

Abstract
Background
As a globally farmed oyster species, Magallana gigas has garnered significant attention due to the contaminated RNA viruses that have caused illness in humans. However, limited knowledge is available on the bioaccumulation status and overall diversity of RNA virome in the M. gigas digestive tissues (DTs). Moreover, there is a lack of understanding regarding the shared community of RNA virome among intertidal mollusks. To address these knowledge gaps, we performed a comprehensive meta-transcriptomic analysis of 173 M. gigas samples from the East China Sea and compared the viral sequences to the meta-transcriptomes of other mollusks and seawater (i.e., the oyster Magallana hongkongensis, bivalves, gastropods, cephalopods, and Yangshan Harbor) through RdRP identification and reads mapping.
Results
Our results indicate that 154 viral RdRPs were confidently identified in the M. gigas DT, with 94% (144/154) showing less than 90% amino acid identity. This indicates the presence of at least 144 putative novel RNA virus species in M. gigas DT. All viruses belonged to the phyla Lenarviricota, Pisuviricota, and Kitrinoviricota, and the marna-like viruses constituted the most diverse assemblage among these newly identified viruses. Furthermore, members of marna-like, picobirna-like, and noda-like virus groups comprise the most prevalent viruses in the M. gigas meta-transcriptome, with 14 RdRP-bearing sequences accounting for at least 1% of the overall aligned reads. M. hongkongensis has been found to harbor the most diverse viruses found in M. gigas, while 37 and 25 newly identified RNA viruses in oysters were discovered in an octopus meta-transcriptome and seawater virome, respectively.
Conclusions
The findings provide insights into the cryptic and hitherto unstudied virus-sharing network across the marine food web. Moreover, the viruses identified in oysters have the potential to serve as indicators for identifying the circulation of marine RNA viruses.
Video Abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s40168-024-01967-x.
Keywords: Magallana gigas, Meta-transcriptome, RNA virus, Diversity, Virus-sharing network
Background
RNA viruses play a significant role in marine environments [1], comprising up to 50% of total counts in ocean virioplankton [2]. The diversity of the RNA virosphere has expanded significantly in the age of metagenomics [3, 4], including in the marine environments. As of this writing, the International Committee on Taxonomy of Viruses (ICTV) has documented 6712 viral species of Riboviria belonging to 139 families with complete or complete coding-region genomes (https://ictv.global/). The persistence of specific RNA viruses in estuarine water has been observed to reach up to 21 days [5], providing an opportunity for further investigation into the circulation of marine RNA viruses in natural environments.
Although the number of RNA-dependent RNA polymerases (RdRPs) of invertebrate-associated RNA viruses [6] is comparatively limited in comparison to those present in subsurface ocean and polar environments [1], the significance of the virome harbored by these organisms should not be underestimated. Invertebrates are considered to be ancient lineages and represent the most diverse ecological group in the ocean [7]. Moreover, RNA viruses identified in invertebrates exhibit sufficient divergence to support the classification of five major phyla within the Riboviria realm [6, 8, 9], and novel lineages of invertebrate RNA viruses have been ratified by ICTV [10]. Nevertheless, as the cupped oyster M. gigas (previously known as Crassostrea gigas [29], https://www.marinespecies.org/aphia.php?p=taxdetails&id=138297) is one of the most prevalent aquaculture species in bivalves, the phylogenetic diversity and the relatively abundance of RNA viruses in these oysters remain poorly understood.
The oyster is capable of efficiently pumping seawater through its gills at a rate of 5 L/h [11]. Previous studies have demonstrated the presence of a diverse RNA virome and replication-associated protein (Rep)-encoding single-stranded (CRESS) DNA viruses in oysters [12, 13]. It is noteworthy that oysters have been observed to tolerate a final concentration of 1010 of reovirus [14], which demonstrates their capacity to accumulate virus particles from marine environments. It has been documented that oysters are frequently associated with human noroviruses (NoVs) worldwide, as evidenced by studies conducted in various locations, including [15–19]. Extensive research efforts have confirmed that histo-blood group antigens like (HBGAs-like) substances secreted by oysters and bacteria residing in their guts actively bind with NoVs [20–23]. Apart from active binding of RNA viruses, we are intrigued by the potential benefits of oyster bioaccumulation for passively enriched viruses such as the family Marnaviridae, which target marine eukaryotic microalgae and fungoid protists [24–27]. Current investigations into oyster-associated viromes are primarily focused on the identification of DNA viromes [13, 28]. The role of oysters in the transmission or circulation of RNA viruses and their ecological connections, as revealed by the virus-sharing network among intertidal mollusks, may provide insights into the potential exchanges of RNA virome between oysters and their predators and prey.
Here, we present a comprehensive study involving the analysis of a meta-transcriptomic dataset derived from 173 oyster digestive tissues collected in distinct batches over the course of a year. By identifying RNA viruses within the virome of M. gigas, we have established an analytical description of the RNA viromes associated with M. gigas, which we have compared to data publicly available for marine mollusk species from the classes of Bivalvia (e.g., M. hongkongensis [28]), Gastropoda, and Cephalopoda [6], and for the seawater virome of Yangshan Harbor, Shanghai, China, which has documented nearly 4500 novel marine RNA viruses [30]. The investigation yielded evidence indicating the existence of a shared RNA virosphere between mollusks and their surrounding aquatic environments. These findings significantly contribute to our understanding of the diversity of RNA viruses harbored by filter-feeding bivalves and provide a foundation for investigating the circulation and enrichment of the RNA viruses in intertidal mollusks.
Methods
Collection and processing of oyster samples
A total of 173 oysters of farmed origin (M. gigas, approximately 150 g each) were purchased from a seafood market in Luchao Port, Shanghai, China, between June 2016 and July 2017. Following the harvesting process, the oysters were stored at 5 to 10 °C for 1 to 2 weeks prior to their transportation to the market. Further details regarding the oysters sampled in each batch are provided in Table S1. Given the nature of filter-feeding behavior observed in oysters, the digestive tissues (DTs), which exclusively encompass the digestive gland, were considered to represent a reservoir for viruses [31, 32]. The dissection of the DTs was completed within a 2-h window following the initial sampling. Following this, the samples were preserved in RNA stabilizer (RNAlater, ThermoFisher, USA) and stored at − 80 °C until use.
RNA extraction and sequencing
Before RNA extraction, the DTs of M. gigas collected over the course of 1 year were combined and processed in a FastPrep-24 (MP, USA) homogenizer with two rounds of homogenization (6 m/s, 30 s). Subsequently, total RNA was extracted using TRIzol (Invitrogen, USA) and purified with a total RNA rapid extraction kit (cat. GK3015, Generay, China). The NEBNext Ultra Directional RNA Library Prep Kit (cat. E7420, Illumina, USA) was utilized for the construction of library with a preference for positive-sense RNA, employing a random primer. The prokaryotic ribosomal RNA (rRNA) was removed using the Ribo-Zero rRNA Removal Kit (cat. MRZMB126, Illumina, USA). Subsequently, three repetitive libraries were established and subjected to sequencing on the Illumina HiSeq platform (Illumina, USA), resulting in three datasets with 150-bp paired-end reads.
Contig assembly and ORF annotation
Adapters and low-quality reads were trimmed using Fastp (v0.23.2) [33] in default parameters, with the exception that the Phred quality threshold was set to 20. The M. gigas reference genome (10 chromosomes, NC_047559.1 to NC_047568.1) was downloaded from GenBank. Oyster-related reads were then removed by using an alignment-based tool, HoCoRT (v1.0.0) [34], with bowtie2 (v.2.5.1) [35] mode enabled. The qualified reads were then assembled by MEGAHIT (v1.2.9) [36], with contigs > 1000 nt retained (option –min-contig-len 1000). Prodigal (v2.6.3) [37] was employed for open reading frame (ORF) prediction with the standard genetic code (parameters: -n -p meta).
