

Abstract
Background
Recent research has postulated that the activation of cGAS-STING-interferon signalling pathways could be implicated in the pathogenesis of Alzheimer’s disease (AD). However, the precise types of interferons and related cytokines, both from the brain and periphery, responsible for cognitive impairment in patients with AD remain unclear.
Methods
A total of 131 participants (78 [59.5%] female and 53 [40.5%] male; mean [SD] age, 61.5 [7.6] years) with normal cognition and cognitive impairment from the China Aging and Neurodegenerative Initiative cohort were included. CSF and serum IFNα-2a, IFN-β, IFN-γ, TNF-α, IL-6, IL-10, MCP-1and CXCL-10 were tested. The correlation between these interferons and related cytokines with AD core biomarkers in the CSF and plasma, cognition performance, and brain MRI measures were analysed.
Results
We found that only CSF IFN-β levels were significantly elevated in Alzheimer’s disease compared to normal cognition. Furthermore, CSF IFN-β levels were significantly associated with AD core biomarkers (CSF P-tau and Aβ42/Aβ40 ratio) and cognitive performance (MMSE and CDR score). Additionally, the CSF IFN-β levels were significantly correlated with the typical pattern of brain atrophy in AD (such as hippocampus, amygdala, and precuneus). In contrast, CSF IL-6 levels were significantly elevated in non-AD cognitively impaired patients compared to other groups. Moreover, CSF IL-6 levels were significantly associated with cognitive performance in non-AD individuals and correlated with the vascular cognitive impairment-related MRI markers (such as white matter hyperintensity).
Conclusion
Our findings demonstrate that distinct inflammatory molecules are associated with different cognitive disorders. Notably, CSF IFN-β levels are significantly linked to the pathology and cognitive performance of AD, identifying this interferon as a potential target for AD therapy.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13195-024-01644-z.
Keywords: Alzheimer’s disease, Interferons, Innate immune, Biomarkers, Neuroinflammation
Introduction
The aging population is expanding globally, leading to a surge in the prevalence of dementia. It is estimated that the number of individuals with dementia will rise from 57.4 million in 2019 to 152.8 million by 2050 [1]. Alzheimer’s disease (AD), a prevalent form of dementia, is marked by progressive cognitive decline that impacts various aspects of daily life [2], and is characterized by pathological hallmark amyloid plaques and tau-associated neurofibrillary tangles. Mounting evidence indicates that aberrant neuro-immunological mechanisms play a role in the onset, progression, and severity of AD [3]. However, the key inflammatory cytokines involved in AD remain a topic of debate.
Recent studies have highlighted on sterile inflammation caused by Cyclic GMP-AMP (cGAMP) synthase (cGAS) -stimulator of interferon genes (STING) signaling pathway in AD mouse models. cGAS recognizes abnormal double – stranded DNA from perturbed mitochondria [4] and synthesizes cGAMP to activate the STING. The activation of STING triggers reactive microglia type I interferon (IFN) response, which disturb synapse integrity and plasticity and induced cognitive impairment [5, 6].
Our recent findings have demonstrated that peripherally dysfunctional CD4 + T cells can induce neuroinflammation by secreting pro-inflammatory cytokines in AD patients [7]. Additionally, activated IFN pathways have been reported in the brains of various AD mouse models [8, 9], and have been confirmed in autopsied brains with AD, potentially leading to further neuroinflammation and eventual synaptic loss [8]. The blockade of interferon-α/β receptor (IFNAR) and microglia-specific Ifnar1 knockout alleviate memory deficits in an AD model [10]. Recent research has revealed that effectors of the interferon signalling pathway can regulate Aβ production [11], while hyperphosphorylated tau protein mediates cognitive impairment through activation of interferon signalling [9].
The IFN protein family constitutes an intricate system, encompassing biological or chemical inducers, toll-like receptors, IFN types, signal transporters, and IFN-stimulated genes (ISGs) [12]. IFNs are classified into three subtypes: type I IFN (IFN-α, IFN-β), type II IFN (IFN-γ), and type III IFN (IFN-λ), which are produced by distinct cell types and serve as crucial regulators of antiviral responses, antigen presentation, autoimmunity, and inflammation [13–16]. Prior in vitro and in vivo studies on AD-related animal models have shown that type I and type II IFNs contribute to the development of AD pathology and cognitive deficits through their effects on microglia [17–21] and neurons [11, 22–24]. Nevertheless, emerging evidence suggests that activated type I IFN signalling with diminished type II IFN signalling in the choroid plexus correlates with cognitive decline in aged mice [25] and AD mouse models [26]. A recent study found that anti-interferon treatment led to reduced brain atrophy in a tauopathy model [27], further supporting the idea that interferon is causally linked to neurodegeneration.
While basic research has highlighted the pathological significance of IFN pathway activation in AD animal models, the specific types and sources of IFNs and related cytokines associated with AD in clinical practice remain uncertain. A literature review revealed that various cytokines participate in distinct cellular interferon signalling responses and may be implicated in the pathogenesis of cognitive impairment (Supplementary Fig. S1). Consequently, further investigations are needed to determine the primary types of IFNs and related cytokines that contribute to AD core biomarkers and cognitive impairment in patients with AD.
In this study, we leveraged an established AD cohort in China [28] to analyse paired CSF and serum levels of IFNs and related cytokines, as well as their associations with AD pathology, cognitive performance, and brain MRI measures in participants at different stages of AD, in comparison with normal cognition and cognitively impaired participants without AD (Supplementary Fig. S2).
Methods
CANDI cohort participants and study design
All participants of the study were recruited from a single cohort, the China Aging and Neurodegenerative Initiative (CANDI) [28] from the First Affiliated Hospital of USTC, which was supported by the Chinese Academy of Sciences. The CANDI cohort was established as a research platform, following the 2018 National Institute on Aging—Alzheimer’s Association (NIA-AA) recommended β amyloid (Aβ) deposition, pathologic tau, and neurodegeneration (ATN) research framework [29], to characterize neurodegeneration-related cognitive impairment diseases, including Alzheimer’s disease (AD), mild cognitive impairment (MCI), vascular dementia (VaD), dementia with Lewy bodies (DLB), frontotemporal dementia (FTD), and normal cognition (NC) individuals aged between 45 and 85. In the CANDI study, participants were followed up longitudinally and underwent a more comprehensive evaluation every year.
