

Abstract
Cancer has become one of the major diseases threatening human health in the twenty-first century due to its incurability. In 2022, new cases of esophageal and gastrointestinal cancers accounted for 17.1% of all newly diagnosed cancer cases worldwide. Despite significant improvements in early cancer screening, clinical diagnostics, and treatments in recent years, the overall prognosis of digestive system cancer patients remains poor. The DEAD-box helicase family, a crucial member of the RNA helicase family, participates in almost every aspect of RNA metabolism, including transcription, splicing, translation, and degradation, and plays a key role in the occurrence and progression of various cancers. This article aims to summarize and discuss the role and potential clinical applications of DEAD-box helicase family proteins in digestive system cancers. The discussion includes the latest progress in the occurrence, development, and treatment of esophageal and gastrointestinal tumors; the main functions of DEAD-box helicase family proteins; their roles in digestive system cancers, including their relationships with clinical factors; effects on cancer proliferation, migration, and invasion; and involved signaling pathways; as well as the existing inhibitory strategies targeting DDX family proteins, are discussed. Additionally, outlooks on future research directions are provided.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-024-05930-0.
Keywords: DEAD-box helicase, Esophageal cancer, Gastrointestinal cancer
Introduction
Cancer is one of the leading chronic non-communicable diseases worldwide, accounting for approximately 16.8% of global deaths, with esophageal and gastrointestinal cancers contributing significantly to this burden. In 2022, new cases of esophageal and gastrointestinal cancers represented 17.1% of all cancers globally. Colorectal cancer ranked third (9.6%), gastric cancer fifth (4.9%), and esophageal cancer eleventh (2.6%). Among cancer-related deaths, these cancers accounted for 20.7%, with colorectal cancer ranked second (9.3%), gastric cancer fifth (6.8%), and esophageal cancer seventh (4.6%) [1]. Despite advancements in treatment, the mortality rates remain high, underscoring the urgent need for new therapeutic targets.
The transmission of genetic information in RNA relies on the changes in its secondary structure [2], with RNA helicases playing a crucial role in regulating these structural transitions. RNA helicases are widely distributed and highly conserved, participating in multiple biological processes, including transcription, splicing, degradation, and translation [3]. Based on conserved motifs, RNA helicases are categorized into different families (SF1-SF5), among which the DEAD-box helicase family is the largest [4]. DEAD-box helicases are involved in DNA transcription, mRNA splicing, ribosome biogenesis, RNA transport, translation, degradation, cell cycle regulation, and miRNA biogenesis [5], thereby regulating cell proliferation and apoptosis.
The functions of the DEAD-box helicase family have been extensively studied, including their roles in liver cancer [6, 7], pancreatic cancer [8, 9], breast cancer [10], and gastrointestinal tumors [11, 12]. Notably, a substantial proportion of research focuses on the involvement of this protein family in tumors of the digestive system and its accessory organs. Through a thorough review of the literature and database analyses (TIMER2.0, GEPIA), it has been observed that DEAD-box helicase family proteins are generally expressed at higher levels in gastrointestinal tumors. This may be closely related to their critical role in viral proliferation. For instance, DDX5 promotes viral dissemination by enhancing RNA methylation of antiviral transcripts, acting as a negative regulator of innate immunity [13]. Similarly, DDX39A facilitates RNA virus escape from innate immunity by transporting specific viral mRNAs to the nucleus, thereby enhancing viral proliferation [14]. DDX46, on the other hand, suppresses antiviral innate immune responses by removing m6A methylation modifications from antiviral mRNAs, leading to their nuclear retention [15]. Meanwhile, viral infections are recognized as significant causative factors in gastrointestinal tumors. For example, Helicobacter pylori plays a critical role in the development of gastric cancer [1]. These findings highlight the exceptional importance and research potential of DEAD-box helicase family proteins in gastrointestinal tumors. However, as our understanding of cellular functions and protein–protein interaction mechanisms continues to deepen, the specific mechanisms underlying their roles remain to be further elucidated.
There has been progress in the development of drugs targeting DEAD-box helicase family proteins, especially DDX3 and DDX5, which are overexpressed in various cancers and exhibit pro-oncogenic roles [16]. Hundreds of ATPase/helicase inhibitors have been developed, but small molecule inhibitors targeting other DEAD-box helicases are still limited, and their molecular mechanisms remain unclear.
This article combines the latest research techniques and optimization algorithms, such as the Moth Flame Optimization (MFO) algorithm [17], Quantum Approximate Optimization Algorithm (QAOA) [18], Spider Monkey Optimization (SMO) [19], Marine Predators Algorithm (MPA) [20], Whale Optimization Algorithm (WOA) [21], and Arithmetic Optimization Algorithm (AOA) [22], for data collection, analysis, and article conceptualization. These advanced algorithms effectively support the in-depth exploration of the topic and the establishment of the research framework, thereby making the research outcomes more comprehensive and systematic.
This paper first provides a comprehensive overview of the latest global incidence, mortality, risk factors, and advances in the diagnosis and treatment of esophageal, gastric, and colorectal cancers. It systematically discusses the role of DEAD-box helicase family proteins in these digestive targets system cancers, incorporating recent research findings that highlight their potential as therapeutic. Furthermore, this paper summarizes the latest progress in drug development targeting DEAD-box helicase family proteins, emphasizing the current research gaps and limitations, and proposes directions for future research. Finally, the paper examines the limitations and gaps in the study of DEAD-box helicase family proteins, aiming to offer unique perspectives and insights for future investigations.
Recent advances in the occurrence, development, and treatment of esophageal and gastrointestinal cancers
In 2022, there were 511,000 new cases of esophageal cancer globally, ranking 11th in terms of incidence worldwide. The incidence rate in men (3.5%) was significantly higher than in women (below 2.5%). The disease caused 445,000 deaths, ranking 7th globally in terms of mortality. The mortality rate in men (5.9%) was substantially higher than in women (2.9%). Geographically, East Asia has the highest incidence rate. According to the age-standardized incidence rate per 100,000, the rates are 12.2 in men and 3.4 in women, making it the highest among major regions globally [1]. The incidence and etiology of esophageal cancer differ significantly between its two histological subtypes: squamous cell carcinoma and adenocarcinoma. In regions with higher human development index, adenocarcinoma is the predominant type and is associated with obesity [23], gastroesophageal reflux disease, and Barrett’s esophagus [24]. Smoking and alcohol consumption are major risk factors for squamous cell carcinoma [25], along with dietary nitrosamines and habitual consumption of hot beverages, which can cause thermal injury and increase the risk of this cancer [26]. Endoscopic resection, including EMR and ESD, is a standard minimally invasive treatment for early esophageal cancer (Tis and T1a), with ESD (endoscopic submucosal dissection) providing precise pathological diagnosis and complete resection with fewer local recurrences [27]. Except for early-stage esophageal cancer, locally advanced unresectable esophageal cancer and the presence of distant metastases, radical surgical resection remains the main treatment. Common and safe surgical types include Ivor-Lewis and Sweet esophagectomy [28]. Neoadjuvant chemotherapy or chemoradiotherapy has become standard strategy for locally advanced esophageal cancer [29], aiming to improve the chances of complete surgical resection. Adjuvant chemotherapy [30], radiotherapy [31], and immunotherapy [32] are commonly used as postoperative adjunctive treatments to prolong patient survival. For unresectable locally advanced and metastatic esophageal cancer, definitive chemoradiotherapy [33], immunotherapy [34, 35], tyrosine kinase inhibitors [36] and vascular endothelial growth factor receptor inhibitors [37, 38], can improve survival.
Gastric cancer ranks fifth in both global incidence and mortality rates. In 2022, there were over 968,000 new cases of gastric cancer worldwide, with a higher incidence rate in men (10.4%) compared to women (8.9%). The disease resulted in nearly 660,000 deaths, with the mortality rate in women (9.4%) slightly higher than that in men (9.2%). Geographically, the highest incidence is observed in East Asia. According to the age-standardized incidence rate per 100,000, the rates are 23.0 in men and 9.7 in women, making it the highest in major regions worldwide [1]. Gastric cancer is classified into cardia (Esophagogastric junction) and non-cardia types. Chronic Helicobacter pylori infection is considered a major cause of non-cardia gastric cancer [39, 40], with other risk factors including advanced age, smoking, alcohol consumption, previous gastric surgery, and pernicious anemia [41, 42]. High-salt diets may also increase the risk of H. pylori infection and synergistically promote gastric cancer development [43]. Cardia cancer is mainly associated with gastroesophageal reflux disease, and about one-fifth of cardia cancers worldwide can also be attributed to H. pylori infection [44]. Similar to the epidemiologic features of colorectal cancer incidence, gastric cancer incidence is also rising among younger populations [45, 46]. Endoscopic resection, including endoscopic mucosal resection (EMR) and endoscopic submucosal dissection (ESD), has become the main and well-defined treatment for early gastric cancer [47, 48], with ESD being the preferred method due to its capability for complete resection and minimal local recurrence [48, 49]. For clinical stages T1 with lymph node positivity or T2-T4a with any lymph node (Nx) involvement without distant metastasis (M0), radical surgical resection is the main treatment, often combined with perioperative chemotherapy [50]. For locally advanced unresectable or metastatic (M1) gastric cancer, chemotherapy [51] can improve survival and quality of life. Treatments such as anti-HER2 monoclonal antibodies [52], anti-VEGFR antibodies [53], and immune checkpoint inhibitors [54] also benefit survival.
In 2022, there were over 1.9 million new cases of colorectal cancer globally, ranking third. The incidence rate was significantly higher in men (6.1%) compared to women (3.5%). The disease caused 904,000 deaths, making it the second leading cause of cancer-related deaths, with the mortality rate in men (7.9%) substantially higher than in women (5.4%). Geographically, rectal cancer is more aggressive, with an age-standardized incidence rate per 100,000 of 12.9 in men and 7.1 in women, ranking sixth globally among major regions. Currently, the incidence of colorectal cancer is primarily concentrated in European countries [1]. Despite a stable or declining overall incidence, colorectal cancer rates among individuals under 50 are increasing annually by 1%-4% in many high-income countries [55]. Overweight, obesity, alcohol consumption, smoking, and red meat consumption increase the risk of colorectal cancer [56, 57], along with long-term inflammatory bowel disease and a history of colorectal adenomas [58, 59]. For early-stage (T1) colorectal cancer patients, endoscopic resection, including endoscopic mucosal resection, endoscopic submucosal dissection, and endoscopic full-thickness resection, is a minimally invasive and safer method [60], with further surgical resection and mesenteric lymphadenectomy determined based on pathological analysis. Traditional surgical complete resection remains the primary treatment for colorectal cancer [56], except for advanced cases. For locally advanced or metastatic colorectal cancer, treatments like chemotherapy, radiotherapy [61], local metastasis resection, or radiofrequency ablation [56] can extend survival. Biological agents, including anti-VEGF monoclonal antibodies [62], EGFR-targeted therapy [63], and PD-1 inhibitors [64], also benefit colorectal cancer patients.
Despite significant advancements in the prevention and treatment of esophageal and gastrointestinal cancers, particularly with the application of immune checkpoint inhibitors significantly improving survival rates, the global incidence and mortality rates of these tumors remain high. This highlights the need for further exploration of the underlying mechanisms of cancer development, identification of new therapeutic targets and biomarkers, and promotion of personalized treatment and early diagnosis to advance the treatment of esophageal and gastrointestinal cancers.
DEAD-box helicase family
Structure of DEAD-box helicase proteins
DEAD-box helicases are composed of two RecA-like domains connected by covalent bonds, forming the core of the helicase, which contains approximately 12 conserved motifs (Fig. 1). The RecA-like domain 1 includes ATP binding and hydrolysis motifs (Q motif, motif I, and motif II), RNA binding motifs (motifs Ia, Ib, and Ic), and a motif III that coordinates ATP and RNA binding sites. The DEAD-box helicase family contains a relatively conserved amino acid sequence Asp(D)-Glu(E)-Ala(A)-Asp(D) located in motif II; this conserved sequence is also the basis for the naming of DEAD-box helicases [4, 65, 66]. RecA-like domain 2 contains RNA binding motifs (motifs IV, IVa, and V), an ATP binding and hydrolysis motif VI, and a motif Va that coordinates ATPase and helicase activities [67]. Besides the RecA-like domains, DEAD-box helicase proteins have variable auxiliary N-terminal and C-terminal regions, which, through interactions with other proteins or RNA, endow DEAD helicases with diverse functions that are essential for their cellular roles [3, 68].
Fig. 1.

Structure of the DEAD-box Helicase Protein. The structure comprises the helicase core formed by RecA-like Domain 1 and RecA-like Domain 2, along with the Amino-terminal and Carboxyl-terminal regions. The helicase core contains motifs for ATP binding and hydrolysis: the Q motif, Motif I, Motif II, and Motif VI; RNA binding motifs: Motif Ia, Motif Ib, Motif Ic, Motif IV, Motif IVa, and Motif V; and motifs coordinating ATP and RNA binding sites: Motif III and Motif Va. The two RecA-like domains are linked by two covalent bonds
Functions of DEAD-box helicase proteins in cells
Transcription
The N-terminal ATPase/helicase domain of DDX1 interacts with the C-terminal transactivation domain of p65 (NF-κB subunit RelA), acting as a cofactor to enhance NF-κB-mediated transcriptional activation, increasing IFN-γ-induced PD-L1 expression in hepatocellular carcinoma cells [69]. In colorectal cancer cells, DDX3 protein can increase the binding of the transcription factor SP1 to the KRAS gene promoter in colorectal cancer cells, thereby enhancing the transcription of KRAS [70]. It also interacts with lncRNA (TCONS_00012883), enhancing the recruitment of transcription factor YY1 to the MMP1 promoter, thereby increasing MMP1 expression and regulating downstream genes [71]. DDX5 occupies the TCF4/LEF binding element (TBE) site on the FOXM1 promoter with β-catenin, acting as a transcriptional co-activator to positively regulate FOXM1 expression [72]. In non-small cell lung cancer cells, DDX5 promotes the transcription of c-Myc and CCND1 [73]. DDX5 also collaborates with β-catenin and NF-κB to occupy the AKT promoter, enhancing AKT transcription [74]. In colorectal cancer cells, DDX20 regulates the progression of colorectal cancer by interacting with the nuclear receptor steroidogenic factor-1, thereby inhibiting its transcriptional activity [75]. The role of DDX21 in colorectal cancer cells is mainly achieved by increasing the expression of CDK1: by interacting with WDR5, promoting the affinity of H3K4me3 to the promoter of cell cycle-dependent protein kinase 1 [76]. In hepatocellular carcinoma cells, DDX27 increases the mRNA and protein expression of major vault protein (MVP) [77]. DDX55 interacts with BRD4, forming a transcriptional regulatory complex that occupies the PIK3CA promoter, positively regulating its transcription in hepatocellular carcinoma cells [78]. DDX56 enhances the transcription of MIST1 by recruiting MECOM (MDS1 and EVI1 complex locus) to the MIST1 promoter, thereby inducing the PTEN-AKT signaling pathway and promoting proliferation in hepatocellular carcinoma cells [79].
Pre-mRNA splicing/alternative splicing
The DDX family proteins often participates in spliceosome assembly, regulating RNA splicing processes. DDX3X binds to KLF4 mRNA in breast cancer cells, regulating the generation of specific KLF4 isoforms by alternative splicing during KLF4 mRNA splicing [80]. DDX5 and DDX17 can also regulate the invasiveness of tumor cells by cascading corresponding signal transduction pathways through selective splicing of RNAs of DNA and chromatin binding factors [81]. DDX17 also induces the retention of intron 3 in lncRNA PXN-AS1 during alternative splicing, producing corresponding transcripts that cascade downstream genes to promote liver cancer metastasis [6]. DDX24 is a component of the 17S U2 snRNP complex, involved in regulating pre-mRNA splicing [12]. DDX27 regulates the expression of LPP protein by modulating the alternative splicing of LPP pre-mRNA, primarily by reducing exon-skipping events, thereby influencing the progression of gastric cancer [82]. In colorectal cancer cells, the DDX39B protein increases the levels of mature FUT3 mRNA by splicing FUT3 pre-mRNA. Additionally, DDX39B also plays a role in the selective splicing of FUT3 [83]. DDX20 interacts with EB virus nuclear proteins. and DDX20 is also a part of the spliceosomal snRNP complex. DDX46 is crucial in pre-mRNA splicing during or before pre-spliceosome assembly [75]. DDX56 can also affect related biological processes by intervening in the synthesis of precursor messenger RNA through alternative splicing [84].
