introduction of XAV-939, a specific antagonist of the β-catenin pathway, further demonstrated that after OGD/R, SIRT3 reduces neuronal apoptosis by activating the β-catenin pathway, and regulates MQC, including enhancing mitochondrial biogenesis, modulating mitochondrial dynamics, enhancing mitophagy, and repairing mitochondrial structure. This cellular validation serves as the rationale for a comprehensive exploration of the impact of exercise training and SIRT3, bridging perspectives from animals to cells and from in vivo to in vitro.
In summary, this study evidences that exercise training enhances the neural functional recovery after brain ischemia in rats by upregulating SIRT3 expression, in which the increased SIRT3 expression alleviates neuronal apoptosis and re-establishes mitochondrial quality control by activating the β-catenin pathway. The findings indicate SIRT3 as a potential therapeutic strategy for injury induced by brain ischemia.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We warmly thank Professor Hongzhi Huang (Fujian Medical University) for proofreading and polishing the manuscript. We also thank assistant researcher Minguang Yang (Fujian University of Traditional Chinese Medicine) for his work in magnetic resonance imaging, and teachers Minxia Wu, Linying Zhou, and Xi Lin (Public Technology Service Center Fujian Medical University) for their contributions to transmission electron microscopy.
Author Contributions
Wenwen Wu, Nan Liu and Hongbin Chen conceived the research design; Wenwen Wu, Zengyu Wei, Zhiyun Wu performed the experiments; Jianmin Chen, Ji Liu, Manli Chen, Jinjin Yuan, Zhijian Zheng contributed new reagents or analytic tools; Zijun Zhao and Qiang Lin analyzed data; Wenwen Wu wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by Excellent Young Scholars Cultivation Project of Fujian Medical University Union Hospital (No. 2022XH026), Joint Funds for the innovation of science and Technology, Fujian province (No. 2020Y9065 and 2021Y9060), and National Natural Science Foundation of China (No. 81802225), Startup Fund for Scientific Research, Fujian Medical University (No. 2023QH2026).
Data Availability
No datasets were generated or analysed during the current study.
Declarations
Competing Interests
The authors declare no competing interests.
Ethical Approval
The protocols adhered to the National Institute of Health guidelines (NIH Publications NO. 80–23, revised 1996). Approval for this experiment was obtained from the Institutional Animal Care and Use Committee (IACUC) of Fujian Medical University, Fujian, China [Approval No. 20220603].
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Wenwen Wu, Zengyu Wei, and Zhiyun Wu contributed equally to this work shared the first authorship.
Contributor Information
Nan Liu, Email: xieheliunan1984@163.com.
Hongbin Chen, Email: hbchen0127@fjmu.edu.cn.
References
- An H, Zhou B, Ji XM. Mitochondrial quality control in acute ischemic stroke. J Cereb Blood Flow Metab. 2021;41(12):3157–70. 10.1177/0271678x211046992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SS, Parvez S, Tabassum H. Progesterone induces neuroprotection following reperfusion-promoted mitochondrial dysfunction after focal cerebral ischemia in rats. Dis Model Mech. 2017;10(6):787–96. 10.1242/dmm.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, Kim KH. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell. 2017;16(1):4–16. 10.1111/acel.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzell AR, Maizy R, Przyklenk K, Sanderson TH. Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury. Mol Neurobiol. 2018;55(3):2547–64. 10.1007/s12035-017-0503-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- Brazzelli M, Saunders DH, Greig CA, Mead GE. Physical fitness training for stroke patients. Cochrane Database Syst Rev. 2011;11:247. 10.1002/14651858.CD003316.pub4. [DOI] [PubMed] [Google Scholar]