A total of 131 CANDI subjects with clinical information, brain MRI, CSF, and plasma biomarkers were rigorously selected for this study (Supplementary Fig. S2). The inclusion criteria for AD and MCI due to AD were as follows: (1) individuals with cognitive complaints, (2) age 50–79 years, (3) evidence of AD pathophysiology through CSF or PET-CT, (4) clinical dementia rating (CDR) score of 0.5, 1, or 2, (5) availability of a caregiver, (6) written informed consent to participate in this study. The exclusion criteria were as follows: (1) other neurological conditions that significantly contributed to the participants’ cognitive impairment, such as vascular dementia, Lewy body dementia, frontotemporal dementia and seizures et al., (2) history of clinically significant organ failure that made it difficult to participate in this study, (3) major depression, bipolar disorder and history of schizophrenia, (4) with acute or chronic immune-mediated inflammatory diseases in recent 6 months. The inclusion criteria for non-AD-related cognitive impairment were as follows: (1) individuals with cognitive complaints, (2) age 50–79 years; (3) without AD pathology detected by CSF or PET-CT, (4) CDR score of 0.5, 1, or 2, (5) availability of a caregiver, (6) written informed consent to participate in this study. The inclusion criteria for NC individuals were as follows: (1) age 50–79 years, (2) absence of cognitive symptoms, as assessed by a specialty physician, (3) CDR score of 0 at the first visit, and (4) written informed consent to participate in this study. The exclusion criteria of non-AD-related cognitive impairment and NC individuals were as follows: (1) history of clinically significant organ failure, such as cardiovascular symptoms, renal dysfunction, or hepatic dysfunction, (2) major depression, bipolar disorder and history of schizophrenia, (3) with acute or chronic immune-mediated inflammatory diseases in recent 6 months.
The neuropsychological assessment, brain MRI, lumbar puncture for AD core biomarkers test and/or 18F-florbetapir (AV45) PET imaging were performed on all participants. In accordance with the 2018 NIA-AA research framework, based on the biomarker profile and cognitive stage, individuals were classified as AD (Aβ+ AD, n = 37), MCI due to AD (Aβ+ MCI, n = 30), non-AD cognitive impairment (Aβ− CI, n = 34), and NC (Aβ− NC, n = 30). Aβ pathology positively (Aβ+) was defined by CSF Aβ42/Aβ40 ratio (cut-off value < 0.0642, n = 60) or AV45-PET (n = 61) imaging, which was elaborated in our recent work [28].
Blood collection, processing, and storage
Blood samples were collected and processed following standard protocols [30]. Regular procedures to obtain plasma and serum were established at the Institute on Aging and Brain Disorder. The blood samples were collected after overnight fasting, separately in EDTA-K2 tubes and serum separator tubes. They were then centrifuged (2000 g, 4 °C) for 10 min. Afterward, plasma and serum were separately aliquoted per 0.2 ml into 1.5 ml low protein binding tubes and stored at -80 °C within 3 h of collection and until measurements were taken.
CSF collection
Lumbar puncture was performed and CSF samples were obtained according to a previously described standardised operating procedure [31]. The CSF samples were collected in the morning in 15 ml sterile tubes after overnight fasting during the baseline inpatient period. They were then centrifuged (2000 g, 4 °C) for 10 min within 2 h and separately aliquoted per 0.2 ml into 1.5 ml low protein binding tubes. CSF aliquots were stored at -80 °C before analysis.
CSF and plasma core biomarker measurements
CSF and plasma Aβ42, Aβ40, total-tau (T-tau), and phospho-tau (181P) (P-tau) were analysed using commercially available Simoa kits (Quanterix, 101995, and 103714). All samples and protocols performed according to the manufacturer’s instructions and were conducted on the same HD-X analyser (Quanterix, USA) at the Neurodegenerative Disorders Research Centre of USTC.
Analysis of IFN-related cytokines
To investigate which subtypes of IFN and IFN-related cytokines were associated with the manifestations of AD, a multiplex panel were created, including IFNα-2a, IFN-β, IFN-γ, TNF-α, interleukin (IL)-6, IL-10, monocyte chemoattractant protein-1 (MCP-1), and C-X-C Motif Chemokine Ligand 10 (CXCL-10) using a multiplex MSD (Mesoscale discoveries) U-plex Biomarker Group 1 (human) assay (K15067L) [32, 33]. The paired CSF and serum from participants were prepared and analysed according to the manufacturer’s instructions, using the same MSD imager. IFN-α-2a in the CSF and serum was not counted because it was lower than the lower limit of detection (LLOD).
Analysis of brain magnetic resonance imaging
The baseline brain MRI of subjects was performed within 1 month after enrollment in CANDI cohort. The MRI protocol was elaborated in our recent work [28]. A three-dimensional (3D) Bravo sequence was processed to analyse the thickness of cortex and volume of brain regions using FreeSurfer 6.0 with standard protocol. Some of scans were excluded due to unqualified raw data. 109 available measures from 104 subjects were included in this study. The measures were listed in Supplementary Table S1.
Human brain samples
Frontal cortices of human brains from patients with AD were obtained from the Brain Bank of USTC, which applied according to the standard procedure approved by the Committee of USTC. The cases used in this study are summarized in Supplementary Table S2. The expression of IFN-β, IFNAR1 with amyloid plaques, and P-Tau (AT8) were detected by immunofluorescence staining and imaged by Confocal Laser Scanning microscope (ZEISS, LSM 800). Primary antibodies included anti-IFN-β (Invitrogen, PA5-20390, 1:200), anti-IFNAR1(Sigma, HPA018015, 1:200), anti-APP (Biolegend, SIG-39320, 1:1000), and anti-P-tau (Invitrogen, MN1020, 1:100) for immunofluorescence. Secondary antibodies used were Alexa Fluor donkey anti-mouse 555 and Alexa Fluor donkey anti-Rabbit 488 (Invitrogen) at a dilution of 1:1000.
The quantification analysis of IFN-β and IFNAR1 was detected by western blot according to standard procedures. Primary antibodies used for western blotting were IFNAR1 (Abcam, ab124764, 1:1000), IFN-β (Invitrogen, PA5-20390, 1:1000) and β-Actin (Proteintech, 66009, 1:1000). Secondaries used were anti-rabbit HRP (Cell signalling technology, 7074, 1:5000) and anti-mouse HRP (Cell signalling technology, 7076, 1:5000).
Statistical analysis
For each CSF and serum cytokine, the normality distribution was checked using the Shapiro-Wilk test and visual inspection via histograms. CSF Aβ42, T-tau, P-tau, Aβ42/Aβ40 ratio, IFN-related cytokines, plasma Aβ40, P-tau, T-tau, and serum TNF-α, IFN-β, IFN-γ, IL-6, IL-10, and CXCL-10 levels did not exhibit a normal distribution, so they were log10 transformed for analysis when needed. Plasma Aβ42 and Aβ42/Aβ40 ratio demonstrated a normal distribution. Outliers with indicator levels greater than 3 standard deviations above the mean were removed from the analysis.
Data are presented as median (interquartile range) or mean ± s.e.m unless otherwise indicated. All the tests were two-tailed, and p < 0.05 was considered statistically significant after adjustment for multiple comparisons. Details of the statistical analysis are provided in the corresponding Figure legends. Analyses were performed using SPSS v.25 (IBM) and Prism 8 (GraphPad). Data were visualised using either Prism 8 or R Studio.
