

Abstract
Porcine Reproductive and Respiratory Syndrome (PRRS) causes huge economic losses to pig farms worldwide. Currently available vaccines do not always offer complete protection, due to the extreme variability of the virus. Therefore, good farming practices must be improved to prevent the disease from spreading across the pig production system. In this study, we inferred the dynamics of PRRSV population in Italy by applying bayesian methods on our ORF7 sequence dataset collected during a 15-years period. Random subsets from the overall dataset were built to reduce analysis runtime. Calculated evolutionary rate was consistent between subsets and with other findings on PRRSV and other RNA viruses (4–7 × 10− 3 substitution/site/year) while Time to the Most Recent Common Ancestor was less consistent (from 1980 to 1990). Despite this, in all population dynamic reconstructions, a massive increase in size calculated in early 2000s lasting until around 2010 was inferred. This spike is followed by very heterogeneous dynamics with some differences between subsets, probably due to the random sampling. Geographical origin was inferred in Emilia-Romagna region despite Lombardy being the region with the highest number of farmed animals and farm size. These findings reflect the choices regarding farm management and biosecurity taken in the last two decades, and not strictly related to PRRS. Phylogeny and phylogeography are powerful tools to better understand microorganisms population dynamics and make appropriate choices for disease control.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12985-024-02569-7.
Keywords: PRRSV, Phylogeny, Phylogeography, Bayesian inference
Introduction
Since its discovery in the early 90s, Porcine Reproductive and Respiratory Syndrome (PRRS) represents a major issue in pig farming and pork meat production, being responsible for consistent economic losses due to reproductive failure in sows and respiratory signs especially in piglets [1, 2], although signs can be observed in infected pigs of all ages [3]. Different studies have described losses in the breeding or growing-pig herds ranging from 12 to 45% of the total economic cost of PRRSV [4, 5]. The disease is caused by two viral species, showing a consistent geographical clustering: Betaarterivirus suid 1 (formerly known as PRRSV type 1), typical of the European continent, and Betaarterivirus suid 2 (formerly PRRSV type 2), spread in North America and Asia but they share only 50–70% nucleotide similarity of their genome [6]. This genetic distance is likely due to their ancestor evolving in different ecological or geographical contexts over an extended period of time, and maybe originating from animal species other than swine [7, 8]. PRRSV-1 is classified into three subtypes, from 1 to 3 [9], while type 2 can be divided into nine lineages, from 1 to 9 [10].
Compared to other production systems in Europe, where pigs are slaughtered at around 110 kg, Italian pig farming is quite peculiar, as it is aimed at raising heavy pigs (∼ 160 Kg) for cured meat production and is characterized by almost no export of animals and import of breeders and finishers. This long production cycle has been suggested to increase PRRSV circulation, explaining the high heterogeneity and the delayed effect of counter-measures on viral population unlike other countries [11]. Meanwhile, limited circulation of animals across national boards could have favoured a noticeable geographical isolation of Italian strains, where typically only PRRSV-1 subtype 1 circulate [8, 11].
PRRSV genome is approximately 15 kb long, with at least 9 Open Reading Frames (ORFs) coding for both non-structural (ORF1a and 1b) and structural proteins (ORF 2–7). In particular, ORF5 and ORF7, which encode for glycoprotein 5 (GP5) and nucleocapsid (N) protein, respectively, have been commonly used for genotyping and phylogenetic analysis due to their genetic variability, especially ORF5 [9, 10, 12, 13]. Being a positive single-stranded RNA virus, PRRSV is characterized by high genetic variability, due to high mutation rate and frequent recombination events [14, 15]. This variability has made disease control challenging, as preventive measures are often ineffective in field conditions. Modified Live Vaccines (MLVs), which currently are the most widely used strategy for PRRS control, are poorly cross-protective against heterologous strains. Moreover, reversion to virulence of MLVs, leading to disease outbreaks, has been reported for PRRSV [16]. In addition, groups of animals within the same farm can be found positive to different PRRSV strains [17, 18]. Exploiting sequence data and metadata, phylodynamic models can provide a picture of viral evolution and transmission by reconstructing the geographic spread patterns and mapping the dynamycs of virus population [19]. The aim of this study was to (i) evaluate the variability of PRRSV-1 in Italy over a 15-years period and (ii) estimate its diffusion across the peninsula.
