

Abstract
INTRODUCTION:
Brain cell–derived small extracellular vesicles (sEVs) in blood offer unique cellular and molecular information related to the onset and progression of Alzheimer’s disease (AD). We simultaneously enriched six specific sEV subtypes from the plasma and analyzed a selected panel of microRNAs (miRNAs) in older adults with/without cognitive impairment.
METHODS:
Total sEVs were isolated from the plasma of participants with normal cognition (CN; n = 11), mild cognitive impairment (MCI; n = 11), MCI conversion to AD dementia (MCI-AD; n = 6), and AD dementia (n = 11). Various brain cell–derived sEVs (from neurons, astrocytes, microglia, oligodendrocytes, pericytes, and endothelial cells) were enriched and analyzed for specific miRNAs.
RESULTS:
miRNAs in sEV subtypes differentially expressed in MCI, MCI-AD, and AD dementia compared to the CN group clearly distinguished dementia status, with an area under the curve (AUC) > 0.90 and correlated with the temporal cortical region thickness on magnetic resonance imaging (MRI).
DISCUSSION:
miRNA analyses in specific sEVs could serve as a novel blood-based molecular biomarker for AD.
Keywords: Alzheimer’s disease, biomarker, brain cells, extracellular vesicles, microRNA
1 |. INTRODUCTION
Alzheimer’s disease (AD) and dementia are global health issues; the population of afflicted individuals is growing annually by ≈10 million new cases, and it is the seventh leading cause of mortality globally.1 Considering the closed anatomic environment of the brain, it is obvious to assume that different brain cells (neurons, astrocytes, microglia, oligodendrocytes, pericytes, endothelial cells, and so on) communicate with each other and may orchestrate the progression or even onset of the disease, as evidenced by the growing literature.2,3 For instance, in addition to the definite pathological lesions such as accumulation of amyloid plaques, tau aggregation in neurofibrillary tangles, and loss of neurons and synapses,4 reactive astrocytes and activated microglia decorating amyloid plaques and oligodendrocytes are known pathological features of the AD brain.5 A comprehensive review by Henstridge et al. compiled the list of multiple AD risk genes and noted that these genes are expressed predominantly in non-neuronal cells.3 Furthermore, vascular risk factors increase the risk of AD.6 Several studies have linked arterial stiffness to cognitive decline, impairment, amyloid beta (Aβ) deposition and its progression, brain atrophy, white matter hyperintensities, and cerebral small vessel disease.7-10 Despite evidence suggesting the involvement of various brain cell types in the progression of AD, the “cause or consequence” relation between different brain cell types and the onset of AD is unclear. Currently, the major limitation in analyzing the role of various brain cell types from AD onset to progression is the inaccessibility of the brain cell type.
Recent development in the field of extracellular vesicles (EVs) has provided a unique opportunity to study the “difficult to access” cell/organ types. EVs are lipid-bound vesicles secreted by all cell types in extracellular space, and their cargos represent the pathophysiological status of the parent cells. EVs are heterogeneous in their size, release pathway, cargos, and function. Exosomes are a subclass of small EVs (sEVs) of ≈30 to 150 nm in diameter and originate from the endocytic pathway, whereas microvesicles (100 nm ≥ 1 μm) bud from the plasma membrane. The possible isolation of cell type–specific sEVs (widely reported as “exosomes”) from systemic biofluids (given their nano size of about 150 nm, they can cross the blood-brain barrier) has been exploited in many studies as a potential biomarker for several diseases, including AD.11-13 Profiling of neuron-derived exosomes (NDEs) for the levels of phosphorylated (p)-T181-tau, p-S396-tau, and Aβ1-42 showed differential levels among cognitively normal (CN), mild cognitive impairment (MCI), and AD individuals and predicted AD development 5 to 10 years before clinical onset.14-16 These studies have clearly highlighted the promise of brain cell–type specific sEVs in the blood as potential liquid biopsies for AD.
MicroRNAs (miRNAs) are a large family of conserved 18- to 25-nucleotide long, single-stranded non-coding RNAs involved in the regulation of gene expression and known to play a key role in the inception and progression of AD.17-19 On histopathologic evaluation, AD is characterized by extracellular amyloid plaques and intracellular tau-containing neurofibrillary tangles,4 leading to synaptic degeneration and hippocampal neuronal loss. The sequential cleavage of amyloid precursor protein (APP) by β- and γ-secretase enzymes generate amyloidogenic Aβ peptide (38 to 43 amino acids), whereas cleavage of APP with α-secretase followed by γ-secretase precludes Aβ formation.20,21 miRNAs regulate the expression of critical genes involved in AD pathogenesis, such as β-site APP-cleaving enzyme 1 (BACE-1), APP, tau, presenilin, and brain-derived neurotrophic factor (BDNF).17,22-26 For example, miR-9, miR-29a, miR-107, miR-125b, and miR-135b target BACE1 mRNA expression,18,25,27-29 whereas miR-106b and miR-132 suppress the expression of APP and are involved in the progression of Aβ and tau pathology.19,30 Furthermore, other AD risk genes like glycogen synthase kinase-3 beta (GSK-3β) and neuron navigator 3 (NAV3) are targeted by miR-9-5p and miR-29a, respectively.31,32 In addition to targeting BACE1, miR-107 also targets a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10)33 and granulin.34 miR-132 was shown to target glycosyltransferase like domain containing 1 (GTDC1) and neuronal nitric oxide synthase (nNOS) to induce tau phosphorylation and apoptosis.30 Disrupted-in-schizophrenia-1 (DISC1, a critical risk factor for many neuropsychiatric phenotypes) and sirtuin 1 (SIRT1, shown protective effects against AD) were reported to be the target of miR-135b.35,36 Due to their crucial role in AD pathogenesis, miRNAs’ expression in brain tissue, cerebrospinal fluid (CSF), and plasma has been examined as a biomarker to predict disease progression. A meta-analysis of 10 studies comprising 770 AD patients and 664 normal controls focused on validating the diagnostic potential of circulatory miRNAs, showed high overall sensitivity, specificity, and diagnostic odds ratio.37 Similarly, a systemic review by Swarbrick et al. reported significantly deregulated 10 miRNAs (hsa-miR-107, hsa-miR-26b, hsa-miR-30e, hsa-miR-34a, hsa-miR-485, hsa-miR-200c, hsa-miR-210, hsa-miR-146a, hsa-miR-34c, and hsa-miR-125b) in the peripheral blood of patients with AD, which were cross-referenced against the miRNAs deregulated in the brain at Braak Stage III (defined by neurofibrillary tangle involvement in limbic regions such as the hippocampus) and hypothesized to be deregulated up to ≈20 years before the clinical onset of AD.38 Although the analysis of circulatory miRNA expression in biofluids (blood or CSF) has shown excellent diagnostic performance, the brain cell specificity of these miRNAs remain in question. In addition, most studies outlining the role of miRNAs in the AD brain have been performed on postmortem brain tissue, which limits the information about dynamic and progressive molecular changes in the brain.
The analysis of various brain cell–derived sEVs from plasma is an excellent tool to study the change in expression of critical miRNAs and provide dynamic brain cell–related molecular information at different disease stages from the same individual. Moreover, analyzing the expression of miRNAs inside sEVs offers several advantages, such as higher stability in biofluids, ease of access, cellular specificity, and predicting the functional consequence related to the change in their expression. Therefore, in this study, we analyzed the expression of a panel of miRNAs, well reported to play a role in AD progression and target major AD risk genes, in various brain cell–derived sEV subtypes simultaneously isolated from the plasma of CN, MCI, MCI conversion to AD dementia (MCI-AD), and AD dementia individuals. The different subtypes were enriched from total sEVs (TEs, isolated from plasma) based on specific markers—L1CAM (sEVL1CAM), GLAST (sEVGLAST), TMEM119 (sEVTMEM119), PDGFRα (sEVPDGFRα), PDGFRβ (sEVPDGFRβ), and CD31 (sEVCD31)—abundantly expressed on the surface of neurons, astrocytes, microglia, oligodendrocytes, pericytes, and endothelial cells, respectively, following methods reported by us earlier.39,40 We showed the diagnostic potential of specific miRNAs loaded in these sEVs to distinguish cognitive impairment (including MCI, MCI-AD, and AD) from healthy individuals. The evidence in this study is a step toward the development of a tool to understand the disease etiology holistically and to predict the disease in the asymptomatic or early MCI stage before the development of symptomatic AD.
2 |. METHODS
2.1 |. Study participants and clinical data
Plasma samples were obtained from the Wake Forest Alzheimer’s Disease Research Center’s (ADRC) Clinical Core cohort participants. The demographic details of the study participants are presented in Table 1. All activities described were approved by the Wake Forest Institutional Review Board and conducted in accordance with the Helsinki Declaration of 1975.
TABLE 1.
Demographics and clinical characteristics of participants.
| Normal cognition (CN) | Mildcognitive impairment (MCI) |
Alzheimer’s disease (AD) | MCI to AD | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (n = 11) | (n = 11) | (n = 11) | (n = 6) | ||||||
| N / Mean | %/SD | N / Mean | %/SD | N / Mean | %/SD | N / Mean | %/SD | ||
| Age (years) | 71.5 | 8.2 | 71.7 | 8.3 | 75.1 | 5.9 | 74.2 | 4.4 | |
| Women | 5 | 45% | 9 | 82% | 6 | 55% | 3 | 50% | |
| Race | White | 8 | 73% | 10 | 91% | 9 | 82% | 5 | 83% |
| African-American | 3 | 27% | 1 | 9% | 2 | 18% | 1 | 17% | |
| Education (years) | 16.5 | 2.7 | 16.1 | 2.6 | 15.5 | 3.1 | 13.8 | 3.1 | |
| Systolic blood pressure (mmHg) | 138.8 | 18.1 | 140.2 | 17.8 | 144.7 | 23.6 | 133.5 | 8.8 | |
| Diastolic blood pressure (mmHg) | 75.4 | 15.7 | 70.8 | 7.2 | 74.1 | 9.2 | 75.3 | 5.5 | |
| Body mass index (kg/m2) | 28.7 | 7.7 | 26.9 | 4.6 | 27.3 | 5.3 | 28.4 | 7.9 | |
| Acetylcholinesterase inhibitor use | 0 | 0% | 0 | 0% | 6 | 55% | 0 | 0% | |
| APOE ε4 carrier | 2 | 18% | 1 | 9% | 7 | 64% | 2 | 33% | |
| Montreal Cognitive Assessment | 27 | 2.2 | 25.8 | 1.5 | 16 | 3.8 | 18.5 | 3.6 | |
| Geriatric Depression Scale | 2 | 3.5 | 0.9 | 1.1 | 2 | 1.9 | 2.5 | 3.6 | |
| Clinical Dementia Rating | 0 | 0.2 | 0 | 0 | 3.9 | 1.5 | 1.2 | 0.9 | |
APOE, apolipoprotein E.; SD: Standard deviation
Adults between the ages of 55 to 85 were recruited into the ADRC Clinical Core from the surrounding community between 2016 and 2020. Participants underwent detailed clinical assessments, including the Uniform Data Set Version 3 (UDSv3) and 3T MRI (magnetic resonance imaging) as described previously.41,42 APOE (apolipoprotein E) genotype was obtained by Taqman using single nucleotide polymorphisms (rs429358 and rs7412) to determine haplotypes of ε2, ε3, and ε4. APOE ε4 was dichotomized to the presence or absence of one or more ε4 alleles. Race was self-reported as a social construct. Exclusion criteria for this cohort included: large vessel stroke (participants with lacunae or small vessel ischemic disease were eligible); other significant neurologic diseases that might affect cognition other than AD; evidence of organ failure, active cancer, uncontrolled clinical depression, psychiatric illness, current use of insulin, or history of substance abuse or heavy alcohol consumption within previous 10 years.