RNA virus identification
The recently published identification methods, parameters, and pre-compiled hidden Markov model (HMM) of the viral RdRP domain profiles [1] were applied to RdRP identification in the oyster meta-transcriptomes in order to detect homologs of the kingdom Orthornavirae. The first-round of identification was conducted using HMMsearch (HMMER v3.1) [38], with ORFs that covered at least 70% of the target HMM profiles and a bit score ≥ 30 considered as candidates for nearly complete RdRP. ORFs with a bit score of < 30 were then subjected to a second round of verification by Palmscan [39] (options: -rt -rdrp -hiconf), during which the reverse transcriptase (RT), RdRP palm-domain-based identification [40], was performed separately. Subsequently, species-level representatives were subjected to further filtration through clustering with CD-HIT (v4.8.1) [41], with a 90% amino acid identity threshold and 75% alignment coverage for shorter sequences (options: -c 0.9 -aS 0.75 -n 5) [40].
Virus clustering network construction
For the identified RdRP-bearing contigs, the closest relatives of which were retrieved through local BLASTp (v2.13.0) [42] against the NCBI nonredundant (nr) database (downloaded in 2023.04.16) with specified parameters (-evalue 1e-10, -max_target_seqs 5). To eliminate redundancies, shorter sequences with over 90% amino acid identity and 75% alignment coverage were removed by CD-HIT [41] as previously described (see the “RNA virus identification”). The combined RdRP representatives were subjected to an all-versus-all BLAST (evalue: 1e-5) analysis using the FASTA option provided in EFI web (https://efi.igb.illinois.edu/efi-est/) [43], resulting in the generation of a representative family-based clustering network, which was subsequently visualized using CytoScape (v3.9.1) (https://cytoscape.org/).
Taxonomic classification based on phylogenetic analysis
In order to infer the phylogenetic lineages of oyster RNA virome, eight order-to-family-level trees were constructed. First, the close relatives in the nr database (see the “Virus clustering network construction”) were incorporated into the inbuilt reference databases. Second, to enhance the reliability of the tree structure, an online BLASTp search was conducted against the NCBI reference protein database (refseq_protein, e-value < 1e-10). The top 10 hits (if available) were retrieved as supplementary data for the reference databases. Third, the RNA viruses were initially clustered based on the clustering network, and the RdRP sequences were extracted based on the identification of conserved domains in the conserved domain database (CDD) [44].
The RdRP footprints were aligned using MAFFT (v7.520) [45] with the sensitive option (–maxiterate 1000 –localpair –thread 1). The gaps were trimmed through the “automated1” mode, which was supplemented by the use of trimAl (v1.4) [46]. The IQ-TREE 2 software [47] was employed for the establishment of unrooted maximum-likelihood phylogenetic trees, with the ultrafast bootstrap method [48] utilized for the estimation of bootstrap values. The optimal model was identified through the application of the ModelFinder algorithm [49], which is integrated into the IQ-TREE 2 software (option: -m MFP -B 1000 -bnni). The phylogenetic trees were subsequently visualized in iToL v6 [50].
Estimation of the relative abundance of RNA viruses
To assess the prevalence of RNA viruses identified in oyster, qualified reads (see the “Contig assembly and ORF annotation”) were aligned to RdRP-bearing contigs using Bowtie2 (v 2.5.1) [35] with the modified “very sensitive” option (–local -D 20 -R 3 -N 0 -L 16 -i S,1,0.50 -I 0 -X 2000 –non-deterministic) [1]. To eliminate the potential overrepresented polyA stretches in abundance estimation, all reads and viral sequences with polyA or polyT ends were trimmed using BBDuk (v 39.01) (https://jgi.doe.gov/data-and-tools/bbtools/) as described by Zayed et al. [1], with the only exception that no iterative trims were performed since no further polyA/polyT reads were removed in the second run. Files containing mapped reads were transformed to sorted BAM in SAMtools (v 1.17) [51], which could be recognized for the calculation of overall genomic coverage and abundance estimation. The files were then filtered with 90% nucleotide identity and 75% read length coverage using CoverM (v 0.6.1) (https://github.com/wwood/CoverM). Transcripts per million (TPM) were employed as an indicator for relative abundance estimation (with the “-m tpm” option enabled in CoverM).
Identification of M. gigas-associated RNA viruses in the meta-transcriptomes of other mollusks and the virome of Yangshan Harbor
To investigate the potential circulation of RNA viruses among seawater-oyster-other intertidal mollusks, we collected a series of meta-transcriptomic datasets from mollusks belonging to the three classes of Bivalvia, Gastropoda, and Cephalopoda [6]. The class Bivalvia includes the following species: M. hongkongensis [28], bivalvia mix Wenzhou (Barbatia virescens, Sinonovacula constricta, Tegillarca granosa, M. ariakensis, Mytilus coruscus), Paphia shell mix Beihai (Paphia undulata and Veneridae sp.), razor shell mix Beihai (Solen strictus), and freshwater shellfish mix Wuhan (Unio douglasiae). The class Gastropoda encompasses a diverse array of gastropod species, including Gastropoda mix Wenzhou (Neverita didyma, Chlorostoma turbinatum, Turritella bacillum, Reishia bronni), murex snail Beihai (Chicoreus asianus), Turritella sea snails mix Beihai (Turritella sp.), and channeled apple snail mix Wenzhou (Pomacea canaliculata). Octopodidae sp. is included in the class Cephalopoda. Furthermore, a coastal-water RNA virome from the East China Sea was included in the analysis [30]. This enabled us to analyze the distribution of RNA viruses among these organisms and their respective environments. To ensure the comparability of the data, we established the following selection criteria for the mollusk transcriptomes. First, the meta-transcriptomes pertain to the RNA viromes. Second, only intestinal samples or mixed samples of whole tissues are selected. Third, samples must be collected from naturally occurring environments and from healthy organisms (e.g., not in a state of dying or subjected to chemical or physical treatments). Following these criteria, a total of 29 datasets were retrieved from public databases (Table S2).
In the context of RNA viruses identified in oyster meta-transcriptome, it is important to note that a relatively high read mapping threshold (option: CoverM -m covered_fraction, 90% nucleotide identity and 75% read coverage) has been applied [1]. Contigs with at least 30% of their length covered by reads will be considered potentially present in either M. hongkongensis or other organisms belonging to the phylum Mollusca. The 30% coverage threshold for the determination of presence was justified by the observation that in 98.5% of mapped contigs with 30% or higher read mapping coverage, reads were mapped to locations within the RdRP-coding region or RdRP polyprotein region. Moreover, among the mapped contigs, 72.4% exhibited at least 50% coverage in the aforementioned region, which is capable of providing identification information (see Figure S1 for the mapping statistics). Moreover, a viral sequence with a minimum of 90% read coverage in publicly available meta-transcriptomes will be considered as a genus- to species-level member shared by M. gigas and specific mollusks [40]. The virome sharing network was visualized using Gephi (v0.10.1).
Results
Quality assessment and statistics of the M. gigas DT meta-transcriptome
To generate high-quality data, three repetitive RNA libraries were sequenced, resulting in raw read counts of 71,910,296, 74,811,948, and 82,238,218 for each dataset, respectively. Following the removal of low-quality bases, the trimming of adapters, and the exclusion of M. gigas-sourced reads, the number of qualified reads obtained was 42,275,528, 44,612,314, and 48,238,492 for the respective datasets, with 40.4 to 41.3% of the total reads discarded. A total of 80,765 contigs (N50 = 1150, L50 = 62,831, N90 = 579, L90 = 194,625, rRNA proportion = 0.32‰) were co-assembled from the three datasets. The longest contig is 31,464 nt in length.
The identification of RdRP based on HMM profiles yielded 236 putative RdRP-bearing contigs. The length of these contigs ranged from 1071 to 9291 nt, with six contigs exceeding 5000 nt in length. A total of 154 of the 236 putative viral contigs contain the palm domain (i.e., motifs A, B, and C). No reverse transcriptases (RTs) were identified. In addition, a total of 144 contigs exhibited less than 90% amino acid identity for the RdRP, suggesting that at least 144 RNA viral species were newly identified in the oyster virome.