Materials and methods
Samples collection and molecular screening
Overall, 7491 PRRSV positive samples collected by the Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia Romagna from 2008 to 2022 were included in the study. A variety of biological matrixes (such as organs, swabs, serum, blood and semen) was tested to confirm PRRSV presence in diseased animals or for monitoring purposes. Over the considered period, the diagnostic approach changed with the adoption of newer, faster and more accurate molecular protocols. Initially, from 2008 to 2011, manual RNA extraction with guanidine, chloroform and phenol was performed, followed by One-Step RT-PCR reaction using a mixture of 3 primers, two type-specific and a one common primer: 5’-ATGGCCAGCCAGTCAATC-3’ (PRRSV type 1), 5’-GATTGCAAGCAGAGGGAGCGTTC-3’(PRRSV type 2), 5’- GGCGCACAGTATGATGCGTAG-3’ (common primer). The thermal protocol was as follows: 1 cycle of reverse transcription at 50 °C for 30 min, 1 cycle of denaturation at 95 °C for 15 min, 40 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s and extension at 72 °C for 30 s, and 1 cycle of final extension at 72 °C for 7 min. Positive samples were identified through agarose gel electrophoresis and the typing was based on the molecular weight of the product: 180 bp for PRRSV-1 and 280 bp for PRRSV-2. After 2011 the molecular screening was updated by replacing both the extraction method and the PCR assay. Manual extraction was replaced with a semi-automatic method based on magnetic beads, using KingFisher Flex (Thermofisher Scientific) with the NucleoMag™ 96Virus Kit (Macherey-Nagel). A commercial qRT-PCR (i.e. XenoRNA (AgPath-ID™ NA and EU PRRSV Multiplex Reagent kit, Applied Biosystems™)) took over from end-point RT-PCR. After 2020 the diagnostic method was updated once again by replacing the previous extraction kit with the MagMAX™ CORE Nucleic Acid Purification Kit (Applied Biosystems™), and the Real Time RT-PCR kit with Virotype® PRRSV RT-PCR kit (Qiagen/Indical), allowing the European and American PRRSV strain identification together with a third Highly Pathogenic American PRRSV strain in the same reaction. Furthermore, the Virotype® kit includes specific primers and probes for the detection of an endogenous gene as internal control.
ORF7 sequencing
A 510 nucleotides (nt) region, including ORF7 (387 ± 6 nt) was the target for Sanger sequencing. A One step RT-PCR Kit (Qiagen) was set up with the specific primers: 5’CTTCGGAGCCTCGTGYTGGGCGGCAA-3’(forward) and 5’-TCGCCCTAATTGAATAGGTGA3’ (reverse) according to the the following protocol: 1 cycle of reverse transcription at 50 °C for 30 min, 1 cycle of initial denaturation at 95 °C for 15 min, 42 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 40 s and extension at 72 °C for 50 s, 1 cycle of final extension at 72 °C for 7 min. Purification of PCR product was carried out with Exonuclease I to remove the amplification primers and Thermosens Phosphatase alkaline (FastAp) to remove unincorporated dNTPs. Sanger sequencing was performed in both directions with the Big Dye® terminator ready reaction v1.1 kit (Applied Biosystems™) using the same PCR primers according to the following protocol: 1 cycle of initial denaturation at 96 °C for 90 s, 25 cycles of denaturation at 96 °C for 10 s and annealing at 55 °C for 5 s and a final extension at 60 °C for 4 min. Sequence purification was performed with BigDye® Xterminator Purification kit (Applied Biosystems™) according to the manufacturer’s instructions. Electropherograms quality analysis was performed on forward and reverse sequences by Sequencing analysis v5.4 software (Applied Biosystems™). Raw data (.ab1 format file) were analyzed by Lasergene software SeqMan module (DNAStar, Madison USA versions from 7 to 17).