Participants completed cognitive testing with the UDSv343 test battery, including the Montreal Cognitive Assessment (MoCA). Subjective questionnaires assessing mood and perceived change in cognitive symptoms were administered at this visit, including the 15-item Geriatric Depression Scale (GDS); the Clinical Dementia Rating (CDR) scale; and the Functional Assessment Questionnaire, which was used to estimate the capacity to manage activities of daily living. UDSv3 cognitive test scores were normalized to create z-scores based on age, race, gender, and education.44 Z-scores were combined to create domain-specific cognitive performance for executive function, memory, language, attention, visuospatial, and phonemic fluency.43 A modified Preclinical Alzheimer’s Cognitive Composite (PACC5)45 was created from five cognitive tests: the Mini-Mental State Examination (MMSE), Free and Cued Selective Reminding Test (FCSRT), Craft Story verbatim recall of the Craft Story, Digit Symbol Substitution Test (DSST), and category fluency.
Detailed MRI acquisition parameters have been described previously.41 MRI was acquired on a 3T Siemens Skyra with a 32-channel head coil (Erlangen, Germany) using the following sequences: high-resolution T1-weighted images obtained with a magnetization-prepared rapid gradient echo (MP-RAGE) sequence; T2 fluid-attenuated inversion recovery (FLAIR) obtained with a three-dimensional (3D) inversion recovery GE sequence. Detailed descriptions of image-processing procedures have also been described previously.41 Briefly, T1 processing included normalization and tissue segmentation using statistical parametric mapping (SPM12) (www.fil.ion.ucl.ac.uk/spm) CAT12. Cortical thickness was calculated on T1 using FreeSurfer v5.3 for a temporal lobe region of interest (shown to be a useful measure of neurodegeneration in regions characteristically affected in AD and related dementias) by averaging surface area–weighted cortical thickness of bilateral entorhinal, inferior/middle temporal, and fusiform regions.46
Adjudication of cognitive diagnosis by expert panel consensus occurred following a review of all available clinical, neuroimaging, and cognitive data in accordance with current National Institute on Aging–Alzheimer’s Association guidelines for the diagnosis of MCI,47 AD, and their subtypes.48 Diagnoses of MCI and dementia at ADRCs are predicated on the National Alzheimer’s Coordinating Center (NACC) coding guide (https://files.alz.washington.edu/documentation/uds3-ivp-guidebook.pdf), with each of the specific dementia syndromes defined based on embedded diagnostic criteria referenced in the NACC coding guide. The panel consisted of investigators with extensive experience assessing the cognitive status and identifying cognitive impairment in older adults, including neuropsychologists, neurologists, and geriatricians.
2.2 |. Isolation of total sEVs (TEs) and specific sEV subtypes
TEs were isolated from the plasma of participants from all the groups using a modified precipitation method described by us previously.49,50 Briefly, plasma samples were centrifuged sequentially at 500 × g for 5 min, 2000 × g for 10 min, and 10,000 × g for 30 min at 4°C to remove any cell debris and large-size vesicles. Finally, TEs were isolated using ExoQuick (System Biosciences, Palo Alto, California, USA) following the manufacturer’s recommendations and dissolved in filtered Dulbecco’s phosphate-buffered saline (DPBS). For the isolation of different sEV subtypes, 1000 μg of TEs were incubated with 5 μg of specific biotin-labeled antibodies—L1CAM (ThermoFisher Scientific, Waltham, MA, USA, Cat. No. 13-1719-82), GLAST (Miltenyi Biotec, Auburn, CA, USA, Cat. No. ACSA-1-Biotin), TMEM119 (BioLegend, San Diego, CA, USA, Cat. No. 853302), PDGFRα (ThermoFisher, Scientific, Cat. No. A15732), PDGFRβ (BioLegend, Cat. No. 323604), and CD31 (BioLegend, Cat. No. 102503)—overnight at 4°C with continuous mixing. TMEM119 antibody was labeled with biotin using FluoReporter Mini-Biotin-XX protein labeling Kit (ThermoFisher, Cat. No. F6347). In addition, 200 μL of streptavidin-coated magnetic beads (ThermoFisher, Cat. No. 10608D) were washed once with 400 μL of washing buffer (0.2 micron filtered PBS + 0.1% bovine serum albumin (BSA)) and resuspended in 100 μL of the same buffer before adding to the sEV-antibody suspension for 1 h incubation at room temperature (RT) with continuous mixing. Beads were then washed 3 times with washing buffer. Beads bound to specific sEV subtypes were either used directly to isolate RNA, or sEVs were eluted in 200 μL of gentle Ag/Ab elution buffer (ThermoFisher, Cat. No 21027, pH 6.6) for further characterization. Beads were magnetized and removed, and eluted sEVs were transferred to a new tube containing 10% v/v 1 M Tris (pH = 9) to neutralize the pH of the elution buffer.
2.3 |. Nanoparticle tracking analysis (NTA)
The size and concentration of sEVs were analyzed with NTA using Nanosight NS300 (Malvern Instruments, UK), as described earlier.39 Five videos of 30 s each were recorded for every sample, and the average of five videos was represented as the final size and concentration count per sample.
2.4 |. Immunogold labeling
For immuno-gold (IG) labeling, TE or sEV subtypes were fixed with 2% paraformaldehyde in PBS buffer (pH 7.4) and then adsorbed for 1 h to a carbon-coated grid. To analyze the typical surface markers on TEs, the sEVs were incubated with CD63 (Abcam, Cat. No. ab59479) primary antibody. sEV subtypes were incubated with primary antibodies, that is, L1CAM (Abcam, Waltham, MA, USA, Cat. No. ab24345), GLAST (ThermoFisher, PA5-80012), TMEM119 (BioLegend, Cat. No. 853302), PDGFRα (Novus Biologicals, Centennial, CO, USA, Cat. No. MAB322-100), PDGFRβ (ThermoFisher, MA5-15143), and CD31 (Abcam, Cat. No. ab28364). As a positive control for each sEV subtype, we used CD63 antibody, and no primary antibody (but gold-labeled secondary antibody) was used as a negative control. A secondary antibody tagged with 10 nm gold particles was further used, and images were captured on a Tecnai T12 transmission electron microscope (TEM).
2.5 |. Exo-check antibody array
TEs were characterized for exosomal biomarkers using Exo-check exosome antibody array (System Biosciences, Palo Alto, CA, USA) following vendor’s protocol.
2.6 |. Flow cytometry
Flow cytometry of the TEs isolated from the plasma of participants from different groups was performed to evaluate the percentage of each sEVs subtype. Briefly, 20 μL of TEs were incubated with 1 μL fluorescently labeled antibodies—L1CAM-PE (BioLegend, Cat. No. 371603), GLAST-APC (Milteny Biotech, Cat. No. 130-123-555), TMEM119 (BioLegend, Cat. No. 853302), PDGFRα-PE (ThermoFisher, Cat. No. A15785), PDGFRβ-PE (BioLegend, Cat. No. 323605), and CD31-PE (BioLegend, Cat. No. 102408)—for 2 h at room temperature in the dark. TMEM119 antibody was fluorescently labeled using APC conjugation kit (Abcam, Cat. No. ab201807) as per the manufacturer’s recommendations. Following antibody incubation, membrane-labeling dye CellBrite steady 488 (CellBrite steady membrane staining Kit, Biotium, Fremont, CA, USA) was diluted 200-fold in 0.1 micron filtered PBS, and 50 uL of diluted dye was added to the TEs for 15 min incubation in the dark. Samples were diluted 100- to 200-fold in 0.1% BSA in PBS filtered through a 0.1 micron filter to achieve an abort ratio of less than 10%. TE samples were acquired on CytoFlex (Beckman Coulter Life Science, Indianapolis, United States) for 60 s at a low flow rate. Filtered PBS was run for 60 s in between the samples. TE without dye was used to set the gate for dye-positive sEVs, and TEs labeled with dye but without fluorescent antibody were used to set the gate for PE/AF647-positive events.
To confirm the purity of sEV subtypes, we used the fluorescently labeled antibody (mentioned above) along with a second validation maker, that is, Synaptophysin-AF647 (Novus Biologicals, Cat No. NBP1-47483AF647) for sEVL1CAM, GFAP-PE (BD Bioscience, Franklin Lake, NJ, USA, Cat. No. 561483) for sEVGLAST, Iba-1-PE (Santa Cruz, Dallas, TX, Cat. No. sc-32725) for sEVTMEM119, Claudin-11-AF647 (R&D systems, Minneapolis, MN, USA, Cat. No. FAB4280RR) for sEVPDGFRα, and Von Willebrand Factor-AF647 (Novus Biologicals, Cat. No. NBP2-34510AF647) for sEVCD31. sEV subtypes were incubated with both markers in separate tubes at RT for 2 h before adding membrane-labeling dye at the same dilution as used for TEs. Samples were diluted further to achieve an abort ratio of less than 10% and acquired for 60 s at a low flow rate. sEV subtypes without membrane-labeling dye were used to set the gate for dye-positive sEVs and without fluorescently labeled antibodies for PE/AF647-positive events.
2.7 |. Analyses of miRNA expression in sEVs
TE and all sEV subtypes were analyzed for the expression of eight miRNAs: miR-9-5p, miR-29a-5p, miR-106b-5p, miR-107, miR-125b-5p, miR-132-5p, miR-135b-5p, and miR-210-3p with real-time polymerase chain reaction (PCR) using TaqMan assays. cel-miR-39-3p was used as an external normalization control. Isolation of total RNA, including miRNA and complementary DNA (cDNA) synthesis was performed with TEs and specific sEV subtypes using our published protocol.39 Prepared cDNA was further diluted 3-fold, and 1 μL of cDNA was used for the quantitative PCR (qPCR) analysis using miRNA-specific Taq-Man Advanced miRNA Assay (20X) (ThermoFisher) in the final 10 μL reaction. An equal amount of total RNA was used for each sEV subtype for cDNA synthesis, and an equal volume of cDNA was used for qPCR expression analysis. Relative expression of different miRNAs in the same sEV subtype was normalized with external control (cel-miR-39-3p) to calculate ΔCt values. Furthermore, the mean value of the CN group was used to calculate the ΔΔCt value for all the samples and data presented as fold change (2−ΔΔCt).
2.8 |. miRNA target analysis
The major AD risk genes were identified from the available database (OMIM: https://www.omim.org/entry/104300), recently published large cohort meta-analysis, and other literature.51-53 The analysis of miRNAs that target these genes (top one-third of miRNAs) was performed using an miRNA data integration portal tool: mirDIP.54 The eight miRNAs included in this study that target these AD risk genes are tabulated (Table S1). In addition, we identified the top 1% target genes (1582 targets) of these eight miRNAs using mirDIP and analyzed the tissue specificity of their targets using Tissue Specific Expression Analysis (TSEA) online platform55 (Figure S1).
2.9 |. Statistical analysis
The change in expression of miRNAs was assessed after calculating fold-change with respect to control using the 2−ΔΔCt method. The statistical significance was calculated with an unpaired multiple t-test. Binary logistic regression models were created to classify participants by overall impairment, defined as having a diagnosis of MCI or dementia status relative to normal cognition and adjusted for age and sex. miRNA fold-changes were selected using a forward selection method for each sEV subtype or from all sEV subtypes, with an entry significance level of 0.05. Receiver-operating characteristic (ROC) plots are reported. Models were created in SAS Studio V.03.05. Participant demographics were compared across cognitive status groups using chi-square tests and one-way analysis of variance (ANOVA) and adjusted for age, gender, race, and education. Multivariable general linear models examined the relationship between brain imaging parameters and cardiometabolic status, adjusting for age, gender, race, and education using SAS 9.4. p-values < 0.05 were considered statistically significant. Graphs were plotted in GraphPad Prism 9.1.2 and R version 4.1.0.