Results
Participants’ characteristics
Table 1 summarizes the demographics, CSF, and plasma core biomarker levels of the participants. The APOE ε4 status prevalence varied between Aβ− (NC and CI) and Aβ+ (MCI and AD) groups, while the MMSE cognitive score decreased in Aβ− CI and Aβ+ groups compared to the Aβ− NC group. However, there were no differences in age, education, and sex distribution among the groups. As anticipated, significant differences were observed in the levels of CSF Aβ42, Aβ42/Aβ40 ratio, P-tau, T-tau, and plasma Aβ42, Aβ42/Aβ40 ratio, P-tau between the Aβ− (NC and CI) and Aβ+ (MCI and AD) groups.
Table 1.
Participants’ characteristics and AD core biomarkers among groups
| Aβ− NC n = 30 |
Aβ− CI n = 34 |
Aβ+ MCI n = 30 |
Aβ+ AD n = 37 |
p values | |
|---|---|---|---|---|---|
| Age, yr | 59 (54–65) | 62 (54–67) | 63 (57–71) | 61 (54–67) | 0.161 |
| Sex, F/M, n | 15/15 | 23/11 | 16/14 | 24/13 | 0.395 |
| Education, yr | 6.5 (4.5–10.3) | 5.5 (3–11) | 9 (7.7–14) | 8 (3–11) | 0.055 |
| APOE ε4 carriers | 2 (6.7%) | 3 (8.8%) | 16 (53.3%)a, b | 22 (59.5%)a, b | < 0.001* |
| MMSE | 27.5 (25–29) | 16 (4.5–21)a | 20 (13.3–23.2)a | 11 (4.8–15.5)a, c | < 0.001* |
| CSF biomarkers 1 | |||||
| Aβ42, pg/ml | 670 (482–871) | 479 (297–604) | 283 (237–337)a | 203 (158–246)a, b | < 0.001* |
| Aβ40, ng/ml | 10.3 (6.1–10.3) | 7.2 (3.6–7.2) | 8.6 (5.8–8.6) | 7.8 (3.7–7.8)a | 0.027* |
| Aβ42/Aβ40 |
0.095 (0.077–0.104) |
0.082 (0.071–0.092) |
0.042 (0.036–0.050)a, b |
0.040 (0.035–0.048)a, b |
< 0.001* |
| P-tau, pg/ml | 31 (28–40) | 35 (28–41) | 102 (79–147)a, b | 118 (94–150)a, b | < 0.001* |
| T-tau, pg/ml | 79 (63–99) | 88 (70–117) | 168 (121–255)a, b | 183 (122–238)a, b | < 0.001* |
| Plasma biomarkers 2 | |||||
| Aβ42, pg/ml | 11.9 (8.3–13.7) | 11.1 (7.9–12.1) | 9.1 (7.6–11.1)a | 9.7 (7.8–11.1)a | < 0.001* |
| Aβ40, pg/ml | 179 (155–199) | 188 (163–207) | 177 (163–218) | 191 (162–209) | 0.860 |
| Aβ42/Aβ40 |
0.067 (0.060–0.076) |
0.060 (0.053–0.068) |
0.048 (0.040–0.059)a, b |
0.053 (0.045–0.057)a, b |
< 0.001* |
| P-tau, pg/ml | 2.1 (1.5–2.6) | 1.9 (1.6–2.8) | 5.0 (2.7–7.3)a, b | 6.6 (4.4-8.0)a, b | < 0.001* |
| T-tau, pg/ml | 2.4 (2.0-2.9) | 2.5 (2.0-3.5) | 2.9 (2.4–4.4)a | 2.9 (2.4–3.6) | 0.038* |
Data are median (interquartile range) except sex and APOE ε4 carriers. p values are derived from ANCOVA adjusted for age and sex and chi-square (sex and APOE ε4 carriers). 1In CSF biomarker, Aβ42, Aβ40, Aβ42/Aβ40 were missing for six participants in Aβ−NC group, five in Aβ− CI group, one in Aβ+ MCI group, three in Aβ+ AD group; p-tau and t-tau were missing for seven participants in Aβ− NC group, three participant in Aβ−CI group, one participant in Aβ+ MCI group, two participants in Aβ+ AD group. 2In plasma biomarker, Aβ42, Aβ40, Aβ42/Aβ40 and p-tau were missing in one participant in Aβ− NC, Aβ− CI, Aβ+ MCI group. *Significant values. Abbreviations: NC, normal cognition; CI; cognitive impairment; F, female; M, male; yr, years; MMSE, Mini-Mental State Examination; P-tau, phosphorylated tau; T-tau, total tau; Aβ42, amyloid-β 42; Aβ40, amyloid-β 40
aSignificant values versus Aβ− NC group
bSignificant values versus Aβ− CI group
cSignificant values versus Aβ+ MCI group
Analysis of IFN subtypes and related cytokines in CSF and serum across groups
Figure 1 displays the differences in CSF IFN-related cytokine levels among groups. Significant differences were found in CSF IFN-β levels between the Aβ− and Aβ+ groups. Specifically, CSF IFN-β levels were significantly higher in the Aβ+ MCI (p = 0.005) and AD (p < 0.001) groups than in the Aβ− NC group. Additionally, the CSF IFN-γ level was significantly higher in Aβ+ AD group (p = 0.046) compared to the Aβ− CI group. Interestingly, IL-6 levels in the CSF (but not in the serum; Supplementary Fig. S3) were significantly elevated in the Aβ− CI individuals compared to Aβ− NC (p = 0.016), Aβ+ MCI (p = 0.016) and Aβ+ AD (p = 0.005) groups. No other cytokines in CSF and serum displayed significant changes between the Aβ− and Aβ+ groups.
Fig. 1.

Comparison of CSF IFN-related cytokines among groups. Dot and violin plots depicting the levels of each IFN-related cytokines in each group. Violin plots depict the median (horizontal bar) and interquartile range (IQR, dashed bars). p values are assessed by one-way analysis of covariance adjusted for age and sex, followed by Bonferroni corrected pairwise multiple comparisons
Relationship between IFN-related cytokines and AD core biomarkers in CSF and serum
To evaluate the impact of IFN-related cytokines on AD pathology, we analysed the association between IFN-related cytokines and AD core biomarkers in CSF (Fig. 2) and peripheral blood (Supplementary Fig. S4). In a partial correlation analysis adjusted for age, sex, and APOE ε4 carriers, among these cytokines, only CSF IFN-β showed a significant positive correlation with P-tau (r = 0.230, p = 0.017) and T-tau (r = 0.237, p = 0.013), and a negative correlation with Aβ42/Aβ40 ratio (r = -0.250, p = 0.010), indicating a connection between IFN-β and AD core pathologies. Additionally, CSF IL-6 and MCP-1 positively correlated with the Aβ42/Aβ40 ratio.
Fig. 2.

