

Abstract
The genome sequencing revolution has revealed that all species possess a large number of unique genes critical for trait variation, adaptation, and evolutionary innovation. One widely used approach to identify such genes consists of detecting protein‐coding sequences with no homology in other genomes, termed orphan genes. These genes have been extensively studied, under the assumption that they represent valid proxies for species‐specific genes. Here, we critically evaluate taxonomic, phylogenetic, and sequence evolution evidence showing that orphan genes belong to a range of evolutionary ages and thus cannot be assigned to a single lineage. Furthermore, we show that the processes generating orphan genes are substantially more diverse than generally thought and include horizontal gene transfer, transposable element domestication, and overprinting. Thus, orphan genes represent a heterogeneous collection of genes rather than a single biological entity, making them unsuitable as a subject for meaningful investigation of gene evolution and phenotypic innovation.
Keywords: gene evolution, lineage‐specific genes, ORFans, phylostratigraphy, species‐specific genes, taxonomically restricted genes
Orphan genes represent genes with no homologs in other species and are widely assumed to represent genuine species‐specific loci. We show that molecular evolution and sequence homology analyses reject this assumption and that orphan genes constitute a patchwork of genes of different evolutionary ages that originate throughout various mechanisms.
INTRODUCTION
One of the most significant discoveries of the genomic era is that the gene content varies substantially between individuals and among closely related species. For example, humans and chimpanzees on average differ by 270 genes that are present in either species.[ 1 ] The notion that new genes could arise during evolution emerged early in modern biology. A duplication of the BAR gene in the fruit fly Drosophila was recognized and characterized before the discovery that DNA is the molecule responsible for genetic inheritance.[ 2 , 3 ] However, only the advent of genomic data allowed for estimating how many genes are unique to each species.
The analysis of the first sequenced eukaryotic genome from the budding yeast Saccharomyces cerevisiae led to a surprising finding: about 30% of all genes in this species shared no homology with known DNA and protein sequences from other organisms. The term orphan genes was then coined to describe these apparent lineage‐specific loci.[ 4 ] Soon after, the alternative but conceptually equivalent term ORFans was introduced for genes without shared homology in bacteria and archaea.[ 5 ] When genes from a broader taxonomic unit are considered, the more general terminology of taxonomically restricted genes, or TRGs, is currently preferred.[ 6 , 7 , 8 ] Hereafter, we will use the term orphan genes in a broad sense to also encompass TRGs.
After their discovery, it was immediately recognized that the number of orphan genes should decrease substantially with new genomes becoming available, because more homologous genes would become detectable in newly sequenced species. However, dozens to hundreds of orphan genes are typically found in virtually any newly sequenced genome. Furthermore, evolutionary and experimental investigations across the tree of life have progressively reinforced the view that new genes profoundly affect phenotypic variation and adaptation.[ 9 ] The combination of these findings has propelled a vigorous interest in the study of orphan genes that has led to more than 340 peer‐reviewed articles on this subject (PubMed searches for “orphan genes,” “ORFans,” and “taxonomically restricted genes” after removing redundant papers and excluding papers containing “orphan receptor”).
The appeal of orphan genes. There are several reasons why orphan gene analyses are common, particularly in new genomes. First, their identification relies on bioinformatic approaches that are easy to implement and deliver “yes or no” results, given that homology is either found or it is not. Second, orphan genes are often used to inform on the evolutionary trends of new genes. For instance, younger orphans tend to be shorter, less complex, and more rapidly evolving than older orphan genes, a.k.a. TRGs, and ancestral non‐orphan genes.[ 10 , 11 , 12 , 13 , 14 , 15 ]
Furthermore, orphan genes are often considered equivalent to species‐ or lineage‐specific genes, and as such, they are expected to represent genomic innovations involved in recent phenotypic changes. Although young (species‐specific) orphan genes are sometimes considered annotation errors due to the lack of identifiable folds and recognizable domains in their proteins,[ 12 , 16 ] striking examples of biologically functional cases have been documented. Recent reports have shown that the hominoid‐specific orphan gene ENSG00000205704 is involved in brain development and neocortex size. Overexpression of the ENSG00000205704 gene in an organoid model resulted in a larger organoid size and longer maturation time, whereas the opposite phenotype was observed in knock‐out experiments. In vivo functionality of ENSG00000205704 has also been determined in transgenic mice.[ 17 ] In Drosophila melanogaster, a large proportion of young orphan genes showed an embryo‐lethal phenotype when knocked‐down,[ 18 ] although subsequent work has shown that most of these genes are non‐essential.[ 19 ] Among plants, the A. thaliana‐specific orphan QQS regulates carbon and nitrogen allocation.[ 20 ]
The convergent origin of genes encoding antifreeze glycoproteins (AFGPs) in Arctic codfishes and Antarctic notothenioid fish is a further example worth mentioning. AFGPs proteins contain several repeats of the tripeptide unit (Thr‐Ala‐Ala) implicated in the binding of ice crystals that prevent freezing damage. In Antarctic notothenioids, afgp genes evolved from the chimeric assembly of a partially duplicated trypsinogen‐like protease (TLP) gene fused with nearby expanded Thr‐Ala‐Ala repeats into a single locus[ 21 ]. In Arctic codfishes, the afgp gene originated entirely from non‐coding DNA.[ 22 , 23 ] These remarkable findings exemplify the impact of new genes on adaptation and the type of “evolutionary tinkering”[ 24 ] that can independently lead to similar solutions across different lineages. Several other studies have shown that young orphan genes as a group tend to be associated with adaptation, responses to environmental stressors and speciation[ 11 , 13 , 23 , 25 , 26 , 27 ].
In this perspective, we argue that despite their continuing appeal, orphan genes represent a problematic category that has led to several misconceptions concerning the evolution of both genes and traits. Some of the flaws discussed here have been previously recognized.[ 8 , 28 , 29 , 30 , 31 , 32 ] Notably, Schlotterer proposed to shift the focus from orphan genes to de novo genes.[ 29 ] While de novo genes represent a distinct evolutionary category of genes,[ 33 ] we maintain, as Schlotterer also suggested, that the variety of mechanisms responsible for the formation of new genes should be comprehensively addressed. As discussed below, a few studies have followed this line of thinking, which we suggest should be applied consistently. Here, the full range of issues associated with the concept of orphan genes and its (mis‐)use in evolutionary biology is fully addressed for the first time. Altogether, a large body of evidence suggests that orphan genes represent a heterogeneous group of genes, both by age and by evolutionary origin, thus calling into question their appropriateness as proper biological units.
PITFALLS OF ORPHAN GENES: (I) INHERENT BIASES
Orphan gene inferences are influenced by sampling, taxonomic diversity, and genome coverage
Orphan genes are typically detected throughout the approach known as phylostratigraphy, which determines the age of a given gene based on its distribution along a phylogeny of species.[ 34 ] In phylostratigraphy, the evolutionary distribution of genes from a focal species is assessed in other taxa using sequence homology searches implemented with tools like BLAST[ 35 ] or DIAMOND.[ 36 ] The gene age can thus be approximated as the age range of the ancestral branch, or phylostratum, of the phylogeny that includes all the species containing that gene. For example, a primate orphan gene must have evolved after the separation of primates from their sister lineage of colugos (order Dermoptera) but before the divergence of primates into multiple lineages (Figure 1A). Primate‐specific genes will occur in both lineages of existing primates, Haplorrhines and Strepsirrhini[ 37 ] (Figure 1A). Further similarity searches are necessary across databases of other non‐primate species to avoid false positives due to gene loss in the sister group colugos. However, genome data availability, gene annotation quality, species diversity per lineage, rate of gene losses, and other factors can affect the accuracy of phylostratigraphy.[ 38 ]
FIGURE 1.