3 |. RESULTS
3.1 |. Characterization of sEVs
Table 1 describes the analytic sample of 39 ADRC participants selected for this study, which included those adjudicated as CN (n = 11, women = 5/11, 45%), MCI (n = 11, women = 9/11, 82%), MCI-AD (N = 6, women = 3/6, 50%), and AD dementia (N = 11, women = 6/11, 55%). Overall, the sample had a mean age of 74 ± 6 years; 59% were women and 18% self-reported as Black/African American. As expected, participants adjudicated with AD differed according to clinical characteristics, in addition to being older, more likely to carry the APOE ε4 allele, and to be treated with acetylcholinesterase inhibitors.
The TE and sEV subtypes were characterized for their size, concentration, purity, and specificity. NTA analysis showed that the average size of TE was 100–120 nm ± 3.70 (Figure S2A), and ExoCheck arrays confirmed the presence of typical exosomal markers (ICAM, Alix, CD81, CD63, EpCAM, ANXA5, and TSG101) in TEs (Figure S2B). Furthermore, flow cytometry confirmed the relative expression of various biomarkers on the surface of TEs: L1CAM (4%–9%), GLAST (7%–12%), TMEM119 (3%–7%), PDGFRα (1%–2%), PDGFRβ (7%–32%), and CD31 (15%–23%) (Figure S2C and S2D). Of interest, the percentage of L1CAM+ TE decreased in the MCI group; PDGFRβ+ TE decreased in MCI-AD and AD groups, whereas CD31+ TE decreased in the MCI, MCI-AD, and AD groups (Figure S2C and S2D). Immunogold labeling followed by TEM confirmed the presence of tetraspanin CD63 on the surface of TEs (Figure S2E). Finally, analysis of expression of eight miRNAs in TEs was performed which showed increased expression of miR-107 in MCI, MCI-AD and AD groups, while miR-135b-5p and miR-210-3p showed increased expression in AD and MCI groups, respectively (Figure S3A). Further, the expression of miR-29a-5p and miR-135b-5p correlated significantly with temporal lobe cortical thickness.
NTA analysis of different brain cell–derived sEVs confirmed the size of less than 150 nm (Figures S4-S9). The specificity of isolated sEVs was confirmed by immuno-gold labeling and TEM for the marker used to pull out each sEV subtype, and CD63 was used as the positive control (Figures S4-S9). Furthermore, flow cytometry analysis was performed to assess the purity of sEV using a fluorescently labeled antibody against the surface marker used to isolate each sEV subtype. The purity was further authenticated using a second validation marker abundantly expressed on the surface of specific sEV subtype (Figures S4-S9).
3.2 |. sEVL1CAM characterization and miRNA expression analysis
After establishing the purity of sEVL1CAM (Figure S4), the expression of eight miRNAs in sEVL1CAM was analyzed using real-time PCR. We observed significant overexpression of miR-9-5p (p = 0.0010) and miR-106b-5p (p < 0.0001) and a decreased expression of miR-29a-5p (p = 0.036) in sEVL1CAM in the AD group compared to the CN group. The expression of miR-125b-5p and miR-132-5p was higher in adults with MCI-AD (p = 0.013 and 0.0010) and AD dementia (p = 0.0015 and p = 0.0014) compared to CN (Figure 1A). Of interest, we observed a higher expression of hypoxia-responsive miR-210-3p (5.8-fold average increase, p = 0.003) selectively in the MCI group compared to CN, whereas no change in expression was observed in the MCI-AD and AD groups (Figure 1A). We did not observe any significant difference in the expression of miR-135b-5p in the MCI, MCI-AD, and AD groups compared to CN. Moreover, the expression of miR-107 was not detected in EVL1CAM.
FIGURE 1.
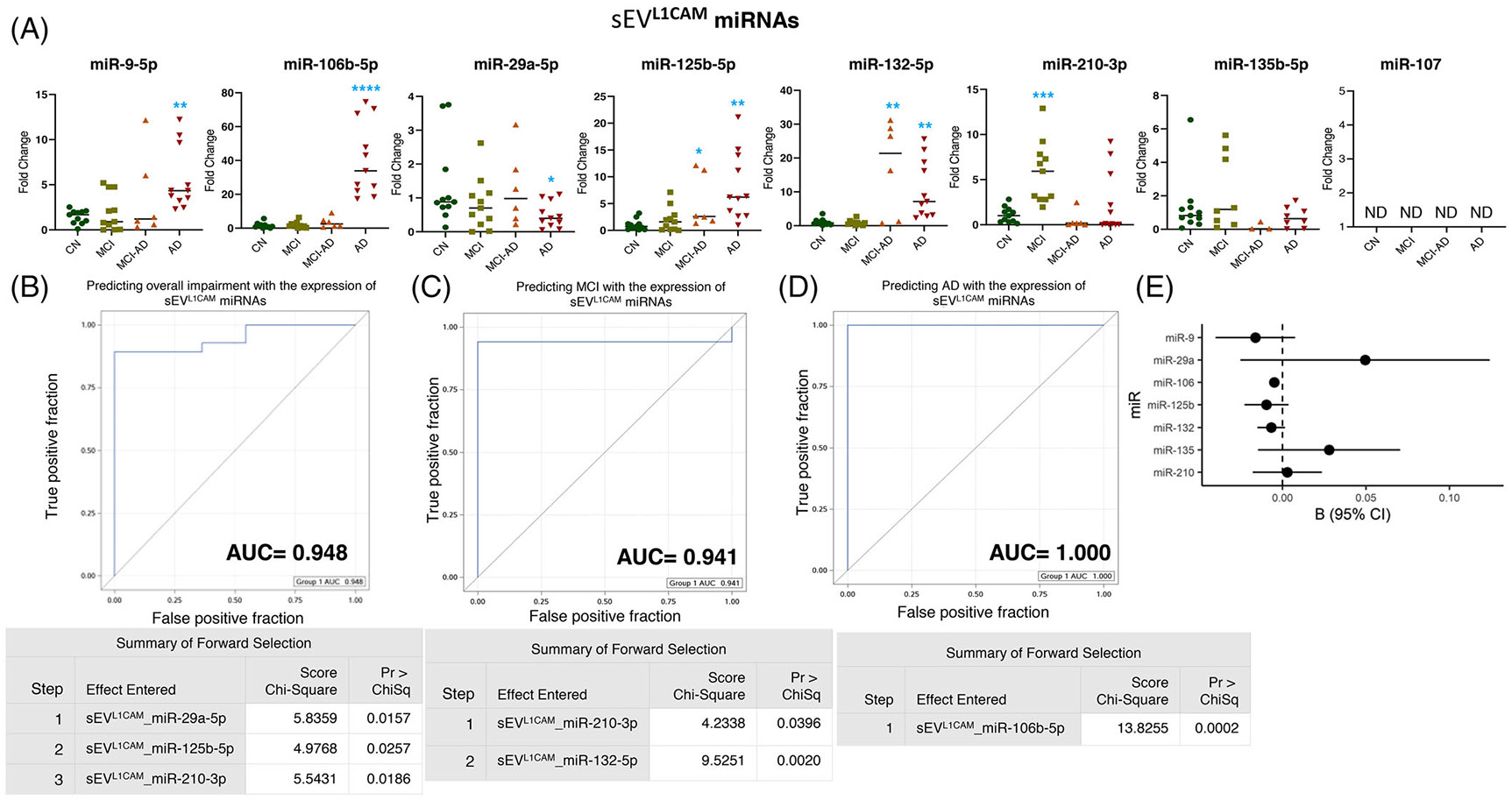
Expression profiling of specific microRNAs (miRNAs) in small extracellular vesicle (sEV)L1CAM and their correlation with cognitive impairment and temporal cortical thickness. (A) The expression of eight miRNAs was analyzed in sEVL1CAM in subjects with normal cognition (CN; n = 11), mild cognitive impairment (MCI; n = 11), MCI-AD (Alzheimer’s disease) (n = 6), or AD (n = 11) by quantitative real-time polymerase chain reaction (PCR). Fold-change in the expression of miRNA showing detectable expression was calculated by the ΔΔCt method (as mentioned in Methods) after normalization with cel-miR-39-3p. Fold-change of all the samples is plotted by calculating 2−ΔΔCt. ND = “not detectable.” *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001. (B–D) Receiver-operating characteristic (ROC) curves curated from logistic regression classifiers of sEVL1CAM for overall impairment (B), MCI (C), or AD (D) outcomes using the forward selection method with alpha = 0.05. All models were adjusted for age and sex. Summary of the forward selection sEVL1CAM miRNA shown under each ROC curve. (E) Correlation of detected miRNAs in sEVL1CAM with the temporal cortical thickness on magnetic resonance imaging (MRI).
Furthermore, logistic regression models were built using forward selection and an alpha cutoff of 0.05 to generate ROC plots. The differential expression of miR-29a-5p, miR-125b-5p, and miR-210-3p in sEVL1CAM was shown to predict overall cognitive impairment (including MCI, MCI-AD, and AD dementia) with an area under the curve (AUC) = 0.948 (Figure 1B). The expression of miR-210-3p and miR-132-5p showed AUC = 0.941 in predicting the MCI, and miR-106-5p expression showed AUC = 1.000 for predicting AD (Figure 1C and 1D). Importantly, the expression of miR-106b-5p in sEVL1CAM showed a significant negative correlation with cortical thickness in regions prone to age-related dementias as imaged in MRI (Figure 1E and Table 2).
TABLE 2.
Correlation of sEV miRNAs with temporal cortical thickness.