Correlation analysis of IFN-related cytokines with AD core biomarkers in the CSF. Partial correlations between IFN-related cytokines and AD core biomarkers in the CSF, corrected for age, sex, and APOE ε4 status. Red colour indicates negative correlation and blue indicates positive correlation. The colour intensity and size of the circle are proportional to the correlation coefficient (r). The crossed cells showed no significant correlations
Furthermore, we tested the correlation between serum IFN-related cytokines and plasma AD core biomarkers among the groups (Supplementary Fig. S4). We found that only serum IL-10 (r = 0.235, p = 0.010) and MCP-1 (r = 0.235, p = 0.009) positively correlated with plasma P-tau. These findings suggest that a peripheral AD core biomarker, plasma P-tau, may trigger (or link with) a different set of inflammatory markers (such as IL-10 and MCP-1).
Association of IFN-related cytokines with APOE ε4 carriers
Research has shown that APOE ε4 plays a critical role in neuroinflammation and neurodegeneration in Alzheimer’s disease [34]. To investigate the effect of APOE ε4 status on IFN-related cytokines, we conducted a linear regression analysis to evaluate the association of each IFN-related cytokine with APOE ε4 status (Supplementary Table S3). Interestingly, APOE ε4 carriers were significantly associated with CSF IFN-β (β = 0.194, p = 0.037), IFN-γ (β = 0.232, p = 0.014) and serum IFN-β (β = 0.204, p = 0.023), but not with the other IFN-related cytokines. As there is a significant association between APOE ε4 carriers and AD core biomarkers, and AD core biomarkers with CSF IFN-β, AD core biomarkers could be a mediator of the association between APOE ε4 and CSF IFN-β. Thus, we performed a mediation analysis with CSF Aβ42/Aβ40 ratio and P-tau as mediators. The effect of APOE ε4 carriers on CSF IFN-β was largely mediated by AD core biomarkers, while the direct effect of APOE ε4 carriers on CSF IFN-β was no longer significant (Supplementary Fig. S5A). Similarly, the effect of APOE ε4 status on CSF IFN-γ was partly mediated by AD core biomarkers (Supplementary Fig. S5B). Unlike CSF IFN-β and IFN-γ, the significant association of serum IFN-β with APOE ε4 status was not mediated by AD core biomarkers (Supplementary Fig. S5C). These results indicate that the observed differences in CSF IFN-β and IFN-γ with APOE ε4 status may be driven by AD pathology, while the difference in serum IFN-β with APOE ε4 status may be driven by other factors.
Association of IFN-related cytokines with cognitive performance
In the entire sample, only CSF IFN-β showed a significant negative correlation with MMSE score (β = -0.230, p = 0.011) (Fig. 3A). Furthermore, CSF IFN-β levels were significantly higher in cognitively impaired subjects (CDR ≥ 0.5) compared to those with normal cognition (CDR = 0) (Fig. 3B), suggesting that higher CSF IFN-β levels are associated with worse cognitive performance. When dividing the sample into Aβ− and Aβ+ groups, a significant negative correlation between CSF IFN-β and MMSE score (β = -0.473, p < 0.0001) was observed in the Aβ− groups. However, no significant relationship was found between CSF IFN-β and MMSE score in the Aβ+ group, which is consistent with the lack of difference in CSF IFN-β levels between Aβ+ MCI and Aβ+ AD groups. Additionally, in the Aβ− group, CSF IL-6 showed a significant negative correlation with MMSE score (β = -0.244, p = 0.003), while in the Aβ+ group, MCP-1 was significantly negatively correlated with MMSE score (β = -0.437, p = 0.002) (Fig. 3A).
Fig. 3.

Association of CSF IFN-related cytokines with cognitive performance. Scatter plots and line charts display the association of each IFN-related cytokines in the CSF with MMSE score (A) and CDR score (B) in all individuals. (A) Each point depicts the value of the IFN-related cytokines of an individual and the solid lines indicate the regression line for each index in Aβ− and Aβ+ groups. Red circle indicates Aβ− individuals and green indicates Aβ+ individuals. The standardized regression coefficients (β) and p values are shown and analyzed using a linear regression model adjusted for age, sex, APOE ε4 status and the years of education. (B) The levels of each IFN-related cytokines among subjects with different CDR score. Data are mean ± SEM. p value < 0.05 means significant
Moreover, serum TNF-α levels (β = -0.208, p = 0.015) were also negatively associated with MMSE score (Supplementary Fig. S6A), primarily in Aβ+ group (β = -0.244, p = 0.033). Mediation analysis suggested that the relationship between serum TNF-α and MMSE score was independent of the effect of CSF IFN-β (Supplementary Fig. S7A) and AD core biomarkers (Supplementary Fig. S7B) on MMSE score. Additionally, serum IL-10, an anti-inflammatory cytokine, was significantly higher in dementia subjects (CDR ≥ 1) compared to cognitively normal individuals (CDR = 0) (Supplementary Fig. S6B), which aligns with the previous finding that IL-10 was increased in Aβ+ AD group compared to the Aβ− NC group. These results suggest that changes in different types of cytokines may correspond to various forms of cognitive impairment, particularly CSF IFN-β and IL-6.
Association of CSF IFN-β and IL-6 with brain atrophy
Elevated CSF IFN-β levels were observed in Aβ+ MCI and AD groups, and were correlated with AD core biomarkers and cognitive performance as shown in Figs. 2 and 3. Since neuron loss related brain shrinkage is also a hallmark of AD, we examined the correlation between CSF IFN-β and brain MRI measures in these subjects using partial correlation analysis adjusting for age and sex (data not shown). We found that 13 brain MRI measures, such as thickness about posterior-dorsal part of the cingulate gyrus, supramarginal gyrus, superior parietal lobule, precuneus, hippocampus and amygdala (Fig. 4A), all negatively correlated with CSF IFN-β (Supplementary Table S4). Almost all of these cortical atrophies were significantly greater in Aβ+ MCI and AD individuals compared to Aβ− individuals, except for mid-anterior and anterior corpus callosum volume (Supplementary Fig. S8). As expected, we observed that CSF IFN-β was inversely associated with all selected measures in brain MRI using a linear regression model (Fig. 4B). Furthermore, mediation analysis suggested that the effect of CSF IFN-β on brain atrophy (such as hippocampus and amygdala) was largely mediated by AD core biomarkers (P-tau and Aβ42/Aβ40 ratio) (Supplementary Fig. S9). These findings indicate that increased CSF IFN-β is involved in AD pathology, leading to distinct atrophy.
Fig. 4.