Diverse RNA viral lineages identified in M. gigas
For contigs carrying the RdRP domain, the clustering similarity matrix with hits in nr indicated the identification of at least 14 families/assemblages, representing three major phyla (Lenarviricota, Pisuviricota, and Kitrinoviricota) within the kingdom Orthornavirae (Fig. 1). The lack of members of the phyla Negarnaviricota, Ambiviricota, and Duplornaviricota is likely attributable to the library construction kit employed being biased towards positive-sense RNA. The largest assemblage was that of the Picornavirales order, which accounted for approximately half (78 out of 144) of the M. gigas DT-associated RNA viruses identified. The majority of viruses within this cluster were identified as marna-like viruses. It is noteworthy that Durnavirales constituted another substantial assemblage identified in the oyster. This assemblage included the picobirna-like and partiti-like viruses, which were interspersed with viruses belonging to the families of Marnaviridae, Narnaviridae, Nodaviridae, and Picornaviridae. Four taxonomic groups, namely Fiersviridae (formerly known as Leviviridae), Solemoviridae, Hepeviridae, and Yanvirus, formed distinct clusters, with no close relatives connected to other taxonomic groups. This would suggest that oysters may have played a role in the selection of factors that facilitated the divergent evolution of these RNA viruses.
Fig. 1.

A mega-taxonomy cluster constructed based on the pairwise identity of the viral RdRP sequences from Magallana gigas. The nodes represent the viruses identified in this study or reference sequences from GenBank. Edges indicate pairs of connected nodes that have passed the pairwise BLASTp filter (e-value 1e-10). The color of nodes indicates the origin of the viruses. The corresponding phyla of the different clusters are indicated on the right
Phylogenetic analysis of the RdRP-bearing contigs revealed the presence of at least eight orders, including Norzivirales, Wolframvirales, Durnavirales, Picornavirales, Sobelivirales, Hepelivirales, Nodamuvirales, and Tolivirales. The phylogenetic tree (Fig. 2) was constructed based on order to family scale as part of the clades, such as Weivirus and Yanvirus, were not fully resolved at the family level and have not yet been ratified by the ICTV.
Fig. 2.

Phylogenetic analysis of the viruses (RdRPs) from M. gigas DT and related viruses from the NCBI nonredundant (nr) database. Unrooted phylogenetic trees are constructed using maximum-likelihood estimation and 1000 iterative refinements. Only bootstrap values greater than 0.7 are displayed in the tree. Green lines indicate the RNA viruses identified in this study. The scale bar indicates the average number of amino acid substitutions per site. If a specific virus is identified as the dominant virus in other meta-transcriptomes, it will be represented by a differently colored square on the right. a, b, c, d, e, f, g, h, i The phylogenetic tree of the following viral lineages: Marnaviridae, Durnavirales, Picornavirales, Sobelivirales, Fiersviridae, Wolframvirales, Tolivirales, Hepelivirales, and Nodaviridae, respectively
The phylum Pisuviricota
With regard to the 144 newly identified putative RNA viruses, the largest clade is the Marnaviridae, where 65 viruses are situated and cover all 7 ICTV-approved genera [24] (Fig. 2a). It is noteworthy that with the exception of marna-like viruses, no RNA virus family is shared across bivalves, gastropods, cephalopods, and sea water. Four viral sequences (k141_480947, k141_179532, k141_59248, and k141_443730) were fully covered (> 90%) by meta-transcriptomes of various mollusks. In addition, there are 10 more marna-like viruses that are shared specifically within bivalves. Nine of the viral sequences are at least 90% mapped in M. hongkongensis datasets alone. Two viruses (k141_480947 and k141_179532) form a distinct branch at the base of the Marnaviridae clade and cluster with Beihai mollusks virus 2 and Macrobrachium rosenbergii virus 4. The virus k141_59248, which is a close relative of Beihai mollusks virus 1, forms an interleaved branch in the Kusarnavirus and Locarnavirus, filling the gaps between these two genera. Only the virus k141_443730 exhibits a clear phylogenetic affiliation with salisha-like virus.
The second largest clade was mainly comprised of members affiliated with the families Picobirnaviridae and Partitiviridae (Fig. 2b). Of these, the virus k141_356777 exhibited the closest phylogenetic relationship to the subclade of vertebrate-associated picobirnaviruses, with a 44% amino acid identity and the highest alignment score with the RdRP of Beihai picobirna-like virus 7 (Table S3). It was also identified in a meta-transcriptome of the peanut worm, a Sipunculida species found in the intertidal zone of southern China. Additionally, close relatives of the virus k141_356777 were identified in freshwater macrophytes and river sediments. Adjacent to the vertebrate-sourced Picobirnavirus branch, six newly identified RNA viruses in oyster (k141_508154, k141_496647, k141_13296, k141_466580, k141_309251, and k141_454727) formed a distinct clade (Fig. 2b). Their close relatives were present in various environments or organisms, including mudflats, sediment, farmland, freshwater mussels, peanut worms, and freshwater Isoptera [52]. Of note, four picobirna-like viruses (k141_478039, k141_147375, k141_435056, and k141_33296) had at least 90% of their sequences covered by the M. hongkongensis meta-transcriptome, suggesting a potential association between these viruses and the genus Magallana. However, these RNA viruses are not shared between M. gigas and gastropods, suggesting that they are highly specific to bivalves as carriers or hosts. Conversely, two of the four viruses (k141_478039 and k141_435056) were detected in cephalopods, which may be attributed to the potential for oysters to serve as a food source for octopuses.
A further distinctive virus (k141_166510) is observed in a deep branch of the Partitiviridae family (Fig. 2b). Its close relative was identified as belonging to the Partitiviridae family (USE08392.1), with 57% identity and 98% query coverage. This clade was positioned in close proximity to the plant-targeting genus Deltapartitivirus. K141_166510 could not be read-mapped by any other mollusk meta-transcriptomes, due to the fact that partitiviruses typically lack a lytic phase in their life cycle and have strictly limited target host ranges (i.e., plants, fungi, and protozoa) [53], which suggests that the source of the partiti-like virus could not have originated from a M. gigas-related culture environment.
Viruses belonging to the Dicistroviridae and Picornaviridae exhibited a lower tendency to be found among mollusks. The natural hosts of these viral families are predominantly arthropods and vertebrates [54, 55], which rarely interact with mollusks. Two viruses were shared by bivalves (Fig. 2c). One of these (k141_5361) was a dicistro-like virus, which had genus-level relatives identified in some M. hongkongensis and other bivalves (Table S2). The other virus k141_343322 was identified in the meta-transcriptome of the razor shell and exhibited characteristics consistent with those of picorna-like viruses. It is noteworthy that one of the Caliciviridae (k141_128612) was identified with high confidence (99% nucleotide identity and 96% coverage to norovirus GI). The source of this virus appeared to be wastewater drainage in an urban area. However, a total of 28 molluscan meta-transcriptomes yielded no reads that could be mapped to k141_128612.
A total of six solemo-like viruses were identified in the M. gigas DT RNA virome, five of which clustered with previously discovered aquatic solemo-like viruses (Fig. 2d). Of note, genus-level relatives of the virus k141_19360 were identified in several specimens, including M. hongkongensis, the Paphia shell, the Octopus, and even in Turritella sea snails (with 86% sequence coverage). k141_204896 is present in the Yangshan Harbor virome (Table S2). The solenoviruses are known to infect fire ants [56]. One lineage (Varroa destructor virus) in the phylogenetic clade containing the virus k141_19360 replicates in mite parasite of bees. It is therefore possible that the distribution of specific solenoviruses from terrestrial to aquatic habitats may be facilitated by parasites.
The phylum Lenarviricota
Among the identified viruses, those belonging to the phylum Lenarviricota exhibited a lower degree of diversity than those in Pisuviricota. One potential candidate, the virus k141_98645, was grouped with fiers-like viruses previously identified in pond sediment and barnacles (Fig. 2e) [52].
The family Narnaviridae, which lacks a capsid, has been observed to specifically target fungi, plants, and potentially algae [57–59]. Consequently, the presence of the narna-like virus in oysters is very likely similar to that of Marnaviridae, occurring via the ingestion of fungi and algae by mollusks. Of note, among the eight newly identified narna-like viruses, one was shared between Magallana species, indicating that the circulation of Narnaviridae and their putative hosts in mollusks is limited (Fig. 2f).
The phylum Kitrinoviricota
A total of 20 newly identified viruses were associated with the order Tolivirales. These viruses could be further divided into four assemblages: eight weivirus-like viruses, two yanvirus-like viruses, seven tombus-like viruses, and three viruses belonging to a potentially new clade that interleaves between the Weivirus and Yanvirus assemblages (Fig. 2g). The virus k141_105782 was derived from Weivirus, which has been found in invertebrates [6]. It is closely related to the Beihai weivirus-like viruses and the Barns Ness breadcrumb sponge weivirus-like viruses. However, our understanding of the host and environmental distribution of this virus is incomplete. In the case of the tombus-like virus k141_130289, no plant-related tombusviruses were identified within the same clade. In contrast, all members of this branch are aquatic-related viruses. The virus k141_130289 was also identified in gastropods and cephalopods, indicating the potential for a shared viral lineage among molluscan species.