Phylogenetic analysis
Sequences were univocally identified using the farm code, which is unique for every farm in the Country, and the sampling date. Incomplete sequences, with ambiguous nucleotide or those with missing metadata were excluded from the analysis. Obtained sequences were aligned using MAFFT 7 algorithm implemented in Mega Align PRO® 17.3.1(DNASTAR, Inc. Madison, WI) and a Maximum Likelihood (ML) tree was built with iqtree2 [20]. In both cases, vaccine and prototype sequences (subtype 1, 2 and 3) were included. These strains were excluded from the dataset for the phylodynamic analysis. The presence of a temporal signal was initially assessed using TempEST v1.5.3 [21]. For phylodynamic analysis, the complete dataset was sub-sampled with replacement 10 times. Each subset contained a random 10% of the whole dataset and underwent the same analysis. Subsequently, each subset was aligned once more and analyzed with BEAST v1.10.4 [22]. Each analysis was run twice, and the resulting files were combined through the LogCombiner tool included in the BEAST package, while removing the first 10% as burn-in. The substitution model (GTR + G + I) was selected based on the BIC score, automatically calculated with iqtree2 before the computation of the phylogenetic tree. The relaxed lognormal molecular clock [23] and Bayesian Skyline [24] were selected, as clock and tree models, respectively. Lastly, Markov Chain Monte Carlo (MCMC) was set at 300 millions, with a sample frequency of 30 thousand steps. The resulting log file was evaluated with Tracer 1.6 [25] and maximum clade credibility (MCC) trees were obtained through the TreeAnnotator software included in the BEAST package. A further 10% burn-in was removed when annotating each tree. Phylogeographic analysis was carried out following the same parametres with the addition of latitude and longitude as continouos traits and the implementation of the Brownian random walk migration model. The calculated tree was visualized in R Software [26] using the ggtree package [27] while plots of BEAST outputs were obtained with the ggplot2 package [28]. Visualization of viral population diffusion was performed with SPREAD3 software [29].
Results
A total of 4609 ORF7 sequences out of 7491 (61.8%) were included in the final dataset, originating from 15 Italian Regions and 50 Provinces (Additional File 1), between 2008 and 2022. Among those removed from the dataset, 594 had at least one ambiguous nucleotide while 114 were too short to be included. Out of these, 24 were PRRSV Type 2 and 10 had ambiguous nucletides. The remaining 2174 samples were removed because either identical to other samples in terms of nucleotide sequence, farm and collection date (e.g. a simultaneous sampling of sera from the same farm with 100% sequence similarity) or incomplete additional information. Overall, 1062 farms were sampled at least once throughout the considered period. The ML tree showed that all sequences belong to subtype 1, as none clustered with PRRSV-1 subtype 2 or 3 (data not shown). For this reason, ML tree with vaccine strains only as reference is shown (Fig. 1). Preliminary analysis of the phylogenetic tree in TempEst showed the presence of a proper temporal signal consisting of an evolutionary rate of 2.69 × 10− 3 substitutions/site/year (data not shown).
Fig. 1.

Maximum Likelihood tree with based on the complete ORF7 dataset. Color coded tips are the vaccine sequences. Some clusters containing only sampled sequences are collapsed for graphical purposes. Root was placed through the best fitting root function in TempEst
In the phylodynamic analysis, the mean estimated evolutionary rate was comparable among all runs and ranged from 4.12 × 10− 3 (subset 6, 95% HPD Interval [3.5739*10− 3, 4.7131*10− 3]) to 5.76 × 10− 3 (subset 5, 95% HPD Interval [5.0155*10− 3, 6.5472*10− 3]). Mean Time to the Most Recent Common Ancestor was less consistent throughout datasets and ranged from 1960.24 in subset 7 (95% HPD Interval [1938.3982, 1980.286]) to 1990.18 (95% HPD Interval [1983.249, 1996.0503]) in subset 1 (Figs. 2 and 3).
Fig. 2.

Density plot of the mean evolutionary rate. Values for the representation were obtained from the log file resulted from the BEAST analysis for each PRRSV-1 subset (color-coded)
Fig. 3.

Density plot of Time to the Most Recent Common Ancestor (TMRCA). Values for the representation were obtained from the log file resulted from the BEAST analysis for each PRRSV-1 subset (color-coded)
Bayesian Skyline reconstruction showed a population increase between 2003 and 2010, with slight differences in every run but consistent nonetheless (Fig. 4). A contraction was observed thereafter until around 2012. This was followed by a stabilization period marked by minor fluctuations in timing and magnitude, which varied depending on the dataset considered.
Fig. 4.

Mean effective population size of the Italian PRRSV-1 population. The analysis of virus population through time assessed by Bayesian Skyline reconstruction. The 10 combined subsets are color-coded
The spatial analysis performed with SPREAD3 placed the origin of PRRSV-1 in the province of Parma, Emilia-Romagna region, with the exception of subset 7, which was inferred in South Tuscany, which also showed the lowest evolutionary rate. From this area, across all runs, the common pattern was the spread northwards and southwards from their respective origin. Initially concentrating in Lombardy and Emilia Romagna regions for few years, as expected given the origin sequences included in the dataset, a subsequent radial expansion was observed, especially eastwards and westward (Fig. 5).