Association of measures of interest in brain MRI with CSF IFN-β. (A) Pial view of the regions of measures of interest associated with CSF IFN-β in brain from different perspective. (B) Scatter plots representing the association of each measure of interest in brain MRI with CSF IFN-β in all individuals. Each point depicts the value of the measures of an individual and the solid lines indicate the regression line for each index. The standardised regression coefficients (β) and p values are shown and analysed using a linear regression model adjusted for age and sex. p value < 0.05 means significant
Additionally, CSF IL-6 levels were elevated in the Aβ− CI group and correlated with cognitive performance. Similar analysis was performed to examine the correlation between CSF IL-6 and brain MRI measures using the above method. Eight brain MRI measures, such as thickness of thalamus, pallidum, precentral gyrus and volume of white matter hyperintensity, were correlated with CSF IL-6 (Supplementary Table S5). Interestingly, these measures distinct from those correlating with IFN-β, partially align with the MRI markers indicative of vascular cognitive impairment [35, 36], such as whiter matter hyperintensities and thalamus atrophy. CSF IL-6 was significantly associated with these measures using a linear regression analysis (Supplementary Fig. S10). Unlike CSF IFN-β, mediation analysis found that the effect of CSF IL-6 on these measures was independent of AD core biomarkers (Supplementary Fig. S11).
Pathological association of IFN-β and IFNAR1 in AD brain tissues
Our analysis revealed that CSF IFN-β is significantly associated with AD markers, cognitive decline, and cortical atrophy. To further confirm the expression and distribution of IFN-β in the brains, we examined the expression of IFN-β and IFNAR1 proteins with typical AD pathology, amyloid plaques (6E10) and p-tau (Ser202 and Thr205, AT8), to assess the expression of these proteins in autopsied brain tissues with AD. Alongside the labelling of plaques and neurofibrillary tangles, we observed widespread IFN-β expression in AD brains, especially in some tangles and plaques regions (Fig. 5A) and overexpressed in AD brains (Fig. 5C-D) compared to non-AD brains. Additionally, IFNAR1 was extensively expressed in both non-AD patient control brains and AD patient brains, particularly in neurofibrillary tangles regions and plaques regions (Fig. 5B), but without difference in expression between groups (Fig. 5C-D). This is consistent with IFN pathway activation in human AD brains [8]. Collectively, these data suggest that the activation of IFN-β signalling pathway may represent a prominent feature of neuroinflammation in AD patients.
Fig. 5.
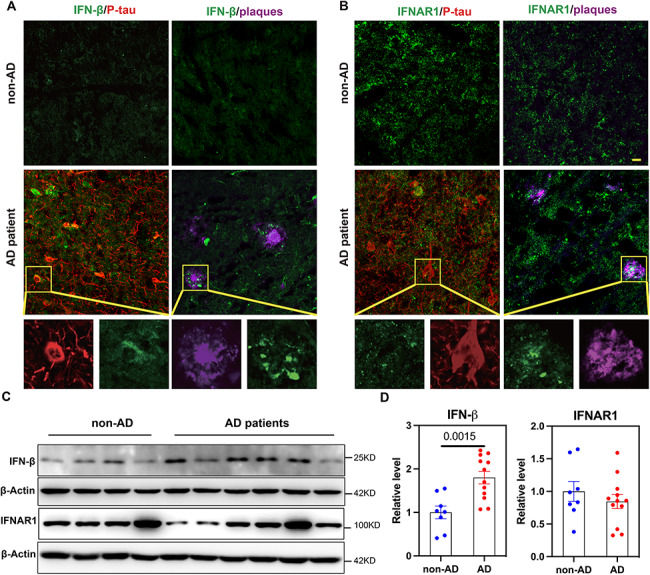
Expression of IFN-β and IFNAR1 in AD patients’ brain tissues. Examination of IFN-β and IFNAR1 in AD and non-AD patients’ brain specimens. Amyloid plaques are stained with Aβ (6E10, magenta) and neurofibrillary tangles with P-tau (AT8, red). Representative amplified confocal images of (A) IFN-β (green) widespread expression in neurofibrillary tangle and plaque regions; (B) IFNAR1 (green) expressed in neurofibrillary tangle and plaque regions; Scale bar: 20 𝛍m. (C) Western blot for IFN-β, IFNAR1 and β-Actin using brain tissue lysates from AD patients (n = 12) and non-AD patients (n = 8). (D) Ratio of IFN-β and IFNAR1 to β-Actin from C. Data are reported as mean ± SEM. Data are analyzed by two-tailed unpaired t-test
Discussion
In this study, we aimed to identify changes in IFN-related cytokines in AD patients using a U-Plex MSD multiplex panel. This was the first study to demonstrate that the main subtype of IFNs, IFN-β, is significantly higher in the CSF of Aβ+ MCI and AD patients compared to cognitively normal Aβ− individuals. Additionally, we demonstrated that CSF IFN-β levels significantly correlate with CSF AD core biomarker levels. Interestingly, we found that CSF IFN-β levels are inversely associated with cognitive performance and AD specific brain atrophy in MRI scans. Consistently, we observed upregulated levels of IFN-β in post-mortem brain tissue of AD patients, particularly in regions with amyloid plaques and neurofibrillary tangles. Overall, our study provides pathological and clinical evidence suggesting that CSF IFN-β may contribute to AD pathology and disease progression.
Prior research has explored the role of type I IFNs in cognitive function in various diseases [4, 5, 8, 37, 38]. Several studies have shown elevated type I IFNs signalling in amyloid related AD models [5, 8], P301S-tau mouse models [38, 39], Down syndrome brain organoids [37] and ageing mice [4], which aligns with our findings of increased IFN-β levels in the brains of AD patients. This elevation might result from abnormal double-stranded DNA accumulation due to disease-induced mitochondrial damage, activating the cGAS-STING-type I interferon pathway [4], promoting neuroinflammation, synaptic loss, and cognitive impairment. Suppressing type I IFNs pathway activation through inhibiting IFNAR or Ifnar1 deletion [10, 38], blocking STING or Cgas deletion [5], ameliorating the synaptic loss and cognitive impairment. Inhibiting age-induced type I IFN-dependent gene expression in the choroid plexus promoted neurogenesis and partially rescued cognitive function [25]. Furthermore, other forms of cognitive impairment have been linked to microbiota-gut-brain axis dysregulation driven by microbiome-dependent intestinal IFN-γ from Th1 cells of the peripheral system [40]. Studies in animal models and our patients highlight that chronic type I IFNs signalling plays a role in cognitive impairment, indicating its potential as a therapeutic target.
In this study, we also measured several pro-inflammatory (TNF-α, IL-6, MCP-1, and CXCL-10) and anti-inflammatory (IL-10) cytokines that can be regulated by IFNs [41]. However, we found no difference in the expression levels of these cytokines between the Aβ+ and Aβ− groups, except for higher CSF IL-6 in the Aβ− CI group compared to Aβ− NC and Aβ+ groups.
Higher CSF IL-6 levels were observed in the Aβ− CI group compared to Aβ− NC and Aβ+ groups, which is consistent with previous findings [42–44]. Longitudinal cohort studies have indicated an association between elevated plasma IL-6 levels and cognitive decline over a 10-year fellow-up period [42]. Our data also showed that CSF IL-6 negatively correlated with cognition in non-AD individuals and was associated with some brain MRI features of VCI/VaD. This is consistent with previous studies that found high levels of IL-6 can cause dysfunction in vascular endothelium [45], higher WMH volumes [46, 47], involvement in the progression of VCI [48, 49]. However, a meta-analysis [50] and recent population-based cohort studies revealed that IL-6 increase the risk of all-cause dementia [51]. Therefore, the cause effect of IL-6 levels on VCI and other types of dementia should be further validated in high-quality longitudinal cohort and basic experimental studies.