- Cai Y, Yang EY, Yao XH, et al. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 2021;38:15. 10.1016/j.redox.2020.101792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos JC, Bozi LHM, Krum B, et al. Exercise preserves physical fitness during aging through AMPK and mitochondrial dynamics. Proc Natl Acad Sci U S A. 2023;120(2):11. 10.1073/pnas.2204750120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CY, Wu CC, Pan PH, et al. Tetramethylpyrazine alleviates mitochondrial abnormality in models of cerebral ischemia and oxygen/glucose deprivation Reoxygenation. Exp Neurol. 2023;367: 114468. 10.1016/j.expneurol.2023.114468. [DOI] [PubMed] [Google Scholar]
- Chen D, Zheng K, Wu H, et al. Lin28a attenuates cerebral ischemia/reperfusion injury through regulating Sirt3-induced autophagy. Brain Res Bull. 2021;170:39–48. [DOI] [PubMed] [Google Scholar]
- Chen H, Liu J, Chen M, et al. SIRT3 facilitates mitochondrial structural repair and functional recovery in rats after ischemic stroke by promoting OPA1 expression and activity. Clin Nutr (Edinburgh, Scotland). 2024a;43(7):1816–31. 10.1016/j.clnu.2024.06.001. [DOI] [PubMed] [Google Scholar]
- Chen M, Liu J, Wu W, et al. SIRT1 restores mitochondrial structure and function in rats by activating SIRT3 after cerebral ischemia/reperfusion injury. Cell Biol Toxicol. 2024b;40(1):31. 10.1007/s10565-024-09869-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Shen W, Jin L, et al. Treadmill exercise promotes neurogenesis and myelin repair via upregulating Wnt/β-catenin signaling pathways in the juvenile brain following focal cerebral ischemia/reperfusion. Int J Mol Med. 2020;45(5):1447–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CS, Chang JC, Cheng FC, Liu KH, Su HL, Liu CS. Modulation of mitochondrial dynamics by treadmill training to improve gait and mitochondrial deficiency in a rat model of Parkinson’s disease. Life Sci. 2017;191:236–44. 10.1016/j.lfs.2017.10.003. [DOI] [PubMed] [Google Scholar]
- Drake JC, Laker RC, Wilson RJ, Zhang M, Yan Z. Exercise-induced mitophagy in skeletal muscle occurs in the absence of stabilization of Pink1 on mitochondria. Cell Cycle. 2019;18(1):1–6. 10.1080/15384101.2018.1559556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigin VL, Stark BA, Johnson CO, et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021;20(10):795–820. 10.1016/s1474-4422(21)00252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JX, Lu C, Dai Q, Sheng JQ, Xu M. SIRT3 Facilitates Amniotic Fluid Stem Cells to Repair Diabetic Nephropathy Through Protecting Mitochondrial Homeostasis by Modulation of Mitophagy. Cell Physiol Biochem. 2018;46(4):1508–24. 10.1159/000489194. [DOI] [PubMed] [Google Scholar]
- Fu WY, Liu Y, Yin H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019;2019:15. 10.1155/2019/9757201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata C, Jamnick NA, Bishop DJ. Training-Induced Changes in Mitochondrial Content and Respiratory Function in Human Skeletal Muscle. Sports Med. 2018;48(8):1809–28. 10.1007/s40279-018-0936-y. [DOI] [PubMed] [Google Scholar]
- Guo T, Chen M, Liu J, et al. Neuropilin-1 promotes mitochondrial structural repair and functional recovery in rats with cerebral ischemia. J Transl Med. 2023;21(1):297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Yongchun L, Lihua C, et al. Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/β-catenin pathway and blocking mitochondrial fission. Cell Stress Chaperones. 2018;23(5):1079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Ning NY, Zhou QX, Khoshnam SE, Farzaneh M. Mitochondria as a therapeutic target for ischemic stroke. Free Radical Biol Med. 2020;146:45–58. 10.1016/j.freeradbiomed.2019.11.005. [DOI] [PubMed] [Google Scholar]