Ribosome biogenesis
DDX21 can form a 7SK small nuclear ribonucleoprotein complex by binding with 7SK RNA, accumulating at the promoter of RNA polymerase II. It regulates RNA polymerase II and affects the synthesis of ribosomal proteins and snoRNA, thereby playing a multifaceted role in various steps of ribosome biogenesis [85]. DDX21 forms ring structures around multiple RNA polymerase I (Pol I) complexes, inhibiting pre-rRNA transcription, which can be counteracted by SLERT (a snoRNA-ended lncRNA) [86]. DDX27 regulates ribosome biogenesis by modulating ribosomal RNA maturation in zebrafish [87].
RNA transport
DDX39B, in colorectal cancer cells, participates in forming the mRNA export complex through protein interactions. This export complex binds to the first exon of FUT3 mRNA, thereby facilitating the export of FUT3 mRNA [83]. Additionally, DDX39B can form a complex with PKM2 and importin α5, accelerating the nuclear translocation of PKM2 through a mechanism independent of ERK1/2-mediated PKM2 phosphorylation [88].
Translation
DDX3 recognizes specific RNA structures and/or sequence elements within the 5′-untranslated region to regulate the translation and protein synthesis of target genes [89]. For instance, DDX3 interacts with the 5′-untranslated region of Rac1 mRNA to promote its translation, affecting β-catenin protein stability in a Rac1-dependent manner [90]. DDX3X mediates cap-independent MITF protein translation by embedding an internal ribosome entry site (IRES) within the MITF mRNA 5′-untranslated region, controlling MITF protein levels and influencing melanoma metastasis potential and response to targeted therapy [91]. DDX6 forms a protein complex with YBX1, binding to stem-loops in the 3′-untranslated region (UTR) of mRNAs of proliferation/self-renewal regulators (such as CDK1, HMGB2, ACTL6a, and EZH2), recruiting them to EIF4E to promote their translation [92]. DDX6 also promotes cap-independent c-myc mRNA translation through an internal ribosome entry site (IRES) in gastric cancer cells [93]. Additionally, DDX6 acts as an RNA-binding protein for HER2 and FGFR2 mRNAs in gastric cancer cells, positively regulating their translation [94]. In pancreatic cancer cells, DDX6 binds to KIFC1 (Kinesin Family Member C1) mRNA, upregulating its protein expression, promoting pancreatic cancer proliferation, and inhibiting apoptosis [8]. However, in breast cancer cells, DDX6 interaction with VEGF mRNA 5′-UTR inhibits translation [95]. This contradiction in promoting and inhibiting translation may be influenced by different intracellular environments of various tumor types.
RNA degradation
DEAD-box helicase family proteins can regulate RNA degradation processes through either promotion or inhibition. In colorectal cancer cells, DDX3X stabilizes GATA2 mRNA by binding to lncRNA GATA2-AS1, maintaining GATA2 mRNA levels [96]. DDX6 mediates mRNA decay of differentiation-inducing factors (such as KLF4) by interacting with the decapping complex protein (EDC3) [92]. EIF4E binding protein 4E-T cooperates with DDX6 and the CCR4-NOT complex, linking the Lsm1-7/Pat1 complex associated with the 3′ end of mRNA to EIF4E bound to the 5′ cap, promoting mRNA decay [97]. The DDX20 and Ago protein (Ago1-4) complex can selectively bind to the guide strand of miRNA, promote RISC formation, and play a role in mRNA inhibition or degradation [98]. In gastric cancer cells, DDX24 regulates HK1 mRNA stability, positively influencing HK1 levels at the transcriptional level [12]. In hepatocellular carcinoma cells, DDX24 binds to LAMB1 (Laminin Subunit Beta-1) mRNA, increasing its stability [99]. In gastric cancer cells, DDX54 binds to lncRNA SNHG10 and PBX3 (Pre-B-cell Leukemia Transcription Factor 3) mRNA, maintaining PBX3 mRNA stability [100].
Cell cycle regulation
DDX1 influences the cell cycle of testicular tumor cells by regulating CCND2, thereby promoting the progression of testicular tumors [101]. Silencing DDX3X slows the proliferation of gastric cancer cells by inducing G1 phase arrest [80]. Downregulation of DDX5 reduces the expression levels of CDK2 (cyclin-dependent kinase 2) and CyclinD1, blocking the G1/S cell cycle checkpoint to inhibit esophageal cancer cell proliferation [102]. DDX21 promotes the G1/S phase transition in gastric cancer cells by upregulating Cyclin D1 and CDK2 levels, participating in cell cycle regulation [103]. It also interacts with CDC5L protein to promote the G2/M transition, thus enhancing colorectal cancer cell proliferation [104]. Downregulation of DDX27 delays the G1/S transition in colorectal cancer cells [105] and regulates the gastric cancer cell cycle through both TP53-dependent and independent mechanisms [106]. Silencing DDX46 causes cell cycle arrest in the G1 phase, regulating the cell cycle of esophageal squamous cell carcinoma cells [107]. Silencing DDX56 increases the expression of FOXO1 and its downstream effector p21 Cip1, which binds to CDK2, leading to the inactivation of the CDK2/cyclin E1 complex at the G1-S DNA damage checkpoint, thereby arresting gastric cancer cells in the G1-S phase [108].
miRNA biogenesis
DDX5, as part of the Drosha microprocessor, is recruited by TGF-β and BMP-specific SMAD signal transducers to form a complex with pri-miR-21, thereby regulating the biogenesis of miRNA-21 [109]. p53 also interferes with the functional assembly between DDX5 and the Drosha complex, affecting its regulation of miRNA synthesis [110]. DDX18 protein can also combine with Drosha, as a component of the complex, to promote the maturation of microRNA-21 [111].
Protein–protein interactions
DDX helicase proteins can exert additional cellular functions through protein–protein interactions. DDX3X binds to USP7, stabilizing itself to prevent degradation by the ubiquitin–proteasome system (UPS), thereby enhancing Wnt/β-catenin signaling and promoting the development of a partial EMT state in colorectal cancer cells [112]. DDX27 protein promotes the interaction between nucleophosmin (NPM1) and p65 within the nucleus, increasing the binding of p65 to NF-κB gene promoters. This enhances transcription and promotes the expression of downstream NF-κB target genes, thereby increasing the proliferation of colorectal cancer cells and inhibiting apoptosis [113]. DDX39B protein can directly bind to the amino terminus of PKM2 protein, reduce its ubiquitination and degradation, and increase the stability of PKM2 protein [88].
The impact on R-Loop metabolism
R-loops, composed of RNA/DNA hybrid duplexes and the displaced non-template strand, play crucial roles in cellular processes. These roles are dualistic: on the one hand, cells tightly regulate RNA/DNA hybrids within R-loops to facilitate critical events such as transcription termination, gene regulation, telomere stability, and DNA repair. On the other hand, dysregulation of R-loop dynamics can lead to transcription-replication conflicts, DNA damage, and genomic instability [114]. Thus, precise regulation of R-loop levels serves as an essential quality control mechanism for maintaining genomic stability. Recent studies have identified the RNA/DNA hybrids within R-loops as immunogenic entities. Aberrant accumulation of R-loops can activate innate immune responses, leading to cell death and contributing to the development of cancer and neurodegenerative diseases [115]. Multiple studies have demonstrated the critical roles of DEAD-box helicase family proteins in R-loop regulation. For instance, DDX5 directly resolves RNA/DNA hybrid structures, thereby preventing R-loop accumulation [116]. DDX18 reduces the formation of R-loops during transcription and participates in the removal of R-loops associated with DNA damage [117]. DDX19 removes R-loops generated during replication-transcription conflicts, thus maintaining genomic stability [118]. DDX21 efficiently unwinds R-loops, reducing their accumulation and mitigating DNA damage [119]. DDX41, enriched in promoter regions, unwinds RNA/DNA hybrids to decrease R-loop accumulation, thereby alleviating inflammatory responses triggered by replication stress and double-strand breaks, and preventing familial acute myeloid leukemia (AML) associated with its mutations [120]. Additionally, DDX1 regulates R-loop formation through interactions with other protein factors [121]. These findings highlight the complexity of DEAD-box helicase family proteins in R-loop metabolism and their diverse roles in cellular functions.
In summary, the DDX helicase family proteins regulate tumorigenesis and progression by participating in various aspects of RNA metabolism in tumor cells. DDX helicases act not only as helicases but also as transcription factors, splicing factors, translational regulators, degradation factors, and cell cycle regulators, making them important targets in tumor research.
Roles of DEAD-box helicases in esophageal cancer
DDX5
DDX5 mRNA is highly expressed in ESCC tissues. IHC results show that DDX5 protein expression is significantly upregulated in ESCC tissues compared to adjacent normal samples; the expression levels of DDX5 in corresponding ESCC cell lines are also significantly upregulated [102, 122]. KM survival curve analysis shows that high DDX5 protein levels lead to lower survival rates. The high expression of DDX5 promotes tumor cell growth, migration, invasion, subcutaneous tumor growth in mice, and metastasis to organs in vivo [102]. Silencing DDX5 significantly inhibits the proliferation of esophageal cancer cells in vitro and the growth of xenografted tumors in vivo [122]. Downregulation of DDX5 reduces the expression levels of CDK2 and CyclinD1, blocking the G1/S cell cycle checkpoint to inhibit esophageal cancer cell proliferation [102]. It also leads to increased E-cadherin expression and decreased Vimentin expression, inhibiting the EMT process in esophageal cancer cells [102, 122]. Silencing DDX5 leads to a significant downregulation of BIP, p-PERK, and p-eIF2α protein levels, as well as a reduction in P62 protein expression. This indicates that DDX5 promotes the progression of esophageal squamous cell carcinoma by inducing autophagic defects and endoplasmic reticulum stress [102]. Additionally, Z. Ma et al. reported that silencing DDX5 significantly inhibited the expression of β-catenin and c-Myc, thereby suppressing the proliferation and invasion of esophageal cancer cells mediated by the Wnt/β-catenin signaling pathway [122].
DDX46
DDX46 mRNA and protein expression are significantly higher in ESCC tissues than in normal tissues. Overexpression of DDX46 promotes colony formation and proliferation abilities of ESCC cells. Silencing DDX46 causes cell cycle arrest in the G1 phase and reduces Akt and IκBα phosphorylation to inhibit NF-κB activity, inducing apoptosis [107].
DDX51
In ESCC, both mRNA and protein levels of DDX51 are higher in tumor tissues compared to adjacent normal tissues. Its high expression is associated with lower tumor differentiation, lymph node metastasis and advanced AJCC stage. KM analysis shows that high DDX51 expression is associated with lower survival rates. High DDX51 expression promotes the proliferation, migration, and invasion of ESCC cells and reduces apoptosis. Low expression of DDX51 inhibits the growth of ESCC tumors in mouse xenograft models. Mechanistically, DDX51 promotes the progression of ESCC by increasing the phosphorylation levels of PTEN, PI3K, AKT, and mTOR, activating the PI3K/AKT signaling pathway [123].
Roles of DEAD-box helicases in gastric cancer
DDX5
In gastric cancer tissues, both the mRNA and protein levels of DDX5 are elevated compared to the adjacent normal tissues. DDX5 expression increases with the progression of gastric cancer. High expression of DDX5 is significantly associated with high Ki-67 expression, indicating poor prognosis and high invasiveness. High expression of DDX5 significantly increases the proliferation and colony formation of gastric cancer cells, as well as the proliferation of gastric cancer in xenograft models. Mechanistically, DDX5 overexpression increases the expression of phosphorylated mTOR (p-mTOR) and S6K1 (p-S6K1), without significantly affecting the total protein levels of mTOR and S6K1, thereby increasing the activity of the mTOR/S6K1 signaling pathway, which is crucial for gastric cancer cell proliferation [124].
DDX6
In gastric cancer tissues, the protein expression level of DDX6 is markedly increased compared to adjacent normal gastric tissues, and it is not correlated with the histopathological type or clinical stage of the cancer. Silencing DDX6 inhibits the growth of gastric cancer cells. DDX6 promotes the growth of gastric cancer cells and cancer progression by binding to c-Myc mRNA and promoting c-Myc protein expression [93].
DDX18
In gastric cancer tissues, DDX18 mRNA and protein levels are significantly elevated. The expression of DDX18 is correlated with cancer location, size, Borrmann type, vascular invasion, differentiation degree, lymph node metastasis, and TNM stage. Cox multivariate regression analysis shows that the expression of DDX18 can be used as an independent factor to predict the overall survival of patients with gastric cancer after surgery. DDX18 promotes the proliferation, migration, and invasion of gastric cancer cells and reduces apoptosis, while also promoting tumor growth in nude mouse models. Mechanistically, DDX18 protein binds to Drosha as a component of the complex, promoting the maturation of microRNA-21, which binds to the 3′UTR of PTEN, promoting PTEN mRNA degradation, affecting AKT phosphorylation, regulating the AKT signaling pathway, and promoting the progression of gastric cancer [111].
DDX20
In gastric cancer tissues, both the mRNA and protein expression levels of DDX20 are significantly higher compared to the adjacent normal tissues. Studies have shown that high expression of DDX20 in gastric cancer cell lines promotes cell proliferation, migration, and invasion. However, log-rank tests indicate that patients with higher levels of DDX20 expression have longer overall survival and disease-free survival compared to those with lower levels. This discrepancy may be due to the elevated DDX20 expression leading to a significant increase in activated CD8 + and CD4 + T cells, thereby resulting in enhanced immune activation against the tumor. The specific mechanisms underlying this phenomenon require further investigation [125].
DDX21
In gastric cancer tissues, both the mRNA and protein levels of DDX21 are significantly upregulated compared to the adjacent normal tissues [103, 126]. DDX21 expression is positively correlated with tumor size, lymph node metastasis, and TNM stage [103]. Analysis of GSE224654 data shows that gastric cancer patients with high DDX21 expression have lower survival rates [126]. High DDX21 expression promotes gastric cancer cell proliferation, colony formation [103], migration [126], and xenograft tumor growth [103]. Mechanistically, DDX21 promotes the G1/S phase transition in gastric cancer cells by upregulating Cyclin D1 and CDK2 levels, promoting gastric cancer growth and progression [103]. Additionally, M. Chang et al. reported that lnc-PLCB1 reduces the stability of DDX21 protein by binding to it, thereby downregulating the expression of CCND1 (proliferation-related factor) and Slug (epithelial-mesenchymal transition-related factor), inhibiting tumor development in gastric cancer [126].
DDX24
In gastric cancer tissues, the mRNA and protein expression levels of DDX24 are significantly higher compared to the adjacent normal tissues. DDX24 expression is significantly correlated with histological grade, tumor size, invasion depth, and lymph node metastasis of gastric cancer. High DDX24 expression leads to poor survival rates in gastric cancer patients. Overexpression of DDX24 promotes the proliferation, migration, and invasion of gastric cancer cells, as well as the growth of gastric cancer in xenograft models. Mechanistically, DDX24 positively regulates the levels of HK1 by stabilizing HK1 mRNA at the transcriptional level. Increased HK1 expression promotes glucose uptake and lactate production in gastric cancer cells, thus promoting tumor progression [12].