Fig. 5.
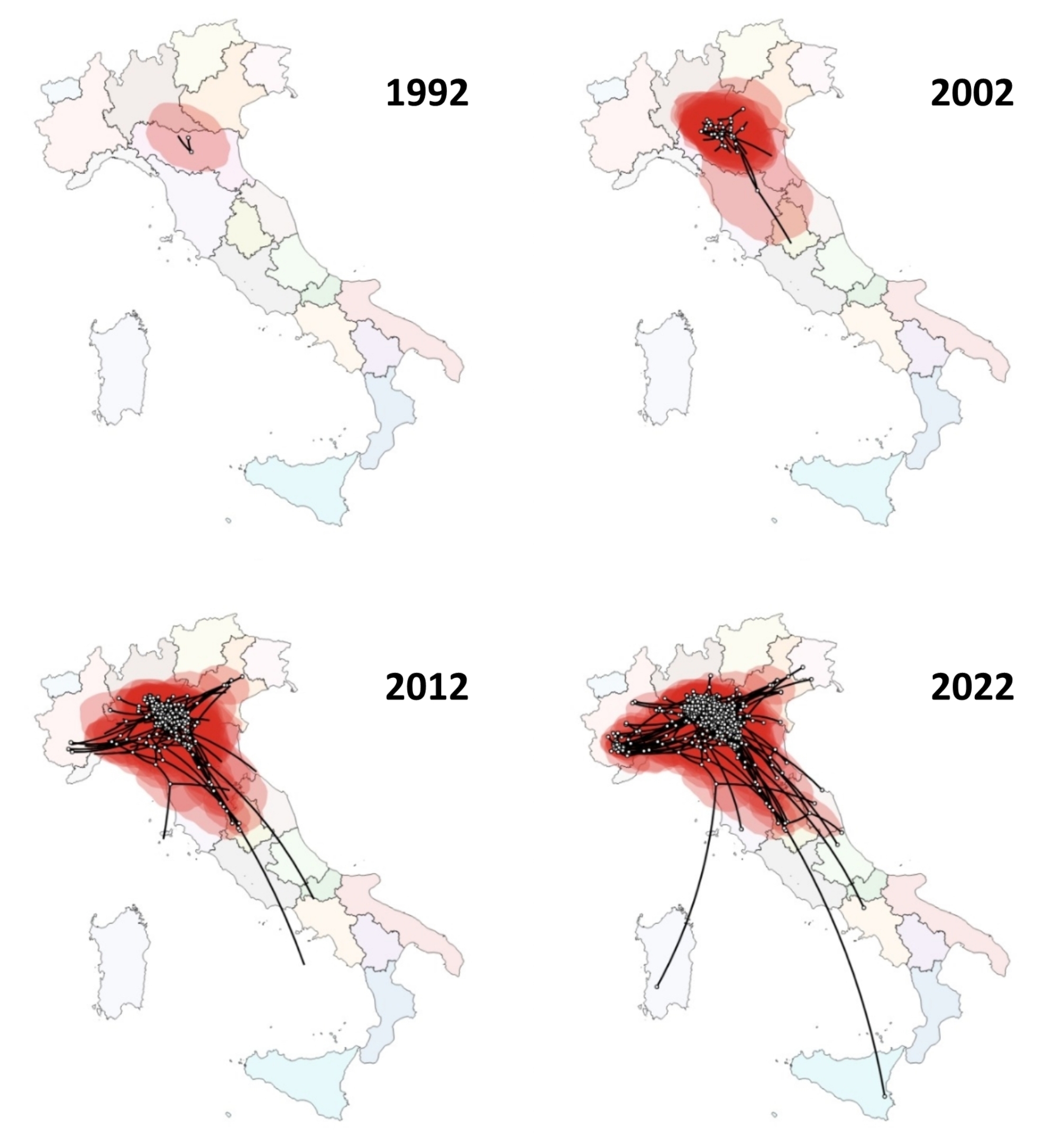
Spatial analysis of PRRSV-1 in Italy performed with SPREAD3. Analysis of subset 2 is showed as an example. White points represent internal nodes and tips of the inferred tree, red polygons represent the uncertainty areas. From 1992 to 2002 a diffusion North-South oriented can be visualized. From 2012 on, the previous spread continues with the addition of the diffusion eastwards and westwards. Every map represents a 10-year progression from the previous one
Discussion
Phylogenetic analysis
In this study, we applied a phylodynamic approach to evaluate the evolutionary dynamics of PRRSV-1 in Italy from 2008 to 2022. Over 15 years, PRRSV-1 population remained composed exclusively by subtype 1 strains as previously observed [9, 11]. As expected, farms located in norther regions provided the majority of the sequences, as Pianura Padana is more suitable for livestock farming rather than southern regions. For example, while holding less than 10% of the total farms in Italy, Lombardy houses almost 50% of pigs farmed [30]. Moreover, the increase of farmed animals and the simultaneous decrease of sequencing cost resulted in a much higher portion of recent sequences rather than older ones both in our dataset and our subsamplings which may have caused a bias in downstream results. BEAST analysis revealed a substitution rate consistent to that already found for PRRSV [31] and for other positive-sense single-stranded RNA viruses as well [32]. As shown in the Skyline reconstruction (Fig. 3), in some cases the tMRCA was inferred in the early 90s, as commonly accepted, while in other cases, the root dated in the early 80s, well before the spread of PRRSV-1 in Europe. This inconsistency may reflect the in silico random sampling, suggesting a certain sampling effect, that might slightly bias the overall estimations, being the sample distribution unbalanced both geographically (most sequences are from Lombardy, Emilia-Romagna and Piedmont Regions) and temporally (most sequences are from 2019 to 2022). Considering that the oldest sequence obtained through our diagnostic activity is from January 2nd, 2008, it is plausible to suggest that the disease remained undetected or misdiagnosed for several years until being reported around the world. When older sequences (from early 2000 to late 90s) could be analyzed, the tMRCA could be inferred even before 1982, as demonstrated previously outside Italy, with slightly different timing but comparable dynamics [33]. Therefore, the estimated tMRCA might have been underestimated due to limited sample availability, as sequences sampled through the study period are not evenly distributed.
Pig farms management changes ripercussions