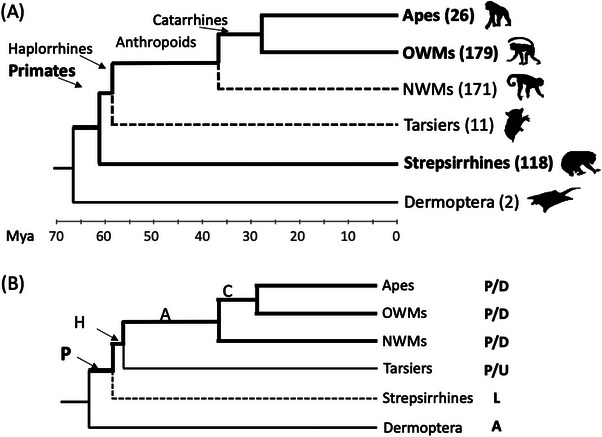
(A) Phylogenetic distribution of a hypothetical gene in primates and their sister group Dermoptera. Although the gene has been lost in NWMs and Tarsiers (dashed lines), it is maintained in some species within the two major lineages Haplorrhines and Strepsirrhines, and should thus be considered primate‐specific. Tree modified from Vanderpool et al.[ 37 ]. OWMs: Old World Monkeys (Cercopithecidae). NWMs: New World Monkeys (Platyrrhines). Number of species in each group is shown in parentheses. Mya: million years ago. (B) Gene loss or lack of genomic data affect the age estimate of a primate‐specific gene. The gene is assigned to the Anthropoid lineage but originated in the Primate lineage. P/D: present and detected. P/U: present and undetected. L: lost. A: absent. Gray lines: lineages where the gene is not present.
In the case of primates, the limited genomic resources and lower gene annotation quality in tarsiers and Strepsirrhines diminish the power to detect genes that are homologous to Anthropoid genes (Figure 1B). As a result, putative Anthropoids‐specific genes may in fact represent Haplorrhines‐ or primate‐specific genes that are yet unreported in tarsiers and Strepsirrhines (Figure 1B). Similar issues emerge when outgroup taxa contain a few species, as is the case with colugos, with only two extant species represented. The sum effect of these limitations is a significant age underestimation for many older genes that will appear as primate‐specific despite having evolved long before the origin of primates.
PITFALLS OF ORPHAN GENES: (II) PHYLOSTRATIGRAPHY LIMITATIONS
Phylostratigraphy has become a widely used approach to estimate gene age because it relies on sequence homology and sequence similarity searches, two fundamental aspects of modern bioinformatics and genomics. The fundamental assumption of phylostratigraphy is that homologous genes will always be detected by sequence similarity searches. It has been repeatedly shown that this view is incorrect. Comparative genomic analyses have confirmed across a wide range of species that many genes, particularly those expressed in some tissues (reproductive organs, especially testis) or involved in specific responses (immune‐related genes), tend to evolve more rapidly than others.[ 39 , 40 , 41 , 42 , 43 ] The sequence of these genes may divergence from their sister species’ orthologous genes—that is, genes that they share a common ancestry with—beyond what can be recognized via phylostratigraphy. As a result, orphan genes will include a number of false positives represented by old, fast evolving genes.
Although BLAST and other tools used to detect sequence similarity are somewhat robust to this issue,[ 44 ] they inevitably fail to detect homology when the sequence divergence is elevated, which occurs over short evolutionary periods in such rapidly evolving genes[ 45 ]. This phenomenon has been recently codified as “homology detection failure,” or HDF[ 46 ]. Recent studies have quantified HDF in several organisms. In a series of sequence simulation analyses, Moyers and Zhang have shown that phylostratigraphy‐based estimates of gene age bear significant rates of missed homology[ 32 , 47 , 48 , 49 ] (for a rebuttal, see[ 50 ]). Error rates of up to 24% among eukaryotes were estimated for genes that evolve faster than expected.[ 32 ] Using a novel sequence homology decay model, Weisman et al.[ 46 ] recently found that the probability of HDF is high in more than half of orphan genes in both the yeast Saccharomyces and Drosophila. Estimates of HDF have been integrated into novel tools developed to assess the age of gene families[ 51 ] and are likely to become increasingly prominent in gene evolution studies.
A further approach to remedy HDF consists of integrating phylostratigraphy with microsynteny analyses, which assess the conservation of the genomic location, or collinearity, of DNA sequences between species. A gene sharing microsynteny between two species is thought to derive from a gene present in the common ancestors of those species. However, microsynteny decreases with evolutionary distance and rates of sequence substitution, thus becoming less useful for age estimates in older genes[ 52 ]. A recent study analyzing microsynteny in budding yeast, fruit fly, and human genes showed that, on average, one‐third of orphan genes are older than inferred by phylostratigraphy alone, probably representing rapidly evolving older genes[ 53 ]. Given that microsynteny analyses or corrections for HDF have rarely been used, it is likely that most estimates of young orphan genes are inflated.
A further indication of phylostratigraphy shortcomings emerges from the significant variation among estimates of these genes between closely related species. For instance, Light et al.[ 28 ] reported that 3.03% of the protein‐coding genes in Mus musculus are orphans versus 0.75% in Rattus norvegicus. Wissler et al.[ 54 ] found high variance of species‐specific orphan genes estimates between 30 arthropod lineages. Among plants, Guo 2013[ 55 ] identified 5.3% and 12.3% orphan genes in the sister species Arabidopsis thaliana and A. lyrata, respectively. While some of this variation can be due to biological processes, changes in gene annotation accuracy are more likely responsible for a larger proportion of such variation. This effect can be accounted for throughout synteny analyses, but it is not addressed by phylostratigraphy alone and can thus lead to the observed discrepancies between species. A review collating estimates from multiple sources also showed varying estimates of orphan gene numbers in A. thaliana from three independent investigations (958, 1324, and 1430), probably the result of different methodological approaches between studies.[ 13 ]
PITFALLS OF ORPHAN GENES: (III) ORIGIN VIA MULTIPLE EVOLUTIONARY PROCESSES
One fundamental yet underappreciated aspect concerning the biology of orphan genes is the complexity of evolutionary mechanisms that contribute to their formation.[ 29 ] Historically, the origin of orphan genes has been attributed to two processes: (1) gene duplication followed by rapid sequence divergence (the duplication‐divergence model) and (2) de novo gene birth.[ 50 , 56 , 57 ] The combination of large genomic datasets, molecular evolution analyses and high‐throughput sequencing technologies have revealed that new genes can originate throughout several other evolutionary processes aside from duplication‐divergence or de novo birth (Figure 2C–H).
FIGURE 2.
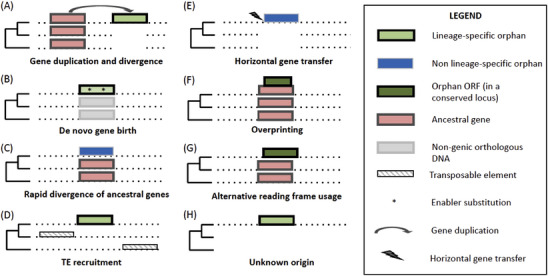
Types of new genes and orphan genes and their genomic signatures. A phylogeny of the focal species (top clade) and two outgroups is shown next to each case. Not all orphans represent lineage‐specific genes. Dotted line: genomic DNA. TE: transposable element.