DDX27
In gastric cancer tissues, DDX27 mRNA and protein are highly expressed [82, 106], and TCGA gene expression profiles show significant elevation in gastric adenocarcinoma. In GC cell lines, the mRNA and protein levels of DDX27 are significantly upregulated compared to normal epithelial cell line GSE-1. KM analysis reveals that patients with high DDX27 expression have poorer overall survival [82]. Moreover, high expression of DDX27 is associated with deeper tumor infiltration [82], lymph node infiltration [82], vascular invasion [106], and distant organ metastasis [82, 106]. Multivariate Cox analysis indicates that high DDX27 expression is an independent prognostic factor for OS [82] and CSS [106] in gastric cancer patients. High DDX27 expression enhances the colony formation ability [106], motility [82], and tumorigenicity [106], and promotes lung metastasis in gastric cancer cells [82]. Mechanistically, Y. Jin et al. reported that DDX27 has a splicing function, regulating the expression of LPP (Lipoma-preferred partner) by reducing the skipping exon event on exon 3 of the full-length LPP transcript, thereby enhancing the translation of functional LPP protein, promoting epithelial-mesenchymal transition (EMT), and advancing gastric cancer progression through the DDX27/LPP/EMT regulatory axis [82]. Additionally, Y. Tsukamoto et al. found that silencing DDX27 leads to an increased proportion of gastric cancer cells in the G1 phase, with a reduction in S phase and G2/M phase cells. DDX27 induces nuclear TP53 accumulation, showing that DDX27 can regulate the cell cycle through both TP53-dependent and independent mechanisms, promoting gastric cancer cell proliferation and tumor progression [106].
DDX46
DDX46 mRNA expression is significantly upregulated in gastric cancer tissues compared to normal gastric tissues. DDX46 mRNA and protein levels are also significantly higher in human gastric cancer cell lines compared to normal gastric epithelial cell line GSE-1. Overexpression of DDX46 enhances the proliferation, invasion, and growth of xenograft tumors in gastric cancer cells. Silencing DDX46 reduces the phosphorylation levels of AKT, subsequently decreasing the phosphorylation of GSK-3β. This results in increased GSK-3β-mediated degradation of β-catenin and suppression of the Wnt signaling pathway. Therefore, DDX46 may mediate the progression of gastric cancer by regulating the Akt/GSK-3β/β-catenin signaling pathway [127].
DDX56
In gastric cancer tissues, both mRNA and protein levels of DDX56 are higher than in adjacent non-cancerous tissues, and KM curve analysis shows that patients with high DDX56 expression have lower survival rates. DDX56 expression is positively correlated with lymph node metastasis. High DDX56 expression promotes the proliferation, migration, and invasion of gastric cancer cells, reduces apoptosis, and promotes the growth of xenograft tumors. Mechanistically, DDX56 promotes gastric cancer progression by inhibiting FOXO1, p21 Cip1 protein expression, thereby activating the downstream cyclin E1/CDK2/c-Myc signaling pathway [108].
Roles of DEAD-box helicases in colorectal cancer
DDX3
In colorectal cancer, high DDX3 expression is positively correlated with phosphorylated DVL2 and nuclear β-catenin expression [128]. High DDX3 expression promotes the invasiveness of colorectal cancer cells [70, 128] and the ability to metastasize to the lungs in nude mice CRC models [128]. KM analysis indicates that high DDX3 expression is associated with shorter overall survival (OS) and relapse-free survival (RFS) in colorectal cancer patients [70, 128], and Cox regression analysis shows that DDX3 is an independent prognostic factor for OS and RFS [128]. Mechanistically, high DDX3 expression enhances the binding of SP1 to the KRAS promoter, increasing KRAS expression, which activates the PI3K/AKT pathway, leading to GSK3β phosphorylation at Ser9, reducing GSK3β-mediated β-catenin degradation, increasing β-catenin levels, and promoting tumor invasion and metastasis through the β-catenin/ZEB1 axis, resulting in poor prognosis in colorectal cancer patients [70]. T. Y. He et al. also revealed that DDX3-mediated β-catenin/TCF activation occurs partially through the CK1ε/Dvl2 axis. DDX3 acts as a regulatory subunit of CK1ε, promoting CK1ε phosphorylation of Dvl2, blocking PP2A-mediated β-catenin degradation, increasing β-catenin stability, and enhancing the binding of nuclear β-catenin to TCF, upregulating downstream β-catenin/TCF signaling pathway genes, promoting tumor invasion and progression in colorectal cancer [128]. Additionally, M. R. Heerma van Voss et al. reported that DDX3 acts on the TCF4 promoter to increase TCF4 expression, and upregulates the mRNA expression of downstream TCF4 target genes. DDX3 knockdown reduces colorectal cancer cell proliferation and causes cell cycle arrest in the G1 phase [129]. These studies collectively highlight the crucial role of β-catenin in DDX3-mediated colorectal cancer progression and the need for further research on the interaction mechanisms between different signaling pathways.
DDX5
In colorectal cancer, DDX5 mRNA expression is significantly elevated, and its transcription levels increase with cancer progression. IHC shows that DDX5 protein is also upregulated in colon cancer tissues compared to normal colon samples. High DDX5 expression promotes the proliferation, migration, and colony formation ability of colorectal cancer cells. DDX5 regulates the colorectal cancer cell cycle by increasing the percentage of cells in the S phase, promoting tumor growth. The mechanism involves the DDX5/β-catenin/FOXM1 axis. DDX5 and β-catenin occupy the TCF4/LEF binding element (TBE) site on the FOXM1 promoter, acting as a transcriptional co-stimulatory factor to enhance FOXM1 transcription, which increases FOXM1 expression. As an oncogene in colorectal cancer, FOXM1 overexpression promotes proliferation, migration, and tumor growth [72]. Additionally, N. Wu et al. reported that O-GlcNAcylation modifies DDX5 protein, increasing its stability, activating the AKT/mTOR signaling pathway, and promoting colorectal cancer progression [130].
DDX10
In colorectal cancer, DDX10 mRNA and protein expression are significantly elevated, correlated with cancer staging. In colon adenocarcinoma, high DDX10 expression is associated with poor prognosis. High expression of DDX10 promotes colorectal cancer cell proliferation, migration, invasion, tumor in vivo and metastasis; reduces colorectal cancer cell apoptosis. Mechanistically, DDX10 selectively splices RPL35 mRNA, increasing the occurrence of alternative termination (AT) in RPL35 mRNA, which then acts on downstream E2F transcription factor-mediated signaling pathways, promoting colorectal cancer progression [131], though further research is needed to elucidate the specific signaling pathways involved.
DDX17
In colorectal cancer, DDX17 mRNA and protein levels are significantly higher than in non-cancerous mucosal tissues. In colorectal liver metastases, DDX17 protein expression is also significantly elevated compared to primary colorectal cancer tissues. DDX17 expression was correlated with tumor size, N stage, M stage, and overall TNM stage. High expression of DDX17 is associated with a shorter OS in CRC patients, and multivariate Cox analysis shows that DDX17 expression is an independent prognostic factor for overall survival. High DDX17 expression promotes CRC cell proliferation, migration, and invasion, as well as liver metastasis in xenograft models. High expression of DDX17 inhibits Claudin-1 and E-cadherin expression while promoting the expression of vimentin and N-cadherin, thereby facilitating the progression of EMT. Mechanistically, DDX17 downregulates miR-149-3p, reducing its inhibitory effect on CYBRD1 (cytochrome b reductase 1) by binding to CYBRD1’s 3′-UTR, leading to increased CYBRD1 expression, promoting CRC cell metastasis and EMT, advancing colorectal cancer metastasis and invasiveness [132].
DDX21
DDX21 mRNA and protein expression are significantly elevated in colorectal cancer tissues compared to adjacent normal tissues [76, 104, 133, 134], and high DDX21 expression is associated with lower overall survival in CRC patients. High DDX21 expression promotes CRC cell proliferation [76, 104] and Ki-67 expression, as well as xenograft tumor growth [104]. Mechanistically, P. Lu et al. reported that DDX21 interacts with WDR5, promoting the affinity of H3K4me3 to the promoter of CDK1, thereby increasing CDK1 transcriptional expression, participating in cell cycle regulation, accelerating the cell cycle, and promoting CRC cell proliferation and tumor growth [76]. Additionally, K. Wang et al. found that DDX21 promotes CRC cell G2/M transition by interacting with cell division cycle 5 protein, enhancing tumor cell proliferation and tumor growth in vivo. DDX21 downregulation is accompanied by decreased MMP-2 and -9 expression, suggesting a potential role for DDX21 in colorectal cancer metastasis [104].
DDX27
DDX27 mRNA and protein levels are significantly higher in colorectal cancer tissues compared to adjacent normal tissues [105, 113]. DDX27 expression is related to tumor location, with lower expression in colon cancer compared to rectal cancer, but both higher than in adjacent normal tissues. KM curves show that high DDX27 expression is significantly associated with poorer relapse-free survival in CRC patients; in colon cancer, high DDX27 expression is related to shorter survival, but no significant difference is observed in rectal cancer. Cox regression analysis shows that high DDX27 expression is an independent risk factor for relapse-free survival. High DDX27 expression promotes CRC cell proliferation [105, 113] and inhibits apoptosis [105]; DDX27 downregulation delays G1/S transition and increases CRC cell sensitivity to 5-fluorouracil [113]. High expression of DDX27 promotes the expression of Slug and vimentin while inhibiting E-cadherin expression to regulate epithelial-mesenchymal transition (EMT), thereby facilitating the migration and invasion of colorectal cells. High DDX27 expression promotes CRC growth and lung metastasis in mice. Mechanistically, DDX27 protein interacts directly with nucleophosmin (NPM1), promoting its interaction with nuclear p65, enhancing p65 binding activity to NF-кB target gene promoters, and increasing the expression of downstream NF-kB target genes (BIRC3, CCL20, CXCL3, NFKBIA, TNF, and TNFAIP3), promoting cell proliferation, inhibiting apoptosis, and advancing CRC tumor metastasis [113]. Additionally, C. Yang et al. reported that DDX27 regulates CRC cell stemness. Downregulation of DDX27 reduced CRC sphere formation, the expression of CD44, CD133, EpCAM, and LGR5, and reduces in vitro and in vivo CSC self-renewal capacity. High DDX27 expression stabilizes the self-renewal phenotype of CRC cells, delaying differentiation into a non-stem cell-like state, leading to CSC overpopulation and promoting tumor initiation and progression [105].
DDX39B
DDX39B exhibits higher mRNA and protein expression levels in colorectal cancer tissues compared to adjacent non-tumor tissues [11, 83, 88]. Its expression is further elevated in CRC liver metastases compared to paired primary tumors. High DDX39B expression correlates with larger tumor size, poorer histological grade, regional lymph node and distant organ metastasis, and advanced AJCC stage. Patients with high DDX39B expression have poorer prognosis compared to those with low expression. Multivariate Cox regression analysis indicates that high DDX39B expression is an independent prognostic factor for overall survival in CRC patients [88]. DDX39B promotes CRC cell proliferation [11, 88], migration, invasion [83, 88], as well as tumor growth in xenograft models [11, 88] and CRC metastasis to the lungs [88] and spleen [83]. Additionally, xenografts with high DDX39B expression exhibit increased Ki-67 protein levels in tumor tissues [11]. Mechanistically, H. Zhang et al. demonstrated that DDX39B directly binds to the first exon of CCND1/CDK6 and facilitates their splicing and export, thus upregulating their expression, promoting G1/S phase transition in CRC cells, and reducing the percentage of CRC cells in the G1 phase, thereby enhancing CRC proliferation [11]. Simultaneously, C. He et al. indicated that DDX39B also plays a role in the alternative splicing of FUT3. DDX39B binds to the splice sites of pre-mRNA of FUT3, significantly increasing the mature FUT3 mRNA levels. Moreover, DDX39B participates in forming the mRNA export complex through protein interactions. This export complex binds to the first exon of FUT3 mRNA, thereby facilitating the export of FUT3 mRNA. High FUT3 expression enhances fucosylation of TGFβR-I, activates TGFβ/SMAD2 signaling, upregulates MMPs and EMT transcription factors (e.g., Snail, Slug, ZEB1), driving epithelial-mesenchymal transition and promoting CRC progression [83]. Additionally, DDX39B interacts with proteins to interfere with protein degradation. G. Zhao et al. reported that The study by G. Zhao, et al. indicated that DDX39B directly binds to PKM2 through protein–protein interactions, competitively inhibiting stub1-mediated ubiquitination and degradation of PKM2, thereby increasing the stability of PKM2. DDX39B recruits importin α5, forming a complex with DDX39B, PKM2, and importin α5 to accelerate nuclear translocation of PKM2 independently of ERK1/2-mediated phosphorylation of PKM2. Subsequently, nuclear PKM2 acts as a protein kinase and transcription co-activator, activating the Warburg effect in CRC, regulating the expression of oncogenes and metabolic genes, and promoting CRC tumor progression [88].
DDX46
DDX46 protein levels are significantly elevated in colorectal cancer (CRC) compared to paired adjacent tissues. Increased DDX46 expression promotes CRC cell proliferation and colony formation. Silencing DDX46 leads to elevated levels of apoptosis markers caspase-3 and PARP-1, resulting in increased apoptosis of CRC cells. Additionally, it induces G0/G1 cell cycle arrest, thereby inhibiting cell proliferation and colony formation [2].
DDX56
In colorectal cancer (CRC), both mRNA and protein levels of DDX56 are significantly higher than in adjacent normal tissues. High DDX56 expression is associated with lymphatic infiltration and distant metastasis in tumors. Elevated DDX56 expression correlates with reduced overall survival in CRC patients. Multivariate Cox analysis indicates that high DDX56 expression is an independent prognostic factor for overall survival. High DDX56 expression promotes in vitro proliferation, migration, and in vivo xenograft tumor growth of CRC cells. Mechanistically, DDX56 affects the selective splicing of the tumor suppressor gene WEE1, increasing the levels of aberrant intron-retained WEE1 mRNA, while reducing the levels of intron-free WEE1, a key component of the normal G2-M cell cycle checkpoint. This reduction diminishes its ability to prevent entry into mitosis upon DNA damage, thereby promoting cell cycle progression and cell proliferation, and advancing CRC progression [81].
Summary of the role of DEAD-box helicase family proteins in digestive system tumors
Through the analysis of DEAD-box helicase family proteins in the development of esophageal, gastric, and colorectal cancers, it becomes evident that these proteins play a crucial biological role in digestive system tumors. DEAD-box helicase family proteins regulate cancer cell gene expression and cellular behavior through multiple mechanisms, contributing to tumor initiation, progression, and metastasis. These functions include regulation of the cell cycle, epithelial-mesenchymal transition (EMT), miRNA biosynthesis, RNA stability control, RNA splicing, stem cell activity, stabilization of target proteins, nuclear import of target proteins, and RNA nuclear export. Among these, the role of DEAD-box helicase family proteins in cell cycle regulation is particularly prominent, especially during tumor cell proliferation. Studies have shown that these proteins regulate cyclins (such as Cyclin D1, CDK2, CCND1, CDK6, and CDC5L) and their interacting partners to control cell cycle progression, thereby promoting tumor cell proliferation and survival. This process provides the necessary conditions for tumor growth and plays a crucial role in cancer resistance and metastasis.
In addition, DEAD-box helicase family proteins are widely involved in the regulation of multiple key signaling pathways during digestive system tumorigenesis, especially the β-catenin-related pathway. β-catenin is a key regulator of tumorigenesis and metastasis, participating in tumor progression through various mechanisms. DEAD-box helicase family proteins regulate β-catenin stability and activity, influencing the signaling pathways that promote the formation and metastasis of digestive system tumors. The main pathways involved include: 1. Wnt/β-catenin signaling pathway: This pathway plays a crucial role in many digestive system cancers, regulating cell proliferation, differentiation, and stem cell fate, thereby promoting tumor initiation and progression. 2. Akt/GSK-3β/β-catenin signaling pathway: Akt and GSK-3β regulate cell proliferation and survival through interactions with β-catenin, supporting tumor cell growth. 3. KRAS/PI3K/AKT/GSK-3β/β-catenin/ZEB1 signaling pathway: This pathway integrates multiple oncogenic signals and regulates tumor invasiveness and metastatic potential. 4. CK1ε/Dvl2/β-catenin/TCF signaling pathway: This pathway promotes EMT in tumor cells, which is a key mechanism for tumor metastasis and invasion. 5. DDX5/β-catenin/FOXM1 axis: Through interactions with β-catenin and FOXM1, DDX5 regulates tumor cell proliferation and drug resistance, further exacerbating the malignancy of the tumor. DEAD-box helicase family proteins regulate the intracellular dynamics of these signaling pathways, thereby modulating the occurrence, development, and metastasis of digestive system tumors. Specifically, these proteins alter the transcription and translation processes of cancer cells through interactions with various key molecules, regulating tumor cell proliferation, survival, metastasis, and drug resistance. These mechanisms provide new targets for developing therapeutic strategies against gastrointestinal tumors. In particular, DEAD-box helicase family proteins, due to their critical roles in cell cycle regulation, RNA processing, and cell signaling, may become novel biomarkers and therapeutic targets for early cancer diagnosis and targeted therapy in the future.