APOE, the strongest genetic risk factor for AD, has been linked to different inflammatory responses in APOE ε4 carriers and noncarriers [52]. Our data showed that CSF IFN-β, IFN-γ and serum IFN-β levels were significantly higher in APOE ε4 carriers than in other genotypes. However, including AD core pathology biomarkers as mediators altered the results, suggesting that the observed differences in CSF IFN-β and IFN-γ between APOE ε4 carriers and non-carriers are driven by Aβ and tau pathology. This aligns with studies showing that APOE ε4 carriers alone doesn’t directly cause neurological damage in AD brain organoids, its interplay with AD status accelerates AD pathological changes [53]. In contrast, changes in serum IFN-β with APOE genotypes were not affected by Aβ or tau-related biomarkers, indicating that this difference was independent of AD pathology. Studies have established a significant association between APOE ε4 and hypercholesterolemia [54]. However, APOE gene knockout-induced hypercholesterolemia still activates the cGAS-STING-type I interferon pathway [55], implying that variations in serum IFN-β with APOE genotypes might be related to hypercholesterolemia. Further studies on the relationship between periphery lipids and IFN-β could strengthen these hypothesis.
We also examined the relationships between IFN-related cytokines and cognition performance using MMSE and CDR scores. After adjusting for age, sex, APOE ε4 status, and education years, we found an inverse association between CSF IFN-β and cognitive performance. The association is consistent with finding from studies animal model studies, which showed that type I IFN signalling activation occurs in AD mouse models [10, 26], while blocking IFNAR can rescue memory and synaptic deficits [10]. The subgroup analysis revealed that CSF IFN-β levels were significantly higher in the Aβ+ group than Aβ− group. However, nonsignificant correlation was observed between CSF IFN-β and MMSE score in Aβ+ group. This aligns with our previous findings, which showed nonsignificant difference in CSF IFN-β levels between Aβ+ MCI and AD groups. These results indicated that while CSF IFN-β may be implicated in cognitive impairment related to AD, it does not significant link to disease progression.
In addition, we found a significant negative correlation between CSF IFN-β levels and MMSE score in the Aβ− group, implying that chronic IFN-β activation may contribute to cognitive decline in non-AD patients. These findings align with recent research indicating that long-term activation of interferon signalling in the brain during aging may lead to cognitive deficits [4]. These observations suggest that CSF IFN-β may play a part in the pathological processes underlying various types of cognitive impairment. Additional studies comparing the activation of interferon signalling pathways across cognitive disorders and exploring pathological mechanisms should be carried out.
We observed an inverse association between CSF IFN-β and typical cortical atrophy in AD patients, such as superior parietal lobule, hippocampus and amygdala, which were similar regions of atrophy to the typical AD atrophy subtype reported in the literature [56]. Moreover, recent finding demonstrated that imaging of cortical neuroinflammation was positively associated with structural and functional network disruption [57]. In contrast, CSF IL-6 is associated with motor cortex atrophy, thalamus and pallidum volume, and white matter hyperintensity volume. These associations are comparable to findings from community volunteers, showing higher peripheral inflammation (IL-6) associated with lower cortical gray and white matter, hippocampus volumes [43].
Our study identified subtypes of interferon and related cytokines in Aβ+ MCI and AD patients. We discovered that CSF IFN-β is causally associated with AD pathology and cognitive impairment. In line with our CSF findings, IFN-β was primarily upregulated, especially in regions near amyloid plaques and neurofibrillary tangles in post-mortem AD brain tissues. This finding suggests that CSF IFN-β may be involved in AD-related cortical atrophy, accelerating cognitive impairment. The monoclonal antibody antagonist of IFNAR, anifrolumab, has been approved for the treatment of systemic lupus erythematosus [58] but have not yet been tested in AD patients. Therefore, we can conduct exploratory clinical trials to evaluate whether inhibiting the type I interferon response could improve AD markers and cognition.
The study has certain limitations. To minimize the impact of confounding factors, we strictly screened the participants in our CANDI cohort. However, the subgroups in this cross-sectional study were relatively small. Further research is needed to confirm the role of CSF IFN-β in AD pathogenesis using large populations and longitudinal cohorts. Additionally, it is crucial to identify the specific cells that contain and secrete high levels of IFN-β. A thorough investigation into how IFN-β impacts AD pathology, such as plaque formation and tau phosphorylation, is also essential for future research.
Our study pinpointed IFN-β as the primary isoform of interferon in the brain and CSF related to AD. We demonstrated that CSF IFN-β level was significantly higher in Aβ+ MCI and AD patients. Importantly, we discovered that IFN-β levels are associated with AD core pathology, cognitive performance, and brain atrophy. Our findings support the hypothesis of targeting the type I IFN response, particularly the IFN-β signalling pathway, in AD patients with high CSF IFN-β concentrations to potentially reverse or slow down cognitive decline.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
This publication is part of the China Aging and Neurodegenerative Initiative (CANDI) study. The authors would like to express their most sincere gratitude to the CANDI project participants. Collaborators of the CANDI study include: Feng Gao, Xinyi Lv, Linbin Dai, Qiong Wang, Peng Wang, Zhaozhao Cheng, Qiang Xie, Ming Ni, Yan Wu, Xianliang Chai, Wenjing Wang, Huaiyu Li, Feng Yu, Yuqin Cao, Fang Tang, Bo Pan, Guoping Wang, Kexue Deng, Shicun Wang, Qiqiang Tang, Jiong Shi, Yong Shen.
Abbreviations
- AD
Alzheimer’s disease
- ANCOVA
One-way analysis of covariance
- CANDI
The China Aging and Neurodegenerative Initiative
- CDR
Clinical dementia rating
- CI
Cognitive impairment
- NC
Normal cognition
- CXCL-10
C-X-C Motif Chemokine Ligand 10
- DLB
Dementia with Lewy bodies
- FTD
Frontotemporal dementia
- IFN
Interferon
- IFNAR
Interferon I receptor
- IL
Interleukin
- ISGs
IFN-stimulated genes
- LLOD
Lower limit of detection
- LPB
Low protein binding
- MCI
Mild cognitive impairment
- MCP-1
Monocyte chemoattractant protein-1
- MSD
Mesoscale Discovery
- NIA-AA
National Institute on Aging-Alzheimer’s Association
- TNF
Tumour necrosis factor
- USTC
University of Science and Technology of China
- VaD
Vascular dementia
Author contributions
WQ contributed to the study design and conception of the presented idea; analyzed and interpreted the data; drafted the manuscript. YSF, WCX, HDT, ZMG and ZYX are major roles in the acquisition of data. GF is major role in revision of the manuscript. SJ is major role in revision of the manuscript for content. LAI contributed to the conception of the presented idea and revised of the manuscript. SY contributed to the study design and conception of the presented idea; drafted the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (92149304, 82030034); the Chinese Academy of Science (XDB39000000); Anhui Provincial Key R&D Project (202304295107020059); Natural Sciences Foundation of Anhui Province (1908085QH321).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval
This study was approved by the Medical Ethics Committee of the First Affiliated Hospital of the USTC in Hefei, China (2019KY-26). All participants provided written informed consent for participating in the study. The Clinical Trial Number: (ChiCTR1900024777||http://www.chictr.org.cn/) with the Clinical Trial Registry (July 27, 2019).