The closest relatives to the Hepelivirales RNA virus identified in M. gigas are not those of vertebrate hosts (Fig. 2h and Table S3), indicating that the virus k141_521148 likely originated from aquatic invertebrates and was not a result of contamination (e.g., the norovirus found in oyster). The presence of a hepe-like virus (k141_521148) exclusively in oysters, without detection in other mollusks, suggests that the oyster M. gigas is likely the primary host for this virus.
The Nodaviridae family of viruses is known to infect insects and fish [60]. No Betanodavirus-like viruses were identified in the M. gigas. The virus k141_99838 represents a quasi-species of Beihai noda-like virus 5 (99% amino acid identity, 100% query coverage) (Fig. 2i), the source of which is the horseshoeing crab [6]. Notably, the viral sequence could be mapped to a significant extent (99% coverage) in the razor shell meta-transcriptome (Table S2). Additionally, the virus k141_248919 is closely related to Beihai noda-like virus 9 (95% amino acid identity, 100% coverage) (Fig. 2i) (Table S3), which was initially identified in razor shell [6] but subsequently found in M. hongkongensis as well. Meanwhile, the close relatives of these viruses have yet to be identified in bivalves (Fig. 2i). These results strongly indicate that different aquatic invertebrates harbor the same quasi-species of nodaviruses, therefore rendering bivalves (e.g., Magallana) a potential intermediate carrier for these nodaviruses. Consequently, the nodavirus in M. gigas may serve as a viral signature for evaluating the extent of virus circulation among intertidal bivalves.
Relative abundance of RNA viruses in M. gigas
The relative abundance of RNA viruses in the oysters exhibits considerable variability. The log-transformed TPM values indicated that 18 viruses exceeded the threshold of 4 (Fig. 3). This indicates that the reads corresponding to these viruses comprise at least 1% of the total aligned reads. The 18 viruses belong to three families: Marnaviridae, Picobirnaviridae, and Nodaviridae. Among the remaining 11 viral lineages, the abundance estimation of the sole member of the hepe-like virus k141_521148 was the highest at 3.71 (Table S2). This finding provides further evidence that M. gigas may serve as a potential reservoir for this specific aquatic hepevirus. In comparison to the hepe-like virus, the other families that are uniquely identified in M. gigas appear to have a lower dominance. For example, calici-like viruses (2.91), fiers-like (3.01), partiti-like (2.73), and yan-like (ranging from 2.98 to 3.07) accounted for less than 0.1% of all aligned reads. This suggests that these viral families may have limited distribution capacity in the aquatic environment or may have a lower persistence level within the digestive tissue of M. gigas. The solemo-like and narna-like viruses exhibited the lowest abundance estimations (2.52–2.56) and were found not only in M. gigas but also in other molluscan species.
Fig. 3.
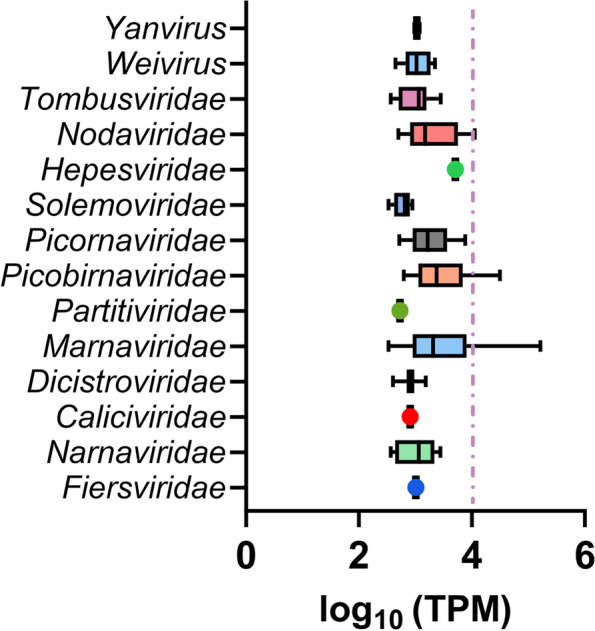
The relative abundance of viruses identified in the M. gigas DT determined based on the log10 transformed TPM. Box plot depicts the relative abundance of the viruses at the family level. The dashed line represents the threshold of 1% overall aligned reads in the M. gigas DT meta-transcriptome
The relative abundance of marna-like viruses in M. gigas is significantly higher than that of any other viral family (Table S2). This suggests that the marna-like viruses may serve as the primary “algae by-products” that accumulate in oyster DT, given that the actual hosts of these viruses, such as marine microalgae, are the major prey consumed by oysters [61]. The most prevalent marna-like virus k141_292756 accounts for 16.43% of the total reads aligned to its sequence. However, this virus is neither a related virus on a genus rank found in other molluscan species nor prevalent in the Yangshan Harbor. It is plausible that the potential host for this virus is closely associated with M. gigas, or that it is highly specific to the surrounding environment, such as algal habitants.
The family Picobirnaviridae represents the second largest assemblage of oyster RNA virome (Fig. 3, Table S2). Four picobirna-like viruses have reached an overall abundance of at least 1% of aligned reads. The putative hosts for the oyster-associated picobirnaviruses may be more specific, potentially related to the dominant prokaryotes inhabiting the DT of M. gigas [62, 63]. The virus k141_147375 is the only common picobirnavirus that is present in both mollusks and the surrounding water environment.
The most prevalent viruses shared within the genus Magallana, and other members of the phylum Mollusca, do not represent the most abundant viruses identified in M. gigas (Fig. 4A, B, Table S2). It suggests that virus-sharing networks are likely characterized by free-living viral particles rather than viruses specifically infecting mollusks. This is further corroborated by the comparison of the relative abundance of viruses in the viromes of M. gigas and Yangshan Harbor. Seven out of the 13 genus-level relatives exhibited higher TPM in seawater (Fig. 4C).
Fig. 4.

The relative abundance of the most prevalent RNA viruses from M. gigas meta-transcriptome. The viruses were classified as “present” (i.e., at least 30% of the sequences were covered by reads in other meta-transcriptomes) in datasets of M. hongkongensis (A), other mollusks (B), and the Yangshan Harbor (C). The color of the dots indicates the origin of the datasets. The size of the dots is proportional to the log10 transformed TPM
Viruses in M. gigas present in other mollusks and seawater
Among the 154 viruses identified, 12% (18/154) were found to be globally present in meta-transcriptomes of M. hongkongensis, other mollusks, and the Yangshan Harbor virome. In contrast, 49% (75/154) were identified specifically in M. hongkongensis, while only 42% (65/154) were found among other bivalves, gastropods, and cephalopods. Additionally, 20% (31/154) were identified in the Yangshan Harbor virome (Fig. 5A). For genus representatives (sequences that have 90% coverage by reads in other datasets), the number of shared members decreased to eight in M. hongkongensis, seven in other mollusks, and four in seawater (Fig. 5B). A total of 52 viruses (34%) exhibited < 30% sequence coverage in either mollusk meta-transcriptomes or the Yangshan Harbor virome. These viruses may not be as frequently shared or present as those previously mentioned.
Fig. 5.

The RNA viruses shared among M. gigas, other mollusks, and seawater. A A Venn diagram illustrating the prevalence of viruses from M. gigas DT in the phylum Mollusca and seawater. The names of the different ecological groups are indicated at the top of the diagram. B Bar plots of identified RNA viruses from M. gigas present or dominant in other datasets based on family scale. C The network structure of the M. gigas DT virome. The nodes represent shared viruses in oysters or meta-transcriptomes of other ecological groups. The edges of the graph represent the viruses that have been identified in the M. gigas dataset and are present in subsequent datasets. The size of the dots represents the number of viruses present in the respective datasets
The viruses present in each meta-transcriptome were depicted through a virus-sharing network diagram (Fig. 5C). It is evident that the meta-transcriptomes of M. hongkongensis exhibit the most diverse range of RNA viruses shared with M. gigas. Datasets of CRR468170, CRR468165, and CRR468169 shared 38, 35, and 33 viruses with M. gigas, respectively. In contrast, two meta-transcriptomes from freshwater mollusks (SRR3401656, channeled apple snail, and SRR3401658, U. douglasiae) share only two viruses with the M. gigas virome, respectively. The disparate prevalence of shared viruses between marine and freshwater mollusks indicates that the primary circulation pathway of RNA viruses in oyster is likely to occur within coastal regions, rather than being transported by river runoff.