Targeted therapies for DEAD box helicases
DEAD box helicases play crucial roles in various cellular functions and cancer progression in numerous types of malignancies. Developing targeted drugs against DEAD box helicase proteins offers a new therapeutic direction and target for cancer treatment. Although progress has been made in the development of drugs targeting DEAD box helicase proteins, research limitations still exist regarding different cancer types and DEAD box helicase proteins. Below is an overview of drugs targeting DEAD box helicase proteins (see Table 1).
Table 1.
Drugs targeting DEAD-box helicase proteins
| Drug Name | Chemical Structure | Targeted DEAD Box Helicase Protein and Mechanism | Effect on Cancers |
|---|---|---|---|
| RK-33 | 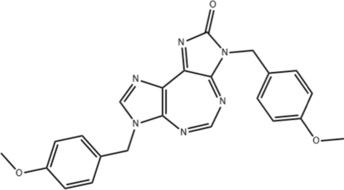 |
DDX3; Binds to ATP-binding site, inhibits RNA helicase activity |
CRC: Inhibits Wnt signaling pathway, suppresses CRC cell growth, and promotes apoptosis; |
| Lung cancer: Inhibits Wnt signaling, induces apoptosis, inhibits NHEJDNA repair pathway, acts as a radiosensitizer; | |||
| DDX5 | CRC: Reduces DDX5 protein levels, unknown mechanism | ||
| NZ51 | 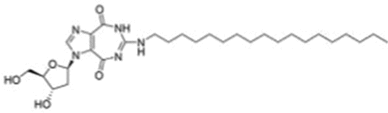 |
DDX3; Binds to ATP-binding site, inhibits RNA helicase activity |
Breast cancer: Inhibits cell viability and motility |
| Ketorolac Salt | 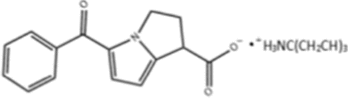 |
DDX3; Binds to P-loop, inhibits RNA helicase activity |
Oral cancer: Inhibits growth of cells and xenograft tumors |
| 7-AID | 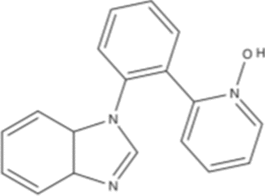 |
DDX3; Binds to ATP-binding domain, inhibits helicase activity |
Cervical squamous cell carcinoma: Anti-proliferative, promotes apoptosis, anti-angiogenesis |
| Ceftriaxone | 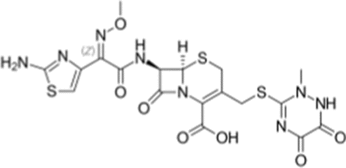 |
DDX3X; Binds to ATP-binding site, inhibits RNA helicase activity |
RB and NB: Inhibits translation of MYCN oncogene, suppresses tumor cell growth |
| RX-5902 | 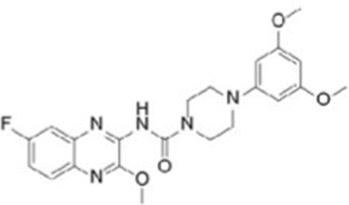 |
DDX5; Inhibits phosphorylated DDX5 binding with β-catenin; Binds to phosphorylated DDX5, inhibits β-catenin-dependent ATPase activity |
Triple-negative breast cancer: Inhibits cell proliferation and EMT transition |
| FL118 | 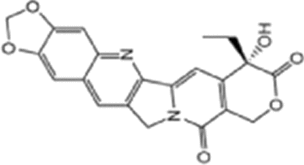 |
DDX5; Inhibits DDX5 expression |
PDAC and CRC: Anti-tumor efficacy |
| EGCG | 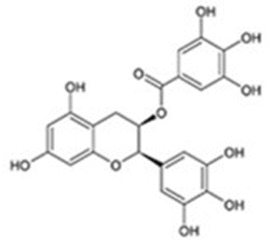 |
DDX5; Dose-dependent degradation of DDX5 protein |
GC: Blocks β-catenin oncogenic signaling |
| Resveratrol | 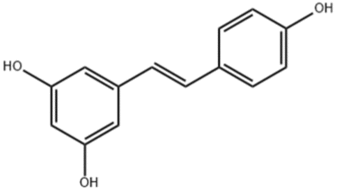 |
DDX5; Promotes metalloprotease-dependent degradation of DDX5 protein |
Prostate cancer: Inhibits mTORC1 signaling in androgen-independent prostate cancer cells, suppresses cell growth |
| Simvastatin | 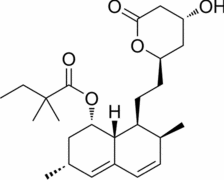 |
DDX5; Inhibits DDX5 expression |
Renal cell carcinoma: Inhibits DDX5 expression and promotes DUSP5 expression, suppresses RCC progression |
| Delphinidin | 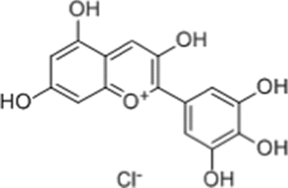 |
DDX17; Inhibits DDX17 expression |
Hepatocellular carcinoma: Inhibits MDR protein and DDX17 expression, increases autophagosome and apoptosis, enhances cisplatin anti-tumor effect |
| Morphine | 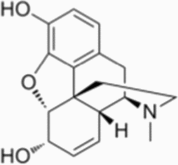 |
DDX49; Inhibits DDX49 expression |
Hepatocellular carcinoma: Reduces phosphorylated MAPK levels and downstream gene expression, suppresses HCC progression |
CRC: Colorectal cancer, GC: Gastric cancer, PDAC: Pancreatic ductal adenocarcinoma, RB: retinoblastoma, NB: neuroblastoma
Drugs targeting DEAD box helicase 3 (DDX3)
DDX3 is highly expressed in various cancers and often functions as an oncogene promoting cancer development. To date, hundreds of different compounds have been developed as potential ATPase/helicase inhibitors of DDX3 [16], with RK-33 being the most widely studied and applied.
RK-33 is a novel small molecule inhibitor synthesized by Kondaskar et al. It binds to the ATP-binding site of DDX3, inhibiting its RNA helicase activity [135]. In colorectal cancer cells, RK-33 specifically binds to DDX3, inhibits its activity, reduces Wnt signaling pathway activation, suppresses CRC cell growth, and promotes apoptosis. Colorectal cancer with wild-type APC and CTNNB1 mutations show higher sensitivity to RK-33 [129]. In lung cancer cells, RK-33 specifically binds to DDX3, inhibits its activity, impairs Wnt signaling, leads to G1 phase cell cycle arrest, and induces apoptosis. Additionally, RK-33 enhances the sensitivity of lung cancer cells and mouse lung cancer models to radiotherapy by inhibiting the non-homologous end joining (NHEJ) DNA repair pathway (superior to radiosensitizer carboplatin) [136]. Further studies have designed small molecule inhibitors targeting DDX3 by simulating RK-33, such as two compounds obtained using computational techniques (PubChem ID: 71,524,060, 71,523,969). These compounds exhibit higher binding affinity than RK-33 and can serve as new drug design scaffolds, though their specific roles in tumors require further research [137].
NZ51 inhibits DDX3’s ATP-dependent helicase activity through thermodynamically favorable hydrophobic and hydrophilic interactions with the amino acid residues at DDX3’s ATP binding site. NZ51 impacts the cell viability and motility of breast cancer cell lines by inhibiting DDX3 helicase activity [138].
Ketorolac salt is a pyrrolizine carboxylic acid derivative that forms stable hydrogen bonds with the P-loop region of DDX3, inhibiting its ATPase activity, and suppresses the in vitro and in vivo growth of oral cancer [139].
Ceftriaxone potentially inhibits ATP-dependent helicase function of DDX3X by binding to the ATP-binding cleft, inhibiting the translation of the MYCN proto-oncogene, thereby suppressing the growth of MYCN-amplified retinoblastoma (RB) and neuroblastoma (NB) cells [140].
Drugs targeting DEAD box helicase 5 (DDX5)
RX-5902 has been shown to inhibit the interaction between phosphorylated DDX5 (p68) and β-catenin, reducing nuclear translocation of β-catenin. RX-5902 directly binds to phosphorylated DDX5 (p68), inhibiting β-catenin-dependent ATPase activity, resulting in decreased levels of downstream proteins c-Myc and cyclin D1, thereby inhibiting cell proliferation and epithelial-mesenchymal transition (EMT) [141, 142]. RX-5902 has undergone phase I and II clinical trials in triple-negative breast cancer [143, 144]. RK-33 also reduces DDX5 protein levels in colorectal cancer cells, though the specific mechanism remains unclear [129]. FL118, whose chemical structure is similar to camptothecin and features a unique “10,11-methylenedioxy” structure, directly targets DDX5. FL118 can inhibit DDX5 expression, exhibiting significant antitumor efficacy in pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC). However, the specific mechanism of FL118’s action on DDX5 remains unclear [145].
Other drugs affecting DEAD box helicase proteins
Apart from the aforementioned drugs targeting DDX3 and DDX5 with elucidated molecular mechanisms, other drugs have also been reported to exert anticancer effects by targeting DEAD box helicase proteins, though their specific mechanisms are not yet clear.
7-Azaindole derivatives(7-AID) compounds, such as (5-[1H-pyrrolo(2,3-b)pyridin-5-yl]pyridin-2-ol), interact with Tyr200 and Arg202 in the Q-motif of DDX3, binding to the ATP-binding domain to exert inhibitory effects. 7-AID exhibits antiproliferative, pro-apoptotic, and anti-angiogenic effects in cervical squamous cell carcinoma, although the precise molecular mechanisms are yet to be clarified. Interestingly, 7-AID compounds can effectively inhibit DDX3 expression in a dose-dependent manner in cervical and breast cancer cells, but the exact reasons remain unknown [146].
(-)-Epigallocatechin-3-gallate (EGCG), the most abundant and biologically active polyphenol in green tea, induces dose-dependent degradation of DDX5 (p68) protein via the proteasome, inhibiting β-catenin oncogenic signaling, thus suppressing the proliferation of gastric cancer cells [147].
Resveratrol, a dietary phytochemical found in grapes and wine, degrades DDX5 protein through metalloproteinase-dependent mechanisms without affecting DDX5 mRNA levels. The reduction of DDX5 protein inhibits mTORC1 signaling in androgen-independent prostate cancer cells, suppressing cancer cell growth and providing a basis for further in vivo studies [148].
Simvastatin inhibits DDX5 expression and promotes DUSP5 expression, inhibiting renal cell carcinoma progression [149].
Delphinidin suppresses the expression of multidrug resistance protein 1 and DDX17 in hepatocellular carcinoma cell lines, leading to increased autophagosome formation and apoptosis. When combined with cisplatin, delphinidin enhances its antitumor effects [150].
Morphine inhibits hepatocellular carcinoma growth and progression by reducing DDX49 expression, thereby decreasing the levels of activated (phosphorylated) MAPK and downstream gene expression changes [151].
Clinical research and application progress of DEAD-box helicase protein targeted drugs
Currently, clinical research on DEAD-box helicase-targeted drugs is relatively limited. The known clinical studies primarily focus on RX-5902, a targeted drug for DDX5. In a Phase I clinical trial, 35 participants were enrolled, of which 15 showed stable disease. The most common adverse events included anorexia, nausea, vomiting, diarrhea, weight loss, and fatigue, indicating that RX-5902 is safe and well-tolerated at the tested dose and schedule [143]. However, most drug research is still in the preclinical stage, which may be attributed to several factors: 1. Diverse and Overlapping Functions of DEAD-Box Helicase Family Members: DEAD-box helicase family members exhibit a wide range of cellular functions, with some functional overlap. This results in a lack of selectivity and specificity for certain inhibitors. Structure-based drug design (SBDD) or virtual screening approaches can be used to develop more specific DEAD-box helicase inhibitors that ensure precise binding to the target helicase. Additionally, high-throughput screening (HTS) methods can be employed to identify compounds with good selectivity, followed by structure optimization using techniques such as crystallography and NMR. 2. Interference with Normal Cellular Functions: DEAD-box helicases are involved in many fundamental cellular processes, and their inhibition may disrupt normal cell functions, leading to toxic reactions. To minimize effects on normal cells, it is crucial to develop more selective inhibitors that specifically target tumor cells or virus-infected cells. Furthermore, combining these inhibitors with other drugs in combination therapy could help reduce the side effects associated with monotherapy. 3. Challenges in Validating Efficacy and Safety in Clinical Trials: DEAD-box helicase inhibitors face challenges in proving their efficacy and safety in clinical trials due to the complexity of their target proteins in cells. Designing reasonable clinical trial protocols to demonstrate the effectiveness and safety of these inhibitors remains a challenge. More preclinical studies are needed to comprehensively evaluate the toxicity, pharmacokinetics, and efficacy of DEAD-box helicase inhibitors. Additionally, multi-phase clinical trials (Phase I, II, III) should be conducted to progressively validate the safety and efficacy of these inhibitors in different patient populations, particularly in the context of personalized medicine.
Future research directions and outlook
The DEAD-box helicase family comprises a vast array of members, each playing roles in various cellular functions and signal transduction pathways, forming a complex and extensive functional network. Due to their multifunctionality, DEAD-box helicases are crucial in the occurrence and progression of various cancers.
Esophageal and gastrointestinal cancers rank high in global cancer incidence, characterized by high malignancy, late diagnosis, and limited treatment options, all of which restrict patient survival. DEAD-box helicase proteins are significantly involved in the development and progression of esophageal and gastrointestinal cancers through various oncogenic molecular mechanisms and regulatory networks (see Tables 2 and 3, Figs. 2, 3, 4 and 5).
Table 2.
Expression, function, relationship with patient survival and prognosis, and relationship with clinical factors of cancer of the DEAD-box helicase family in esophageal cancer, gastric cancer, and colorectal cancer
| DEAD-Box Helicase | Cancer | Expression Status | Role in Cancer | Relationship with Patient Survival and Prognosis | Relevant Clinical Factors |
|---|---|---|---|---|---|
| DDX3 | CRC | NA | Promotive | Independent prognostic factor for OS and RFS | NA |
| DDX5 | EC | High mRNA and protein expression | Promotive | KM analysis: high expression correlates with lower survival | NA |
| GC | High mRNA and protein expression | Promotive | NA | NA | |
| CRC | High mRNA and protein expression | Promotive | NA | NA | |
| DDX6 | GC | High protein expression | Promotive | NA | NA |
| DDX10 | CRC | High mRNA and protein expression | Promotive | NA | TNM stage |
| DDX17 | CRC | High mRNA and protein expression | Promotive | Independent prognostic factor for OS | Tumor size, lymph node metastasis, distant metastasis, TNM stage |
| DDX18 | GC | High mRNA and protein expression | Promotive | Independent prognostic factor for OS | Tumor location, size, Borrmann type, differentiation, vascular invasion, lymph node metastasis, TNM stage |
| DDX20 | GC | High mRNA and protein expression | NA | log-rank test shows high expression correlates with longer OS and PFS | NA |
| DDX21 | GC | High mRNA and protein expression | Promotive | KM analysis: high expression correlates with lower survival | Tumor size, lymph node metastasis, TNM stage |
| CRC | High mRNA and protein expression | Promotive | KM analysis: high expression correlates with lower survival | Lack of research | |
| DDX24 | GC | High mRNA and protein expression | Promotive | KM analysis: high expression correlates with lower survival | Histological grade, tumor size, depth of tumor invasion, lymph node metastasis |
| DDX27 | GC | High mRNA and protein expression | Promotive | Independent prognostic factor for OS and CSS | Depth of tumor invasion, venous invasion, lymph node metastasis, distant metastasis |
| CRC | High mRNA and protein expression | Promotive | Independent prognostic factor for PFS | Tumor location | |
| DDX39B | CRC | High mRNA and protein expression | Promotive | Independent prognostic factor for OS | Tumor size, histological grade, lymph node metastasis, distant metastasis, TNM stage |
| DDX46 | EC | High mRNA and protein expression | Promotive | NA | NA |
| GC | High mRNA and protein expression | Promotive | NA | NA | |
| CRC | High protein expression | Promotive | NA | NA | |
| DDX51 | EC | High mRNA and protein expression | Promotive | KM analysis: high expression correlates with lower survival | Tumor differentiation, N stage, TNM stage |
| DDX56 | GC | High mRNA and protein expression | Promotive | KM analysis: high expression correlates with lower survival | Lymph node metastasis |
| CRC | High mRNA and protein expression | Promotive | Independent prognostic factor for OS | Lymph node metastasis, distant metastasis |
CRC: Colorectal cancer, EC: Esophageal carcinoma, GC: Gastric cancer, “NA” represents Lack of research
Table 3.