Despite many of these processes being known for decades, they have been investigated only in a handful of studies. For example, primate orphan genes have been found to also evolve via transposable element (TE) recruitment.[ 10 ] One important finding of this work is that many orphan genes represent “evolutionary chimeras” (similarly to the notothenioid afgp genes discussed above), wherein pieces that originated from different mechanisms were assembled into novel genetic units. In a thorough evolutionary examination of A. thaliana, species‐specific genes were arranged into multiple categories based on their evolutionary origins.[ 58 ] The comparative analysis of seven ant genomes showed a high number of orphan genes, but given the age of these genes, it could not always discriminate between de novo genes and gene losses. In the model nematode Pristionchus pacificus, the evolutionary history of 29 orphan genes has been characterized, although these genes represent a fraction of all the identified orphans.[ 59 ] A recent survey of 4,644 species in the human gut microbiome has determined the mechanisms of origin of more than 631,000 orphan genes.[ 60 ] In the following paragraphs, we describe the nature of all possible mechanisms generating orphan genes and indicate existing estimates of their frequency based on these five studies, which we also summarize in Table 1.
TABLE 1.
Available estimates of different evolutionary mechanisms’ percentage contribution to orphan genes.
| Mechanism | Primates[ 10 ] | Ants[ 54 ] | Arabidopsis thaliana[ 58 ] a | Pristionchus pacificus[ 59 ] a | Human gut microbiome[ 60 ] |
|---|---|---|---|---|---|
| Duplicated‐divergent | 24.4 | 9.9 | 22.2 | 44.4 | NA |
| De novo | 5.6 | 43.5 | 25.1 | 22.2 | 0.17 |
| TE‐derived | 52.6 | 12.4 | 9.7 | NA | NA |
| HGT | NA | 0.1 | NA | NA | 3.7 b |
| Overprinting | NA | 11.1 | 8.84 c | 25.9 | 1.34 c |
| ARFU | NA | 2.2 | 14.8 | ||
| Unknown | 17.4 | 20.8 | 36.7 | 22.2 | 94.8 |
Abbreviations: ARFU, alternative reading frame usage; HGT, horizontal gene transfer; TE, transposable element.
The sum here is > 100% because some genes originated via multiple evolutionary processes.
These genes represent only HGT from bacteriophages.
Overprinting and ARFU were not separated in the A. thaliana and the human gut microbiome studies.
Gene duplication‐divergence. Following duplication, parent and daughter gene sequences increasingly diverge by accumulating DNA changes (Figure 2A). When extensive, this process can lead to the loss of identifiable sequence similarity between the daughter gene and its homologs in other species.[ 56 , 61 ] The duplication‐divergence model posits that new copies can rapidly gain substitutions in their early evolutionary stages when relaxed (neutral) and adaptive sequence evolution is more likely to occur. Interestingly, a close association between the rate of gene duplication and the rate of sequence evolution has been shown in humans and Drosophila melanogaster,[ 62 , 63 ] suggesting that gene families with rapid copy number turnover may be especially prone to generate orphan genes via duplication‐divergence. As described above, such families typically include genes involved in the immune defense and reproduction; this result from evolutionary conflicts between hosts and pathogens and between sexes that fuel rapid gene turnover and high sequence divergence. The contribution of this mechanism to orphan gene varies from ∼ 10% in ants to 44% in the nematode P. pacificus, with intermediate estimates in A. thaliana and primates (Table 1). In the human gut microbiome, the contribution of gene duplication‐divergence to orphan genes has not been estimated,[ 60 ] although it is possible that a large proportion of orphan genes derive from this process (pers. comm.).
De novo gene birth. De novo genes evolve via enabler substitutions that lead to the transcription and translation of a previously non‐genic DNA sequence (Figure 2B). As well put by Schlotterer,[ 29 ] de novo genes represent protein‐coding loci that truly evolve from scratch! First discovered less than two decades ago in Drosophila,[ 64 , 65 ] de novo gene birth is increasingly recognized as a common process in eukaryotic, bacterial, and viral genomes,[ 33 , 60 , 66 , 67 , 68 , 69 ] with some studies suggesting that it may occur as often as gene duplication.[ 12 , 70 , 71 ] However, current estimates of de novo gene birth vary by 2–3 orders of magnitude within species. For instance, between 3 and 82 protein‐coding de novo genes have been described in human.[ 72 , 73 , 74 , 75 , 76 ] Similarly, de novo genes ranged between ∼ 6%–44% in orphan gene analyses in eukaryotes (Table 1). Among the numerous orphan genes reported in the human gut microbiome, only less than 0.2% originated de novo, possibly due to the scarcity of intergenic DNA in bacteria and archaea.[ 60 ] The lack of standardized methods to identify de novo genes and the challenges associated with the discovery of enabler substitutions using synteny data are likely major factors responsible for the discrepancies in de novo gene birth estimates among eukaryotes. Computational pipelines have been recently developed with the goal of addressing these issues.[ 77 ]
Rapid gene evolution. As precisely estimated by recent works on homology‐detection failure, a high proportion of genes evolve at a pace that erodes their homology with existing orthologs. Importantly, these are mostly formed by old non‐duplicated genes (Figure 2C), rather than highly diverging gene duplicates. In other words, many if not most orphan genes may represent false positives! In a recent account, one of us[ 31 ] has shown that at least 13% of putative rodent‐specific de novo genes were ancestral genes that showed conserved synteny with their primate orthologs.[ 78 ] For example, the lymphocyte antigen‐coding gene CD52,[ 79 ] reported as a mouse‐specific de novo gene,[ 78 ] is in fact shared across mammals.[ 31 ] Thus, the diminished conservation of specific gene sequences can affect orphan and de novo gene estimates. Beyond the handful of species examined in recent HDF and synteny studies, the impact of rapid evolution of ancestral genes to estimates of orphan genes is essentially unknown.
Transposable element domestication. Transposable elements (TEs) contain one to a few protein‐coding sequences that can be ‘domesticated’ and become host genes with cellular function (Figure 2D). Host genes are recognized as TE‐derived when they encode for a protein with high sequence homology with proteins encoded primarily or exclusively by other transposable elements. Genes that evolved from TEs can be identified as orphans within a lineage if the domestication event occurred only in that lineage and the proteins they encode share no homology with other host proteins. Because there are many TE types and their genes evolve fast, the homology of TE‐derived genes with other host genes tend to be low or absent. Overall, TE gene domestication represents an important source of new genes and protein domains across prokaryotes[ 80 ] and eukaryotes.[ 81 , 82 , 83 , 84 , 85 ] A second major route for TEs to contribute to orphan genes is via the recruitment of TE sequence into a novel chimeric gene locus. For example, TE sequences occur in more than half of orphan genes in primates[ 10 ] and rice[ 86 ], and ∼ 30% in silkworm.[ 87 ] Conversely, only ∼ 10% of orphan genes contain TE‐derived DNA in ants and A. thaliana [ 54 , 58 ] (Table 1). In most cases, only part of a transposable element or a noncoding TE region is included in the new gene, therefore gaining the ability to encode for a novel protein sequence with no similarity with known TE or host proteins. Although identifying TE‐derived DNA in coding regions is relatively straightforward, the total contribution of transposable element genes and other sequences to orphan genes is rarely verified and probably vastly underestimated. In prokaryotes, TEs tend to be uncommon and are unlikely to generate a substantial number of orphan genes.