In addition to the viruses identified in the Magallana species, other bivalve RNA viromes analyzed (hereafter referred to as the datasets reported by Shi et al. [6], see Table S2 “Mollusca”) have demonstrated that a significant proportion of sequences are included by the RNA virome of M. gigas. SRR3401916 (razor shell), SRR3401648 (unknown bivalve), and SRR3401917 (Turritella sea snails) collectively contribute to nearly half of the shared RNA viruses in oyster, with 32, 25, and 21 viruses, respectively. The meta-transcriptome of the octopus (SRR3401912) exhibited a remarkable shared diversity with the oyster-associated RNA viruses, with 37 viruses present in this dataset. It suggests a pivotal role played by the food chain in the shunt of viruses among mollusks. The collective findings highlight the interconnectivity of virus distribution between different species within the marine ecosystem.
Discussion
The circulation RNA viruses identified in both mollusks and the surrounding environment
The oyster M. gigas is widely cultivated in the intertidal zone [64], where its filter-feeding behavior allows researchers to identify potentially harmful entities [65], including viruses, in oysters from the surrounding waters and in other farmed species [66]. Consequently, it is possible that the viromes of coastal invertebrates, particularly mollusks, share certain characteristics. This is exemplified by the discovery of ostreid herpesvirus-1 (OsHV-1), which is responsible for the mass mortality of the M. gigas worldwide [67]. Interestingly, OsHV-1 has been detected in the cephalopod Octopus vulgaris at a high prevalence rate of 87.5% [68]. Moreover, the transmission of OsHV-1 has been observed between the fast-moving predator, the shore crab (Carcinus maenas), and M. gigas at culture sites [69]. It would appear that an investigation into the distribution of oyster-associated RNA viruses in both mollusks and the coastal environment will serve to highlight the significant yet often overlooked occurrence of RNA viral circulation events.
Our findings indicate that the RNA viruses in M. gigas disperse nonspecifically through seawater, particularly those belonging to the Marnaviridae family. For example, among the 13 genus-level relatives shared between M. gigas and the Yangshan Harbor, 10 are associated with Marnaviridae (Table S2). This is in contrast to noroviruses of the Caliciviridae family, which are thought to have been concentrated through potential ligand-specific binding [23]. In addition, a number of RNA viruses from the Yangshan Harbor are among those that are frequently found in bivalves, gastropods, and octopus, e.g., the virus k141_59248, k141_480497, k141_121534, and k141_488207. Of note, these samples were collected from a diverse array of geographical locations, encompassing the Yellow Sea and the East China Sea and extending to the South China Sea. Accordingly, the seawater harboring these cosmopolitan mollusks may serve as a primary conduit for the spread of these RNA viruses. In the East China Sea, harmful algae, including Alexandrium catenella and Dinophysis acuminata, periodically form blooms [70]. These blooms not only present a risk to humans through paralytic shellfish poisoning (PSP) and diarrhetic shellfish poisoning (DSP) but also result in mass mortality in shellfish [71]. The discovery of cosmopolitan marnaviruses in M. gigas suggests the possibility that marna-like viruses present in M. gigas may be targeting algal residences or even those poisoning algae. In light of the considerable research efforts devoted to the discovery and analysis of RNA viruses in diatoms and other algae [72–75] and research on the use of oysters to mitigate eutrophication [76, 77], there is an opportunity for further investigation to elucidate the complex interactions between oysters, RNA viruses, and harmful algal blooms.
A discrepancy is observed in the prevalence and abundance of viruses from M. hongkongensis across different meta-transcriptomes (Fig. 4a). The principal dissimilarities between the M. hongkongensis datasets pertain to the sampling periods and locations [28], with the sequencing strategy and oyster health condition remaining consistent. Among the most prevalent viruses identified in M. hongkongensis, only two RNA viruses were found in datasets of CRR468145 and CRR468146, respectively, while the CRR468141, CRR468157, and CRR468158 accounted for 90% (10/11) of the most prevalent RNA viruses in M. hongkongensis (Fig. 4A). The fluctuations in the prevalence of viruses across datasets reflect the heterogeneity of the virome among individual oysters, suggesting potential variations in viral populations within oyster populations. Additionally, it is likely that the preferences in amplification during sequencing may contribute to the overrepresentation of specific viral populations, thereby reinforcing these differences. This is exemplified by the polyA-enriched meta-transcriptome observed in invertebrates (Fig. 4B). Datasets obtained using a polyA-based approach in bivalves and gastropods only shared 1–2 viruses with the M. gigas virome. In contrast, random-priming-based meta-transcriptomes from the same samples exhibited a 2- to fivefold increase in the number of shared viruses (Table S2). Consequently, some viral-related sequences may be overlooked, and the overall prevalence of the M. gigas-associated viruses may be somewhat underestimated. These observations underscore the necessity of considering the constraints and potential distortions associated with disparate sequencing methodologies when interpreting viral prevalence and diversity in oyster RNA viromes.
Protist viruses in M. gigas
The novel marna-like viruses identified in oysters appear to be associated with marine protists. Firstly, since bivalves and gastropods do not prey on each other, if the viruses were species specific, they would not have been shared among such a broad range of mollusks. This suggests that these viruses may be part of the virioplankton community. Secondly, all known hosts of marnaviruses are protists, particularly marine microalgae [24]. Lastly, the considerable number of shared RNA viruses indicates the presence of a substantial viral population, which is sufficient to enable the requisite sequencing depth and coverage. The abundance of viruses observed in these mollusks may be attributed to the daily prey consumed by these mollusks.
The marna-like (1), salisha-like (4), and unclassified Marnaviridae viruses (4) are shared within the genus Magallana. It is important to note that three of these viruses are shared with intertidal gastropods or cephalopods (Table S2), rather than bivalves. These three viruses belong to the Salisharnavirus clade, indicating that their primary host may not be the major prey for M. gigas, or they may be less prevalent in the corresponding environment, such as the benthic algae habitat.
Oyster cultivation is dependent upon the use of algae diets, particularly diatoms [78]. In this study, the presence of shared marna-like viruses among mollusks has permitted the confident identification of the food webs within the ecosystem, which involve seawater, oysters, and octopuses. However, the downstream life cycle of algal viruses that aggregate in oyster DT remains unknown, as does the likelihood of reintroduction to alga hosts. The discovery of a high prevalence of marna-like viruses in mollusks and coastal viromes raises questions about the role that oysters play in protist-related viral bioaccumulation. One may inquire whether marna-like viruses are passively enriched from the surrounding seawater or whether they actively exploit oyster bioaccumulation as a strategy for their next round of infection. It is hypothesized that RNA viruses may derive several benefits from oysters. Firstly, the oyster’s passive filtration process allows it to process seawater at a rate of 5 L/h [11]. During this process, virions, prokaryotes, and protists are efficiently absorbed and well mixed within oyster DT. Secondly, the digestion of algae in oyster digestive tracts may contribute to the accelerated circulation of algal viruses. Finally, it can be posited that RNA viruses may be influencing the bioaccumulation of oysters. It is well documented that noroviruses are specifically bound to oysters with the help of HBGAs-like substances [20, 23, 79–81]. However, this is not the only way RNA viruses could facilitate their transmission. Recent reports have highlighted a unique phenomenon, whereby oyster genes are interfered with by alien RNA in a manner that is analogous to a “Trojan Horse.” In this instance, an engineered double-stranded RNA is introduced into the oyster through an alga vector. Indeed, oyster gene expression is inhibited, including that of the clock gene, which in turn regulates bioaccumulation [82]. Despite the paucity of knowledge regarding the effects of virus-sourced RNA interference on oysters, we posit that investigating the common oyster-associated RNA viruses shared among mollusks would provide valuable insights into validating these effects. It is of particular importance to direct attention to the most dominant species, such as the marna-like and picobirna-like clades found within oysters.