Abnormally expressed DEAD-box helicases in esophageal cancer, gastric cancer, and colorectal cancer and their involved cellular functions and related mechanisms
| Cancer | DEAD-Box Helicase | Cellular Functions | Mechanism |
|---|---|---|---|
| EC | DDX5 |
Regulates cell cycle; Promotes EMT process |
Induces autophagy defect and ER stress; Regulates Wnt/β-catenin signaling pathway |
| DDX46 | Regulates cell cycle | Regulates NF-κB signaling pathway | |
| DDX51 | Regulates target protein phosphorylation | Regulates PTEN/PI3K/AKT signaling pathway | |
| GC | DDX5 | Regulates target protein phosphorylation | Regulates mTOR/S6K1 signaling pathway |
| DDX6 | Regulates translation | Promotes c-Myc protein expression | |
| DDX18 | Regulates miRNA biogenesis | Regulates PTEN/AKT signaling pathway | |
| DDX21 | Regulates cell cycle | Upregulates Cyclin D1 and CDK2 levels | |
| upregulates CCND1 and Slug expression | |||
| DDX24 | Regulates RNA stability | Regulates HK1 mRNA stability, positively regulates HK1 level at transcriptional level | |
| DDX27 |
Regulates RNA alternative splicing; Promotes EMT process |
Affects DDX27/LPP/EMT regulatory axis | |
| Regulates cell cycle | Regulates cell cycle through TP53-dependent and independent mechanisms | ||
| DDX46 | Regulates target protein phosphorylation | Regulates Akt/GSK-3β/β-catenin signaling pathway | |
| DDX56 | Regulates translation | Inhibits FOXO1/p21 Cip1 protein expression, regulates cyclin E1/CDK2/c-Myc signaling pathway | |
| CRC | DDX3 | Regulates transcription | Regulates KRAS/PI3K/AKT/GSK-3β/β-catenin/ZEB1 signaling pathway |
| Regulates target protein phosphorylation | Regulates CK1ε/Dvl2/β-catenin/TCF signaling pathway | ||
|
Regulates transcription; Regulates cell cycle |
Regulates TCF4 mRNA expression, downstream signaling cascade | ||
| DDX5 | Regulates transcription | Regulates DDX5/β-catenin/FOXM1 axis | |
| Regulates target protein phosphorylation | Regulates DDX5/AKT/mTOR signaling pathway | ||
| DDX10 | Regulates RNA selective splicing | Acts on E2F and downstream signaling pathways through RPL35 | |
| DDX17 |
Regulates miRNA biogenesis; promotes EMT process |
Regulates miR-149-3p/CYBRD1/EMT pathway | |
| DDX21 |
Regulates transcription; Regulates cell cycle |
Interacts with WDR5 to enhance CDK1 transcription, regulates cell cycle | |
|
Interacts with CDC5L, regulates cell cycle; Regulates MMP-2/9 expression | |||
| DDX27 |
Regulates transcription; Regulates cell cycle; Promotes EMT process |
Interacts with NPM1 and p65, regulates NF-κB signaling pathway | |
| Regulates stem cell activity | Regulates stem cell self-renewal phenotype | ||
| DDX39B |
Regulates RNA splicing, export; Regulates cell cycle |
Binds to CCND1/CDK6 to assist in splicing and export, and regulates the cell cycle | |
| Regulates RNA alternative splicing and export | selectively splices TUT pre-mRNA, nuclear export of mRNA, regulates TGFβ/SMAD2 signaling pathway | ||
|
Enhances the stability of target protein; Regulates the nuclear import of target protein |
DDX39B binds to PKM2 and inhibits its ubiquitination degradation; DDX39B, PKM2 and importin α5 complex accelerates nuclear translocation of PKM2 |
||
| DDX46 | Regulates cell cycle |
Inhibits caspase-3 and PARP-1 levels, reduces apoptosis; Promotes cell cycle progression |
|
| DDX56 |
Regulates RNA selective splicing; Regulates cell cycle |
Selectively splices WEE1, promotes cell cycle progression |
CRC: Colorectal cancer, EC: Esophageal carcinoma, GC: Gastric cancer
Fig. 2.

The Role of DEAD-Box Helicases in Esophageal Cancer: DDX5: DDX5 enhances the expression of CDK2 and Cyclin D1, promoting the G1/S phase transition in esophageal cancer cells. Additionally, DDX5 mediates increased expression of E-cadherin and decreased expression of Vimentin, facilitating the epithelial-mesenchymal transition (EMT) in esophageal cancer cells. Moreover, DDX5 promotes the expression of BIP, p-PERK, p-eIF2a, and P62, contributing to esophageal cancer progression through the induction of autophagy defects and endoplasmic reticulum stress. DDX5 also facilitates the expression of β-catenin and c-Myc, mediating cell proliferation and invasion in esophageal cancer via the Wnt/β-catenin signaling pathway. DDX46: Silencing DDX46 causes cell cycle arrest at the G1 phase and reduces the phosphorylation of Akt and IκBα, thereby inhibiting NF-κB activity and inducing apoptosis in esophageal cancer cells. DDX51: DDX51 promotes esophageal cancer progression by increasing the phosphorylation levels of PTEN, PI3K, AKT, and mTOR, thereby activating the PI3K/AKT signaling cascade
Fig. 3.

The Role of DEAD-Box Helicases in Gastric Cancer: DDX5: DDX5 increases the expression of p-mTOR and p-S6K1, promoting the growth of gastric cancer. DDX6: DDX6 binds to c-Myc mRNA, enhancing the expression of c-Myc protein, which promotes cell proliferation and the progression of gastric cancer. DDX18: DDX18 interacts with Drosha as a component of the protein complex, promoting the maturation of microRNA-21. MicroRNA-21 binds to the 3´-UTR of PTEN, leading to PTEN mRNA degradation and subsequent AKT phosphorylation, thereby promoting gastric cancer progression. DDX21: DDX21 promotes gastric cancer progression by upregulating Cyclin D1 and CDK2 levels, increasing the G1/S phase transition in gastric cancer cells. Additionally, DDX21 upregulates the expression of Slug, further promoting gastric cancer progression. DDX24: DDX24 positively regulates the level of HK1 by stabilizing HK1 mRNA at the transcriptional level, thereby enhancing glucose uptake and lactate production in gastric cancer cells, contributing to tumor progression. DDX27: DDX27 regulates LPP protein expression by reducing the SE event on the third exon of LPP transcripts, enhancing the translation of functional domain-containing LPP protein. This regulation via the DDX27/LPP/EMT axis promotes gastric cancer progression. DDX27 also reduces the proportion of cells in the G1 phase while increasing the proportion in the S and G2/M phases, thereby regulating the cell cycle and promoting cell proliferation and tumor progression in gastric cancer. DDX46: DDX46 promotes AKT phosphorylation and increases GSK-3β phosphorylation, leading to decreased GSK-3β-mediated β-catenin degradation and promoting gastric cancer progression through the Wnt signaling pathway. DDX56: DDX56 inhibits the expression of FOXO1 and p21 Cip1, thereby activating the downstream cyclin E1/CDK2/c-Myc signaling pathway, which promotes gastric cancer progression
Fig. 4.

The Role of DEAD-Box Helicases in Colorectal Cancer: DDX3: DDX3 enhances the transcription of KRAS by increasing the binding of SP1 to the KRAS promoter, leading to increased KRAS expression and activation of the PI3K/AKT pathway. This results in the phosphorylation of GSK3β at Ser9, reducing GSK3β-mediated degradation of β-catenin, thereby increasing β-catenin levels. This subsequently promotes tumor invasion and metastasis through the β-catenin/ZEB1 axis. Additionally, DDX3 acts as a regulatory subunit of CK1ε, promoting CK1ε-mediated phosphorylation of Dvl2. The increased phosphorylated Dvl2 blocks PP2A-mediated degradation of β-catenin, enhancing β-catenin stability and increasing its binding to TCF in the nucleus. This upregulates the downstream genes of the β-catenin/TCF signaling pathway, promoting tumor invasion and progression in colorectal cancer. Knockdown of DDX3 induces G1 phase cell cycle arrest. DDX5: DDX5 and β-catenin occupy the TCF4/LEF binding elements (TBE) on the FOXM1 promoter, acting as a transcriptional co-stimulatory factor to promote FOXM1 transcription. Overexpression of FOXM1 enhances the proliferation, migration, and tumor growth in colorectal cancer. O-GlcNAcylation of DDX5 protein increases its stability, which activates the AKT/mTOR signaling pathway, promoting colorectal cancer progression. DDX10: DDX10 selectively splices the mRNA of RPL35, increasing the occurrence of AT (alternative terminator) in RPL35 mRNA. This then affects downstream E2F transcription factor-mediated signaling pathways, promoting colorectal cancer progression. DDX17: DDX17 reduces the levels of Claudin-1 and E-cadherin while increasing the levels of Vimentin and N-cadherin, thereby promoting the EMT process. DDX17 downregulates miR-149-3p, reducing its inhibitory effect on CYBRD1 expression by binding to the 3´-UTR of CYBRD1. The resulting increase in CYBRD1 expression promotes CRC cell metastasis and EMT, contributing to the aggressive progression and invasion of colorectal cancer. DDX21: DDX21 interacts with WDR5, a core component of the MLL/SET1 complex, increasing the enrichment of H3K4me3 on the CDK1 promoter and enhancing CDK1 expression at the transcriptional level. This accelerates the cell cycle, promoting colorectal cancer cell proliferation and tumor growth. DDX21 also promotes the G2/M transition in CRC cells and enhances tumor proliferation and growth in vivo. Furthermore, DDX21 promotes the expression of MMP-2 and MMP-9. DDX27: DDX27 regulates EMT by upregulating mesenchymal markers (Slug and Vimentin) and downregulating epithelial markers (E-cadherin), thereby promoting colorectal cell migration and invasion. DDX27 protein directly interacts with NPM1, facilitating its interaction with p65 in the nucleus, which increases the binding activity of p65 to NF-κB target gene promoters. This enhances transcription and upregulates downstream NF-κB target genes, promoting colorectal tumor progression
Fig. 5.

The Role of DEAD-Box Helicases in Colorectal Cancer: DDX27: DDX27 promotes the gene expression of known CSC markers (CD44, CD133, EpCAM, LGR5), stabilizing the self-renewal phenotype of CRC cells and contributing to tumor initiation and development. DDX39B: DDX39B protein directly binds to the first exon of CCND1/CDK6 and upregulates their expression by assisting in their splicing and export, thereby promoting the G1/S phase transition in CRC cells and enhancing CRC proliferation. DDX39B also binds to the splicing site of FUT3 pre-mRNA, significantly increasing the mature FUT3 mRNA. Furthermore, as part of the mRNA export complex (EJC or TREX complex), DDX39B binds to the first exon of FUT3 mRNA, promoting its export. High expression of FUT3 facilitates the fucosylation of TGFβR-I, activating the TGFβ/SMAD2 signaling pathway, and upregulating the expression of MMPs and EMT transcription factors (Snail, Slug, ZEB1), driving epithelial-mesenchymal transition and advancing CRC progression. Additionally, DDX39B directly interacts with the N-terminus of PKM2 and enhances its stability by competitively inhibiting Stub1-mediated ubiquitination and degradation of PKM2. DDX39B recruits importin α5, forming a complex with PKM2 and importin α5 to accelerate PKM2 translocation to the nucleus. Nuclear PKM2 then functions as a protein kinase and transcriptional co-activator, activating the Warburg effect in colorectal cancer, regulating the expression of oncogenes and metabolic genes, and promoting tumor progression. DDX46: DDX46 mediates the reduction of apoptosis markers caspase-3 and PARP-1, decreasing apoptosis in CRC cells, while promoting the G0/G1 phase transition in CRC cells, thereby enhancing cell proliferation. DDX56: DDX56 selectively splices the tumor-suppressing gene WEE1, leading to an increase in WEE1 mRNA with abnormal intron retention. Conversely, the amount of intronless WEE1, a key component of the normal G2-M cell cycle checkpoint, is reduced, diminishing its role in preventing mitosis upon DNA damage in cells. This promotes CRC cell cycle progression and cell proliferation
Despite extensive literature on these molecular mechanisms and regulatory networks, several limitations persist: 1. Sample Size: Most studies involve a small number of cases, lacking large-scale research. 2. Mechanistic Detail: Some studies do not delve deeply into the specific molecular mechanisms of DEAD-box helicase proteins, such as their precise binding sites with RNA or RNP, structural changes in RNA upon binding, and specific roles as splicing factors or transcription coactivators. 3. Pathway Crosstalk: Research on the crosslinking of multiple signaling pathways is limited. A single DEAD-box helicase protein can exhibit similar or different cellular functions in the same or different cancers, involving multiple signaling pathways. Some studies might investigate the macroscopic effects on cellular functions but overlook the influence of a single DEAD-box helicase protein on various signaling pathways within the same tumor cell and their interrelationships. 4. Protein–Protein Interactions: The roles of many DEAD-box helicase family proteins are mediated by protein–protein interactions, yet their precise molecular mechanisms remain unclear. 5. Expression Variability: While DEAD-box helicase family proteins are highly expressed in various cancers, their roles differ across different types of cancer cells, possibly related to the tumor’s intracellular environment and microenvironment, which remain under-researched. 6. Cancer Type Focus: Research on the roles of DEAD-box helicases in gastric and colorectal cancers is abundant, but studies on their roles in esophageal cancer are scarce. 7. Prognostic Potential: There is a lack of studies on whether DEAD-box helicase proteins can serve as independent prognostic factors for cancer and their relationships with clinical factors. Thus, we need more detailed research on the specific binding sites and interaction mechanisms between DEAD-box helicases and RNA or proteins, the structural changes occurring upon binding, and how these changes affect helicase function. Mapping the interactions between helicases and various molecular networks in cancer cells, elucidating the molecular mechanisms of protein–protein interactions affecting cancer cell behavior, understanding the impact of DEAD-box helicases on the tumor microenvironment, and exploring their differential effects in various tumor types are essential. 8. Currently, research on the potential of DEAD-box helicase family proteins as early diagnostic biomarkers for cancer remains insufficient. Most studies primarily focus on analyzing clinical data to evaluate whether these proteins serve as independent prognostic predictors, with limited exploration into their utility for early diagnosis. Additionally, the lack of analyses targeting specific population subgroups hampers a comprehensive understanding of their roles across different demographic groups. 9.In the therapeutic domain, research on inhibitors of DEAD-box helicase family proteins has predominantly centered on the development of small-molecule targeted drugs. However, the exploration of targeted therapies based on immunotherapy and gene-editing technologies remains relatively underdeveloped, and new therapeutic paradigms have yet to be established.