| miRNA | Temporal Cortical Thickness | |||
|---|---|---|---|---|
| N | B | SE | p-value | |
| TE_miR-9-5p | 37 | −0.0295 | 0.0250 | 0.247 |
| TE_miR-29a-5p | 37 | −0.0119 | 0.0057 | 0.045 |
| TE_miR-107 | 37 | −0.0062 | 0.0035 | 0.088 |
| TE_miR-125b-5p | 37 | 0.0161 | 0.0112 | 0.160 |
| TE_miR-135b-5p | 37 | −0.0258 | 0.0073 | 0.001 |
| TE_miR-210-3p | 37 | −0.0062 | 0.0113 | 0.591 |
| sEVL1CAM_miR-9-5p | 37 | −0.0162 | 0.0117 | 0.173 |
| sEVL1CAM_miR-29a-5p | 37 | 0.0495 | 0.0366 | 0.186 |
| sEVL1CAM_miR-106b-5p | 37 | −0.0049 | 0.0014 | 0.002 |
| sEVL1CAM_mi_125b-5p | 37 | −0.0096 | 0.0064 | 0.148 |
| sEVL1CAM_miR-132-5p | 37 | −0.0067 | 0.0041 | 0.107 |
| sEVL1CAM_miR-135b-5p | 29 | 0.0279 | 0.0206 | 0.188 |
| sEVGLAST_miR-210-3p | 37 | 0.0029 | 0.0101 | 0.775 |
| sEVGLAST_miR-29a-5p | 37 | −0.0053 | 0.0127 | 0.680 |
| sEVGLAST_miR-107 | 37 | −0.0033 | 0.0011 | 0.004 |
| sEVGLAST_mi_125b-5p | 37 | −0.0150 | 0.0134 | 0.273 |
| sEVGLAST_miR-132-5p | 37 | −0.0180 | 0.0083 | 0.038 |
| sEVGLAST_miR-210-3p | 37 | 0.0082 | 0.0121 | 0.504 |
| sEVTMEM119_miR-29a-5p | 37 | 0.0014 | 0.0309 | 0.965 |
| sEVTMEM119_miR-106b-5p | 34 | −0.0171 | 0.0074 | 0.028 |
| sEVTMEM119_miR-125b-5p | 37 | −0.0115 | 0.0164 | 0.489 |
| sEVTMEM119_miR-132-5p | 37 | −0.0053 | 0.0024 | 0.034 |
| sEVTMEM119_miR-210-3p | 37 | 0.0108 | 0.0100 | 0.290 |
| sEVPDGFRα_miR-29a-5p | 37 | −0.0272 | 0.0105 | 0.015 |
| sEVPDGFRα_miR-107 | 37 | −0.0035 | 0.0022 | 0.115 |
| sEVPDGFRα_miR-125b-5p | 37 | −0.0074 | 0.0053 | 0.176 |
| sEVPDGFRα_miR-135b-5p | 37 | −0.0195 | 0.0133 | 0.154 |
| sEVPDGFRα_miR-210-3p | 37 | 0.0061 | 0.0183 | 0.740 |
| sEVPDGFRβ_miR-9-5p | 37 | 0.0108 | 0.0417 | 0.797 |
| sEVPDGFRβ_miR-125b-5p | 37 | 0.0151 | 0.0385 | 0.697 |
| sEVPDGFRβ_miR-132-5p | 37 | −0.0323 | 0.0427 | 0.455 |
| sEVPDGFRβ_miR-135b-5p | 37 | 0.0008 | 0.0023 | 0.739 |
| sEVPDGFRβ_miR-210-3p | 37 | 0.0557 | 0.0453 | 0.228 |
| sEVCD31_miR-29a-5p | 37 | −0.0093 | 0.0062 | 0.143 |
| sEVCD31_miR-125b-5p | 37 | −0.0047 | 0.0026 | 0.075 |
| sEVCD31_miR-132-5p | 37 | −0.0009 | 0.0021 | 0.673 |
| sEVCD31_miR-135b-5p | 37 | −0.0005 | 0.0011 | 0.624 |
| sEVCD31_miR-210-3p | 37 | −0.0210 | 0.0101 | 0.045 |
Number of participants (N), Regression Coefficient (B), Standard error (SE), rows highlighted with bold text represent statistically significant correlation (p < 0.05).
3.3 |. sEVGLAST characterization and miRNA expression analysis
The miRNA expression analysis of sEVGLAST displayed significant over-expression of miR-29a-5p (p = 0.0016), miR-125b-5p (p < 0.0010), and miR-132-5p (p = 0.0085) selectively in the AD group, although no change in expression in the MCI and MCI-AD groups was noted compared to CN (Figure 2A). Furthermore, compared to the CN, the expression of miR-107 showed increased expression in sEVGLAST from MCI (average increase 2.6-fold, p = 0.045), MCI-AD (average increase 20.35-fold, p = 0.0002) to AD dementia (average increase 32.88-fold, p < 0.0001). Of interest, like sEVL1CAM, sEVGLAST also showed increased expression of hypoxic miR-210-3p (p = 0.026) selectively in the MCI group. We could not detect the expression of miR-9-5p, miR-106b-5p, and miR-135b-5p in sEVGLAST (Figure 2A).
FIGURE 2.
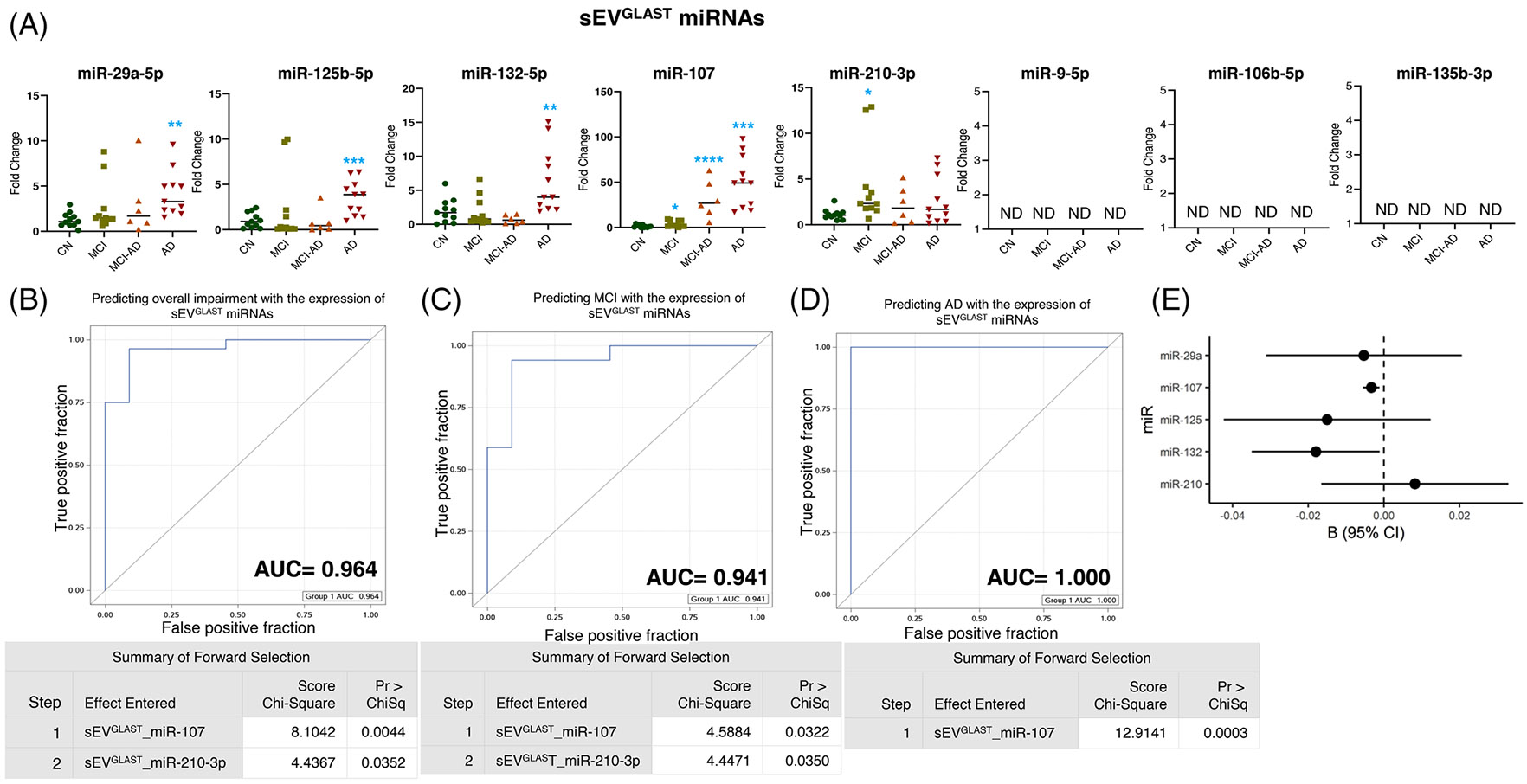
Expression profiling of specific microRNAs (miRNAs) in small extracellular vesicle (sEV)GLAST and their correlation with cognitive impairment and temporal cortical thickness. (A) The expression of eight miRNAs was analyzed in sEVGLAST in subjects with normal cognition (CN; n = 11), mild cognitive impairment (MCI; n = 11), MCI-AD (Alzheimer’s disease) (n = 6), or AD (n = 11) by quantitative real-time polymerase chain reaction (PCR). Fold-change in the expression of miRNA showing detectable expression was calculated by the ΔΔCt method after normalization with cel-miR-39-3p. Fold-change of all the samples is plotted by calculating 2−ΔΔCt. ND = “not detectable.” *p < 0.05, **p < 0.005. (B–D) Receiver-operating characteristic (ROC) curves curated from logistic regression classifiers of sEVGLAST for overall impairment (B), MCI (C), or AD (D) outcomes using the forward selection method with alpha = 0.05. All models were adjusted for age and sex. Summary of the forward selection sEVGLAST miRNA shown under each ROC curve. (E) Correlation of detected miRNAs in sEVGLAST with the temporal cortical thickness on magnetic resonance imaging (MRI).
Next, the logistic regression model showed that the expression of miR-107 and miR-210-3p in sEVGLAST can predict overall cognitive impairment (including MCI, MCI-AD, and AD dementia) and MCI with AUCs = 0.964 and 0.941, respectively (Figure 2B and 2C). While the expression of miR-107 in sEVGLAST can perfectly predict the incidence of AD dementia (AUC = 1.000) (Figure 2D) and significantly negatively correlated with the cortical thickness (p = 0.004) along with miR-132-5p (p = 0.038) (Figure 2E and Table 2).
3.4 |. sEVTMEM119 characterization and miRNA expression analysis
The expression of miR-29a-5p showed a significant reduction in all groups compared to CN, with the most significant reduction in AD dementia (MCI ≈40% average reduction p = 0.022, MCI-AD showed ≈32% average reduction p = 0.018, and AD ≈65% average reduction p = 0.008) (Figure 3A). Similarly, we observed a reduction of miR-125b-5p (p = 0.017), although only in sEVTMEM119 from the MCI group. A slight but statistically significant decrease in the expression of miR-106b-5p (≈58% average decrease p = 0.047) was observed in the MCI group; however, the expression increased significantly in the MCI-AD (p = 0.002) and AD (p = 0.0016) groups (Figure 3A). The expression of miR-132-5p indicated a significant increase in the MCI-AD (p = 0.016) and AD (p = 0.0006) groups. Of interest, sEVTMEM119 showed an increased expression of miR-210-3p in MCI-AD (p = 0.024) (Figure 3A). We could not detect the expression of miR-9-5p, miR-107, and miR-135b-5p in sEVTMEM119 (Figure 3A).
FIGURE 3.