The Bayesian Skyline reconstruction showed a peak around 2010, with several fluctuations observed in the subsequent years. This result is likely due to the transition from closed-farm to multi-site production system, that took place from late 90s to early 2000s in Italy [34]. This transition significantly increased animal movements over longer distances, leading to a massive spread of the virus among different farms and/or among different production sites of the same farms. After 2010s, there was an improvement in farming practices and a thorough application of biosecurity procedures due to the spread of Aujeszky disease and Swine vescicular disease (SVD) outbreaks in North Italy [35, 36]. These measures, which are still in place, could have had a positive and indirect effect in limiting PRRSV-1 on the territory as well. From 2015 to nowadays, the viral population has shown a “roller-coaster” dynamic probably caused by different components, such as prolonged viremia, persistent infection in lymphoid tissue [37], and the seasonal introduction of new animals in farms to sustain the national pig meat demand [38]. In addition, PRRSV can spread through various routes in farms, not necessarly via animals. Non-porous common materials and feed ingredients have also been shown to transmit PRRSV, with higher transmission rates at lower temperatures [39]. It has been also demonstrated that certain PRRSV strains can be airborne transmitted for several kilometers [40], which is critical in high density farm areas like the Pianura Padana in our study. This variety of risk factors, through which PRRSV can spread between farms, emphasize the difficulties in control and/or eradication of the disease, which have different outcomes even when abovementioned biosecurity protocols are applied with consistency.
Spatial analysis
A further analysis in SPREAD3 reconstructed PRRSV-1 movements within the Italian peninsula. The diffusion appears to have originated in north-central Italy, in mid-to-late of the 20th century, specifically in the province of Parma in Emilia-Romagna, despite Lombardy being the Italian region with the highest number of animals and farm density. From this origin, after initially migrating northward for a few kilometers, Lombardy became the main geographic area from where the virus spread in multiple directions, including southern Italy, where pigs are less intensively raised. Indeed, it has been demonstrated that while larger farms, even if in low numbers, play a major potential role in the diffusion of infectious diseases in pigs, acting as super-spreaders, also small facilities can be relevant in PRRSV-1 epidemiology when featured by low biosecurity levels, as often occurs in rural southern settings [38, 41]. On a side note, Modified Live Vaccines could have played a minor but still significant role both in the initial spread of PRRSV-1 and in the most recent years, since they were constantly introduced in Europe starting from early 2000s [16]. However, there are risks associated with these vaccines, such as reversion to virulence and recombination between vaccine strains, potentially leading to severe outcomes [42].