Currently, several small molecule inhibitors targeting DEAD-box helicase family proteins exist, but most target DEAD-box helicase 3 (DDX3) and DEAD-box helicase 5 (DDX5), particularly DDX3, due to its significant roles in tumor and viral replication. These inhibitors primarily target the ATP binding site, the Q motif, affecting corresponding enzyme activities, which poses certain limitations. This provides insight for new drug development, focusing on small molecule drugs targeting other binding sites. Although some reported drugs exhibit inhibitory effects on DEAD-box helicase family proteins, the specific mechanisms remain unclear, highlighting a future research focus. Furthermore, the clinical application of DEAD-box helicase protein inhibitors requires more research, particularly on the inhibitors’ impacts on normal cellular metabolism and signaling pathways. This necessitates clinical studies on the safety and side effects of more inhibitors. Additionally, developing inhibitors targeting other DEAD-box helicase proteins, such as DDX46 and DDX56, highly expressed in gastric and colorectal cancers, may enable DEAD-box helicase family inhibitors to exert inhibitory effects across various cancers. Furthermore, it is essential to further elucidate the potential value of DEAD-box helicase family proteins in the early diagnosis of cancer, with the goal of achieving more efficient diagnostic methods and early interventions. At the same time, treatment strategies should be expanded to explore novel targeted drugs based on immunotherapy and gene-editing technologies, paving the way for the development of a more comprehensive therapeutic framework.
In conclusion, summarizing the roles and molecular mechanisms of DEAD-box helicase family proteins in digestive system cancers and elucidating their inhibitory drugs further underscores their importance in cancer progression. Future research should focus more on their specific molecular mechanisms, regulatory networks in cancer, and potential as drug targets to develop more effective therapeutic strategies against gastrointestinal cancers.
Supplementary Information
Abbreviations
- DEAD-BOX
Helicase proteins in the article
- DDX1
DEAD-BOX helicase 1
- DDX3
DEAD-BOX helicase 3
- DDX3X
DEAD-BOX helicase 3 X-Linked
- DDX5
DEAD-BOX helicase 5
- DDX6
DEAD-BOX helicase 6
- DDX10
DEAD-BOX helicase 10
- DDX17
DEAD-BOX helicase 17
- DDX18
DEAD-BOX helicase 18
- DDX20
DEAD-BOX helicase 20
- DDX21
DEAD-BOX helicase 21
- DDX24
DEAD-BOX helicase 24
- DDX27
DEAD-BOX helicase 27
- DDX39B
DEAD-BOX helicase 39B
- DDX46
DEAD-BOX helicase 46
- DDX49
DEAD-BOX helicase 49
- DDX51
DEAD-BOX helicase 51
- DDX54
DEAD-BOX helicase 54
- DDX55
DEAD-BOX helicase 55
- DDX56
DEAD-BOX helicase 56
- ACTL6a
Actin Like 6A
- AKT
Protein kinase B
- BRD4
Bromodomain containing 4
- BIRC3
Baculoviral IAP Repeat Containing 3
- CCND1
Cyclin D1
- CCND2
Cyclin D2
- CDK1
Cyclin-dependent kinase 1
- CDK2
Cyclin-dependent kinase 1
- CDC5L
Cell Division Cycle 5 Like
- CRC
Colorectal cancer
- CSS
Cancer-specific survival
- CYBRD1
Cytochrome B Reductase 1
- CCL20
Chemokine Ligand 20
- CXCL3
Chemokine (C-X-C motif) ligand 3
- DDX
DEAD-Box
- EMR
Endoscopic mucosal resection
- ESD
Endoscopic submucosal dissection
- EZH2
Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit
- EIF4E
Eukaryotic initiation factor 4E
- EC
Esophageal carcinoma
- EMT
Epithelial-Mesenchymal Transition
- EpCAM
Epithelial cell adhesion molecule
- FOXM1
Forkhead box protein M1
- FUT3
Fucosyltransferase 3
- FGFR2
Fibroblast growth factor receptor 2
- FOXO1
Forkhead box O1
- GATA2
GATA binding protein 2
- GC
Gastric cancer
- HMGB2
High Mobility Group Box 2
- HDI
Human development index
- HER2
Human epidermalgrowth factor receptor-2
- HK1
Hexokinase 1
- IHC
Immunohistochemistry
- lncRNA
Long non-coding RNA
- IRES
Internal ribosome entry site
- IκBα
Inhibitor of NF-κB
- KM
Kaplan-Meier
- KLF4
Krüppel-like factor 4
- KRAS
Kirsten rat sarcoma viral oncogene
- KIFC1
Kinesin family member C1
- LPP
Lipoma-Preferred Partner
- LAMB1
Laminin subunit beta-1
- LGR5
Leucine Rich Repeat Containing G Protein-Coupled Receptor 5
- MITF
Melanocyte inducing transcription factor
- MMP1
Matrix Metallopeptidase 1
- MLL
Mixed lineage leukemia
- MVP
Major Vault Protein
- MIST1
Macrophage Stimulating 1
- mTOR
Mammalian target of rapamycin
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NPM1
Nucleophosmin 1
- NPM1
Nucleophosmin 1
- NFKBIA
NFKB Inhibitor Alpha
- PBX3
PBX Homeobox 3
- PKM2
Pyruvate kinase isozyme type M2
- PIK3CA
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PTEN
Phosphatase and tensin homologue
- PXN-AS1
PXN Antisense RNA 1
- PARP-1
Poly(ADP-Ribose) Polymerase 1
- rDNA
Ribosomal DNA
- rRNA
Ribosomal RNA
- Rac1
Ras-related C3 botulinum toxin substrate 1
- RISC
RNA-induced silencing complex
- SE
Skipping exon
- SP1
Specificity protein 1
- snoRNA
Small nucleolar RNA
- snRNP
Small nuclear ribonucleoproteins
- Stub1
STIP1 Homology And U-Box Containing Protein 1
- SNHG10
Small nucleolar RNA host gene 10
- TGF-β
Transforming growth factor β
- TNF
Tumor Necrosis Factor
- TNFAIP3
TNF Alpha Induced Protein 3
- USP7
Ubiquitin Specific Peptidase 7
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
- WDR5
WD Repeat Domain 5
- YBX1
Y-Box Binding Protein 1
- YY1
Yin-Yang1
Author contributions
Xiaochao Ma conducted the literature review, summarized the content, wrote the paper, and completed the figures and tables. Da Qin provided additional literature support. Tianyu Lu, Yue Yang, Ze Tang, Rui Wang, and Youbin Cui supervised the writing and revision of the manuscript.
Funding
This work was supported by grants from China scholarship council.
Availability of data and materials
Data availability is not applicable to this article as no new data were created or analyzed in this study.
Declarations
Competing interests
The authors declare that there are no competing interests associated with the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–63. 10.3322/caac.21834. (publishedOnlineFirst:20240404). [DOI] [PubMed] [Google Scholar]
- 2.Li M, Ma Y, Huang P, et al. Lentiviral DDX46 knockdown inhibits growth and induces apoptosis in human colorectal cancer cells. Gene. 2015;560(2):237–44. 10.1016/j.gene.2015.02.020. (publishedOnlineFirst:20150211). [DOI] [PubMed] [Google Scholar]
- 3.Ali MAM. The DEAD-box protein family of RNA helicases: sentinels for a myriad of cellular functions with emerging roles in tumorigenesis. Int J Clin Oncol. 2021;26(5):795–825. 10.1007/s10147-021-01892-1. (publishedOnlineFirst:20210303). [DOI] [PubMed] [Google Scholar]
- 4.Cordin O, Banroques J, Tanner NK, et al. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. 10.1016/j.gene.2005.10.019. (publishedOnlineFirst:20051207). [DOI] [PubMed] [Google Scholar]
- 5.Sarkar M, Ghosh MK. DEAD box RNA helicases: crucial regulators of gene expression and oncogenesis. Front Biosci (Landmark Ed). 2016;21(2):225–50. 10.2741/4386. (publishedOnlineFirst:20160101). [DOI] [PubMed] [Google Scholar]
- 6.Zhou HZ, Li F, Cheng ST, et al. DDX17-regulated alternative splicing that produced an oncogenic isoform of PXN-AS1 to promote HCC metastasis. Hepatology. 2022;75(4):847–65. 10.1002/hep.32195. (publishedOnlineFirst:20211221). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao H, Xie Z, Tang G, et al. Knockdown of terminal differentiation induced ncRNA (TINCR) suppresses proliferation and invasion in hepatocellular carcinoma by targeting the miR-218-5p/DEAD-box helicase 5 (DDX5) axis. J Cell Physiol. 2020;235(10):6990–7002. 10.1002/jcp.29595. (publishedOnlineFirst:20200129). [DOI] [PubMed] [Google Scholar]
- 8.Deng X, Liu Z, Wang B, et al. The DDX6/KIFC1 signaling axis, as regulated by YY1, contributes to the malignant behavior of pancreatic cancer. FASEB J. 2024;38(7): e23581. 10.1096/fj.202400166R. [DOI] [PubMed] [Google Scholar]
- 9.Lin C, Li T, Wang Y, et al. METTL3 enhances pancreatic ductal adenocarcinoma progression and gemcitabine resistance through modifying DDX23 mRNA N6 adenosine methylation. Cell Death Dis. 2023;14(3):221. 10.1038/s41419-023-05715-1. (publishedOnlineFirst:20230328). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Z, Song J, Hua Y, et al. The role of DDX46 in breast cancer proliferation and invasiveness: a potential therapeutic target. Cell Biol Int. 2023;47(1):283–91. 10.1002/cbin.11930. (publishedOnlineFirst:20221006). [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, He C, Guo X, et al. DDX39B contributes to the proliferation of colorectal cancer through direct binding to CDK6/CCND1. Cell Death Discov. 2022;8(1):30. 10.1038/s41420-022-00827-7. (publishedOnlineFirst:20220119). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ni Y, Zhuang Z. DDX24 promotes tumor progression by mediating hexokinase-1 induced glycolysis in gastric cancer. Cell Signal. 2024;114: 110995. 10.1016/j.cellsig.2023.110995. (publishedOnlineFirst:20231202). [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Cai Y, Ma Z, et al. The RNA helicase DDX5 promotes viral infection via regulating N6-methyladenosine levels on the DHX58 and NFκB transcripts to dampen antiviral innate immunity. PLoS Pathog. 2021;17(4): e1009530. 10.1371/journal.ppat.1009530. (publishedOnlineFirst:20210428). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi P, Guo Y, Su Y, et al. SUMOylation of DDX39A Alters Binding and Export of Antiviral Transcripts to Control Innate Immunity. J Immunol. 2020;205(1):168–80. 10.4049/jimmunol.2000053. (publishedOnlineFirst:20200511). [DOI] [PubMed] [Google Scholar]
- 15.Zheng Q, Hou J, Zhou Y, et al. The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat Immunol. 2017;18(10):1094–103. 10.1038/ni.3830. (publishedOnlineFirst:20170828). [DOI] [PubMed] [Google Scholar]
- 16.Kukhanova MK, Karpenko IL, Ivanov AV. DEAD-box RNA Helicase DDX3: Functional Properties and Development of DDX3 Inhibitors as Antiviral and Anticancer Drugs. Molecules. 2020. 10.3390/molecules25041015. (published Online First: 20200224). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abualigah L, Al-Abadi L, Ikotun AM, et al. 15 - A review of mothflame optimization algorithm: analysis and applications. In: Abualigah L, ed. Metaheuristic Optimization Algorithms: Morgan Kaufmann 2024:205–19.
- 18.Abualigah L, AlNajdawi S, M. Ikotun A, et al. 11 - Quantum approximate optimization algorithm: a review study and problems. In: Abualigah L, ed. Metaheuristic Optimization Algorithms: Morgan Kaufmann 2024:147–65.
- 19.Abualigah L, Alshatti SM, Ikotun AM, et al. 9 - Spider monkey optimizations: application review and results. In: Abualigah L, ed. Metaheuristic Optimization Algorithms: Morgan Kaufmann 2024:117–31.
- 20.Abualigah L, Odah S, M. Ikotun A, et al. 10 - Marine predator’s algorithm: a survey of recent applications. In: Abualigah L, ed. Metaheuristic Optimization Algorithms: Morgan Kaufmann 2024:133–45.
- 21.Abualigah L, Abualigah Ra, Ikotun AM, et al. 8 - Whale optimization algorithm: analysis and full survey. In: Abualigah L, ed. Metaheuristic Optimization Algorithms: Morgan Kaufmann 2024:105–15.
- 22.Abualigah L, Abusaleem A, Ikotun AM, et al. 6 - Arithmetic optimization algorithm: a review and analysis. In: Abualigah L, ed. Metaheuristic Optimization Algorithms: Morgan Kaufmann 2024:73–87.