- He MT, Kittur FS, Hung CY, et al. A Novel Plant-Produced Asialo-rhuEPO Protects Brain from Ischemic Damage Without Erythropoietic Action. Transl Stroke Res. 2022;13(2):338–54. 10.1007/s12975-021-00943-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YH, Zhan Q, Zhang HB, et al. Increased Susceptibility to Ischemic Brain Injury in Neuroplastin 65-Deficient Mice Likely via Glutamate Excitotoxicity. Front Cell Neurosci. 2017a;11:9. 10.3389/fncel.2017.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZY, Li N, Chen BY, Gong ZK, Wang QH, Fan LG. Rehabilitation Nursing for Cerebral Stroke Patients within a Suitable Recovery Empty Period. Iran J Public Health. 2017b;46(2):180–5. [PMC free article] [PubMed] [Google Scholar]
- Hwang DJ, Koo JH, Kwon KC, et al. Neuroprotective effect of treadmill exercise possibly via regulation of lysosomal degradation molecules in mice with pharmacologically induced Parkinson’s disease. J Physiol Sci. 2018;68(5):707–16. 10.1007/s12576-017-0586-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Piña-Crespo J, Li Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol Brain. 2019;12(1):104. 10.1186/s13041-019-0525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larcher T, Lafoux A, Tesson L, et al. Characterization of Dystrophin Deficient Rats: A New Model for Duchenne Muscular Dystrophy. PLoS One. 2014;9(10):13. 10.1371/journal.pone.0110371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Leung JCK, Yiu WH, et al. Tubular β-catenin alleviates mitochondrial dysfunction and cell death in acute kidney injury. Cell Death Dis. 2022;13(12):1061. 10.1038/s41419-022-05395-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SX, Lin YJ, Lin BB, et al. Resting-state Functional Magnetic Resonance Imaging Analysis of Brain Functional Activity in Rats with Ischemic Stroke Treated by Electro-acupuncture. J Stroke Cerebrovasc Dis. 2017;26(9):1953–9. 10.1016/j.jstrokecerebrovasdis.2017.06.018. [DOI] [PubMed] [Google Scholar]
- Marchetti B. Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease. Int J Mol Sci. 2018;19(12):3743. 10.3390/ijms19123743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–21. 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouwei Z, Ronghua C, Hongbin C, et al. Netrin-1 Promotes Synaptic Formation and Axonal Regeneration via JNK1/c-Jun Pathway after the Middle Cerebral Artery Occlusion. Front Cell Neurosci. 2018;12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz A, Corrêa CL, Lopez-Lopez A, Costa-Besada MA, Diaz-Ruiz C, Labandeira-Garcia JL. Physical Exercise Improves Aging-Related Changes in Angiotensin, IGF-1, SIRT1, SIRT3, and VEGF in the Substantia Nigra. J Gerontol Ser A-Biol Sci Med Sci. 2018;73(12):1594–601. 10.1093/gerona/gly072. [DOI] [PubMed] [Google Scholar]
- Nogueira-Ferreira R, Ferreira R, Padrao AI, et al. One year of exercise training promotes distinct adaptations in right and left ventricle of female Sprague-Dawley rats. J Physiol Biochem. 2019;75(4):561–72. 10.1007/s13105-019-00705-4. [DOI] [PubMed] [Google Scholar]
- Pan GY, Zhang HM, Zhu AQ, et al. Treadmill exercise attenuates cerebral ischaemic injury in rats by protecting mitochondrial function via enhancement of caveolin-1. Life Sci. 2021;264:10. 10.1016/j.lfs.2020.118634. [DOI] [PubMed] [Google Scholar]
- Park J, Kim J, Mikami T. Exercise-Induced Lactate Release Mediates Mitochondrial Biogenesis in the Hippocampus of Mice <i>via</i> Monocarboxylate Transporters. Front Physiol. 2021;12:14. 10.3389/fphys.2021.736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrino C, Gargiulo G, Pironti G, et al. Cardiovascular effects of treadmill exercise in physiological and pathological preclinical settings. Am J Physiol-Heart Circ Physiol. 2011;300(6):H1983–9. 10.1152/ajpheart.00784.2010. [DOI] [PubMed] [Google Scholar]