Horizontal gene transfer. Horizontal gene transfer, or HGT, is a potential source of novel genes, particularly in bacteria and archaea, although a growing number of HGT cases have been documented across eukaryotes (Figure 2E). HGT is more likely to produce orphans when the event is ancient, donor and acceptor species are distantly related, and the transferred gene evolves rapidly.[ 29 ] It is also possible that many horizontally transferred genes are removed from genome assemblies and thus go unidentified, because software that recognizes possible contaminant DNA could label and remove sequenced reads from these genes. Despite these limitations, extensive homology searches and genome analyses could reveal more HGT‐related orphans, as reported in ants.[ 54 ] Notably, the HGT contribution to orphan genes correlates with the availability of genome sequences between donor and acceptor species. In prokaryotes, HGT mostly involve closely related strains and will therefore produce more orphans in less well‐sequenced taxa.[ 88 ] However, a substantial number or prokaryotic orphan genes might derive from overlooked sources, particularly viruses. For instance, at least 3.7% of orphan genes detected in the human gut microbiome showed a likely viral origin.[ 60 ]
Overprinting and alternative reading frame usage. Most eukaryotic genes are characterized by a coding region broken down into exons separated by introns. Different combinations of exons can be assembled into mRNAs via alternative splicing, thus coding for multiple similar isoforms of the same protein. This 20th century view of the eukaryotic gene organization has received a considerable “upgrade” owing to findings from high‐throughput sequencing experiments. First, transcriptomic data have shown that many genes generate antisense transcripts encoded by the DNA strand opposite to the one used as a template to synthesize mRNA.[ 89 , 90 ] Second, the sequencing of RNAs bound to ribosomes, or Ribo‐seq, has shown that multiple frames on the same mRNA molecule can be translated into entirely different proteins. This additional layer of gene expression is known as overprinting (Figure 2F) or alternative reading frame usage (ARFU, Figure 2G), depending on whether transcription occur on a fully or partially overlapping coding region of the main coding sequence. Sequences originating from these processes represent orphan proteins, rather than orphan genes, because they are encoded from the same DNA regions of existing genes.[ 91 ] Originally described in viral genomes,[ 66 , 92 ] overprinting and ARFU are now associated with the emergence of hundreds to thousands of novel proteins in bacteria[ 93 ] and eukaryotes.[ 94 , 95 ] Overprinting and alternative reading frame usage have also been identified as sources of orphan proteins in ants,[ 54 ] nematodes[ 59 ] prokaryotes[ 60 , 93 ] and A. thaliana‐specific genes,[ 58 ] but despite the rapidly increasing Ribo‐seq datasets they are mostly uncharacterized in orphan gene analyses (Table 1).
Orphan genes of unknown origin. The specific evolutionary mechanisms involved in the formation of some orphan genes cannot always be ascertained due to the lack of both homology to any known sequence (genes, TEs) and microsynteny (Figure 2H). The well‐studied and functionally important QQS gene is an example of an orphan gene that appears to have literally evolved out of nowhere in A. thaliana.[ 20 ] Quite surprisingly, genes from DNA with no explainable origins form 37%, 17%, 21%, and 95% of orphans in Brassicaceae,[ 58 ] primates,[ 10 ] ants,[ 54 ] and the human gut microbiome[ 60 ] respectively (Table 1). Functionally, these orphans could be regarded as de novo genes, because they encode proteins with no apparent homology to other proteins. Indeed, in the literature, QQS is often referred to as a de novo gene. It is possible, and should be investigated, if genes of unknown origin represent a functionally distinct category from genes that emerged de novo.
Functional differences between orphan genes with distinct evolutionary origins. Evidence suggests that new genes originated through distinct evolutionary processes are likely going to affect different organismal processes. Orphan gene that originate from rapidly evolving ancestral genes are likely to maintain the same function over evolutionary time. Conversely, de novo genes and chimeric genes with both de novo and TE‐derived sequences encode for proteins with entirely novel structure and may play a role in various cellular pathways and adaptive traits.[ 17 , 22 , 23 , 96 ] Genes that contain TE coding sequences will likely be involved in gene expression regulation, DNA excision, DNA integration, reverse transcription and cell fusion.[ 84 , 85 , 97 ] Although several cases of functional genes evolved through overprinting and alternative reading frames usage have been documented in bacteria, eukaryotes, and especially viruses,[ 91 , 98 , 99 ] the biological role of most such genes remains obscure. Horizontally transferred genes in prokaryotes are common but appear biased in favor of specific groups of genes.[ 100 , 101 , 102 ] In eukaryotes, HGT is less common and often involves bacteria or fungi donors that provide genes encoding enzymes with metabolic or bactericidal functions.[ 103 ] Thus, functional analyses of orphan genes as a single unit cannot accurately reflect the complex functional diversity of new genes.
DIFFERENCES BETWEEN ORPHAN AND NON‐ORPHAN GENES CAN BE EXPLAINED BY ORPHAN GENE ORIGINATION MECHANISMS
The distinction between orphan and non‐orphan genes has often been examined through features such as expression level, evolutionary rate, and biological function. While these comparisons offer valuable insights, they can be misleading without considering the specific mechanisms responsible for the origin of orphan genes. This section addresses how the mechanisms of orphan gene origination impact some of these features and explains the observed differences between orphan and non‐orphan genes.
Expression levels. Overall, orphan genes tend to be expressed at lower levels than other genes.[ 13 , 56 , 87 ] In prokaryotes, HGT is rampant and is known to largely involve genes with diminished expression compared to endogenous genes.[ 104 ] Thus, HGT might explain the lower expression levels of orphan genes in bacteria and archaea. In eukaryotes, where HGT is rare, low expression levels might be associated with de novo genes, which are known for their limited transcription rates.[ 11 , 29 , 105 , 106 ] Moreover, rapidly‐evolving genes are often associated with lower expression levels,[ 107 ] possibly due to their involvement in species‐specific adaptations that do not require high constitutive expression. Conversely, orphan genes evolved via overprinting and alternative reading frame usage might be highly expressed due to their proximity to established regulatory sequences. Therefore, orphan genes expression levels reflect the complex interplay between mechanisms of origin and their varying frequency across major taxa.
Evolutionary rates. It has been repeatedly shown that all orphan genes combined evolve at a faster pace compared to non‐orphan genes.[ 10 , 14 , 61 ] This finding aligns with the expectation for both the duplication‐divergence and the rapid evolution origination mechanisms. Similarly, horizontally transferred genes exhibit higher substitution rates than core (conserved) genes in E. coli,[ 108 ] and de novo genes overall have been found to evolve more rapidly than non‐orphan genes.[ 15 , 109 , 110 ] However, strong sequence conservation has been reported in genes encoding TE‐derived domains,[ 84 , 111 ] and substantial sequence changes in orphan genes evolved via overprinting and ARFU should be selected against, because of the overlap between these genes and conserved non‐orphan genes.
Functional impact. A central question in the study of orphan genes pertains to their possible biological function. Sequence homology remains a key predictor of the function of a gene that has not been experimentally characterized,[ 112 ] but by definition, orphan genes share no homology with other genes. However, the expectations concerning the functionality of orphan genes vary depending on their mechanism of origin. De novo genes, overprinting‐derived genes and genes evolved via alternative reading frame usage encode for proteins with unknown function. On the other hand, orphan genes that originated via duplication‐divergence, rapid sequence evolution and HGT are expected to share a similar function with that of their “parent” genes. Additionally, gene duplication events are more frequent in genes that are less essential to core biological processes.[ 113 ] Orphan genes that evolved via duplication‐divergence might thus appear less functionally important than non‐orphan genes. Finally, orphan genes encoding complete protein domains derived from transposable elements also share partial or full functional overlap with their parent TEs, as documented for several transcription factors in vertebrates.[ 84 ]
Overall, fundamental gene features vary substantially between orphan genes that originated via different mechanisms, obfuscating the biological meaning of comparisons between orphan and non‐orphan genes. Therefore, taking into account the different types of orphan genes is essential to understanding the unique properties of lineage‐specific genes and their role in species adaptation and evolutionary innovation.