- 23.Lindkvist B, Johansen D, Stocks T, et al. Metabolic risk factors for esophageal squamous cell carcinoma and adenocarcinoma: a prospective study of 580,000 subjects within the Me-Can project. BMC Cancer. 2014;14:103. 10.1186/1471-2407-14-103. (publishedOnlineFirst:20140218). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Zheng R, Zeng H, et al. Annual report on status of cancer in China, 2011. Chin J Cancer Res. 2015;27(1):2–12. 10.3978/j.issn.1000-9604.2015.01.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCormack VA, Menya D, Munishi MO, et al. Informing etiologic research priorities for squamous cell esophageal cancer in Africa: a review of setting-specific exposures to known and putative risk factors. Int J Cancer. 2017;140(2):259–71. 10.1002/ijc.30292. (publishedOnlineFirst:20160824). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steffen A, Schulze MB, Pischon T, et al. Anthropometry and esophageal cancer risk in the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2009;18(7):2079–89. 10.1158/1055-9965.EPI-09-0265. (publishedOnlineFirst:20090630). [DOI] [PubMed] [Google Scholar]
- 27.Zhu H, Ma X, Ye T, et al. Esophageal cancer in China: practice and research in the new era. Int J Cancer. 2023;152(9):1741–51. 10.1002/ijc.34301. (publishedOnlineFirst:20221005). [DOI] [PubMed] [Google Scholar]
- 28.Li B, Xiang J, Zhang Y, et al. Comparison of Ivor-Lewis vs Sweet esophagectomy for esophageal squamous cell carcinoma: a randomized clinical trial. JAMA Surg. 2015;150(4):292–8. 10.1001/jamasurg.2014.2877. [DOI] [PubMed] [Google Scholar]
- 29.Yang H, Liu H, Chen Y, et al. Neoadjuvant chemoradiotherapy followed by surgery versus surgery alone for locally advanced squamous cell carcinoma of the esophagus (NEOCRTEC5010): a Phase III Multicenter, Randomized. Open-Label Clinical Trial J Clin Oncol. 2018;36(27):2796–803. 10.1200/JCO.2018.79.1483. (publishedOnlineFirst:20180808). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ni W, Yu S, Xiao Z, et al. Postoperative adjuvant therapy versus surgery alone for stage IIB-III esophageal squamous cell carcinoma: a Phase III Randomized Controlled Trial. Oncologist. 2021;26(12):e2151–60. 10.1002/onco.13914. (publishedOnlineFirst:20210819). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng W, Yang J, Ni W, et al. Postoperative Radiotherapy in Pathological T2–3N0M0 Thoracic Esophageal Squamous Cell Carcinoma: Interim Report of a Prospective, Phase III. Randomized Controll Study Oncol. 2020;25(4):e701–8. 10.1634/theoncologist.2019-0276. (publishedOnlineFirst:20200221). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly RJ, Ajani JA, Kuzdzal J, et al. Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N Engl J Med. 2021;384(13):1191–203. 10.1056/NEJMoa2032125. [DOI] [PubMed] [Google Scholar]
- 33.Cooper JS, Guo MD, Herskovic A, et al. Chemoradiotherapy of locally advanced esophageal cancer: long-term follow-up of a prospective randomized trial (RTOG 85–01). Radiat Therapy Oncol Group JAMA. 1999;281(17):1623–7. 10.1001/jama.281.17.1623. [DOI] [PubMed] [Google Scholar]
- 34.Huang J, Xu J, Chen Y, et al. Camrelizumab versus investigator’s choice of chemotherapy as second-line therapy for advanced or metastatic oesophageal squamous cell carcinoma (ESCORT): a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 2020;21(6):832–42. 10.1016/s1470-2045(20)30110-8. (publishedOnlineFirst:20200513). [DOI] [PubMed] [Google Scholar]
- 35.Luo H, Lu J, Bai Y, et al. Effect of Camrelizumab vs Placebo Added to Chemotherapy on Survival and Progression-Free Survival in Patients With Advanced or Metastatic Esophageal Squamous Cell Carcinoma: The ESCORT-1st Randomized Clinical Trial. JAMA. 2021;326(10):916–25. 10.1001/jama.2021.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, Xiao J, Fang W, et al. Anlotinib for previously treated advanced or metastatic esophageal squamous cell carcinoma: a double-blind randomized phase 2 trial. Cancer Med. 2021;10(5):1681–9. 10.1002/cam4.3771. (publishedOnlineFirst:20210214). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanwei L, Feng H, Ren P, et al. Safety and Efficacy of Apatinib Monotherapy for Unresectable, Metastatic Esophageal Cancer: a Single-Arm, Open-Label Phase II Study. Oncologist. 2020;25(10):e1464–72. 10.1634/theoncologist.2020-0310. (publishedOnlineFirst:20200529). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang B, Qi L, Wang X, et al. Phase II clinical trial using camrelizumab combined with apatinib and chemotherapy as the first-line treatment of advanced esophageal squamous cell carcinoma. Cancer Commun (Lond). 2020;40(12):711–20. 10.1002/cac2.12119. (publishedOnlineFirst:20201212). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Martel C, Georges D, Bray F, et al. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. 2020;8(2):e180–90. 10.1016/S2214-109X(19)30488-7. (publishedOnlineFirst:20191217). [DOI] [PubMed] [Google Scholar]
- 40.Thrift AP, Wenker TN, El-Serag HB. Global burden of gastric cancer: epidemiological trends, risk factors, screening and prevention. Nat Rev Clin Oncol. 2023;20(5):338–49. 10.1038/s41571-023-00747-0. (publishedOnlineFirst:20230323). [DOI] [PubMed] [Google Scholar]
- 41.Murphy G, Dawsey SM, Engels EA, et al. Cancer Risk After Pernicious Anemia in the US Elderly Population. Clin Gastroenterol Hepatol 2015;13(13):2282–9 e1–4. 10.1016/j.cgh.2015.05.040published Online First: 20150614 [DOI] [PMC free article] [PubMed]
- 42.Morgagni P, Gardini A, Marrelli D, et al. Gastric stump carcinoma after distal subtotal gastrectomy for early gastric cancer: experience of 541 patients with long-term follow-up. Am J Surg. 2015;209(6):1063–8. 10.1016/j.amjsurg.2014.06.021. (publishedOnlineFirst:20140807). [DOI] [PubMed] [Google Scholar]
- 43.Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer. 2007;10(2):75–83. 10.1007/s10120-007-0420-0. (publishedOnlineFirst:20070625). [DOI] [PubMed] [Google Scholar]
- 44.Wu AH, Tseng CC, Bernstein L. Hiatal hernia, reflux symptoms, body size, and risk of esophageal and gastric adenocarcinoma. Cancer. 2003;98(5):940–8. 10.1002/cncr.11568. [DOI] [PubMed] [Google Scholar]
- 45.Anderson WF, Rabkin CS, Turner N, et al. The Changing Face of Noncardia Gastric Cancer Incidence Among US Non-Hispanic Whites. J Natl Cancer Inst. 2018;110(6):608–15. 10.1093/jnci/djx262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnold M, Park JY, Camargo MC, et al. Is gastric cancer becoming a rare disease? A global assessment of predicted incidence trends to 2035. Gut. 2020;69(5):823–9. 10.1136/gutjnl-2019-320234. (publishedOnlineFirst:20200130). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hatta W, Gotoda T, Koike T, et al. History and future perspectives in Japanese guidelines for endoscopic resection of early gastric cancer. Dig Endosc. 2020;32(2):180–90. 10.1111/den.13531. (publishedOnlineFirst:20190917). [DOI] [PubMed] [Google Scholar]
- 48.Pimentel-Nunes P, Libanio D, Bastiaansen BAJ, et al. Endoscopic submucosal dissection for superficial gastrointestinal lesions: European Society of Gastrointestinal Endoscopy (ESGE) Guideline - Update 2022. Endoscopy. 2022;54(6):591–622. 10.1055/a-1811-7025. (publishedOnlineFirst:20220506). [DOI] [PubMed] [Google Scholar]
- 49.Park YM, Cho E, Kang HY, et al. The effectiveness and safety of endoscopic submucosal dissection compared with endoscopic mucosal resection for early gastric cancer: a systematic review and metaanalysis. Surg Endosc. 2011;25(8):2666–77. 10.1007/s00464-011-1627-z. (publishedOnlineFirst:20110318). [DOI] [PubMed] [Google Scholar]
- 50.Smyth EC, Nilsson M, Grabsch HI, et al. Gastric cancer. Lancet. 2020;396(10251):635–48. 10.1016/S0140-6736(20)31288-5. [DOI] [PubMed] [Google Scholar]
- 51.Smyth EC. Chemotherapy for resectable microsatellite instability-high gastric cancer? Lancet Oncol. 2020;21(2):204. 10.1016/S1470-2045(20)30025-5. [DOI] [PubMed] [Google Scholar]
- 52.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–97. 10.1016/S0140-6736(10)61121-X. (publishedOnlineFirst:20100819). [DOI] [PubMed] [Google Scholar]
- 53.Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383(9911):31–9. 10.1016/S0140-6736(13)61719-5. (publishedOnlineFirst:20131003). [DOI] [PubMed] [Google Scholar]
- 54.Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10111):2461–71. 10.1016/S0140-6736(17)31827-5. (publishedOnlineFirst:20171006). [DOI] [PubMed] [Google Scholar]
- 55.Araghi M, Soerjomataram I, Bardot A, et al. Changes in colorectal cancer incidence in seven high-income countries: a population-based study. Lancet Gastroenterol Hepatol. 2019;4(7):511–8. 10.1016/S2468-1253(19)30147-5. (publishedOnlineFirst:20190516). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dekker E, Tanis PJ, Vleugels JLA, et al. Colorectal cancer. Lancet. 2019;394(10207):1467–80. 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 57.Ma X, Lu T, Qin D, et al. Analysis of pulmonary artery variation based on 3D reconstruction of CT angiography. Front Physiol. 2023. 10.3389/fphys.2023.1156513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jess T, Rungoe C, Peyrin-Biroulet L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin Gastroenterol Hepatol. 2012;10(6):639–45. 10.1016/j.cgh.2012.01.010. (publishedOnlineFirst:20120128). [DOI] [PubMed] [Google Scholar]
- 59.Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015;110(2):223–62; quiz 63. 10.1038/ajg.2014.435published Online First: 20150203. [DOI] [PMC free article] [PubMed]
- 60.Law R, Das A, Gregory D, et al. Endoscopic resection is cost-effective compared with laparoscopic resection in the management of complex colon polyps: an economic analysis. Gastrointest Endosc. 2016;83(6):1248–57. 10.1016/j.gie.2015.11.014. (publishedOnlineFirst:20151201). [DOI] [PubMed] [Google Scholar]
- 61.Glynne-Jones R, Wyrwicz L, Tiret E, et al. Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv263. 10.1093/annonc/mdy161. [DOI] [PubMed] [Google Scholar]
- 62.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 63.Pietrantonio F, Cremolini C, Petrelli F, et al. First-line anti-EGFR monoclonal antibodies in panRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Crit Rev Oncol Hematol. 2015;96(1):156–66. 10.1016/j.critrevonc.2015.05.016. (publishedOnlineFirst:20150605). [DOI] [PubMed] [Google Scholar]
- 64.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91. 10.1016/S1470-2045(17)30422-9. (publishedOnlineFirst:20170719). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Linder P, Jankowsky E. From unwinding to clamping-the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12(8):505–16. 10.1038/nrm3154. (publishedOnlineFirst:20110722). [DOI] [PubMed] [Google Scholar]
- 66.Linder P, Fuller-Pace FV. Looking back on the birth of DEAD-box RNA helicases. Biochim Biophys Acta. 2013;1829(8):750–5. 10.1016/j.bbagrm.2013.03.007. (publishedOnlineFirst:20130329). [DOI] [PubMed] [Google Scholar]
- 67.Jarmoskaite I, Russell R. DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip Rev RNA. 2011;2(1):135–52. 10.1002/wrna.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang L, Li X. DEAD-Box RNA helicases in cell cycle control and clinical therapy. Cells. 2021. 10.3390/cells10061540. (published Online First: 20210618). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu J, Yang T, Luo Y, et al. DEAD-box helicase 1 inhibited CD8(+) T cell antitumor activity by inducing PD-L1 expression in hepatocellular carcinoma. Cancer Sci. 2024;115(3):763–76. 10.1111/cas.16076. (publishedOnlineFirst:20240119). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu DW, Lin PL, Cheng YW, et al. DDX3 enhances oncogenic KRAS‑induced tumor invasion in colorectal cancer via the beta‑catenin/ZEB1 axis. Oncotarget 2016;7(16):22687–99. 10.18632/oncotarget.8143. [DOI] [PMC free article] [PubMed]
- 71.Yang P, Li J, Peng C, et al. TCONS_00012883 promotes proliferation and metastasis via DDX3/YY1/MMP1/PI3K-AKT axis in colorectal cancer. Clin Transl Med. 2020;10(6): e211. 10.1002/ctm2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tabassum S, Basu M, Ghosh MK. The DEAD-box RNA helicase DDX5 (p68) and beta-catenin: The crucial regulators of FOXM1 gene expression in arbitrating colorectal cancer. Biochim Biophys Acta Gene Regul Mech. 2023;1866(2): 194933. 10.1016/j.bbagrm.2023.194933. (publishedOnlineFirst:20230329). [DOI] [PubMed] [Google Scholar]
- 73.Wang Z, Luo Z, Zhou L, et al. DDX5 promotes proliferation and tumorigenesis of non-small-cell lung cancer cells by activating beta-catenin signaling pathway. Cancer Sci. 2015;106(10):1303–12. 10.1111/cas.12755. (publishedOnlineFirst:20150903). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarkar M, Khare V, Guturi KK, et al. The DEAD box protein p68: a crucial regulator of AKT/FOXO3a signaling axis in oncogenesis. Oncogene. 2015;34(47):5843–56. 10.1038/onc.2015.42. (publishedOnlineFirst:20150309). [DOI] [PubMed] [Google Scholar]
- 75.Hobani YH, Almars AI, Alelwani W, et al. Genetic Variation in DEAD-Box Helicase 20 as a putative marker of recurrence in propensity-matched colon cancer patients. Genes (Basel). 2022. 10.3390/genes13081404. (published Online First: 20220807). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu P, Yu Z, Wang K, et al. DDX21 Interacts with WDR5 to promote colorectal cancer cell proliferation by activating CDK1 expression. J Cancer. 2022;13(5):1530–9. 10.7150/jca.69216. (publishedOnlineFirst:20220228). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xiaoqian W, Bing Z, Yangwei L, et al. DEAD-box Helicase 27 promotes hepatocellular carcinoma progression through ERK signaling. Technol Cancer Res Treat. 2021;20:15330338211055952. 10.1177/15330338211055953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu B, Zhou S, Long D, et al. DDX55 promotes hepatocellular carcinoma progression by interacting with BRD4 and participating in exosome-mediated cell-cell communication. Cancer Sci. 2022;113(9):3002–17. 10.1111/cas.15393. (publishedOnlineFirst:20220610). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou H, Du Y, Wei X, et al. DDX56 transcriptionally activates MIST1 to facilitate tumorigenesis of HCC through PTEN-AKT signaling. Theranostics. 2022;12(14):6069–87. 10.7150/thno.72471. (publishedOnlineFirst:20220815). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cannizzaro E, Bannister AJ, Han N, et al. DDX3X RNA helicase affects breast cancer cell cycle progression by regulating expression of KLF4. FEBS Lett. 2018;592(13):2308–22. 10.1002/1873-3468.13106. (publishedOnlineFirst:20180621). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kouyama Y, Masuda T, Fujii A, et al. Oncogenic splicing abnormalities induced by DEAD-Box Helicase 56 amplification in colorectal cancer. Cancer Sci. 2019;110(10):3132–44. 10.1111/cas.14163. (publishedOnlineFirst:20190829). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin Y, Yang S, Gao X, et al. DEAD-Box Helicase 27 triggers epithelial to mesenchymal transition by regulating alternative splicing of lipoma-preferred partner in gastric cancer metastasis. Front Genet. 2022;13: 836199. 10.3389/fgene.2022.836199. (publishedOnlineFirst:20220504). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He C, Li A, Lai Q, et al. The DDX39B/FUT3/TGFbetaR-I axis promotes tumor metastasis and EMT in colorectal cancer. Cell Death Dis. 2021;12(1):74. 10.1038/s41419-020-03360-6. (publishedOnlineFirst:20210112). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bourgeois CF, Mortreux F, Auboeuf D. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat Rev Mol Cell Biol. 2016;17(7):426–38. 10.1038/nrm.2016.50. (publishedOnlineFirst:20160602). [DOI] [PubMed] [Google Scholar]
- 85.Calo E, Flynn RA, Martin L, et al. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature. 2015;518(7538):249–53. 10.1038/nature13923. (publishedOnlineFirst:20141124). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xing YH, Yao RW, Zhang Y, et al. SLERT Regulates DDX21 Rings Associated with Pol I Transcription. Cell 2017;169(4):664–78 e16. 10.1016/j.cell.2017.04.011. [DOI] [PubMed]
- 87.Bennett AH, O’Donohue MF, Gundry SR, et al. RNA helicase, DDX27 regulates skeletal muscle growth and regeneration by modulation of translational processes. PLoS Genet. 2018;14(3): e1007226. 10.1371/journal.pgen.1007226. (publishedOnlineFirst:20180308). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao G, Yuan H, Li Q, et al. DDX39B drives colorectal cancer progression by promoting the stability and nuclear translocation of PKM2. Signal Transduct Target Ther. 2022;7(1):275. 10.1038/s41392-022-01096-7. (publishedOnlineFirst:20220817). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen HH, Yu HI, Tarn WY. DDX3 modulates neurite development via translationally activating an RNA regulon involved in Rac1 Activation. J Neurosci. 2016;36(38):9792–804. 10.1523/JNEUROSCI.4603-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen HH, Yu HI, Cho WC, et al. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene. 2015;34(21):2790–800. 10.1038/onc.2014.190. (publishedOnlineFirst:20140721). [DOI] [PubMed] [Google Scholar]