- Rezaee Z, Marandi SM, Alaei H, Esfarjani F, Feyzollahzadeh S. Effects of Preventive Treadmill Exercise on the Recovery of Metabolic and Mitochondrial Factors in the 6-Hydroxydopamine Rat Model of Parkinson’s Disease. Neurotox Res. 2019;35(4):908–17. 10.1007/s12640-019-0004-x. [DOI] [PubMed] [Google Scholar]
- Salmina AB, Kharitonova EV, Gorina YV, et al. Blood-Brain Barrier and Neurovascular Unit In Vitro Models for Studying Mitochondria-Driven Molecular Mechanisms of Neurodegeneration. Int J Mol Sci. 2021;22(9):22. 10.3390/ijms22094661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahid M, Cobo ER, Chen LB, et al. <i>Prototheca zopfii</i> genotype II induces mitochondrial apoptosis in models of bovine mastitis. Sci Rep. 2020;10(1):10. 10.1038/s41598-020-57645-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Zheng YR, Wu JY, et al. PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window. Autophagy. 2017;13(3):473–85. 10.1080/15548627.2016.1274596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MY, Zhou Y, Fan X. Mitochondrial Quality and Quantity Control: Mitophagy Is a Potential Therapeutic Target for Ischemic Stroke. Mol Neurobiol. 2022;59(5):3110–23. 10.1007/s12035-022-02795-6. [DOI] [PubMed] [Google Scholar]
- Sp N, Kang DY, Jo ES, et al. Tannic Acid Promotes TRAIL-Induced Extrinsic Apoptosis by Regulating Mitochondrial ROS in Human Embryonic Carcinoma Cells. Cells. 2020;9(2):17. 10.3390/cells9020282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinear CM, Lang CE, Zeiler S, Byblow WD. Advances and challenges in stroke rehabilitation. Lancet Neurol. 2020;19(4):348–60. 10.1016/s1474-4422(19)30415-6. [DOI] [PubMed] [Google Scholar]
- Tang CY, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol. 2021;17(5):299–318. 10.1038/s41581-020-00369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashi T, Otsuka S, Takada S, et al. Neuroprotective effects of different frequency preconditioning exercise on neuronal apoptosis after focal brain ischemia in rats. Neurol Res. 2019;41(6):510–8. 10.1080/01616412.2019.1580458. [DOI] [PubMed] [Google Scholar]
- van der Kooij MA, Ohl F, Arndt SS, Kavelaars A, van Bel F, Heijnen CJ. Mild neonatal hypoxia-ischemia induces long-term motor- and cognitive impairments in mice. Brain Behav Immun. 2010;24(5):850–6. 10.1016/j.bbi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Wang Z, Sun R, Wang G, et al. SIRT3-mediated deacetylation of PRDX3 alleviates mitochondrial oxidative damage and apoptosis induced by intestinal ischemia/reperfusion injury. Redox Biol. 2020;28:101343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guan X, Gao CL, et al. Medioresinol as a novel PGC-1α activator prevents pyroptosis of endothelial cells in ischemic stroke through PPARα-GOT1 axis. Pharmacol Res. 2021;169: 105640. 10.1016/j.phrs.2021.105640. [DOI] [PubMed] [Google Scholar]
- Wu MM, Gu XP, Ma ZL. Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury. Mol Neurobiol. 2021;58(10):5253–71. 10.1007/s12035-021-02494-8. [DOI] [PubMed] [Google Scholar]
- Xin T, Lu CZ. SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging-US. 2020;12(16):16224–37. 10.18632/aging.103644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Geng KY, Zhang YS, et al. Sirt3 deficiency impairs neurovascular recovery in ischemic stroke. CNS Neurosci Ther. 2018a;24(9):775–83. 10.1111/cns.12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhang X, Ge HF, et al. Epothilone B Benefits Nigrostriatal Pathway Recovery by Promoting Microtubule Stabilization After Intracerebral Hemorrhage. J Am Heart Assoc. 2018b;7(2):21. 10.1161/jaha.117.007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XY, Feng P, Zhang XJ, et al. The diabetes drug semaglutide reduces infarct size, inflammation, and apoptosis, and normalizes neurogenesis in a rat model of stroke. Neuropharmacology. 2019;158:14. 10.1016/j.neuropharm.2019.107748. [DOI] [PubMed] [Google Scholar]