Human viruses in M. gigas
Norovirus has long been recognized as a food contaminant in shellfish worldwide [15, 83]. Moreover, other viruses have been identified in oysters, including hepatitis A virus (HAV), enterovirus (EV), rotavirus (RV), and astrovirus (AV). These viruses are known to be major causes of human acute infectious gastroenteritis [84]. However, they were absent in our oyster RNA virome, suggesting that either the virus particles or their genomic fragments are not captured by the meta-transcriptome. A number of potential explanations for this phenomenon have been put forth. Firstly, the prevalence of HAV, EV, RV, and AV in oysters is not as high as that of NoV, resulting from fewer viral particles in the environment. For example, a quantitative-PCR-based study revealed that HAV was not detected in 253 bivalves, while a higher prevalence of noroviruses was confirmed, with at least six genotypes identified in the same samples [85]. Another study demonstrated that noroviruses were present in much greater numbers than human adenovirus, human bocavirus, and RV, with a ratio of over 100-fold [86]. Moreover, it has been reported that noroviruses exhibit a high prevalence rate (96.6%) in oyster-associated human gastroenteritis cases [87], indicating the advantages of norovirus circulation in bivalves over the other human enteric viruses. Indeed, amplicon-based sequencing may be a more efficient method for monitoring enteric RNA viruses than meta-transcriptome-based sequencing [88]. Furthermore, the specific absorptive activities of noroviruses in oysters play a crucial role in facilitating their identification in meta-transcriptome analyses. This notion is supported by several aspects. Firstly, carbohydrates present in oyster digestive ducts have been demonstrated to specifically bind to viral particles [21, 22]. Secondly, certain bacteria present in the oyster DT are capable of producing HBGA-like components, such as extracellular polysaccharides, in the form of binding ligands that could interact with noroviruses [20, 23]. This binding mechanism probably enables the enrichment of NoVs in the M. gigas DT, thereby increasing the likelihood of their detection in meta-transcriptome analyses.
The recent pandemic of SARS-CoV-2 has prompted interest in investigating the potential presence of this virus in oysters as far back as 2016. It has been demonstrated that the spike protein of SARS-CoV-2 can be readily bioaccumulated in the oyster DT [31]. Meanwhile, virus particles have been detected in the feces of patients diagnosed with COVID-19 [89]. Moreover, experimental evidence has demonstrated that SARS-CoV-2 can remain viable in river and wastewater [90]. This suggests that the virus is capable of persisting in an aquatic environment. Consequently, during the SARS-CoV-2 outbreak, oysters exposed to contaminated water through fecal contamination may readily accumulate the virus in their digestive gland. It is, however, important to note that the absence of Coronaviridae, including SARS-CoV-2, in the M. gigas RNA virome throughout the entire year suggests that the virus was not expected to be present during that time.
Conclusion
The identification of the RNA virome has enabled the creation of a comprehensive catalog of over 140 viral species within the digestive tissues of M. gigas. This catalog establishes links to both abiotic and biotic environments. Given the filter-feeding nature of the oyster, our findings indicate that it may serve as a reservoir for virus accumulation and a conduit for potential virus dispersal from the marine environment to mollusks. Notably, we observe the predominance and diversity of marna-like and picobirna-like assemblages, which underscores their significance in the oyster virome. Our research indicates that diverse networks of RNA viruses are present in oysters, providing insight into the circulation mechanisms of microbiomes and offering a valuable resource for monitoring potential human pathogenic microorganisms in marine environments.
Supplementary Information
Supplementary Material 1: Table S1. The sampling status of the M. gigas, including the date, number of sampled individuals, and the sampling location.
Supplementary Material 2: Table S2. The relative abundance of viruses from the oyster meta-transcriptome (calculated as log10-transformed TPM). The proposed taxonomy for each virus is presented on page 1. The prevalence of oyster-associated RNA viruses among different datasets is presented, including M. hongkongensis (Page 2), parts of the organisms from the phylum Mollusca (Page 3), and seawater (Yangshan Harbor virome, Page 4). The representatives of RNA viruses identified at the order and family levels in this study will be considered present in a given dataset if at least 30% of the sequence is covered by reads in that dataset.
Supplementary Material 3: Table S3. Top hits of viral RdRPs from the oyster meta-transcriptome in the NCBI non-redundant dataset.
Supplementary Material 4. Sequences of identified viral RdRPs from the oyster meta-transcriptome.
Supplementary Material 5. Original alignment files of phylogenetic trees in Fig. 2.
{kind=link}
Supplementary Material 6: Figure S1. The distribution of the percentage of genes encoding RNA-dependent RNA polymerase (RdRP) or RdRP polyprotein covered in reads mapping is presented for contigs with a minimum coverage of 30%. The x-axis depicts the databases utilized for recruitment, while the y-axis represents the percentage of coverage.
Acknowledgements
Not applicable.
Abbreviations
- ICTV
International Committee on Taxonomy of Viruses
- NoV
Norovirus
- HBGAs
Histo-blood group antigens
- DT
Digestive tissue
- HMM
Hidden Markov model
- RdRP
RNA-dependent RNA polymerase
- RT
Reverse transcriptase
- CDD
Conserved Domain Database
- WTA
Whole Transcriptomic Amplification
- TPM
Transcripts per million
- YSH
Yangshan Harbor
- HAV
Hepatitis A virus
- EV
Enterovirus
- RV
Rotavirus
- AV
Astrovirus
Authors’ contributions
SW conducted a comprehensive analysis and interpretation of the sequence data, and was a major contributor in writing the manuscript. YN edited and visualized the figures. SY provided resources. YY collected the samples. YW contributed to the conceptualization of the study, provided supervision, reviewed and edited the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (32370151, 41376135).
Data availability
The dataset supporting the conclusions of this article is available for download from the NCBI Sequence Read Archive, BioProject PRJNA1121899 (http://www.ncbi.nlm.nih.gov/bioproject/1121899).
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zayed AA, Wainaina JM, Dominguez-Huerta G, Pelletier E, Guo J, Mohssen M, et al. Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science. 1979;2022(376):156–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steward GF, Culley AI, Mueller JA, Wood-Charlson EM, Belcaid M, Poisson G. Are we missing half of the viruses in the ocean? ISME J. 2013;7:672–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simmonds P, Adams MJ, Benk M, Breitbart M, Brister JR, Carstens EB, et al. Consensus statement: virus taxonomy in the age of metagenomics. Nat Rev Microbiol. 2017;15:161–8. [DOI] [PubMed] [Google Scholar]
- 4.Koonin EV, Kuhn JH, Dolja VV, Krupovic M. Megataxonomy and global ecology of the virosphere. ISME J. 2024;18:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rexin D, Rachmadi AT, Hewitt J. Persistence of infectious human Norovirus in estuarine water. Food Environ Virol. 2024;16:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, et al. Redefining the invertebrate RNA virosphere. Nature. 2016;540:539–43. [DOI] [PubMed] [Google Scholar]
- 7.Lopez JV, Kamel B, Medina M, Collins T, Baums IB. Multiple facets of Marine Invertebrate Conservation Genomics. Annu Rev Anim Biosci. 2019;7:473–97. [DOI] [PubMed] [Google Scholar]
- 8.Zhang YY, Chen Y, Wei X, Cui J. Viromes in marine ecosystems reveal remarkable invertebrate RNA virus diversity. Sci China Life Sci. 2022;65:426–37. [DOI] [PubMed] [Google Scholar]