Consent for publication
No applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Qiong Wang, Email: qiongw@ustc.edu.cn.
Yong Shen, Email: yongshen@ustc.edu.cn.
References
- 1.Collaborators GF. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7(2):e105–25. 10.1016/S2468-2667(21)00249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, Cummings J. Flier Alzheimer’s disease Lancet. 2021;397(10284):1577–90. 10.1016/S0140-6736(20)32205-4. van der. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gulen MF, Samson N, Keller A, Schwabenland M, Liu C, Gluck S, et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature. 2023;620(7973):374–80. 10.1038/s41586-023-06373-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie X, Ma G, Li X, Zhao J, Zhao Z, Zeng J. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5xFAD mice. Nat Aging. 2023;3(2):202–12. 10.1038/s43587-022-00337-2. [DOI] [PubMed] [Google Scholar]
- 6.Udeochu JC, Amin S, Huang Y, Fan L, Torres ERS, Carling GK, et al. Gan. Tau activation of microglial cGAS-IFN reduces MEF2C-mediated cognitive resilience. Nat Neurosci. 2023;26(5):737–50. 10.1038/s41593-023-01315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai L, Wang Q, Lv X, Gao F, Chen Z, Shen Y. Elevated beta-secretase 1 expression mediates CD4(+) T cell dysfunction via PGE2 signalling in Alzheimer’s disease. Brain Behav Immun. 2021;98:337–48. 10.1016/j.bbi.2021.08.234. [DOI] [PubMed] [Google Scholar]
- 8.Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020;130(4):1912–30. 10.1172/jci133737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Udeochu JC, Amin S, Huang Y, Fan L, Torres ERS, Carling GK, et al. Tau activation of microglial cGAS-IFN reduces MEF2C-mediated cognitive resilience. Nat Neurosci. 2023. 10.1038/s41593-023-01315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy ER, Chiu G, Li S, Propson NE, Kanchi R, Wang B, Coarfa C, Zheng H. Cao. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid beta plaques. Immunity. 2022. 10.1016/j.immuni.2022.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hur JY, Frost GR, Wu X, Crump C, Pan SJ, Wong E, et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature. 2020;586(7831):735–40. 10.1038/s41586-020-2681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR. Stark. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6(12):975–90. 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. 2019;20(12):1574–83. 10.1038/s41590-019-0466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubel P, Urban C, Bergant V, Schneider WM, Knauer B, Stukalov A, et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat Immunol. 2019;20(4):493–502. 10.1038/s41590-019-0323-3. [DOI] [PubMed] [Google Scholar]
- 15.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45. 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazear HM, Schoggins JW, Diamond MS. Shared and Distinct Functions of Type I and Type III Interferons. Immunity. 2019;50(4):907–23. 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore Z, Mobilio F, Walker FR, Taylor JM, Crack PJ. Abrogation of type-I interferon signalling alters the microglial response to Aβ(1–42). Sci Rep. 2020;10(1):3153. 10.1038/s41598-020-59917-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minter MR, Moore Z, Zhang M, Brody KM, Jones NC, Shultz SR, et al. Deletion of the type-1 interferon receptor in APPSWE/PS1∆E9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol Commun. 2016;4(1):72. 10.1186/s40478-016-0341-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue F, Tian J, Yu C, Du H, Guo L. Type I interferon response-related microglial Mef2c deregulation at the onset of Alzheimer’s pathology in 5×FAD mice. Neurobiol Dis. 2021;152:105272doi. 10.1016/j.nbd.2021.105272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones RS, Minogue AM, Fitzpatrick O, Lynch MA. Inhibition of JAK2 attenuates the increase in inflammatory markers in microglia from APP/PS1 mice. Neurobiol Aging. 2015;36(10):2716–24. 10.1016/j.neurobiolaging.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170(2):680–92. 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging. 2014;35(5):1012–23. 10.1016/j.neurobiolaging.2013.10.089. [DOI] [PubMed] [Google Scholar]
- 23.Minter MR, Main BS, Brody KM, Zhang M, Taylor JM, Crack PJ. Soluble amyloid triggers a myeloid differentiation factor 88 and interferon regulatory factor 7 dependent neuronal type-1 interferon response in vitro. J Neuroinflammation. 2015;12:71. 10.1186/s12974-015-0263-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279(47):49523–32. 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- 25.Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346(6205):89–93. 10.1126/science.1252945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mesquita SD, Ferreira AC, Gao F, Coppola G, Geschwind DH, Sousa JC, et al. The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer’s disease. Brain Behav Immun. 2015;49:280–92. 10.1016/j.bbi.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023. 10.1038/s41586-023-05788-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao F, Lv X, Dai L, Wang Q, Wang P, Cheng Z, et al. A combination model of AD biomarkers revealed by machine learning precisely predicts Alzheimer’s dementia: China Aging and Neurodegenerative Initiative (CANDI) study. Alzheimer’s & Dementia; 2022. 10.1002/alz.12700. [DOI] [PubMed]
- 29.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al Contributors, NIA-AA Research Framework. Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–62. 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Bryant SE, Gupta V, Henriksen K, Edwards M, Jeromin A, Lista S, et al. and B.w. groups. Guidelines for the standardization of preanalytic variables for blood-based biomarker studies in Alzheimer’s disease research. Alzheimers Dement. 2015;11(5):549–60. 10.1016/j.jalz.2014.08.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattsson N, Andreasson U, Persson S, Arai H, Batish SD, Bernardini S, et al. The Alzheimer’s Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011;7(4):386–95. 10.1016/j.jalz.2011.05.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nawroth JC, Petropolis DB, Manatakis DV, Maulana TI, Burchett G, Schlunder K, et al. Modeling alcohol-associated liver disease in a human Liver-Chip. Cell Rep. 2021;36(3):109393. 10.1016/j.celrep.2021.109393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng N, Capelle CM, Baron A, Kobayashi T, Cire S, Tslaf V, et al. DJ-1 depletion prevents immunoaging in T-cell compartments. EMBO Rep. 2022;23(3):e53302. 10.15252/embr.202153302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang ZH, Xia Y, Liu P, Liu X, Edgington-Mitchell L, Lei K, Yu SP, Wang XC, Ye K. ApoE4 activates C/EBPbeta/delta-secretase with 27-hydroxycholesterol, driving the pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2021;202:102032doi. 10.1016/j.pneurobio.2021.102032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Flier WM, Skoog I, Schneider JA, Pantoni L, Mok V, Chen CLH. Scheltens. Vascular cognitive impairment. Nat Rev Dis Primers. 2018;4:18003doi. 10.1038/nrdp.2018.3. [DOI] [PubMed] [Google Scholar]