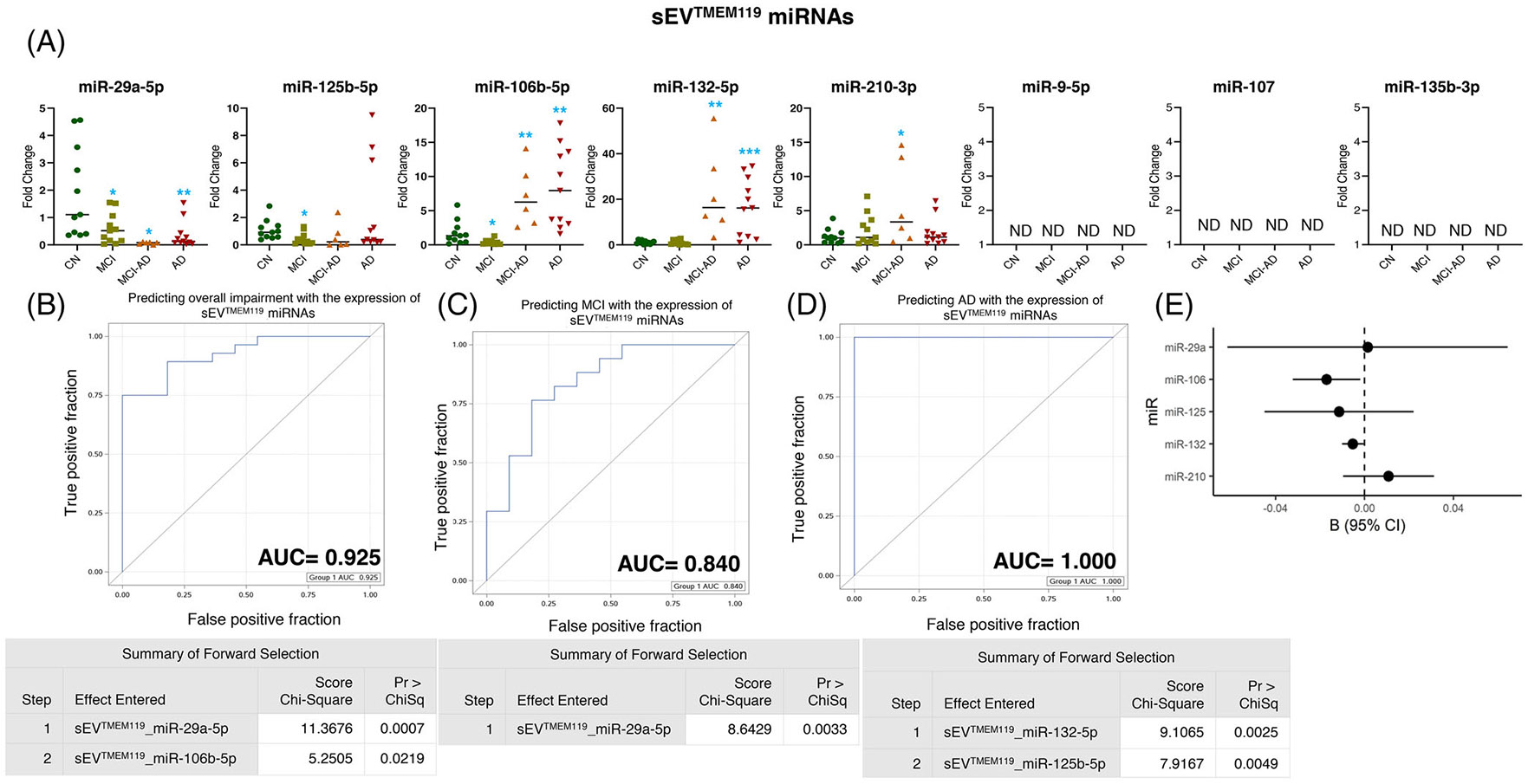
Expression profiling of specific microRNAs (miRNAs) in small extracellular vesicle (sEV)TMEM119 and their correlation with cognitive impairment and temporal cortical thickness. (A) The expression of eight miRNAs was analyzed in sEVTMEM119 in subjects with normal cognition (CN; n = 11), mild cognitive impairment (MCI; n = 11), MCI-AD (Alzheimer’s disease) (n = 6), or AD (n = 11) by quantitative real-time polymerase chain reaction (PCR). Fold-change in the expression of miRNA showing detectable expression was calculated by the ΔΔCt method (as mentioned in methods) after normalization with cel-miR-39-3p. Fold-change of all the samples is plotted by calculating 2−ΔΔCt. ND = “not detectable.” *p < 0.05, **p < 0.005. (B–D) Receiver-operating characteristic (ROC) curves curated from logistic regression classifiers of sEVTMEM119 for overall impairment (B), MCI (C), or AD (D) outcomes using the forward selection method with alpha = 0.05. All models were adjusted for age and sex. Summary of the forward selection sEVTMEM119 miRNA shown under each ROC curve. (E) Correlation of detected miRNAs in sEVTMEM119 with the temporal cortical thickness on magnetic resonance imaging (MRI).
The expression of miR-29a-5p and miR-106-5p in sEVTMEM119 predicted the overall cognitive impairment with AUC = 0.925 (Figure 3B), whereas the expression of miR-29a-5p showed AUC = 0.840 in predicting MCI (Figure 3C). The expression of miR-132-5p and miR-125b-5p could predict the AD group compared to CN with AUC = 1.000 (Figure 3D). Notably, the expression of miR-106b-5p (p = 0.028) and miR-132-5p (p = 0.034) in sEVTMEM119 showed a significant negative correlation with the temporal cortical thickness (Figure 3E and Table 2).
3.5 |. sEVPDGFRα characterization and miRNA expression analysis
The profiling for eight miRNAs in sEVPDGFRα showed significant over-expression of miR-29a-5p (p = 0.002), miR-107 (p = 0.002), and miR-135b-5p (p = 0.033) in AD cohort, whereas no change in the expression of these miRNAs was observed in MCI or MCI-AD groups (Figure 4A). Furthermore, miR-125b-5p showed increased expression in both the MCI-AD (p = 0.017) and AD groups (p = 0.004), and miR-210-3p did not show a change in expression across groups (Figure 4A). We could not detect the expression of miR-9-5p, miR-106b-5p, and miR-132-5p in sEVPDGFRα (Figure 4A).
FIGURE 4.
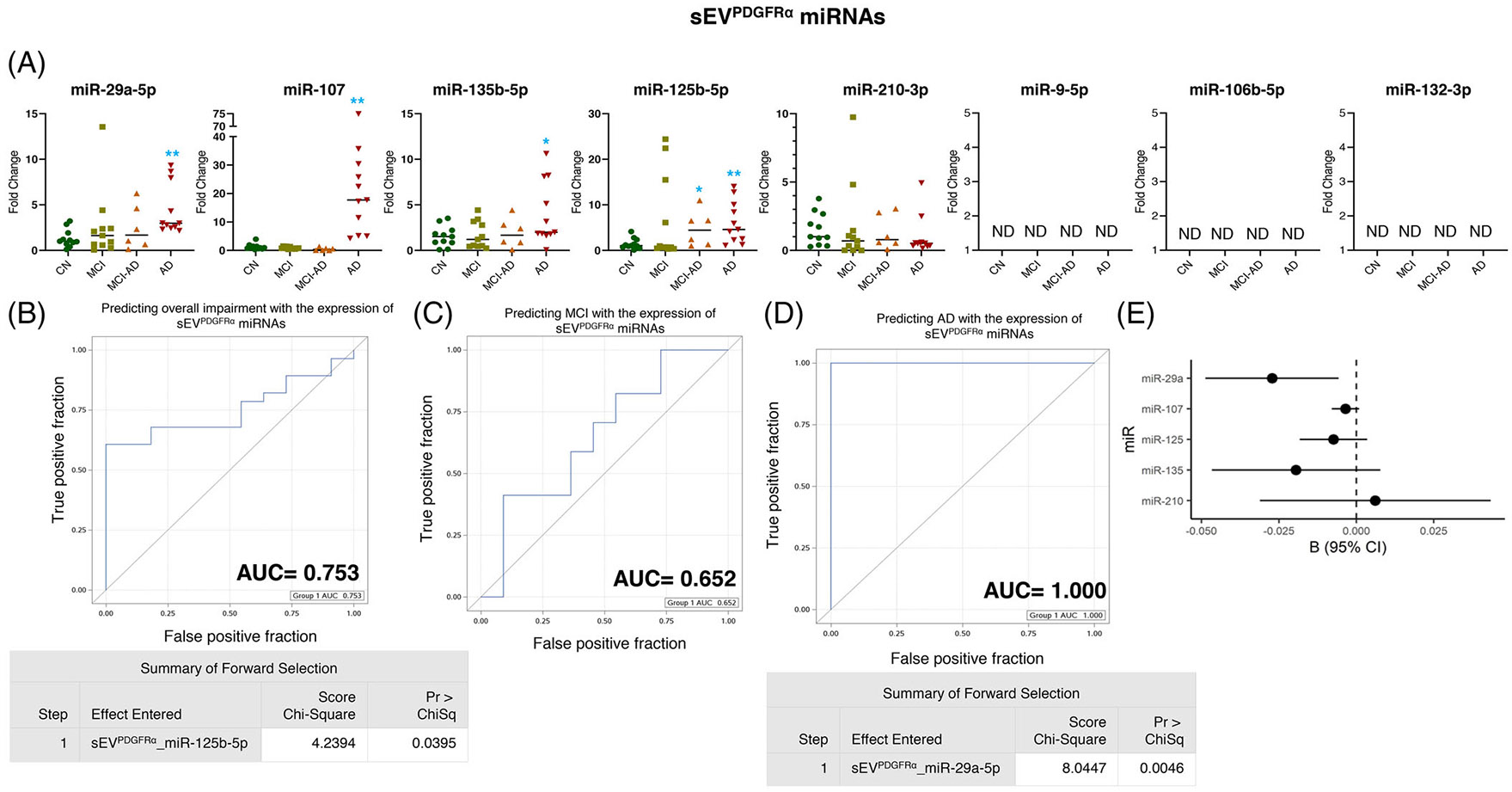
Expression profiling of specific microRNAs (miRNAs) in small extracellular vesicle (sEV)PDGFRα and their correlation with cognitive impairment and temporal cortical thickness. (A) The expression of eight miRNAs was analyzed in sEVPDGFRα in subjects with normal cognition (CN; n = 11), mild cognitive impairment (MCI; n = 11), MCI-AD (Alzheimer’s disease) (n = 6), or AD (n = 11) by quantitative real-time polymerase chain reaction (PCR). Fold-change in the expression of miRNA showing detectable expression was calculated by the ΔΔCt method (as mentioned in methods) after normalization with cel-miR-39-3p. Fold-change of all the samples is plotted by calculating 2−ΔΔCt. ND = “not detectable.” *p < 0.05, **p < 0.005. (B–D) Receiver-operating characteristic (ROC) curves curated from logistic regression classifiers of sEVPDGFRα for overall impairment (B), MCI (C), or AD (D) outcomes using the forward selection method with alpha = 0.05. All models were adjusted for age and sex. Summary of the forward selection sEVPDGFRα miRNA shown under each ROC curve except for the MCI group, where no miRNA was identified to distinguish the MCI from the CN group. (E) Correlation of detected miRNAs in sEVPDGFRα with the temporal cortical thickness on magnetic resonance imaging (MRI).
The expression of miR-125b-5p in sEVPDGFRα displayed AUC = 0.753 in predicting the overall impairment (Figure 4B), although none of the miRNAs could be noted in predicting MCI (Figures 4C). Furthermore, miR-29a-5p in sEVPDGFRα showed AUC = 1.000 in predicting AD incidence (Figure 4D) and negatively correlated with the temporal cortical thickness (Figure 4E and Table 2).
3.6 |. sEVPDGFRβ characterization and miRNA expression analysis
The profiling for eight miRNAs in sEVPDGFRβ showed that the expression of miR-9-5p, miR-125b-5p, and miR-132-5p decreased in the MCI (p = 0.005, p = 0.025, and p = 0.002 respectively) and AD (p = 0.002, p = 0.013, and p = 0.041 respectively) groups. In addition, the expression of miR-9-5p (p = 0.015) and miR-132-5p (p = 0.032) decreased in the MCI-AD group also (Figure 5A). Furthermore, the expression of miR-135b increased in all three groups: MCI (p = 0.004), MCI-AD (p = 0.0001), and AD (p = 0.049). We observed a statistically significant reduction in the expression of miR-210-3p in the MCI-AD (p = 0.015) and AD (p = 0.0135) groups, whereas a slight but statistically non-significant reduction in the MCI group was also observed (Figure 5A). We could not detect the expression of miR-29a-5p, miR-106-5p, and miR-107 in sEVPDGFRβ.
FIGURE 5.