- 9.Rosani U, Shapiro M, Venier P, Allam B. A needle in a haystack: tracing bivalve-associated viruses in high-throughput transcriptomic data. Viruses. 2019;11:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koonin EV, Dolja VV, Krupovic M, Varsani A, Wolf YI, Yutin N, et al. Global organization and proposed megataxonomy of the virus world. Microbiol Mol Biol Rev. 2020;84:e00061-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olalemi A, Baker-Austin C, Ebdon J, Taylor H. Bioaccumulation and persistence of faecal bacterial and viral indicators in Mytilus edulis and Crassostrea gigas. Int J Hyg Environ Health. 2016;219:592–8. [DOI] [PubMed] [Google Scholar]
- 12.Zhu P, Liu GF, Liu C, Yang LL, Liu M, Xie KM, et al. Novel RNA viruses in oysters revealed by virome. iMeta. 2022;1:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu P, Liu C, Liu GF, Liu H, Xie KM, Zhang HS, et al. Unveiling CRESS DNA virus diversity in oysters by virome. Viruses. 2024;16:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bedford AJ, Williams G, Bellamy AR. Virus accumulation by the rock oyster Crassostrea glomerata. Appl Environ Microbiol. 1978;35:1012–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y, Cai H, Hu L, Lei R, Pan Y, Yan S, et al. Molecular epidemiology of oyster-related human noroviruses and their global genetic diversity and temporal-geographical distribution from 1983 to 2014. Appl Environ Microbiol. 2015;81:7615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mozgovoj M, Miño S, Barbieri ES, Tort FL, Victoria-Montero M, Frydman C, et al. GII.4 human norovirus and G8P[1] bovine-like rotavirus in oysters (Crassostrea gigas) from Argentina. Int J Food Microbiol. 2022;365:109553. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Xue L, Gao J, Cai W, Zhang Z, Meng L, et al. A systematic review and meta-analysis indicates a substantial burden of human noroviruses in shellfish worldwide, with GII.4 and GII.2 being the predominant genotypes. Food Microbiol. 2023;109:104140. [DOI] [PubMed] [Google Scholar]
- 18.Rajko-Nenow P, Keaveney S, Flannery J, McIntyre A, Doré W. Norovirus genotypes implicated in two oyster-related illness outbreaks in Ireland. Epidemiol Infect. 2014;142:2096–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loury P, Le Guyader FS, Le Saux JC, Ambert-Balay K, Parrot P, Hubert B. A norovirus oyster-related outbreak in a nursing home in France, January 2012. Epidemiol Infect. 2015;143:2486–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu Y, Han F, Yang M, Zhang X, Chen Y, Yu M, et al. Pseudomonas composti isolate from oyster digestive tissue specifically binds with norovirus GII.6 via Psl extracellular polysaccharide. Int J Food Microbiol. 2023;406:110369. [DOI] [PubMed] [Google Scholar]
- 21.Le Guyader FS, Loisy F, Atmar RL, Hutson AM, Estes MK, Ruvoën-Clouet N, et al. Norwalk virus-specific binding to oyster digestive tissues. Emerg Infect Dis. 2006;12:931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Guyader FS, Atmar RL, Le Pendu J. Transmission of viruses through shellfish: when specific ligands come into play. Curr Opin Virol. 2012;2:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.EshaghiGorji M, Tan MTH, Li D. Influence of fucosidase-producing bifidobacteria on the HBGA antigenicity of oyster digestive tissue and the associated norovirus binding. Int J Food Microbiol. 2021;340:109058. [DOI] [PubMed] [Google Scholar]
- 24.Lang AS, Vlok M, Culley AI, Suttle CA, Takao Y, Tomaru Y, et al. ICTV virus taxonomy profile: Marnaviridae 2021. J Gen Virol. 2021;102:001633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munke A, Kimura K, Tomaru Y, Okamoto K. Capsid structure of a marine algal virus of the order Picornavirales. J Virol. 2020;94:e01855-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Munke A, Li S, Tomaru Y, Okamoto K. Structural insights into common and host-specific receptor-binding mechanisms in algal picorna-like viruses. Viruses. 2022;14:2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takao Y, Mise K, Nagasaki K, Okuno T, Honda D. Complete nucleotide sequence and genome organization of a single-stranded RNA virus infecting the marine fungoid protist Schizochytrium sp. J Gen Virol. 2006;87(Pt 3):723–33. [DOI] [PubMed] [Google Scholar]
- 28.Jiang JZ, Fang YF, Wei HY, Zhu P, Liu M, Yuan WG, et al. A remarkably diverse and well-organized virus community in a filter-feeding oyster. Microbiome. 2023;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvi D, Mariottini P. Revision shock in Pacific oysters taxonomy: the genus Magallana (formerly Crassostrea in part) is well-founded and necessary. Zool J Linn Soc. 2021;192:43–58. [Google Scholar]
- 30.Wolf YI, Silas S, Wang Y, Wu S, Bocek M, Kazlauskas D, et al. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat Microbiol. 2020;5:1262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyu C, An R, Liu C, Shi Z, Wang Y, Luo G, et al. Bioaccumulation pattern of the SARS-CoV-2 spike proteins in Pacific oyster tissues. Appl Environ Microbiol. 2023;89:e0210622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao M, Zhang Z, Zhao X, Geng H, Xue L, Liu D. Spatial distribution and enrichment dynamics of foodborne Norovirus in oyster tissues. Foods. 2023;13:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rumbavicius I, Rounge TB, Rognes T. HoCoRT: host contamination removal tool. BMC Bioinformatics. 2023;24:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6. [DOI] [PubMed] [Google Scholar]
- 37.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babaian A, Edgar R. Ribovirus classification by a polymerase barcode sequence. PeerJ. 2022;10:e14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature. 2022;602:142–7. [DOI] [PubMed] [Google Scholar]
- 41.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oberg N, Zallot R, Gerlt JA. EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme Function Initiative (EFI) Web Resource for Genomic Enzymology Tools. J Mol Biol. 2023;435:168018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoang DT, Chernomor O, Von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024;52:W78-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen YM, Sadiq S, Tian JH, Chen X, Lin XD, Shen JJ, et al. RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat Microbiol. 2022;7:1312–23. [DOI] [PubMed] [Google Scholar]
- 53.Vainio EJ, Chiba S, Ghabrial SA, Maiss E, Roossinck M, Sabanadzovic S, et al. ICTV virus taxonomy profile: Partitiviridae. J Gen Virol. 2018;99:17–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zell R, Delwart E, Gorbalenya AE, Hovi T, King AMQ, Knowles NJ, et al. ICTV virus taxonomy profile: Picornaviridae. J Gen Virol. 2017;98:2421–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valles SM, Chen Y, Firth AE, Guérin DMA, Hashimoto Y, Herrero S, et al. ICTV virus taxonomy profile: Dicistroviridae. J Gen Virol. 2017;98:355–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sõmera M, Fargette D, Hébrard E, Sarmiento C. ICTV virus taxonomy profile: Solemoviridae 2021. J Gen Virol. 2021;102:001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hillman BI, Cai G. The family Narnaviridae. Simplest of RNA viruses. Adv Virus Res. 2013;86:149–76. [DOI] [PubMed] [Google Scholar]
- 58.Cai G, Myers K, Fry WE, Hillman BI. A member of the virus family Narnaviridae from the plant pathogenic oomycete Phytophthora infestans. Arch Virol. 2012;157:165–9. [DOI] [PubMed] [Google Scholar]
- 59.Charon J, Murray S, Holmes EC. Revealing RNA virus diversity and evolution in unicellular algae transcriptomes. Virus Evol. 2021;7:veab070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sahul Hameed AS, Ninawe AS, Nakai T, Chi SC, Johnson KL. ICTV virus taxonomy profile: Nodaviridae. J Gen Virol. 2019;100:3–4. [DOI] [PubMed] [Google Scholar]
- 61.Vignier J, Laroche O, Rolton A, Wadsworth P, Kumanan K, Trochel B, et al. Dietary exposure of Pacific oyster (Crassostrea gigas) larvae to compromised microalgae results in impaired fitness and microbiome shift. Front Microbiol. 2021;12:706214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghosh S, Malik YS. The true host/s of picobirnaviruses. Front Vet Sci. 2021;7:615293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang D. The enigma of picobirnaviruses: viruses of animals, fungi, or bacteria? Curr Opin Virol. 2022;54:101232. [DOI] [PubMed] [Google Scholar]
- 64.Corporeau C, Petton S, Vilaça R, Delisle L, Quéré C, Le Roy V, et al. Harsh intertidal environment enhances metabolism and immunity in oyster (Crassostrea gigas) spat. Mar Environ Res. 2022;180:105709. [DOI] [PubMed] [Google Scholar]