CONCLUSIONS AND FUTURE DIRECTIONS
In this perspective, we propose that the practical benefits of considering orphan genes equivalent to lineage‐specific genes are greatly outweighed by the limitations of this theoretical category of genes. While previous studies have highlighted individual flaws in sequence homology searches,[ 8 , 28 , 29 , 30 , 32 , 46 , 53 ] here we summarized for the first time the numerous issues associated with orphan genes, from uncertainties due to taxonomic under‐sampling and low diversity to homology search error rates and finally to the evolutionary and functional heterogeneity of young genes that originated via multiple processes. In our view, these flaws provide cause to reject the widespread assumption that orphan genes represent a biologically meaningful entity (see also ref. [28]). A fundamental goal of our perspective is to point out the importance of ascertaining specific evolutionary processes that lead to lineage‐specific genes. This step is indispensable to our understanding of both how new genes emerge and, even more critically, how they contribute to phenotypic change and species adaptation. Comparisons of fundamental gene features between orphan and non‐orphan genes, which are broadly investigated to determine new genes' evolution and function, also show great variation depending on the origin mechanism of orphan genes. We have highlighted recently developed tools to assess and correct HDF errors[ 32 , 46 , 51 , 114 ] and to identify specific type of genes, such as de novo genes,[ 77 ] in the hope that they will be increasingly essential in future analyses of new genomes and will help build accurate catalogs of lineage‐specific genes. The accelerating pace of sequencing and assembly of high‐quality genomes[ 115 ] will generate in a short time vast resources to address some of the inherent biases associated with correctly detecting lineage‐specific genes. Similarly, improved tools for genome alignments will enable advanced assessments of synteny conservation across genomes to verify orthology and identify de novo genes.[ 116 ] While orphan genes have been valuable in sustaining the interest around the evolution and function of new genes, the large body of evidence presented here shows that time is ripe to replace this conceptual category with evolutionary and functional gene types reflecting actual biological entities.
AUTHOR CONTRIBUTIONS
Andres Barboza Pereira and Matthew Marano: literature review, writing and preparation of figures. Ramya Bathala, Rigoberto Ayala Zaragoza, Andres Neira, Alex Samano and Adekola Owoyemi: literature review, writing. Claudio Casola: Conceptualization, literature review and synthesis, writing, visualization and preparation of figures.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
The authors are grateful to Alan Pepper and Nikolaos Vakirlis for helpful comments on early versions of the manuscript. This work is supported by the Texas A&M AgriLife Research, the Texas A&M Forest Service and the USDA‐NIFA award TEX0‐1‐9599.
Pereira, A. B. , Marano, M. , Bathala, R. , Zaragoza, R. A. , Neira, A. , Samano, A. , Owoyemi, A. , & Casola, C. (2025). Orphan genes are not a distinct biological entity. BioEssays, 47, e2400146. 10.1002/bies.202400146
Andres Barboza Pereira and Matthew Marano contributed equally to this work.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Cheng, Z. , Ventura, M. , She, X. , Khaitovich, P. , Graves, T. , Osoegawa, K. , Church, D. , Dejong, P. , Wilson, R. K. , Pääbo, S. , Rocchi, M. , & Eichler, E. E. (2005). A genome‐wide comparison of recent chimpanzee and human segmental duplications. Nature, 437, 88–93. [DOI] [PubMed] [Google Scholar]
- 2. Muller, H. J. (1936). Bar duplication. Science (New York, NY), 83, 528–530. [DOI] [PubMed] [Google Scholar]
- 3. Sturtevant, A. H. (1925). The effects of unequal crossing over at the bar locus in Drosophila. Genetics, 10, 117–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dujon, B. (1996). The yeast genome project: What did we learn? Trends in Genetics: TIG, 12, 263–270. [DOI] [PubMed] [Google Scholar]
- 5. Fischer, D. , & Eisenberg, D. (1999). Finding families for genomic ORFans. Bioinformatics, 15, 759–762. [DOI] [PubMed] [Google Scholar]
- 6. Wilson, G. A. , Bertrand, N. , Patel, Y. , Hughes, J. B. , Feil, E. J. , & Field, D. (2005). Orphans as taxonomically restricted and ecologically important genes. Microbiology (N YReading), 151, 2499–2501. [DOI] [PubMed] [Google Scholar]
- 7. Khalturin, K. , Hemmrich, G. , Fraune, S. , Augustin, R. , & Bosch, T. C. G. (2009). More than just orphans: Are taxonomically‐restricted genes important in evolution? Trends in Genetics, 25, 404–413. [DOI] [PubMed] [Google Scholar]
- 8. Klasberg, S. , Bitard‐Feildel, T. , & Mallet, L. (2016). Computational identification of novel genes: Current and future perspectives. Bioinformatics and Biology Insights, 10, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen, S. , Krinsky, B. H. , & Long, M. (2013). New genes as drivers of phenotypic evolution. Nature Reviews Genetics, 14, 645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toll‐Riera, M. , Bosch, N. , Bellora, N. , Castelo, R. , Armengol, L. , Estivill, X. , & Mar Alba, M. (2009). Origin of primate orphan genes, a comparative genomics approach. Molecular Biology and Evolution, 26, 603–612. [DOI] [PubMed] [Google Scholar]
- 11. Carvunis, A. R. , Rolland, T. , Wapinski, I. , Calderwood, M. A. , Yildirim, M. A. , Simonis, N. , Charloteaux, B. , Hidalgo, C. A. , Barbette, J. , Santhanam, B. , Brar, G. A. , Weissman, J. S. , Regev, A. , Thierry‐Mieg, N. , Cusick, M. E. , & Vidal, M. (2012). Proto‐genes and de novo gene birth. Nature, 487, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neme, R. , & Tautz, D. (2013). Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics [Electronic Resource], 14, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arendsee, Z. W. , Li, L. , & Wurtele, E. S. (2014). Coming of age: Orphan genes in plants. Trends in Plant Science, 19, 698–708. [DOI] [PubMed] [Google Scholar]
- 14. Palmieri, N. , Kosiol, C. , & Schlotterer, C. (2014). The life cycle of Drosophila orphan genes. Elife, 3, e01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heames, B. , Schmitz, J. , & Bornberg‐Bauer, E. (2020). A Continuum of evolving de novo genes drives protein‐coding novelty in Drosophila. Journal of Molecular Evolution, 88, 382–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo, W. J. , Li, P. , Ling, J. , & Ye, S. P. (2007). Significant comparative characteristics between orphan and nonorphan genes in the rice (Oryza sativa L.) genome. Comparative and Functional Genomics, 2007, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. An, N. A. , Zhang, J. , Mo, F. , Luan, X. , Tian, L. , Shen, Q. S. , Li, X. , Li, C. , Zhou, F. , Zhang, B. , Ji, M. , Qi, J. , Zhou, W. Z. , Ding, W. , Chen, J. Y. , Yu, J. , Zhang, L. , Shu, S. , Hu, B. , & Li, C. Y. (2023). De novo genes with an lncRNA origin encode unique human brain developmental functionality. Nature Ecology & Evolution, 7, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen, S. D. , Zhang, Y. E. , & Long, M. Y. (2010). New genes in drosophila quickly become essential. Science (New York, NY), 330, 1682–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kondo, S. , Vedanayagam, J. , Mohammed, J. , Eizadshenass, S. , Kan, L. , Pang, N. , Aradhya, R. , Siepel, A. , Steinhauer, J. , & Lai, E. C. (2017). New genes often acquire male‐specific functions but rarely become essential in Drosophila. Genes & Development, 31, 1841–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li, L. , Foster, C. M. , Gan, Q. , Nettleton, D. , James, M. G. , Myers, A. M. , & Wurtele, E. S. (2009). Identification of the novel protein QQS as a component of the starch metabolic network in Arabidopsis leaves. Plant Journal, 58, 485–498. [DOI] [PubMed] [Google Scholar]