Final considerations
Phylodynamic and phylogeographic analysis play an important role in studying the epidemiology of diseases that are challenging to eradicate and/or manage once introduced into farm or groups of farms. These information can support animal health decisions to contain virus spread. Moreover, by combining genetic and geographic data, these approaches provide a dynamic view of viral movements, which is even more critical for zoonotic viruses. Despite being an extremely useful tool, the application of phylodynamics with the use of only fragments of the entire genome does not allow to be applied correctly on recombinant strains or may require different parametres if different portion are considered. Although, applying these approaches on a whole genome scale, combined with comprehensive metadata, complex statistical methods and computing capabilities, can increase exponentially their potential to provide accurate viral population dynamics estimations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Matteo Tonni, Cristian Salogni, Alberto Tiraboschi and Gloria Garbin for their help with the project activities.
Author contributions
Conceived and designed the experiments: G. P., T. S., I. B., G. L. A., M. B. B. Performed laboratory experiments: V. C., S. M., D. C. Analyzed the data: G. P., V. C., S. M., D. C., G. F. Writing—original draft preparation: G. P., G. F., G. V., M. B. B. Writing—review and editing: G. P., S. F., G. F., T. S., I. B., M. B. B. Supervision: I. B., G. L. A., M.B.B. All authors read and approved the final manuscript.
Funding
This research received no external funding.
Data availability
The dataset supporting the conclusions of this article has been deposited in the NCBI GenBank archive with the BioProject ID PRJNA1153755.
Declarations
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Ethical approval
was not required for this work, as samples were part of routine disease surveillance activities carried out by Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia Romagna (IZSLER). All the activities were performed in compliance with state and local regulations.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Holtkamp DJ. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. JSHAP. 2013;21:72–84. [Google Scholar]
- 2.Nathues H, Alarcon P, Rushton J, Jolie R, Fiebig K, Jimenez M, Geurts V, Nathues C. Cost of porcine reproductive and respiratory syndrome virus at individual farm level – an economic disease model. Prev Vet Med. 2017;142:16–29. 10.1016/j.prevetmed.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Zimmerman JJ, Dee SA, Holtkamp DJ, Murtaugh MP, Stadejek T, Stevenson GW, Torremorell M, Yang H, Zhang J. Porcine Reproductive and Respiratory Syndrome viruses (Porcine Arteriviruses). In: Zimmerman JJ, Karriker LA, Ramirez A, Schwartz KJ, Stevenson GW, Zhang J, editors. Diseases of Swine. 11th ed. Hoboken NJ USA: Wiley-Blackwell; 2019. pp. 685–708. [Google Scholar]
- 4.Montaner-Tarbes S, del Portillo HA, Montoya M, Fraile L. 2019. Key gaps in the knowledge of the Porcine Respiratory Reproductive Syndrome Virus (PRRSV). Frontiers in Veterinary Science 6. [DOI] [PMC free article] [PubMed]
- 5.Nieuwenhuis N, Duinhof TF, van Nes A. 2012. Economic analysis of outbreaks of porcine reproductive and respiratory syndrome virus in nine sow herds. Vet Rec. 2012;170(9):225. 10.1136/vr.100101 [DOI] [PubMed]
- 6.Murtaugh MP, Elam MR, Kakach LT. 1995. Comparison of the structural protein coding sequences of the VR-2332 and Lelystad virus strains of the PRRS virus. Arch Virol. 1995;140(8):1451-60. 10.1007/BF01322671 [DOI] [PMC free article] [PubMed]
- 7.Plagemann PG. Porcine reproductive and respiratory syndrome virus: origin hypothesis. Emerg Infect Dis. 2003;9(8):903–8. 10.3201/eid0908.030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi M, Lam TT, Hon CC, Hui RK, Faaberg KS, Wennblom T, Murtaugh MP, Stadejek T, Leung FC. Molecular epidemiology of PRRSV: a phylogenetic perspective. Virus Res Dec. 2010;154(1–2):7–17. Epub 2010 Sep 15. [DOI] [PubMed] [Google Scholar]