- 36.Tan L, Xing J, Wang Z, Du X, Luo R, Wang J, Zhao J, Zhao W, Yin C. Study of gray matter atrophy pattern with subcortical ischemic vascular disease-vascular cognitive impairment no dementia based on structural magnetic resonance imaging. Front Aging Neurosci. 2023;15:1051177. 10.3389/fnagi.2023.1051177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin M, Xu R, Wang L, Alam MM, Ma Z, Zhu S, et al. Type-I-interferon signaling drives microglial dysfunction and senescence in human iPSC models of Down syndrome and Alzheimer’s disease. Cell Stem Cell. 2022;29(7):1135–53. 10.1016/j.stem.2022.06.007. e8.doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanford SAI, Miller LVC, Vaysburd M, Keeling S, Tuck BJ, Clark J, et al. The type-I interferon response potentiates seeded tau aggregation and exacerbates tau pathology. Alzheimers Dement. 2024;20(2):1013–25. 10.1002/alz.13493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naguib S, Torres ER, Lopez-Lee C, Fan L, Bhagwat M, Norman K, et al. Gan. APOE3-R136S mutation confers resilience against tau pathology via cGAS-STING-IFN inhibition. bioRxiv, 2024.10.1101/2024.04.25.591140
- 40.Olson CA, Iniguez AJ, Yang GE, Fang P, Pronovost GN, Jameson KG, et al. Alterations in the gut microbiota contribute to cognitive impairment induced by the ketogenic diet and hypoxia. Cell Host Microbe. 2021;29(9):1378–e13926. 10.1016/j.chom.2021.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolivar S, Anfossi R, Humeres C, Vivar R, Boza P, Munoz C, Pardo-Jimenez V, Olivares-Silva F. Diaz-Araya. IFN-beta Plays Both Pro- and Anti-inflammatory Roles in the Rat Cardiac Fibroblast Through Differential STAT Protein Activation. Front Pharmacol. 2018;9:1368doi. 10.3389/fphar.2018.01368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh-Manoux A, Dugravot A, Brunner E, Kumari M, Shipley M, Elbaz A, Kivimaki M. Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology. 2014;83(6):486–93. 10.1212/WNL.0000000000000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marsland AL, Gianaros PJ, Kuan DC, Sheu LK, Krajina K, Manuck SB. Brain morphology links systemic inflammation to cognitive function in midlife adults. Brain Behav Immun. 2015;48:195–204. 10.1016/j.bbi.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kipinoinen T, Toppala S, Rinne JO, Viitanen MH, Jula AM, Ekblad LL. Association of Midlife Inflammatory Markers With Cognitive Performance at 10-Year Follow-up. Neurology. 2022;99(20):e2294–302. 10.1212/WNL.0000000000201116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang S, Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. 2021;53(7):1116–23. 10.1038/s12276-021-00649-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satizabal CL, Zhu YC, Mazoyer B, Dufouil C, Tzourio C. Circulating IL-6 and CRP are associated with MRI findings in the elderly: the 3 C-Dijon Study. Neurology. 2012;78(10):720–7. 10.1212/WNL.0b013e318248e50f. [DOI] [PubMed] [Google Scholar]
- 47.Gertje EC, Janelidze S, van Westen D, Cullen N, Stomrud E, Palmqvist S, Hansson O. Mattsson-Carlgren. Associations Between CSF Markers of Inflammation, White Matter Lesions, and Cognitive Decline in Individuals Without Dementia. Neurology. 2023;100(17):e1812–24. 10.1212/WNL.0000000000207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Custodero C, Ciavarella A, Panza F, Gnocchi D, Lenato GM, Lee J, Mazzocca A, Sabba C, Solfrizzi V. Role of inflammatory markers in the diagnosis of vascular contributions to cognitive impairment and dementia: a systematic review and meta-analysis. Geroscience. 2022;44(3):1373–92. 10.1007/s11357-022-00556-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmitz M, Hermann P, Oikonomou P, Stoeck K, Ebert E, Poliakova T, Schmidt C, Llorens F, Zafar S. Zerr. Cytokine profiles and the role of cellular prion protein in patients with vascular dementia and vascular encephalopathy. Neurobiol Aging. 2015;36(9):2597–606. 10.1016/j.neurobiolaging.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 50.Darweesh SKL, Wolters FJ, Ikram MA, de Wolf F, Bos D, Hofman A. Inflammatory markers and the risk of dementia and Alzheimer’s disease: A meta-analysis. Alzheimers Dement. 2018;14(11):1450–9. 10.1016/j.jalz.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 51.Zhao Z, Zhang J, Wu Y, Xie M, Tao S, Lv Q, Wang Q. Plasma IL-6 levels and their association with brain health and dementia risk: A population-based cohort study. Brain Behav Immun. 2024;120:430–8. 10.1016/j.bbi.2024.06.014. [DOI] [PubMed] [Google Scholar]
- 52.Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30(9):1350–60. 10.1016/j.neurobiolaging.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao J, Fu Y, Yamazaki Y, Ren Y, Davis MD, Liu CC, et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat Commun. 2020;11(1):5540. 10.1038/s41467-020-19264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lumsden AL, Mulugeta A, Zhou A, Hypponen E. Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine. 2020;59:102954doi. 10.1016/j.ebiom.2020.102954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pham PT, Fukuda D, Nishimoto S, Kim-Kaneyama JR, Lei XF, Takahashi Y, et al. Sata. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur Heart J. 2021;42(42):4336–48. 10.1093/eurheartj/ehab249. [DOI] [PubMed] [Google Scholar]
- 56.Poulakis K, Pereira JB, Muehlboeck JS, Wahlund LO, Smedby O, Volpe G, et al. Japanese Alzheimer’s Disease Neuroimaging, B. Australian Imaging, and s. Lifestyle. Multi-cohort and longitudinal Bayesian clustering study of stage and subtype in Alzheimer’s disease. Nat Commun. 2022;13(1):4566. 10.1038/s41467-022-32202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leng F, Hinz R, Gentleman S, Hampshire A, Dani M, Brooks DJ, Edison P. Neuroinflammation is independently associated with brain network dysfunction in Alzheimer’s disease. Mol Psychiatry. 2023;28(3):1303–11. 10.1038/s41380-022-01878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deeks ED, Anifrolumab. First Approval Drugs. 2021;81(15):1795–802. 10.1007/s40265-021-01604-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.