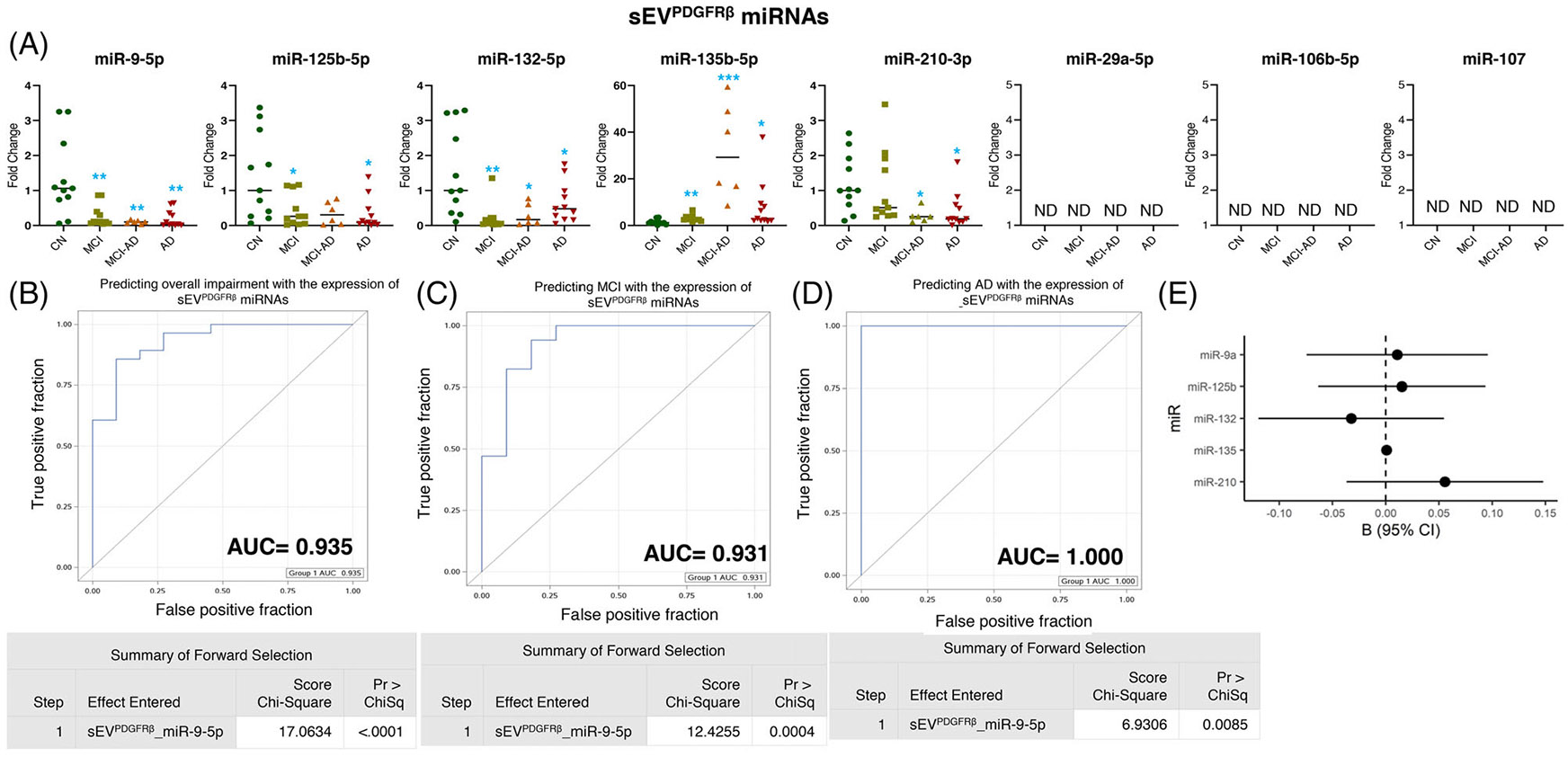
Expression profiling of specific microRNAs (miRNAs) in small extracellular vesicle (sEV)PDGFRβ and their correlation with cognitive impairment and temporal cortical thickness. (A) The expression of eight miRNAs was analyzed in sEVPDGFRβ in subjects with normal cognition (CN; n = 11), mild cognitive impairment (MCI; n = 11), MCI-AD (Alzheimer’s disease) (n = 6), or AD (n = 11) by quantitative real-time polymerase chain reaction (PCR). Fold-change in the expression of miRNA showing detectable expression was calculated by the ΔΔCt method (as mentioned in methods) after normalization with cel-miR-39-3p. Fold-change of all the samples is plotted by calculating 2−ΔΔCt. ND = “not detectable.: *p < 0.05, **p < 0.005. (B–D) Receiver-operating characteristic (ROC) curves curated from logistic regression classifiers of sEVPDGFRβ for overall impairment (B), MCI (C), or AD (D) outcomes using forward selection technique with alpha = 0.05. All models were adjusted for age and sex. Summary of the forward selection sEVPDGFRβ miRNA shown under each ROC curve. (E) Correlation of detected miRNAs in sEVPDGFRβ with the temporal cortical thickness on magnetic resonance imaging (MRI).
Of interest, miR-9-5p expression in sEVPDGFRβ strongly predicted overall cognitive impairment (AUC = 0.935), MCI (AUC = 0.931), and AD (AUC = 1.000) (Figure 5B-5D). However, none of the miRNAs in sEVPDGFRβ showed a statistically significant correlation with temporal cortical thickness (Figure 5E and Table 2).
3.7 |. sEVCD31 characterization and miRNA expression analysis
The miRNAs profiled in sEVCD31 revealed an overexpression of miR-29a-5p, miR-125b-5p, and miR-132-5p in the MCI (p = 0.004, p = 0.031, and p = 0.0001, respectively), MCI-AD (p = 0.005, p = 0.0002, and p < 0.0001, respectively), and AD (p = 0.002, p = 0.0004 and p < 0.0001 respectively) groups. Furthermore, the expression of miR-135b-5p in sEVCD31 was overexpressed in the MCI (p = 0.018) and AD (p < 0.0001) groups, although no significant change was observed in the MCI-AD group. In addition, the expression of miR-210-3p in sEVCD31 showed higher expression in the MCI (p = 0.011) and AD (p = 0.010) cohorts (Figure 6A).
FIGURE 6.
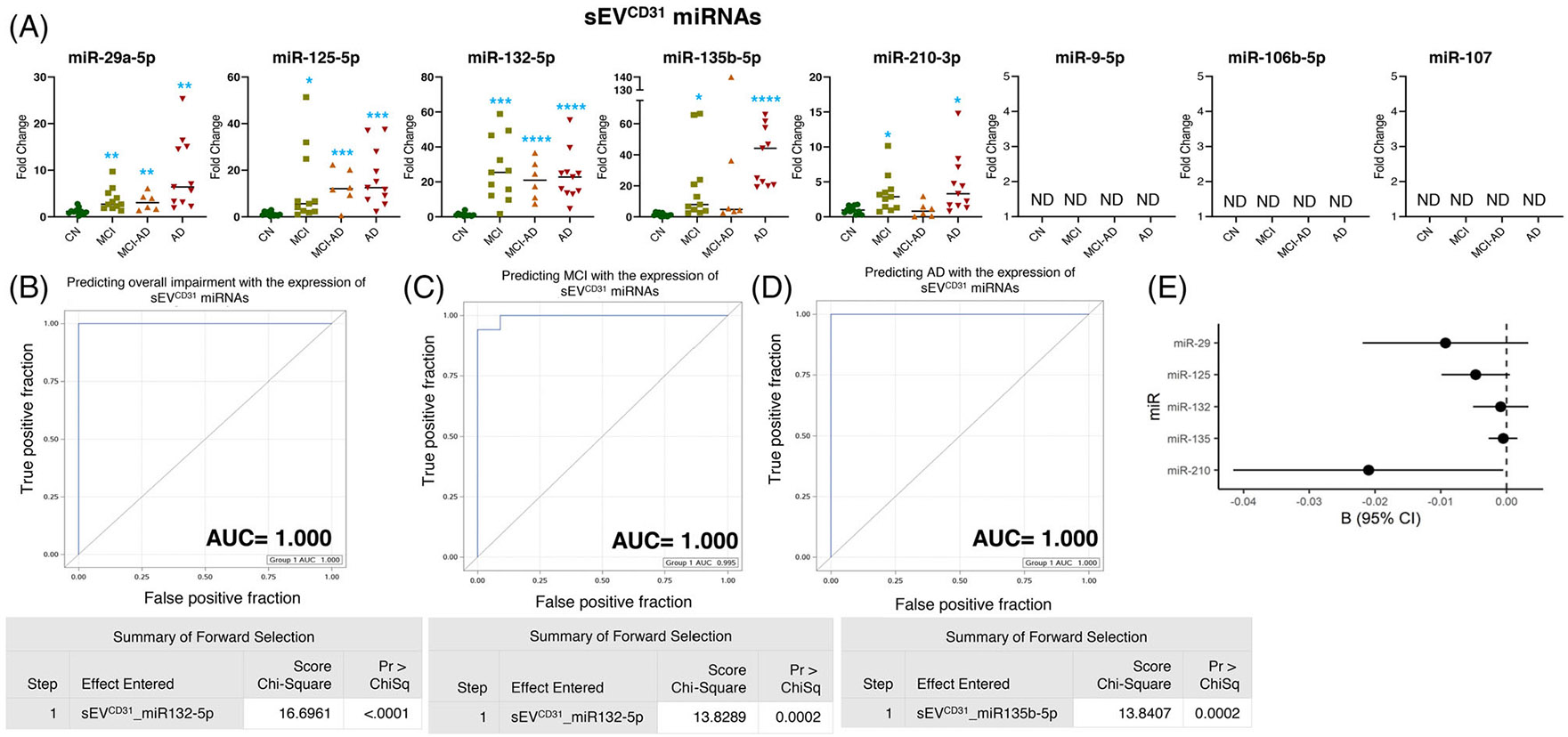
Expression profiling of specific microRNAs (miRNAs) in small extracellular vesicle (sEV)CD31 and their correlation with cognitive impairment and temporal cortical thickness. (A) The expression of eight miRNAs was analyzed in sEVCD31 in subjects with normal cognition (CN; n = 11), mild cognitive impairment (MCI; n = 11), MCI-AD (Alzheimer’s disease) (n = 6), or AD (n = 11) by quantitative real-time polymerase chain reaction (PCR). Fold-change in the expression of miRNA showing detectable expression was calculated by the ΔΔCt method (as mentioned in methods) after normalization with cel-miR-39-3p. Fold-change of all the samples is plotted by calculating 2−ΔΔCt. ND = “not detectable.” *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001. (B–D) Receiver-operating characteristic (ROC) curves curated from logistic regression classifiers of sEVCD31 for overall impairment (B), MCI (C), or AD (D) outcomes using the forward selection technique with alpha = 0.05. All models were adjusted for age and sex. Summary of the forward selection sEVCD31miRNA shown under each ROC curve. (E) Correlation of detected miRNAs in sEVCD31 with the temporal cortical thickness on magnetic resonance imaging (MRI).
The expression of miR-132-5p in sEVCD31could predict the overall impairment and MCI, and the expression of miR-135b-5p in sEVCD31 showed a prediction of AD with 100% efficiency (AUC = 1.000) (Figure 6B-6D). Moreover, the expression of miR-210-3p in sEVCD31 showed a significant negative correlation with cortical thickness (Figure 6E and Table 2).
Of interest, the miRNA expression in sEVPDGFRβ (miR-9-5p) and sEVCD31 (miR-132-5p) could perfectly predict the overall cognitive impairment (AUC = 1.000) (Figure S10A). Similarly, the expression of miR-132-5p in sEVCD31 and the expression of miR-135b-5p in sEVPDGFRβ showed almost absolute prediction for both MCI and AD (Figure S10B and S10C).