- 65.Oyanedel D, Rojas R, Brokordt K, Schmitt P. Crassostrea gigas oysters from a non-intensive farming area naturally harbor potentially pathogenic vibrio strains. J Invertebr Pathol. 2023;196:107856. [DOI] [PubMed] [Google Scholar]
- 66.Vazquez-Boucard C, Escobedo-Fregoso C, Duran-Avelar MDJ, Mercier L, Llera-Herrera R, Escobedo-Bonilla C, et al. Crassostrea gigas oysters as a shrimp farm bioindicator of white spot syndrome virus. Dis Aquat Organ. 2012;98:201–7. [DOI] [PubMed] [Google Scholar]
- 67.Trancart S, Tweedie A, Liu O, Paul-Pont I, Hick P, Houssin M, et al. Diversity and molecular epidemiology of Ostreid herpesvirus 1 in farmed Crassostrea gigas in Australia: geographic clusters and implications for “microvariants” in global mortality events. Virus Res. 2023;323:198994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prado-Alvarez M, García-Fernández P, Faury N, Azevedo C, Morga B, Gestal C. First detection of OsHV-1 in the cephalopod Octopus vulgaris. Is the octopus a dead-end for OsHV-1? J Invertebr Pathol. 2021;183:107553. [DOI] [PubMed] [Google Scholar]
- 69.Bookelaar BE, O’Reilly AJ, Lynch SA, Culloty SC. Role of the intertidal predatory shore crab Carcinus maenas in transmission dynamics of ostreid herpesvirus-1 microvariant. Dis Aquat Organ. 2018;130:221–33. [DOI] [PubMed] [Google Scholar]
- 70.Gu H, Wu Y, Lü S, Lu D, Tang YZ, Qi Y. Emerging harmful algal bloom species over the last four decades in China. Harmful Algae. 2022;111:102059. [DOI] [PubMed] [Google Scholar]
- 71.Pease SKD, Brosnahan ML, Sanderson MP, Smith JL. Effects of two toxin-producing harmful algae, Alexandrium catenella and Dinophysis acuminata (Dinophyceae), on activity and mortality of larval shellfish. Toxins (Basel). 2022;14:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomaru Y, Toyoda K, Kimura K, Takao Y, Sakurada K, Nakayama N, et al. Isolation and characterization of a single-stranded RNA virus that infects the marine planktonic diatom Chaetoceros sp. (SS08-C03). Phycological Res. 2013;61:27–36. [Google Scholar]
- 73.Lang AS, Culley AI, Suttle CA. Genome sequence and characterization of a virus (HaRNAV) related to picorna-like viruses that infects the marine toxic bloom-forming alga Heterosigma akashiwo. Virology. 2004;320:206–17. [DOI] [PubMed] [Google Scholar]
- 74.Tomaru Y, Toyoda K, Kimura K, Hata N, Yoshida M, Nagasaki K. First evidence for the existence of pennate diatom viruses. ISME J. 2012;6(7):1445–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shirai Y, Tomaru Y, Takao Y, Suzuki H, Nagumo T, Nagasaki K. Isolation and characterization of a single-stranded RNA virus infecting the marine planktonic diatom Chaetoceros tenuissimus meunier. Appl Environ Microbiol. 2008;74:4022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silva C, Ferreira JG, Bricker SB, DelValls TA, Martín-Díaz ML, Yáñez E. Site selection for shellfish aquaculture by means of GIS and farm-scale models, with an emphasis on data-poor environments. Aquaculture. 2011;318:444–57. [Google Scholar]
- 77.Pan K, Lan W, Li T, Hong M, Peng X, Xu Z, et al. Control of phytoplankton by oysters and the consequent impact on nitrogen cycling in a subtropical bay. Sci Total Environ. 2021;796:149007. [DOI] [PubMed] [Google Scholar]
- 78.van Houcke J, Medina I, Maehre HK, Cornet J, Cardinal M, Linssen J, et al. The effect of algae diets (Skeletonema costatum and Rhodomonas baltica) on the biochemical composition and sensory characteristics of Pacific cupped oysters (Crassostrea gigas) during land-based refinement. Food Res Int. 2017;100:151–60. [DOI] [PubMed] [Google Scholar]
- 79.Morozov V, Hanisch FG, Wegner KM, Schroten H. Pandemic GII.4 Sydney and epidemic GII.17 Kawasaki308 noroviruses display distinct specificities for histo-blood group antigens leading to different transmission vector dynamics in Pacific oysters. Front Microbiol. 2018;9:2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Auger A, Yu SY, Guu SY, Quéméner A, Euller-Nicolas G, Ando H, et al. Species-specific N-glycomes and methylation patterns of oysters Crassostrea gigas and Ostrea edulis and their possible consequences for the Norovirus–HBGA interaction. Mar Drugs. 2023;21:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Su L, Ma L, Liu H, Zhao F, Su Z, Zhou D. Presence and distribution of histo-blood group antigens in pacific oysters and the effects of exposure to noroviruses Gi.3 and Gii.4 on their expression. J Food Prot. 2018;81:1783–90. [DOI] [PubMed] [Google Scholar]
- 82.Payton L, Perrigault M, Bourdineaud JP, Marcel A, Massabuau JC, Tran D. Trojan horse strategy for non-invasive interference of clock gene in the oyster Crassostrea gigas. Mar Biotechnol. 2017;19:361–71. [DOI] [PubMed] [Google Scholar]
- 83.Analysis of the European baseline survey of norovirus in oysters. EFSA J. 2019;17. [DOI] [PMC free article] [PubMed]
- 84.Le Guyader F, Haugarreau L, Miossec L, Dubois E, Pommepuy M. Three-year study to assess human enteric viruses in shellfish. Appl Environ Microbiol. 2000;66:3241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.La Bella G, Martella V, Basanisi MG, Nobili G, Terio V, La Salandra G. Food-borne viruses in shellfish: investigation on Norovirus and HAV presence in Apulia (SE Italy). Food Environ Virol. 2017;9:179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.do Nascimento LG, Sarmento SK, Leonardo R, Gutierrez MB, Malta FC, de Oliveira JM, et al. Detection and molecular characterization of enteric viruses in bivalve mollusks collected in Arraial do Cabo, Rio de Janeiro, Brazil. Viruses. 2022;14:2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iritani N, Kaida A, Abe N, Kubo H, Sekiguchi JI, Yamamoto SP, et al. Detection and genetic characterization of human enteric viruses in oyster-associated gastroenteritis outbreaks between 2001 and 2012 in Osaka city. Japan J Med Virol. 2014;86:2019–25. [DOI] [PubMed] [Google Scholar]
- 88.Oshiki M, Miura T, Kazama S, Segawa T, Ishii S, Hatamoto M, et al. Microfluidic PCR amplification and MiSeq amplicon sequencing techniques for high-throughput detection and genotyping of human pathogenic RNA viruses in human feces, sewage, and oysters. Front Microbiol. 2018;9:830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spencer EA, Heneghan CJ, Brassey J, Plüddemann A, Onakpoya IJ, Evans DH, et al. SARS-CoV-2 and the role of orofecal transmission: a systematic review. F1000Res. 2021;10:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.de Oliveira LC, Torres-Franco AF, Lopes BC, da Santos BSÁS, Costa EA, Costa MS, et al. Viability of SARS-CoV-2 in river water and wastewater at different temperatures and solids content. Water Res. 2021;195:117002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1: Table S1. The sampling status of the M. gigas, including the date, number of sampled individuals, and the sampling location.
Supplementary Material 2: Table S2. The relative abundance of viruses from the oyster meta-transcriptome (calculated as log10-transformed TPM). The proposed taxonomy for each virus is presented on page 1. The prevalence of oyster-associated RNA viruses among different datasets is presented, including M. hongkongensis (Page 2), parts of the organisms from the phylum Mollusca (Page 3), and seawater (Yangshan Harbor virome, Page 4). The representatives of RNA viruses identified at the order and family levels in this study will be considered present in a given dataset if at least 30% of the sequence is covered by reads in that dataset.
Supplementary Material 3: Table S3. Top hits of viral RdRPs from the oyster meta-transcriptome in the NCBI non-redundant dataset.
Supplementary Material 4. Sequences of identified viral RdRPs from the oyster meta-transcriptome.
Supplementary Material 5. Original alignment files of phylogenetic trees in Fig. 2.
Supplementary Material 6: Figure S1. The distribution of the percentage of genes encoding RNA-dependent RNA polymerase (RdRP) or RdRP polyprotein covered in reads mapping is presented for contigs with a minimum coverage of 30%. The x-axis depicts the databases utilized for recruitment, while the y-axis represents the percentage of coverage.
Data Availability Statement
The dataset supporting the conclusions of this article is available for download from the NCBI Sequence Read Archive, BioProject PRJNA1121899 (http://www.ncbi.nlm.nih.gov/bioproject/1121899).