- Yang MX, He Y, Deng SX, et al. Mitochondrial Quality Control: A Pathophysiological Mechanism and Therapeutic Target for Stroke. Front Mol Neurosci. 2022;14:19. 10.3389/fnmol.2021.786099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingqiong X, Yan W, Guangyun W, et al. YiQiFuMai Powder Injection Protects against Ischemic Stroke via Inhibiting Neuronal Apoptosis and PKCδ/Drp1-Mediated Excessive Mitochondrial Fission. Oxid Med Cell Longev. 2017;2017:1832093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Tian Y, Jiang H, et al. Mechanism of PGC-1α-mediated mitochondrial biogenesis in cerebral ischemia-reperfusion injury. Front Mol Neurosci. 2023;16:7. 10.3389/fnmol.2023.1224964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Sliter DA, Bleck CKE, Ding SZ. Fis1 deficiencies differentially affect mitochondrial quality in skeletal muscle. Mitochondrion. 2019;49:217–26. 10.1016/j.mito.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Schwade M, Smith Y, Wood R, Young LF. Exercise-based interventions for post-stroke social participation: A systematic review and network meta-analysis. Int J Nurs Stud. 2020a;111:10. 10.1016/j.ijnurstu.2020.103738. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cao MY, Wu YM, et al. Improvement in mitochondrial function underlies the effects of ANNAO tablets on attenuating cerebral ischemia-reperfusion injuries. J Ethnopharmacol. 2020b;246:10. 10.1016/j.jep.2019.112212. [DOI] [PubMed] [Google Scholar]
- Zhang TWY, Xia Q, Tu Z, Sun J, Jing Q, Chen P, Zhao X. Propofol Mediated Protection of the Brain From Ischemia/Reperfusion Injury Through the Regulation of Microglial Connexin 43. Front Cell Dev Biol. 2021;9:1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DJ, Sun Y, Tan YZ, et al. Short-Duration Swimming Exercise after Myocardial Infarction Attenuates Cardiac Dysfunction and Regulates Mitochondrial Quality Control in Aged Mice. Oxidative Med Cell Longev. 2018;2018:16. 10.1155/2018/4079041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Bai J, Li N, et al. Protective effects of sirtuin 3 on titanium particle-induced osteogenic inhibition by regulating the NLRP3 inflammasome via the GSK-3β/β-catenin signalling pathway. Bioact Mater. 2021;6(10):3343–57. 10.1016/j.bioactmat.2021.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Bai Y, Sun X, et al. Resveratrol Reestablishes Mitochondrial Quality Control in Myocardial Ischemia/Reperfusion Injury through Sirt1/Sirt3-Mfn2-Parkin-PGC-1α Pathway. Molecules (Basel, Switzerland). 2022;27(17):5545. 10.3390/molecules27175545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Li LY, Chen BW, et al. Chlorogenic acid exerts neuroprotective effect against hypoxia-ischemia brain injury in neonatal rats by activating Sirt1 to regulate the Nrf2-NF-κB signaling pathway. Cell Commun Signal. 2022b;20(1):20. 10.1186/s12964-022-00860-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong HH, Song R, Pang QN, et al. Propofol inhibits parthanatos via ROS-ER-calcium-mitochondria signal pathway in vivo and vitro. Cell Death Dis. 2018;9:14. 10.1038/s41419-018-0996-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Peng L, Li Y, Zhao Y. Silent information regulator 1 ameliorates oxidative stress injury via PGC-1α/PPARγ-Nrf2 pathway after ischemic stroke in rat. Brain Res Bull. 2022;178:37–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.