4 |. DISCUSSION
In the last few decades, there have been increasing efforts to develop blood-based biomarkers to supplement the expensive neuroimaging and invasive CSF-based measures for detection of AD.56 These additional measures are also required, as nearly 20% of cognitively normal elderly individuals have shown evidence of amyloid neuropathology.57,58 Similarly, ≈12% of adults with clinically diagnosed AD have shown to be negative for amyloid positron emission tomography (PET),59 although the rate of Aβ negativity varied across different study populations and APOE genotypes.60-62 Moreover, the severity of AD neuropathological changes does not correlate well with the degree of amyloidosis.63 These ambiguities suggest the vital need to expand the choice of AD biomarkers beyond Aβ and tau for better diagnosis and prognosis of AD. The analysis of miRNAs has shown much promise as a biomarker to predict AD progression and to identify potential drug target genes.64,65 The expression of miRNAs is dynamically and precisely regulated during brain development and neuronal maturation to maintain cell-type specificity and cellular function; therefore, any aberrant change in these miRNAs may affect brain function and pathologies.66-68 However, most of the information about the miRNA-mediated regulation of AD-related processes is either from biofluids (blood, CSF, etc.), which cannot confer the brain cell–type specificity of their expression change or from postmortem brain tissue with a significant risk of artifacts. To overcome these limitations, in the present study, we isolated multiple sEV subtypes from the plasma of a well-characterized spectrum of samples based on unique surface markers expressed by different brain cell types, and following extensive characterization, we analyzed the expression of specific miRNAs in these sEVs. Results showed that the miRNA expression in sEV subtypes changed significantly in adults with MCI, MCI-AD, and AD dementia compared to the CN group, clearly distinguishing the dementia status and correlated with the cortical thickness of brain regions susceptible to AD and other age-related dementias.
Downregulation of serum miR-106b was suggested as a potential biomarker for the early detection of AD with high specificity and sensitivity.69 In the present study, however, we observed significant overexpression of miR-106b-5p, specifically in neuronal-enriched sEVs (sEVL1CAM) from the AD cohort, and showed absolute accuracy in distinguishing AD dementia from normal adults or those with MCI. In addition, the expression of miR-106b-5p also negatively correlated with cortical thickness, which has been suggested previously as the key signature of AD pathology.70 Of interest, we observed significant overexpression of miR-132-5p in sEVL1CAM from the MCI-AD and AD groups. MiR-132 is considered as “neurimmiR” because of its involvement in numerous neurophysiological and pathological processes, including the progression of Aβ and tau pathology.30 However, an inconsistent expression pattern of miR-132 has been reported in serum, brain tissue, or in neurally derived plasma exosomes.71-73 Furthermore, miR-107 was identified to target BACE1 mRNAs, and its reduced expression has been reported in human brain tissue, particularly cerebral cortical laminas, and temporal cortex in AD cases.25,74 We noted undetectable expression of miR-107 in neuronal sEVs, although an increased expression of miR-107 in astrocyte-enriched sEVs (sEVGLAST) was observed, which showed predictability for overall impairment in adults with MCI or AD and also correlated with temporal cortical thickness. Moreover, miR-107 targets granulin and decreased expression and mutation in granulin/progranulin have been implicated directly in frontotemporal dementias and other neurodegenerative diseases, including AD.34,75 Therefore, our results indicate that miRNA expression in brain cell–derived sEVs from plasma could serve as a highly reliable tool compared to their analysis in CSF, plasma, or brain tissue for early diagnosis of AD with high specificity and sensitivity.
Upregulation of miR-106b in the microglia has been linked with their activation in the status epilepticus mouse model,76 whereas a reduced level of miR-29a was reported to exacerbate neurological damage by promoting M1-type microglia polarization.77 In line with these observations, we noted increased expression of miR-106b-5p and decreased expression of miR-29a-5p in microglia-enriched sEVs (sEVTMEM119) in MCI-AD and AD groups, which could dictate the M1 polarization of microglia. Although further studies are needed to assess whether sEVTMEM119 are also enriched for biomarkers of M1-type microglia polarization; but, the expression of these miRNAs in sEVTMEM119 distinguished overall impairment and/or MCI from control participants. Similarly, the expression of miR-132-5p and miR-125b-5p, which regulate cytokine and chemokine release from microglia and are involved in their activation,78,79 in sEVTMEM119 predicted AD in our cohort. The higher expression of miR-125b-5p and miR-29a-5p in oligodendrocyte-enriched sEVs (sEVPDGFRα) distinguished between overall impairment and AD from control participants. Peripheral myelin protein 22 (PMP22), expressed predominantly by myelinating Schwann cells and correlating with the completion of myelination and Schwann cell differentiation, has been identified as the target of miR-29a. The expression of miR-29a was reported to be reduced by 7-fold when cultured Schwann cells were promoted to differentiate.80 The increased expression of miR-29a-5p in AD sEVPDGFRα suggests its inhibitory effect on Schwann cell differentiation and their myelination in the AD condition. Furthermore, the expression of miR-132-5p and miR-135b-5p in sEVCD31 could distinguish between adults with MCI and AD with nearly 100% predictability (Figure S10), indicating the dysfunction of vascular components. These results, for the first time, show the utility of simultaneously characterizing multiple brain cell–derived sEVs from the blood, providing a less-invasive way to comprehend the complex microenvironmental situation and understand the interplay of different brain cells in AD pathogenesis.
Hypoxia and hyperperfusion are the known contributing factors to the vascular damage leading to the breakdown of the blood–brain barrier (BBB), which results in the accumulation of neurotoxic proteins, inflammation, synaptic dysfunction, defects in Aβ/tau clearance, ultimately causing cerebral amyloid angiopathy.81 In addition, hypoxia can lead to memory impairment and neuronal death by shifting APP processing toward the amyloidogenic pathway and downregulating the function of α-secretase.82 Strikingly, we observed an increased level of hypoxic miR-210-3p in sEVL1CAM and sEVGLAST selectively in MCI (prodromal dementia) but not in the MCI-AD or AD group (established dementia). It is possible that in the prodromal stage of dementia, neurons, and astrocytes experience hypoxia, possibly because of vascular dysfunction. In addition, hypoxia was also shown to increase the expression of miR-132 in mouse and human brain microvascular endothelial cells, and its higher expression was observed in the capillaries of mouse brain after traumatic brain injury and in isolated hippocampal neurons.83,84 Of interest, claudin-1, junctional adhesion molecule 3, and tight junction-associated protein 1, which are crucial in maintaining BBB integrity, have been identified as targets of miR-212/132.83 The higher expression of miR-132-5p in sEVCD31 in the present study suggests a possible disruption of the BBB, which correlates well with the hypoxia signature reflected as an increase in miR-210-3p in sEVL1CAM and sEVGLAST (in neuron and astrocytes). In summary, a panel of miRNAs from different sEV subtypes, namely, miR-106b-5p in sEVL1CAM, miR-107 in sEVGLAST, miR-132-5p in sEVTMEM119, miR-29a-5p in sEVPDGFRα, miR-9-5p in sEVPDGFRβ, and miR135b-5p in sEVCD31, can be further validated in a larger cohort for the development of efficient and reliable biomarkers for the diagnosis of AD and also to understand the molecular state of different brain cells during AD pathogenesis.
Despite the potential of miRNAs from different brain cell–derived sEVs in predicting dementia and suggesting the involvement of these brain cells in AD pathogenesis, our study has some limitations. First, we cannot claim the uniqueness of the sEV subtypes, as the markers selected to isolate different sEV are not unique to each brain cell type, their multiple possible states, or the CNS. Nevertheless, these markers can certainly enrich specific brain cell–derived sEVs and represent improved sensitivity along with analysis of TEs from biofluids. Second, this initial work had a limited sample of well-characterized participants based on cognitive status. Consistent with the parent cohort from which they were drawn,41,42 these cognitive groups differed based on important clinical characteristics, including age, APOE ε4 genotype, and treatment (e.g., acetylcholinesterase inhibitors). Therefore, the sample size may have limited statistical power resulting in type 2 error and the ability to adjust for potential confounding factors within the larger group of impaired individuals. Thus the expression profile of the selected miRNAs needs to be further validated in a larger cohort. Regardless of these limitations, the study successfully demonstrated a less-invasive approach of simultaneously isolating multiple brain cell–enriched sEV subtypes from the peripheral circulatory system and their usefulness in offering biomarkers for detecting cognitive impairment. Moreover, the differential expression of miRNA in various sEV subtypes also reflected molecular changes associated with multiple brain cells and microenvironmental situations (e.g., microglia activation, hypoxia), as well as their potential contribution to AD pathogenesis.
Supplementary Material
Highlights.
Multiple brain cell–derived small extracellular vesicles (sEVs) could be isolated simultaneously from blood.
MicroRNA (miRNA) expression in sEVs could detect Alzheimer’s disease (AD) with high specificity and sensitivity.
miRNA expression in sEVs correlated with cortical region thickness on magnetic resonance imaging (MRI).
Altered expression of miRNAs in sEVCD31 and sEVPDGFRβ suggested vascular dysfunction.
miRNA expression in sEVs could predict the activation state of specific brain cell types.
RESEARCH IN CONTEXT.
Systematic Review: We carefully reviewed the existing literature using PubMed and cited relevant publications. We noted that most studies focused on characterizing circulatory Alzheimer’s disease (AD) markers are centered around identifying pathological proteins aberrantly overexpressed in AD brain; or microRNA (miRNA) expression in cerebrospinal fluid (CSF), blood, small extracellular vesicles (sEVs), and postmortem AD brain, but they lack clear information about the cellular origin of these biomarkers.
Interpretation: Our findings establish the methodology for simultaneously isolating several brain cell–enriched sEVs (from neurons, astrocytes, microglia, oligodendrocytes, pericytes, and endothelial cells) from the same plasma sample. For the first time, miRNA expression in these brain cell–derived sEVs showed their usefulness in distinguishing MCI and AD from control subjects, and their expression correlated well with AD signature region thickness, suggesting vascular disruption as the initial event in AD onset.
Future Directions: These findings are promising and need further validation in multiple larger cohorts.
ACKNOWLEDGMENTS
The authors are thankful to Ms Karlyn Donohoe for coordinating the sample storage, retrieval, and transport for this study. The authors also would like to acknowledge the Wake Forest Alzheimer’s Disease Research Center (supported by National Institutes of Health, National Institute on Aging P30 AG072947 [NIH/NIA]) for providing the samples for this study. Wake Forest Baptist Comprehensive Cancer Center Cellular Imaging Shared Resource is supported by NCI (P30CA012197, PI: Dr. Ruben A. Mesa). This work was supported by NIA RF1AG068629 (TH and GD), R01 AG061805 (GD), and P30 AG072947 (SC, TH).