- 21. Chen, L. , DeVries, A. L. , & Cheng, C. H. (1997). Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. PNAS, 94, 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baalsrud, H. T. , Torresen, O. K. , Solbakken, M. H. , Salzburger, W. , Hanel, R. , Jakobsen, K. S. , & Jentoft, S. (2018). De novo gene evolution of antifreeze glycoproteins in codfishes revealed by whole genome sequence data. Molecular Biology and Evolution, 35, 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhuang, X. , Yang, C. , Murphy, K. R. , & Cheng, C. C. (2019). Molecular mechanism and history of non‐sense to sense evolution of antifreeze glycoprotein gene in northern gadids. PNAS, 116, 4400–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jacob, F. (1977). Evolution and tinkering. Science (New York, NY), 196, 1161–1166. [DOI] [PubMed] [Google Scholar]
- 25. Beike, A. K. , Lang, D. , Zimmer, A. D. , Wüst, F. , Trautmann, D. , Wiedemann, G. , Beyer, P. , Decker, E. L. , & Reski, R. (2015). Insights from the cold transcriptome of Physcomitrella patens: Global specialization pattern of conserved transcriptional regulators and identification of orphan genes involved in cold acclimation. The New Phytologist, 205, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang, M. , Dong, X. , Lang, H. , Pang, W. , Zhan, Z. , Li, X. , & Piao, Z. (2018). Mining of Brassica‐Specific Genes (BSGs) and their induction in different developmental stages and under Plasmodiophora brassicae stress in Brassica rapa. International Journal of Molecular Sciences, 19, 2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang, Z. , Wang, Y. , Kasuga, T. , Lopez‐Giraldez, F. , Zhang, Y. , Zhang, Z. , Diaz, R. , Dong, C. , Sil, A. , Trail, F. , Yarden, O. , & Townsend, J. P. (2022). Orphan genes are clustered with allorecognition loci and may be involved in incompatibility and speciation in Neurospora. BioRxiv. [Google Scholar]
- 28. Light, S. , Basile, W. , & Elofsson, A. (2014). Orphans and new gene origination, a structural and evolutionary perspective. Current Opinion in Structural Biology, 26, 73–83. [DOI] [PubMed] [Google Scholar]
- 29. Schlotterer, C. (2015). Genes from scratch–the evolutionary fate of de novo genes. Trends in Genetics: TIG, 31, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith, S. A. , & Pease, J. B. (2017). Heterogeneous molecular processes among the causes of how sequence similarity scores can fail to recapitulate phylogeny. Brief Bioinform, 18, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casola, C. (2018). From de novo to “de nono”: The majority of novel protein‐coding genes identified with phylostratigraphy are old genes or recent duplicates. Genome Biology and Evolution, 10, 2906–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moyers, B. A. , & Zhang, J. (2018). Toward reducing phylostratigraphic errors and biases. Genome Biology and Evolution, 10, 2037–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Oss, S. B. , & Carvunis, A. R. (2019). De novo gene birth. PLos Genet, 15, e1008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Domazet‐Loso, T. , Brajkovic, J. , & Tautz, D. (2007). A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends in Genetics: TIG, 23, 533–539. [DOI] [PubMed] [Google Scholar]
- 35. Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics [Electronic Resource], 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buchfink, B. , Xie, C. , & Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- 37. Vanderpool, D. , Minh, B. Q. , Lanfear, R. , Hughes, D. , Murali, S. , Harris, R. A. , Raveendran, M. , Muzny, D. M. , Hibbins, M. S. , Williamson, R. J. , Gibbs, R. A. , Worley, K. C. , Rogers, J. , & Hahn, M. W. (2020). Primate phylogenomics uncovers multiple rapid radiations and ancient interspecific introgression. Plos Biology, 18, e3000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weisman, C. M. , Murray, A. W. , & Eddy, S. R. (2022). Mixing genome annotation methods in a comparative analysis inflates the apparent number of lineage‐specific genes. Current Biology, 32, 2632–2639.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clark, N. L. , Aagaard, J. E. , & Swanson, W. J. (2006). Evolution of reproductive proteins from animals and plants. Reproduction (Cambridge, England), 131, 11–22. [DOI] [PubMed] [Google Scholar]
- 40. Enard, D. , Cai, L. , Gwennap, C. , & Petrov, D. A. (2016). Viruses are a dominant driver of protein adaptation in mammals. Elife, 5, e12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kasinathan, B. , Colmenares, S. U., 3rd , McConnell, H. , Young, J. M. , Karpen, G. H. , & Malik, H. S. (2020). Innovation of heterochromatin functions drives rapid evolution of essential ZAD‐ZNF genes in Drosophila. Elife, 9, e63368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Demuth, J. P. , De Bie, T. , Stajich, J. E. , Cristianini, N. , & Hahn, M. W. (2006). The evolution of mammalian gene families. PLoS ONE, 1, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ngou, B. P. M. , Heal, R. , Wyler, M. , Schmid, M. W. , & Jones, J. D. G. (2022). Concerted expansion and contraction of immune receptor gene repertoires in plant genomes. Nature Plants, 8, 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alba, M. M. , & Castresana, J. (2007). On homology searches by protein Blast and the characterization of the age of genes. BMC Evolutionary Biology, 7, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Elhaik, E. , Sabath, N. , & Graur, D. (2006). The “inverse relationship between evolutionary rate and age of mammalian genes” is an artifact of increased genetic distance with rate of evolution and time of divergence. Molecular Biology and Evolution, 23, 1–3. [DOI] [PubMed] [Google Scholar]
- 46. Weisman, C. M. , Murray, A. W. , & Eddy, S. R. (2020). Many, but not all, lineage‐specific genes can be explained by homology detection failure. Plos Biology, 18, e3000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moyers, B. A. , & Zhang, J. (2015). Phylostratigraphic bias creates spurious patterns of genome evolution. Molecular Biology and Evolution, 32, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moyers, B. A. , & Zhang, J. (2016). Evaluating phylostratigraphic evidence for widespread de novo gene birth in genome evolution. Molecular Biology and Evolution, 33, 1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moyers, B. A. , & Zhang, J. Z. (2017). Further simulations and analyses demonstrate open problems of phylostratigraphy. Genome Biology and Evolution, 9, 1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Domazet‐Loso, T. , Carvunis, A. R. , Alba, M. M. , Sestak, M. S. , Bakaric, R. , Neme, R. , & Tautz, D. (2017). No evidence for phylostratigraphic bias impacting inferences on patterns of gene emergence and evolution. Molecular Biology and Evolution, 34, 843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barrera‐Redondo, J. , Lotharukpong, J. S. , Drost, H. G. , & Coelho, S. M. (2023). Uncovering gene‐family founder events during major evolutionary transitions in animals, plants and fungi using GenEra. Genome Biology, 24, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shao, Y. , Chen, C. , Shen, H. , He, B. Z. , Yu, D. , Jiang, S. , Zhao, S. , Gao, Z. , Zhu, Z. , Chen, X. , Fu, Y. , Chen, H. , Gao, G. , Long, M. , & Zhang, Y. E. (2019). GenTree, an integrated resource for analyzing the evolution and function of primate‐specific coding genes. Genome Research, 29, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vakirlis, N. , Carvunis, A. R. , & McLysaght, A. (2020). Synteny‐based analyses indicate that sequence divergence is not the main source of orphan genes. Elife, 9, e53500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wissler, L. , Gadau, J. , Simola, D. F. , Helmkampf, M. , & Bornberg‐Bauer, E. (2013). Mechanisms and dynamics of orphan gene emergence in insect genomes. Genome Biology and Evolution, 5, 439–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guo, Y. L. (2013). Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant Journal, 73, 941–951. [DOI] [PubMed] [Google Scholar]