- 9.Stadejek T, Stankevicius A, Murtaugh MP, Oleksiewicz MB. Molecular evolution of PRRSV in Europe: current state of play. Veterinary Microbiology, Special Issue: one world. One Health One Virol. 2013;165:21–8. 10.1016/j.vetmic.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 10.Le Gall A, Legeay O, Bourhy H, Arnauld C, Albina E, Jestin A. Molecular variation in the nucleoprotein gene (ORF7) of the porcine reproductive and respiratory syndrome virus (PRRSV). Virus Res. 1998;54:9–21. 10.1016/S0168-1702(97)00146-9. [DOI] [PubMed] [Google Scholar]
- 11.Forsberg R, Storgaard T, Nielsen HS, Oleksiewicz MB, Cordioli P, Sala G, Hein J, Bøtner A. The genetic diversity of European type PRRSV is similar to that of the north American type but is geographically skewed within Europe. Virology. 2002;2002(2991):38–47. 10.1006/viro.2002.1450. [DOI] [PubMed] [Google Scholar]
- 12.Stadejek T, Oleksiewicz MB, Scherbakov AV, Timina AM, Krabbe JS, Chabros K, Potapchuk D. Definition of subtypes in the European genotype of porcine reproductive and respiratory syndrome virus: nucleocapsid characteristics and geographical distribution in Europe. Arch Virol. 2008;153:1479–88. 10.1007/s00705-008-0146-2. [DOI] [PubMed] [Google Scholar]
- 13.Hao X, Lu Z, Kuang W, Sun P, Fu Y, Wu L, Zhao Q, Bao H, Fu Y, Cao Y, Li P, Bai X, Li D, Liu Z. 2011. Polymorphic genetic characterization of the ORF7 gene of porcine reproductive and respiratory syndrome virus (PRRSV) in China. Virol J. 2011;8:73. 10.1186/1743-422X-8-73 [DOI] [PMC free article] [PubMed]
- 14.Meng XJ. Heterogeneity of porcine reproductive and respiratory syndrome virus: implications for current vaccine efficacy and future vaccine development. Vet Microbiol. 2000;74:309–29. 10.1016/S0378-1135(00)00196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kappes MA, Faaberg KS. PRRSV structure, replication and recombination: origin of phenotype and genotype diversity. Virol 60th Anniversary Issue. 2015;479–480:475–86. 10.1016/j.virol.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chae C. Commercial PRRS modified-live virus vaccines. Vaccines. 2021;9:185. 10.3390/vaccines9020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dee SA, Torremorell M, Rossow K, Mahlum C, Otake S, Faaberg K. 2001. Identification of genetically diverse sequences (ORF 5) of porcine reproductive and respiratory syndrome virus in a swine herd. Can J Vet Res. 2001;65(4):254 – 60. [PMC free article] [PubMed]
- 18.Cheng TY, Campler MR, Schroeder DC, Yang M, Mor SK, Ferreira JB, Arruda AG. 2022. Detection of Multiple Lineages of PRRSV in Breeding and Growing Swine Farms. Front Vet Sci. 2022;9:884733. 10.3389/fvets.2022.884733 [DOI] [PMC free article] [PubMed]
- 19.Alkhamis MA, Perez AM, Murtaugh MP, Wang X, Morrison RB. Applications of bayesian phylodynamic methods in a recent U.S. Porcine Reproductive and Respiratory Syndrome Virus Outbreak. Front Microbiol. 2016;2016(7):67. 10.3389/fmicb.2016.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. 2020. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol. 2020;37(5):1530–1534. 10.1093/molbev/msaa015. Erratum in: Mol Biol Evol. 2020;37(8):2461. [DOI] [PMC free article] [PubMed]
- 21.Rambaut A, Lam TT, Max Carvalho L, Pybus OG. 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2(1):vew007. 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed]
- 22.Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. 2018. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4(1):vey016. [DOI] [PMC free article] [PubMed]
- 23.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4(5):e88. 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences, Molecular Biology and Evolution, Volume 22, Issue 5, May 2005, Pages 1185–1192, 10.1093/molbev/msi103 [DOI] [PubMed]
- 25.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior summarisation in bayesian phylogenetics using Tracer 1.7. Systematic Biology. syy032. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed]
- 26.R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2024. [Google Scholar]
- 27.Yu G, Smith DK, Zhu H, Guan Y, Tsan-Yuk Lam T. T.T.Y., 2017. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 2017, 8, pages 28–36, 10.1111/2041-210X.12628
- 28.H. Wickham. ggplot2: elegant graphics for data analysis. Springer- New York, 2016.