Footnotes
CONFLICT OF INTEREST STATEMENT
GD is the founder of LiBiCo, which has no influence or contribution to the work presented in this manuscript. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Gauthier SR-NP, Morais JA, Webster C, World Alzheimer Report 2021: Journey through the diagnosis of dementia. 2021. [Google Scholar]
- 2.DeStrooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164(4):603–615. [DOI] [PubMed] [Google Scholar]
- 3.Henstridge CM, Hyman BT, Spires-Jones TL. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat Rev Neurosci. 2019;20(2):94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swerdlow RH. Pathogenesis of Alzheimer’s disease. Clin Interv Aging. 2007;2(3):347–359. [PMC free article] [PubMed] [Google Scholar]
- 5.Dzamba D, Harantova L, Butenko O, Anderova M. Glial cells - The key elements of Alzheimer s disease. Curr Alzheimer Res. 2016;13(8):894–911. [DOI] [PubMed] [Google Scholar]
- 6.Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113):2673–2734. [DOI] [PubMed] [Google Scholar]
- 7.Hughes TM, Kuller LH, Barinas-Mitchell EJ, McDade EM, et al. Arterial stiffness and beta-amyloid progression in nondemented elderly adults. JAMA neurology. 2014;71(5):562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes TM, Kuller LH, Barinas-Mitchell EJ, et al. Pulse wave velocity is associated with beta-amyloid deposition in the brains of very elderly adults. Neurology. 2013;81(19):1711–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchell GF, van Buchem MA, Sigurdsson S, et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility–Reykjavik study. Brain : journal of neurology. 2011;134(Pt 11):3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabkin SW. Arterial stiffness: detection and consequences in cognitive impairment and dementia of the elderly. Journal of Alzheimer’s disease : JAD. 2012;32(3):541–549. [DOI] [PubMed] [Google Scholar]
- 11.Lane RE, Korbie D, Hill MM, Trau M. Extracellular vesicles as circulating cancer biomarkers: opportunities and challenges. Clin Transl Med. 2018;7(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciferri MC, Quarto R, Tasso R. Extracellular vesicles as biomarkers and therapeutic tools: from pre-clinical to clinical applications. Biology (Basel). 2021;10(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson LS, Hamlett ED, Stone TD, Sims-Robinson C. Neuronally derived extracellular vesicles: an emerging tool for understanding Alzheimer’s disease. Mol Neurodegener. 2019;14(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winston CN, Goetzl EJ, Akers JC, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement (Amst). 2016;3:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers Dement. 2015;11(6):600–7e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia L, Zhu M, Kong C, et al. Blood neuro-exosomal synaptic proteins predict Alzheimer’s disease at the asymptomatic stage. Alzheimers Dement. 2021;17(1):49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramakrishna S, Muddashetty RS. Emerging role of microRNAs in dementia. J Mol Biol. 2019;431(9):1743–1762. [DOI] [PubMed] [Google Scholar]
- 18.Xie H, Zhao Y, Zhou Y, et al. MiR-9 regulates the expression of BACE1 in dementia induced by chronic brain hypoperfusion in rats. Cell Physiol Biochem. 2017;42(3):1213–1226. [DOI] [PubMed] [Google Scholar]
- 19.Hebert SS, Horre K, Nicolai L, et al. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol Dis. 2009;33(3):422–428. [DOI] [PubMed] [Google Scholar]
- 20.Chow VW, Mattson MP, Wong PC, Gleichmann M An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang YW, Thompson R, Zhang H, Xu H APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith PY, Hernandez-Rapp J, Jolivette F, et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet. 2015;24(23):6721–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banzhaf-Strathmann J, Benito E, May S, et al. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014;33(15):1667–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keifer J, Zheng Z, Ambigapathy G. A MicroRNA-BDNF negative feedback signaling loop in brain: implications for Alzheimer’s Disease. Microrna. 2015;4(2):101–108. [DOI] [PubMed] [Google Scholar]
- 25.Wang WX, Rajeev BW, Stromberg AJ, et al. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28(5):1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Zhang T, Zhou YX, et al. A Presenilin/Notch1 pathway regulated by miR-375, miR-30a, and miR-34a mediates glucotoxicity induced-pancreatic beta cell apoptosis. Sci Rep. 2016;6:36136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebert SS, Horre K, Nicolai L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(17):6415–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li P, Xu Y, Wang B, Huang J, Li Q. miR-34a-5p and miR-125b-5p attenuate Abeta-induced neurotoxicity through targeting BACE1. J Neurol Sci. 2020;413:116793. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Xing H, Guo S, Zheng Z, Wang H, Xu D. MicroRNA-135b has a neuroprotective role via targeting of beta-site APP-cleaving enzyme 1. Exp Ther Med. 2016;12(2):809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang M, Bian Z. Alzheimer’s disease and microRNA-132: a widespread pathological factor and potential therapeutic target. Front Neurosci. 2021;15:687973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shioya M, Obayashi S, Tabunoki H, et al. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. 2010;36(4):320–330. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Zuo X, Han J, et al. MiR-9-5p inhibits mitochondrial damage and oxidative stress in AD cell models by targeting GSK-3beta. Biosci Biotechnol Biochem. 2020;84(11):2273–2280. [DOI] [PubMed] [Google Scholar]
- 33.Augustin R, Endres K, Reinhardt S, et al. Computational identification and experimental validation of microRNAs binding to the Alzheimer-related gene ADAM10. BMC Med Genet. 2012;13:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang WX, Wilfred BR, Madathil SK, et al. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am J Pathol. 2010;177(1):334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossi M, Kilpinen H, Muona M,et al. Allele-specific regulation of DISC1 expression by miR-135b-5p. Eur J Hum Genet. 2014;22(6):840–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li R, Hu J, Cao S. The clinical significance of miR-135b-5p and its role in the proliferation and apoptosis of hippocampus neurons in children with temporal lobe epilepsy. Dev Neurosci. 2020;42(5-6):187–194. [DOI] [PubMed] [Google Scholar]
- 37.Zhang YH, Bai SF, Yan JQ. Blood circulating miRNAs as biomarkers of Alzheimer’s disease: a systematic review and meta-analysis. Biomark Med. 2019;13(12):1045–1054. [DOI] [PubMed] [Google Scholar]
- 38.Swarbrick S, Wragg N, Ghosh S, Stolzing A. Systematic review of miRNA as biomarkers in Alzheimer’s disease. Mol Neurobiol. 2019;56(9):6156–6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A, Kim S, Su Y, et al. Brain cell-derived exosomes in plasma serve as neurodegeneration biomarkers in male cynomolgus monkeys self-administrating oxycodone. Ebiomedicine. 2021;63:103192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar A, Sharma M, Su Y, et al. Small extracellular vesicles in plasma reveal molecular effects of modified Mediterranean-ketogenic diet in participants with mild cognitive impairment. Brain Commun. 2022;4(6):fcac262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coffin C, Suerken CK, Bateman JR, et al. Vascular and microstructural markers of cognitive pathology. Alzheimers Dement (Amst). 2022;14(1):e12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hughes TM, Lockhart SN, Suerken CK, et al. Hypertensive aspects of cardiometabolic disorders are associated with lower brain microstructure, perfusion, and cognition. J Alzheimers Dis. 2022;90(4):1589–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weintraub S, Besser L, Dodge HH, et al. Version 3 of the Alzheimer disease Centers’ Neuropsychological Test Battery in the Uniform Data Set (UDS). Alzheimer Dis Assoc Disord. 2018;32(1):10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sachs BC, Steenland K, Zhao L, et al. Expanded demographic norms for Version 3 of the Alzheimer disease Centers’ Neuropsychological Test Battery in the Uniform Data Set. Alzheimer Dis Assoc Disord. 2020;34(3):191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer’s cognitive composite with semantic processing: the PACC5. Alzheimers Dement (N Y). 2017;3(4):668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwarz CG, Gunter JL, Wiste HJ, et al. A large-scale comparison of cortical thickness and volume methods for measuring Alzheimer’s disease severity. Neuroimage Clin. 2016;11:802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramteke A, Ting H, Agarwal C, et al. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol Carcinog. 2015;54(7):554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patterson SA, Deep G, Brinkley TE. Detection of the receptor for advanced glycation endproducts in neuronally-derived exosomes in plasma. Biochem Biophys Res Commun. 2018;500(4):892–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giri M, Zhang M, Lu Y. Genes associated with Alzheimer’s disease: an overview and current status. Clin Interv Aging. 2016;11:665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bertram L, Tanzi RE. Alzheimer disease risk genes: 29 and counting. Nat Rev Neurol. 2019;15(4):191–192. [DOI] [PubMed] [Google Scholar]
- 54.Tokar T, Pastrello C, Rossos AEM, et al. mirDIP 4.1-integrative database of human microRNA target predictions. Nucleic Acids Res. 2018;46(D1):D360–D70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jia P, Dai Y, Hu R, Pei G, Manuel AM, Zhao Z. TSEA-DB: a trait-tissue association map for human complex traits and diseases. Nucleic Acids Res. 2020;48(D1):D1022–D30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma N, Singh AN. Exploring biomarkers for Alzheimer’s disease. J Clin Diagn Res. 2016;10(7):KE01–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. [DOI] [PubMed] [Google Scholar]
- 58.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45(3):358–368. [DOI] [PubMed] [Google Scholar]
- 59.Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA. 2015;313(19):1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landau SM, Horng A, Fero A, Jagust WJ. Alzheimer’s Disease Neuroimaging I. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology. 2016;86(15):1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson KA, Sperling RA, Gidicsin CM, et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer’s disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement. 2013;9(5 Suppl):S72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fleisher AS, Chen K, Liu X, et al. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging. 2013;34(1):1–12. [DOI] [PubMed] [Google Scholar]
- 63.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Angelucci F, Cechova K, Valis M, Kuca K, Zhang B, Hort J. MicroRNAs in Alzheimer’s disease: diagnostic markers or therapeutic agents. Front Pharmacol. 2019;10:665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei W, Wang ZY, Ma LN, Zhang TT, Cao Y, Li H. MicroRNAs in Alzheimer’s disease: function and potential applications as diagnostic biomarkers. Front Mol Neurosci. 2020;13:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He M, Liu Y, Wang X, Zhang MQ, Hannon GJ, Huang ZJ. Cell-type-based analysis of microRNA profiles in the mouse brain. Neuron. 2012;73(1):35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jovicic A, Roshan R, Moisoi N, et al. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci. 2013;33(12):5127–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149(3):515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Madadi S, Saidijam M, Yavari B, Soleimani M. Downregulation of serum miR-106b: a potential biomarker for Alzheimer disease. Arch Physiol Biochem. 2020:1–5. [DOI] [PubMed] [Google Scholar]
- 70.Busovaca E, Zimmerman ME, Meier IB, et al. Is the Alzheimer’s disease cortical thickness signature a biological marker for memory. Brain Imaging Behav. 2016;10(2):517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hadar A, Milanesi E, Walczak M, et al. SIRT1, miR-132 and miR-212 link human longevity to Alzheimer’s. Disease Sci Rep. 2018;8(1):8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cha DJ, Mengel D, Mustapic M, et al. miR-212 and miR-132 are downregulated in neurally derived plasma exosomes of Alzheimer’s patients. Front Neurosci. 2019;13:1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie B, Zhou H, Zhang R, Song M, Yu L, Wang L, et al. Serum miR-206 and miR-132 as potential circulating biomarkers for mild cognitive impairment. J Alzheimers Dis. 2015;45(3):721–731. [DOI] [PubMed] [Google Scholar]
- 74.Nelson PT, Wang WX. MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. J Alzheimers Dis. 2010;21(1):75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perry DC, Lehmann M, Yokoyama JS, et al. Progranulin mutations as risk factors for Alzheimer disease. JAMA Neurol. 2013;70(6):774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu T, Fu H, Sun JJ, Ding DR, Wang H. miR-106b-5p upregulation is associated with microglial activation and inflammation in the mouse hippocampus following status epilepticus. Exp Brain Res. 2021;239(11):3315–3325. [DOI] [PubMed] [Google Scholar]
- 77.Zhao F, Zhao H, Fan J,et al. MiR-29a knockout aggravates neurological damage by pre-polarizing M1 microglia in experimental rat models of acute stroke. Front Genet. 2021;12:642079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wallach T, Wetzel M, Dembny P, et al. Identification of CNS injury-related microRNAs as novel toll-like receptor 7/8 signaling activators bySmall RNA Sequencing. Cells. 2020;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parisi C, Napoli G, Amadio S, et al. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016;23(3):531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Verrier JD, Lau P, Hudson L, Murashov AK, Renne R, Notterpek L. Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia. 2009;57(12):1265–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hassan H, Chen R. Hypoxia in Alzheimer’s disease: effects of hypoxia inducible factors. Neural Regen Res. 2021;16(2):310–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burek M, Konig A, Lang M, et al. Hypoxia-induced microRNA-212/132 alter blood-brain barrier integrity through inhibition of tight junction-associated proteins in human and mouse brain microvascular endothelial cells. Transl Stroke Res. 2019;10(6):672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun ZZ, Lv ZY, Tian WJ, Yang Y MicroRNA-132 protects hippocampal neurons against oxygen-glucose deprivation-induced apoptosis. Int J Immunopathol Pharmacol. 2017;30(3):253–263. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.