- 56. Tautz, D. , & Domazet‐Loso, T. (2011). The evolutionary origin of orphan genes. Nature Reviews Genetics, 12, 692–702. [DOI] [PubMed] [Google Scholar]
- 57. Rodelsperger, C. , Prabh, N. , & Sommer, R. J. (2019). New gene origin and deep taxon phylogenomics: Opportunities and challenges. Trends in Genetics: TIG, 35, 914–922. [DOI] [PubMed] [Google Scholar]
- 58. Donoghue, M. T. , Keshavaiah, C. , Swamidatta, S. H. , & Spillane, C. (2011). Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana. BMC Evolutionary Biology, 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Prabh, N. , & Rodelsperger, C. (2019). De novo, divergence, and mixed origin contribute to the emergence of orphan genes in Pristionchus Nematodes. G3 Genes|Genomes|Genetics, 9, 2277–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vakirlis, N. , & Kupczok, A. (2024). Large‐scale investigation of species‐specific orphan genes in the human gut microbiome elucidates their evolutionary origins. Genome Research, 34, 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Domazet‐Loso, T. , & Tautz, D. (2003). An evolutionary analysis of orphan genes in Drosophila. Genome Research, 13, 2213–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. O'Toole, A. N. , Hurst, L. D. , & McLysaght, A. (2018). Faster evolving primate genes are more likely to duplicate. Molecular Biology and Evolution, 35, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vance, Z. , Niezabitowski, L. , Hurst, L. D. , & McLysaght, A. (2022). Evidence from Drosophila supports higher duplicability of faster evolving genes. Genome Biology and Evolution, 14, evac003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Begun, D. J. , Lindfors, H. A. , Thompson, M. E. , & Holloway, A. K. (2006). Recently evolved genes identified from Drosophila yakuba and D. erecta accessory gland expressed sequence tags. Genetics, 172, 1675–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Begun, D. J. , Lindfors, H. A. , Kern, A. D. , & Jones, C. D. (2007). Evidence for de novo evolution of testis‐expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics, 176, 1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Keese, P. K. , & Gibbs, A. (1992). Origins of genes: “big bang” or continuous creation? PNAS, 89, 9489–9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McLysaght, A. , & Hurst, L. D. (2016). Open questions in the study of de novo genes: What, how and why. Nature Reviews Genetics, 17, 567–578. [DOI] [PubMed] [Google Scholar]
- 68. Bornberg‐Bauer, E. , Hlouchova, K. , & Lange, A. (2021). Structure and function of naturally evolved de novo proteins. Current Opinion in Structural Biology, 68, 175–183. [DOI] [PubMed] [Google Scholar]
- 69. Karlowski, W. M. , Varshney, D. , & Zielezinski, A. (2023). Taxonomically restricted genes in bacillus may form clusters of homologs and can be traced to a large reservoir of noncoding sequences. Genome Biology and Evolution, 15, evad023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ruiz‐Orera, J. , Hernandez‐Rodriguez, J. , Chiva, C. , Sabido, E. , Kondova, I. , Bontrop, R. , Marqués‐Bonet, T. , & Albà, M. M (2015). Origins of de novo genes in human and chimpanzee. PLos Genet, 11, e1005721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang, L. , Ren, Y. , Yang, T. , Li, G. , Chen, J. , Gschwend, A. R. , Yu, Y. , Hou, G. , Zi, J. , Zhou, R. , Wen, B. , Zhang, J. , Chougule, K. , Wang, M. , Copetti, D. , Peng, Z. , Zhang, C. , Zhang, Y. , Ouyang, Y. , …, & Long, M. (2019). Rapid evolution of protein diversity by de novo origination in Oryza. Nature Ecology & Evolution, 3, 679–690. [DOI] [PubMed] [Google Scholar]
- 72. Knowles, D. G. , & McLysaght, A. (2009). Recent de novo origin of human protein‐coding genes. Genome Research, 19, 1752–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xie, C. , Zhang, Y. E. , Chen, J. Y. , Liu, C. J. , Zhou, W. Z. , Li, Y. , Zhang, M. , Zhang, R. , Wei, L. , & Li, C. Y. (2012). Hominoid‐specific de novo protein‐coding genes originating from long non‐coding RNAs. PLos Genetics, 8, e1002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen, J. Y. , Shen, Q. S. , Zhou, W. Z. , Peng, J. , He, B. Z. , Li, Y. , Liu, C. J. , Luan, X. , Ding, W. , Li, S. , Chen, C. , Tan, B. C. M. , Zhang, Y. E. , He, A. , & Li, C. Y. (2015). Emergence, retention and selection: A trilogy of origination for functional de novo proteins from ancestral LncRNAs in primates. PLos Genetics, 11, e1005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Broeils, L. A. , Ruiz‐Orera, J. , Snel, B. , Hubner, N. , & Van Heesch, S. (2023). Evolution and implications of de novo genes in humans. Nature Ecology & Evolution, 7(6), 804–815. [DOI] [PubMed] [Google Scholar]
- 76. Guerzoni, D. , & McLysaght, A. (2016). De novo genes arise at a slow but steady rate along the primate lineage and have been subject to incomplete lineage sorting. Genome Biology and Evolution, 8, 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Roginski, P. , Grandchamp, A. , Quignot, C. , & Lopes, A. (2024). DE Novo emerged gene SEarch in Eukaryotes with DENSE. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wilson, B. A. , Foy, S. G. , Neme, R. , & Masel, J. (2017). Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth. Nature Ecology & Evolution, 1, 0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Domagala, A. , & Kurpisz, M. (2001). CD52 antigen–a review. Medical Science Monitor, 7, 325–331. [PubMed] [Google Scholar]
- 80. Wang, J. Y. , Pausch, P. , & Doudna, J. A. (2022). Structural biology of CRISPR‐Cas immunity and genome editing enzymes. Nature Reviews Microbiology, 20, 641–656. [DOI] [PubMed] [Google Scholar]
- 81. Casola, C. , Lawing, A. M. , Betran, E. , & Feschotte, C. (2007). PIF‐like transposons are common in drosophila and have been repeatedly domesticated to generate new host genes. Molecular Biology and Evolution, 24, 1872–1888. [DOI] [PubMed] [Google Scholar]
- 82. Joly‐Lopez, Z. , Forczek, E. , Hoen, D. R. , Juretic, N. , & Bureau, T. E. (2012). A gene family derived from transposable elements during early angiosperm evolution has reproductive fitness benefits in Arabidopsis thaliana. PLos Genet, 8, e1002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rajaei, N. , Chiruvella, K. K. , Lin, F. , & Astrom, S. U. (2014). Domesticated transposase Kat1 and its fossil imprints induce sexual differentiation in yeast. PNAS, 111, 15491–15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cosby, R. L. , Judd, J. , Zhang, R. , Zhong, A. , Garry, N. , Pritham, E. J. , & Feschotte, C. (2021). Recurrent evolution of vertebrate transcription factors by transposase capture. Science (New York, NY), 371, eabc6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mi, S. , Lee, X. , Li, X. , Veldman, G. M. , Finnerty, H. , Racie, L. , Lavallie, E. , Tang, X. Y. , Edouard, P. , Howes, S. , Keith, J. C. , & Mccoy, J. M. (2000). Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature, 403, 785–789. [DOI] [PubMed] [Google Scholar]