- 29.Bielejec F, Baele G, Vrancken B, Suchard MA, Rambaut A, Lemey P. 2016. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol Biol Evol. 2016;33(8):2167-9. 10.1093/molbev/msw082. Epub 2016 Apr 23. [DOI] [PMC free article] [PubMed]
- 30.National Data Bank Statistics. (swine): https://www.vetinfo.it/j6_statistiche/#/report-pbi/31
- 31.Yoon SH, Kim H, Park B, Kim H. 2012. Tracing the genetic history of porcine reproductive and respiratory syndrome viruses derived from the complete ORF 5–7 sequences: a Bayesian coalescent approach. Arch Virol. 2012;157(11):2143-51. [DOI] [PubMed]
- 32.Duffy S. 2018. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018;16(8):e3000003. 10.1371/journal.pbio.3000003 [DOI] [PMC free article] [PubMed]
- 33.Franzo G, Faustini G, Legnardi M, Cecchinato M, Drigo M, Tucciarone CM. Phylodynamic and phylogeographic reconstruction of porcine reproductive and respiratory syndrome virus (PRRSV) in Europe: patterns and determinants. Transbound Emerg Dis. 2022;69(5):e2175–84. 10.1111/tbed.14556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris DL. Multi-site Pig production. Ames, Iowa State University; 2000.
- 35.Chiari M, Ferrari N, Bertoletti M, Avisani D, Cerioli M, Zanoni M, Alborali LG, Lanfranchi P, Lelli D, Moreno Martin AM, Antonio L. 2015. Long-Term Surveillance of Aujeszky’s Disease in the Alpine Wild Boar (Sus scrofa). Ecohealth. 2015;12(4):563 – 70. 10.1007/s10393-015-1064-x [DOI] [PubMed]
- 36.Nassuato C, Boender GJ, Eblé PL, Alborali L, Bellini S, Hagenaars TJ. 2013. Spatial transmission of Swine Vesicular Disease virus in the 2006–2007 epidemic in Lombardy. PLoS One. 2013;8(5):e62878. 10.1371/journal.pone.0062878 [DOI] [PMC free article] [PubMed]
- 37.Albina E, Piriou L, Hutet E, Cariolet R, L’Hospitalier R. 1998. Immune responses in pigs infected with porcine reproductive and respiratory syndrome virus (PRRSV). Vet Immunol Immunopathol. 1998;61(1):49–66. 10.1016/s0165-2427(97)00134-7 [DOI] [PMC free article] [PubMed]
- 38.Crescio MI, Mastrantonio G, Bertolini S, Maurella C, Adkin A, Ingravalle F, Simons RRL, DeNardi M, Stark K, Estrada-Peña A, Ru G. Using network analysis to identify seasonal patterns and key nodes for risk-based surveillance of pig diseases in Italy. Transbound Emerg Dis. 2020;2021(686):3541–51. 10.1111/tbed.13960. [DOI] [PubMed] [Google Scholar]
- 39.Lugo Mesa V, Quinonez Munoz A, Sobhy NM, Corzo CA, Goyal SM. 2024. Survival of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) in the Environment. Vet Sci. 2024;11(1):22. 10.3390/vetsci11010022 [DOI] [PMC free article] [PubMed]
- 40.Otake S, Dee S, Corzo C, Oliveira S, Deen J. 2010. Long-distance airborne transport of infectious PRRSV and Mycoplasma hyopneumoniae from a swine population infected with multiple viral variants. Vet Microbiol. 2010;145(3–4):198–208. 10.1016/j.vetmic.2010.03.028 [DOI] [PubMed]
- 41.Relun A, Grosbois V, Sánchez-Vizcaíno JM, Alexandrov T, Feliziani F, Waret-Szkuta A, Molia S, Etter EM, Martínez-López B. 2016. Spatial and Functional Organization of Pig Trade in Different European Production Systems: Implications for Disease Prevention and Control. Front Vet Sci. 2016;3:4. 10.3389/fvets.2016.00004 [DOI] [PMC free article] [PubMed]
- 42.Kvisgaard LK, Kristensen CS, Ryt-Hansen P, Pedersen K, Stadejek T, Trebbien R, Andresen LO, Larsen LE. 2020. A recombination between two Type 1 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV-1) vaccine strains has caused severe outbreaks in Danish pigs. Transbound Emerg Dis. 2020;67(5):1786–1796. 10.1111/tbed.13555 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset supporting the conclusions of this article has been deposited in the NCBI GenBank archive with the BioProject ID PRJNA1153755.