- 91.Phung B, Ciesla M, Sanna A, et al. The X-Linked DDX3X RNA helicase dictates translation reprogramming and metastasis in melanoma. Cell Rep 2019;27(12):3573–86 e7. 10.1016/j.celrep.2019.05.069. [DOI] [PubMed]
- 92.Wang Y, Arribas-Layton M, Chen Y, et al. DDX6 Orchestrates Mammalian Progenitor Function through the mRNA Degradation and Translation Pathways. Mol Cell. 2015;60(1):118–30. 10.1016/j.molcel.2015.08.014. (publishedOnlineFirst:20150924). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Taniguchi K, Iwatsuki A, Sugito N, et al. Oncogene RNA helicase DDX6 promotes the process of c-Myc expression in gastric cancer cells. Mol Carcinog. 2018;57(5):579–89. 10.1002/mc.22781. (publishedOnlineFirst:20180130). [DOI] [PubMed] [Google Scholar]
- 94.Tajirika T, Tokumaru Y, Taniguchi K, et al. DEAD-Box Protein RNA-Helicase DDX6 Regulates the Expression of HER2 and FGFR2 at the Post-Transcriptional Step in Gastric Cancer Cells. Int J Mol Sci. 2018. 10.3390/ijms19072005. (published Online First: 20180709). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Vries S, Naarmann-de Vries IS, Urlaub H, et al. Identification of DEAD-box RNA helicase 6 (DDX6) as a cellular modulator of vascular endothelial growth factor expression under hypoxia. J Biol Chem. 2013;288(8):5815–27. 10.1074/jbc.M112.420711. (publishedOnlineFirst:20130104). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pan Y, Zhu Y, Zhang J, et al. A feedback loop between GATA2-AS1 and GATA2 promotes colorectal cancer cell proliferation, invasion, epithelial-mesenchymal transition and stemness via recruiting DDX3X. J Transl Med. 2022;20(1):287. 10.1186/s12967-022-03483-8. (publishedOnlineFirst:20220625). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nishimura T, Padamsi Z, Fakim H, et al. The eIF4E-Binding Protein 4E-T Is a Component of the mRNA Decay Machinery that Bridges the 5’ and 3’ Termini of Target mRNAs. Cell Rep. 2015;11(9):1425–36. 10.1016/j.celrep.2015.04.065. (publishedOnlineFirst:20150528). [DOI] [PubMed] [Google Scholar]
- 98.Slaby O, Bienertova-Vasku J, Svoboda M, et al. Genetic polymorphisms and microRNAs: new direction in molecular epidemiology of solid cancer. J Cell Mol Med. 2012;16(1):8–21. 10.1111/j.1582-4934.2011.01359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu T, Gan H, He S, et al. RNA Helicase DDX24 Stabilizes LAMB1 to Promote Hepatocellular Carcinoma Progression. Cancer Res. 2022;82(17):3074–87. 10.1158/0008-5472.CAN-21-3748. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Y, Guo H, Zhang H. SNHG10/DDX54/PBX3 feedback loop contributes to gastric cancer cell growth. Dig Dis Sci. 2021;66(6):1875–84. 10.1007/s10620-020-06488-9. (publishedOnlineFirst:20200725). [DOI] [PubMed] [Google Scholar]
- 101.Tanaka K, Okamoto S, Ishikawa Y, et al. DDX1 is required for testicular tumorigenesis, partially through the transcriptional activation of 12p stem cell genes. Oncogene. 2009;28(21):2142–51. 10.1038/onc.2009.89. (publishedOnlineFirst:20090427). [DOI] [PubMed] [Google Scholar]
- 102.Ma L, Zhao X, Wang S, et al. Decreased expression of DEAD-Box helicase 5 inhibits esophageal squamous cell carcinomas by regulating endoplasmic reticulum stress and autophagy. Biochem Biophys Res Commun. 2020;533(4):1449–56. 10.1016/j.bbrc.2020.10.026. (publishedOnlineFirst:20201024). [DOI] [PubMed] [Google Scholar]
- 103.Cao J, Wu N, Han Y, et al. DDX21 promotes gastric cancer proliferation by regulating cell cycle. Biochem Biophys Res Commun. 2018;505(4):1189–94. 10.1016/j.bbrc.2018.10.060. (publishedOnlineFirst:20181012). [DOI] [PubMed] [Google Scholar]
- 104.Wang K, Li B, Fan P, et al. Downregulation of DEAD-box helicase 21 (DDX21) inhibits proliferation, cell cycle, and tumor growth in colorectal cancer via targeting cell division cycle 5-like (CDC5L). Bioengineered. 2021;12(2):12647–58. 10.1080/21655979.2021.2011636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang C, Li D, Bai Y, et al. DEAD-box helicase 27 plays a tumor-promoter role by regulating the stem cell-like activity of human colorectal cancer cells. Onco Targets Ther. 2019;12:233–41. 10.2147/OTT.S190814. (publishedOnlineFirst:20181228). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tsukamoto Y, Fumoto S, Noguchi T, et al. Expression of DDX27 contributes to colony-forming ability of gastric cancer cells and correlates with poor prognosis in gastric cancer. Am J Cancer Res 2015;5(10):2998–3014. published Online First: 20150915. [PMC free article] [PubMed]
- 107.Li B, Li YM, He WT, et al. Knockdown of DDX46 inhibits proliferation and induces apoptosis in esophageal squamous cell carcinoma cells. Oncol Rep. 2016;36(1):223–30. 10.3892/or.2016.4803. (publishedOnlineFirst:20160511). [DOI] [PubMed] [Google Scholar]
- 108.Wang J, Wang Y, Wang J, et al. DEAD-box helicase 56 functions as an oncogene promote cell proliferation and invasion in gastric cancer via the FOXO1/p21 Cip1/c-Myc signaling pathway. Bioengineered. 2022;13(5):13970–85. 10.1080/21655979.2022.2084235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Davis BN, Hilyard AC, Lagna G, et al. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454(7200):56–61. 10.1038/nature07086. (publishedOnlineFirst:20080611). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Suzuki HI, Yamagata K, Sugimoto K, et al. Modulation of microRNA processing by p53. Nature. 2009;460(7254):529–33. 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 111.Zhang Y, Yu F, Ni B, et al. The RNA-Binding Protein DDX18 Promotes Gastric Cancer by Affecting the Maturation of MicroRNA-21. Front Oncol. 2020;10: 598238. 10.3389/fonc.2020.598238. (publishedOnlineFirst:20210108). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Basu B, Karmakar S, Basu M, et al. USP7 imparts partial EMT state in colorectal cancer by stabilizing the RNA helicase DDX3X and augmenting Wnt/beta-catenin signaling. Biochim Biophys Acta Mol Cell Res. 2023;1870(4): 119446. 10.1016/j.bbamcr.2023.119446. (publishedOnlineFirst:20230213). [DOI] [PubMed] [Google Scholar]
- 113.Tang J, Chen H, Wong CC, et al. DEAD-box helicase 27 promotes colorectal cancer growth and metastasis and predicts poor survival in CRC patients. Oncogene. 2018;37(22):3006–21. 10.1038/s41388-018-0196-1. (publishedOnlineFirst:20180314). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chedin F, Benham CJ. Emerging roles for R-loop structures in the management of topological stress. J Biol Chem. 2020;295(14):4684–95. 10.1074/jbc.REV119.006364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Crossley MP, Song C, Bocek MJ, et al. R-loop-derived cytoplasmic RNA–DNA hybrids activate an immune response. Nature. 2022;613(7942):187–94. 10.1038/s41586-022-05545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mersaoui SY, Yu Z, Coulombe Y, et al. Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. Embo j 2019;38(15):e100986. 10.15252/embj.2018100986published Online First: 20190621. [DOI] [PMC free article] [PubMed]
- 117.Lin WL, Chen JK, Wen X, et al. DDX18 prevents R-loop-induced DNA damage and genome instability via PARP-1. Cell Rep. 2022;40(3): 111089. 10.1016/j.celrep.2022.111089. [DOI] [PubMed] [Google Scholar]
- 118.Hodroj D, Recolin B, Serhal K, et al. An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. Embo j 2017;36(9):1182–98. 10.15252/embj.201695131published Online First: 20170317. [DOI] [PMC free article] [PubMed]
- 119.Song C, Hotz-Wagenblatt A, Voit R, et al. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 2017;31(13):1370–81. 10.1101/gad.300624.117. (publishedOnlineFirst:20170808). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mosler T, Conte F, Longo GMC, et al. R-loop proximity proteomics identifies a role of DDX41 in transcription-associated genomic instability. Nat Commun. 2021;12(1):7314. 10.1038/s41467-021-27530-y. (publishedOnlineFirst:20211216). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ribeiro de Almeida C, Dhir S, Dhir A, et al. RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol Cell 2018;70(4):650–62.e8. 10.1016/j.molcel.2018.04.001published Online First: 20180503. [DOI] [PMC free article] [PubMed]
- 122.Ma Z, Feng J, Guo Y, et al. Knockdown of DDX5 Inhibits the Proliferation and Tumorigenesis in Esophageal Cancer. Oncol Res. 2017;25(6):887–95. 10.3727/096504016X14817158982636. (publishedOnlineFirst:20161215). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hu DX, Sun QF, Xu L, et al. Knockdown of DEAD-box 51 inhibits tumor growth of esophageal squamous cell carcinoma via the PI3K/AKT pathway. World J Gastroenterol. 2022;28(4):464–78. 10.3748/wjg.v28.i4.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Du C, Li DQ, Li N, et al. DDX5 promotes gastric cancer cell proliferation in vitro and in vivo through mTOR signaling pathway. Sci Rep. 2017;7:42876. 10.1038/srep42876. (publishedOnlineFirst:20170220). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang Q, Ye Y, Lin R, et al. Analysis of the expression, function, prognosis and co-expression genes of DDX20 in gastric cancer. Comput Struct Biotechnol J. 2020;18:2453–62. 10.1016/j.csbj.2020.09.002. (publishedOnlineFirst:20200914). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chang M, Cui X, Sun Q, et al. Lnc-PLCB1 is stabilized by METTL14 induced m6A modification and inhibits Helicobacter pylori mediated gastric cancer by destabilizing DDX21. Cancer Lett. 2024;588: 216746. 10.1016/j.canlet.2024.216746. (publishedOnlineFirst:20240221). [DOI] [PubMed] [Google Scholar]
- 127.Chen L, Xu M, Zhong W, et al. Knockdown of DDX46 suppresses the proliferation and invasion of gastric cancer through inactivating Akt/GSK-3beta/beta-catenin pathway. Exp Cell Res. 2021;399(1): 112448. 10.1016/j.yexcr.2020.112448. (publishedOnlineFirst:20201219). [DOI] [PubMed] [Google Scholar]
- 128.He TY, Wu DW, Lin PL, et al. DDX3 promotes tumor invasion in colorectal cancer via the CK1epsilon/Dvl2 axis. Sci Rep. 2016;6:21483. 10.1038/srep21483. (publishedOnlineFirst:20160219). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Heerma van Voss MR, Vesuna F, Trumpi K, et al. Identification of the DEAD box RNA helicase DDX3 as a therapeutic target in colorectal cancer. Oncotarget 2015;6(29):28312–26. 10.18632/oncotarget.4873 [DOI] [PMC free article] [PubMed]
- 130.Wu N, Jiang M, Han Y, et al. O-GlcNAcylation promotes colorectal cancer progression by regulating protein stability and potential catcinogenic function of DDX5. J Cell Mol Med. 2019;23(2):1354–62. 10.1111/jcmm.14038. (publishedOnlineFirst:20181128). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou X, Liu Z, He T, et al. DDX10 promotes the proliferation and metastasis of colorectal cancer cells via splicing RPL35. Cancer Cell Int. 2022;22(1):58. 10.1186/s12935-022-02478-1. (publishedOnlineFirst:20220202). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhao G, Wang Q, Zhang Y, et al. DDX17 induces epithelial-mesenchymal transition and metastasis through the miR-149-3p/CYBRD1 pathway in colorectal cancer. Cell Death Dis. 2023;14(1):1. 10.1038/s41419-022-05508-y. (publishedOnlineFirst:20230102). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vasaikar S, Huang C, Wang X, et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell. 2019;177(4):1035-49.e19. 10.1016/j.cell.2019.03.030. (publishedOnlineFirst:20190425). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tanaka A, Wang JY, Shia J, et al. DEAD-box RNA helicase protein DDX21 as a prognosis marker for early stage colorectal cancer with microsatellite instability. Sci Rep. 2020;10(1):22085. 10.1038/s41598-020-79049-9. (publishedOnlineFirst:20201216). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kondaskar A, Kondaskar S, Kumar R, et al. Novel, broad spectrum anti-cancer agents containing the tricyclic 5:7:5-Fused Diimidazodiazepine ring system. ACS Med Chem Lett. 2010;2(3):252–6. 10.1021/ml100281b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bol GM, Vesuna F, Xie M, et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol Med 2015;7(5):648–69. 10.15252/emmm.201404368 [DOI] [PMC free article] [PubMed]
- 137.Rampogu S, Lemuel MR, Lee KW. Virtual screening, molecular docking, molecular dynamics simulations and free energy calculations to discover potential DDX3 inhibitors. Adv Cancer Biol Metastasis. 2022;4: 100022. 10.1016/j.adcanc.2021.100022. [Google Scholar]
- 138.Xie M, Vesuna F, Botlagunta M, et al. NZ51, a ring-expanded nucleoside analog, inhibits motility and viability of breast cancer cells by targeting the RNA helicase DDX3. Oncotarget 2015;6(30):29901–13. 10.18632/oncotarget.4898 [DOI] [PMC free article] [PubMed]
- 139.Samal SK, Routray S, Veeramachaneni GK, et al. Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci Rep. 2015;5:9982. 10.1038/srep09982. (publishedOnlineFirst:20150428). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chittavanich P, Saengwimol D, Roytrakul S, et al. Ceftriaxone exerts antitumor effects in MYCN-driven retinoblastoma and neuroblastoma by targeting DDX3X for translation repression. Mol Oncol. 2024;18(4):918–38. 10.1002/1878-0261.13553. (publishedOnlineFirst:20231127). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Capasso A, Bagby SM, Dailey KL, et al. First-in-Class Phosphorylated-p68 Inhibitor RX-5902 Inhibits beta-Catenin Signaling and Demonstrates Antitumor Activity in Triple-Negative Breast Cancer. Mol Cancer Ther. 2019;18(11):1916–25. 10.1158/1535-7163.MCT-18-1334. (publishedOnlineFirst:20190905). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kost GC, Yang MY, Li L, et al. A Novel Anti-Cancer Agent, 1-(3,5-Dimethoxyphenyl)-4-[(6-Fluoro-2-Methoxyquinoxalin-3-yl)Aminocarbonyl] Piperazine (RX-5902), Interferes With β-Catenin Function Through Y593 Phospho-p68 RNA Helicase. J Cell Biochem. 2015;116(8):1595–601. 10.1002/jcb.25113. [DOI] [PubMed] [Google Scholar]
- 143.Diamond J, Eckhardt G, Gluck L, et al. Phase 1 study of RX-5902, a novel orally bioavailable inhibitor of phosphorylated P68, which prevents β-catenin translocation in advanced solid tumors. Ann Oncol. 2017. 10.1093/annonc/mdx365.021. [Google Scholar]
- 144.Diamond JR, Andreopoulou E, Favret AM, et al. Phase Ib/IIa study of RX-5902, a novel orally bioavailable inhibitor of phosphorylated P68, which prevents nuclear β-catenin translocation in patients with triple negative breast cancer. Ann Oncol. 2018. 10.1093/annonc/mdy272.351. [Google Scholar]
- 145.Li F, Fountzilas C, Puzanov I, et al. Multiple functions of the DEAD-box RNA helicase, DDX5 (p68), make DDX5 a superior oncogenic biomarker and target for targeted cancer therapy. Am J Cancer Res 2021;11(10):5190–213. published Online First: 20211015. [PMC free article] [PubMed]
- 146.Doneti R, Pasha A, Botlagunta M, et al. Molecular docking, synthesis, and biological evaluation of 7-azaindole-derivative (7AID) as novel anti-cancer agent and potent DDX3 inhibitor:-an in silico and in vitro approach. Med Oncol. 2022;39(11):179. 10.1007/s12032-022-01826-5. (publishedOnlineFirst:20220901). [DOI] [PubMed] [Google Scholar]
- 147.Tanaka T, Ishii T, Mizuno D, et al. (-)-Epigallocatechin-3-gallate suppresses growth of AZ521 human gastric cancer cells by targeting the DEAD-box RNA helicase p68. Free Radic Biol Med. 2011;50(10):1324–35. 10.1016/j.freeradbiomed.2011.01.024. (publishedOnlineFirst:20110126). [DOI] [PubMed] [Google Scholar]
- 148.Taniguchi T, Iizumi Y, Watanabe M, et al. Resveratrol directly targets DDX5 resulting in suppression of the mTORC1 pathway in prostate cancer. Cell Death Dis. 2016;7(5): e2211. 10.1038/cddis.2016.114. (publishedOnlineFirst:20160505). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Qiu Y, Zhao Y, Wang H, et al. Simvastatin suppresses renal cell carcinoma cells by regulating DDX5/DUSP5. Scand J Urol. 2021;55(4):337–43. 10.1080/21681805.2021.1876163. (publishedOnlineFirst:20210201). [DOI] [PubMed] [Google Scholar]
- 150.Sun S, Xu K, Yan M, et al. Delphinidin induces autophagic flux blockage and apoptosis by inhibiting both multidrug resistance gene 1 and DEAD-box helicase 17 expressions in liver cancer cells. J Pharm Pharmacol. 2023;75(2):253–63. 10.1093/jpp/rgac037. [DOI] [PubMed] [Google Scholar]
- 151.Dai H, Feng J, Nan Z, et al. Morphine may act via DDX49 to inhibit hepatocellular carcinoma cell growth. Aging (Albany NY) 2021;13(9):12766–79. 10.18632/aging.202946published Online First: 20210505. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data availability is not applicable to this article as no new data were created or analyzed in this study.