- 86. Jin, G. H. , Zhou, Y. L. , Yang, H. , Hu, Y. T. , Shi, Y. , Li, L. , Siddique, A. N. , Liu, C. N. , Zhu, A. D. , Zhang, C. J. , & Li, D. Z. (2019). Genetic innovations: Transposable element recruitment and de novo formation lead to the birth of orphan genes in the rice genome. Jnl of Sytematics Evolution, 59, 341–351. [Google Scholar]
- 87. Sun, W. , Zhao, X. W. , & Zhang, Z. (2015). Identification and evolution of the orphan genes in the domestic silkworm, Bombyx mori. Febs Letters, 589, 2731–2738. [DOI] [PubMed] [Google Scholar]
- 88. Smillie, C. S. , Smith, M. B. , Friedman, J. , Cordero, O. X. , David, L. A. , & Alm, E. J. (2011). Ecology drives a global network of gene exchange connecting the human microbiome. Nature, 480, 241–244. [DOI] [PubMed] [Google Scholar]
- 89. Ardern, Z. , Neuhaus, K. , & Scherer, S. (2020). Are antisense proteins in prokaryotes functional? Frontiers in Molecular Biosciences, 7, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blevins, W. R. , Ruiz‐Orera, J. , Messeguer, X. , Blasco‐Moreno, B. , Villanueva‐Cañas, J. L. , Espinar, L. , Díez, J. , Carey, L. B. , & Albà, M. M (2021). Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications, 12, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ardern, Z. (2023). Alternative reading frames are an underappreciated source of protein sequence novelty. Journal of Molecular Evolution, 91, 570–580. [DOI] [PubMed] [Google Scholar]
- 92. Grasse, P. P. (1977). [Overlapping genes: A priority]. C R Acad Hebd Seances Acad Sci D, 284, 141–142. [PubMed] [Google Scholar]
- 93. Watson, A. K. , Lopez, P. , & Bapteste, E. (2022). Hundreds of out‐of‐frame remodeled gene families in the Escherichia coli pangenome. Molecular Biology and Evolution, 39, msab329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mudge, J. M. , Ruiz‐Orera, J. , Prensner, J. R. , Brunet, M. A. , Calvet, F. , Jungreis, I. , Gonzalez, J. M. , Magrane, M. , Martinez, T. F. , Schulz, J. F. , Yang, Y. T. , Albà, M. M. , Aspden, J. L. , Baranov, P. V. , Bazzini, A. A. , Bruford, E. , Martin, M. J. , Calviello, L. , Carvunis, A. R. , …, & Van Heesch, S. (2022). Standardized annotation of translated open reading frames. Nature Biotechnology, 40, 994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zheng, E. B. , & Zhao, L. (2022). Protein evidence of unannotated ORFs in Drosophila reveals diversity in the evolution and properties of young proteins. Elife, 11, e78772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li, D. , Yan, Z. , Lu, L. , Jiang, H. , & Wang, W. (2014). Pleiotropy of the de novo‐originated gene MDF1. Scientific Reports, 4, 7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wells, J. N. , & Feschotte, C. (2020). A field guide to eukaryotic transposable elements. Annual Review of Genetics, 54, 539–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chung, B. Y. , Miller, W. A. , Atkins, J. F. , & Firth, A. E. (2008). An overlapping essential gene in the Potyviridae. PNAS, 105, 5897–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Pavesi, A. , Vianelli, A. , Chirico, N. , Bao, Y. , Blinkova, O. , Belshaw, R. , Firth, A. , & Karlin, D. (2018). Overlapping genes and the proteins they encode differ significantly in their sequence composition from non‐overlapping genes. PLoS ONE, 13, e0202513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cohen, O. , Gophna, U. , & Pupko, T. (2011). The complexity hypothesis revisited: Connectivity rather than function constitutes a barrier to horizontal gene transfer. Molecular Biology and Evolution, 28, 1481–1489. [DOI] [PubMed] [Google Scholar]
- 101. Kanhere, A. , & Vingron, M. (2009). Horizontal Gene Transfers in prokaryotes show differential preferences for metabolic and translational genes. BMC Evolutionary Biology, 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jain, R. , Rivera, M. C. , & Lake, J. A. (1999). Horizontal gene transfer among genomes: The complexity hypothesis. PNAS, 96, 3801–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Husnik, F. , & McCutcheon, J. P. (2018). Functional horizontal gene transfer from bacteria to eukaryotes. Nature Reviews Microbiology, 16, 67–79. [DOI] [PubMed] [Google Scholar]
- 104. Park, C. , & Zhang, J. (2012). High expression hampers horizontal gene transfer. Genome Biology and Evolution, 4, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lombardo, K. D. , Sheehy, H. K. , Cridland, J. M. , & Begun, D. J. (2023). Identifying candidate de novo genes expressed in the somatic female reproductive tract of Drosophila melanogaster. G3 Genes|Genomes|Genetics, 13, jkad122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li, Z. W. , Chen, X. , Wu, Q. , Hagmann, J. , Han, T. S. , Zou, Y. P. , Ge, S. , & Guo, Y. L. (2016). On the origin of de novo genes in arabidopsis thaliana populations. Genome Biology and Evolution, 8, 2190–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhang, J. , & Yang, J. R. (2015). Determinants of the rate of protein sequence evolution. Nature Reviews Genetics, 16, 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Davids, W. , & Zhang, Z. (2008). The impact of horizontal gene transfer in shaping operons and protein interaction networks–direct evidence of preferential attachment. BMC Evolutionary Biology, 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Peng, J. , & Zhao, L. (2024). The origin and structural evolution of de novo genes in Drosophila. Nature Communications, 15, 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vakirlis, N. N. , Hebert, A. S. , Opulente, D. A. , Achaz, G. , Hittinger, C. T. , Fischer, G. , Coon, J. J. , & Lafontaine, I. (2017). A molecular portrait of de novo genes in yeasts. Molecular Biology and Evolution, 35, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Markova, D. N. , Ruma, F. B. , Casola, C. , Mirsalehi, A. , & Betrán, E. (2022). Recurrent co‐domestication of PIF/Harbinger transposable element proteins in insects. Mob DNA, 13, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ashburner, M. , Ball, C. A. , Blake, J. A. , Botstein, D. , Butler, H. , Cherry, J. M. , Davis, A. P. , Dolinski, K. , Dwight, S. S. , Eppig, J. T. , Harris, M. A. , Hill, D. P. , Issel‐Tarver, L. , Kasarskis, A. , Lewis, S. , Matese, J. C. , Richardson, J. E. , Ringwald, M. , Rubin, G. M. , & Sherlock, G. (2000). Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nature Genetics, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. He, X. , & Zhang, J. (2006). Higher duplicability of less important genes in yeast genomes. Molecular Biology and Evolution, 23, 144–151. [DOI] [PubMed] [Google Scholar]
- 114. Kuchibhatla, D. B. , Sherman, W. A. , Chung, B. Y. , Cook, S. , Schneider, G. , Eisenhaber, B. , & Karlin, D. G. (2014). Powerful sequence similarity search methods and in‐depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. Journal of Virology, 88, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Blaxter, M. , Archibald, J M. , Childers, A K. , Coddington, J A. , Crandall, K. A. , Di Palma, F. , Durbin, R. , Edwards, S. V. , Graves, J A. M. , Hackett, K. J. , Hall, N. , Jarvis, E. D. , Johnson, R. N. , Karlsson, E. K. , Kress, W. J , Kuraku, S. , Lawniczak, M. K. N. , Lindblad‐Toh, K. , Lopez, J. V. , …, & Lewin, H. A. (2022). Why sequence all eukaryotes? PNAS, 119, e2115636118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Armstrong, J. , Hickey, G. , Diekhans, M. , Fiddes, I. T. , Novak, M. , Deran, A. , Fang, Q. , Xie, D. , Feng, S. , Stiller, J. , Genereux, D. , Johnson, J. , Marinescu, V. D. , Alföldi, J. , Harris, R. S. , Lindblad‐Toh, K. , Haussler, D. , Karlsson, E. , Jarvis, E. D. , …, & Paten, B. (2020). Progressive Cactus is a multiple‐genome aligner for the thousand‐genome era. Nature, 587, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.