

Abstract
Lipid metabolism imbalance combined with over-activated inflammation are two key factors for hepatic stestosis. However, on-demand anchoring inflammation and lipid metabolism disorder for hepatic stestosis treatment has yet to be realized. Here we propose a charge reversal fullerene based nano-assembly to migrate hepatic steatosis via inhibiting macrophage-mediated inflammation and normalizing hepatocellular lipid metabolism in obesity mice. Our nano-assembly (abbreviated as FPPD) is comprised of electropositive polyetherimide (PEI), charge-shielded dimethylmaleic anhydride (DMA), and poly(lactic-co-glycolic acid) (PLGA), which provides hydrophobic chains for self-assembly with anti-oxidative dicarboxy fullerene poly(ethylene glycol) molecule (FP). The obtained FPPD nano-assembly owns a charge reversal ability that switches to a positive charge in an acidic environment that targets the electronegative mitochondria both in pro-inflammatory macrophages and steatosis hepatocytes. We demonstrate that the anti-oxidative and mitochondria-targeting FPPD notably reduces inflammation in macrophages and lipid accumulation in hepatocytes by quenching excessive reactive oxygen species (ROS) and improving mitochondrial function in vitro. Importantly, FPPD nano-assembly reveals a superior anti-hepatic steatosis effect via migrating inflammation and facilitating lipid transport in obesity mice. Overall, the charge reversal nano-assembly reduces over-activated inflammation and promotes lipid metabolism that provides an effectiveness of a multi-target strategy for hepatic steatosis treatment.
Keywords: Fullerene nano-assembly, Charge reversal, Inflammation, lipid metabolism, hepatic steatosis
Graphical abstract
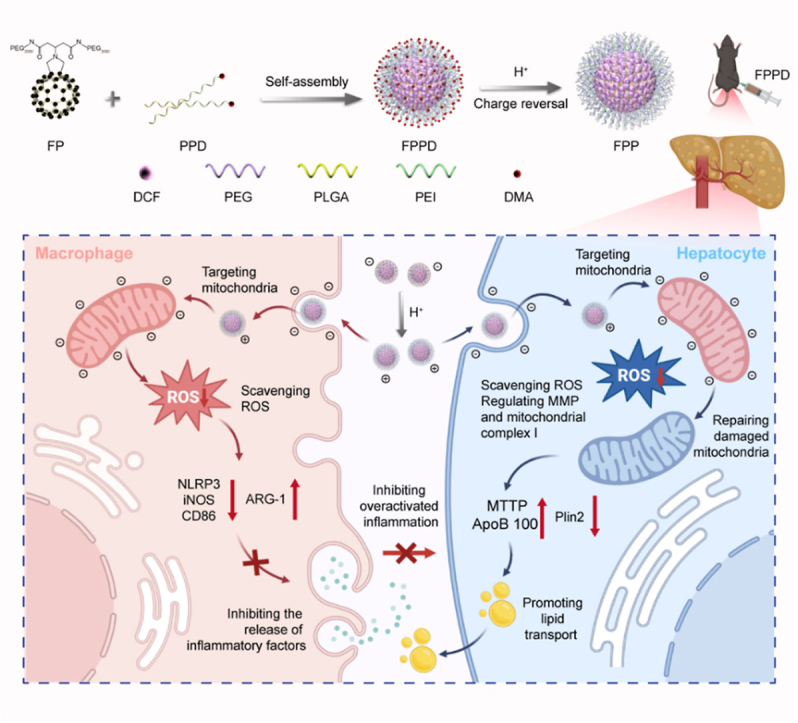
Briefs: The FPPD nano-assembly was comprised of electropositive PEI, PLGA, charge-shielded DMA and free radical scavenging FP. The obtained FPPD nano-assembly owns a charge reversal ability that switches to a positive charge in an acidic environment and targets the electronegative mitochondria both in pro-inflammatory macrophages and steatosis hepatocytes, resulting in sequentially reducing inflammation and lipid accumulation by quenching excessive ROS and improving mitochondrial function.
Highlights
-
•
The antioxidant nanoassembly FPPD is prepared self-assembly method.
-
•
The surface charge of FPPD can change from negative to positive in a slightly acidic environment.
-
•
FPPD targets mitochondria of macrophages and hepatocytes.
-
•
FPPD can clear ROS and inhibit macrophage-mediated inflammation.
-
•
FPPD can repair damaged mitochondria and promote lipid transport in hepatocytes.
-
•
FPPD can mitigate hepatic stestosis by sequentially resolving inflammation and improving lipid metabolism disorder.
1. Introdution
Hepatic steatosis, a common metabolic syndrome, is one type of nonalcoholic fatty liver diseases (NAFLD) [1,2], and may further develop into nonalcoholic steatohepatitis (NASH), progressive fibrosis and hepatocellular carcinoma [[3], [4], [5]]. It causes by excessive lipid deposition in hepatocytes, which leading to hepatic inflammation [6]. Particularly, the liver resident macrophages (Kupffer cells) are the most abundant innate immune cells in the liver and play important roles in the occurrence of inflammation in hepatic steatosis [7]. Generally, the macrophages could be divided into pro-inflammatory M1 macrophage phenotype and anti-inflammatory M2 macrophage phenotype [8]. M1-type macrophages secrete pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which promote hepatic insulin resistance (IR) and lipid metabolism dysfunction [9,10]. Specifically, the lipid metabolism includes lipid synthesis, lipid decomposition and lipid transport, all of which are disrupted in hepatocytes of hepatic steatosis [[11], [12], [13]]. Regrettably, it is an acute shortage on effective drugs to treat hepatic steatosis in clinic. Though the single treatment strategy on anti-inflammation or regulating hepatocellular lipid metabolism has already shown the promises on relieving hepatic steatosis, it could not fundamentally reverse hepatic steatosis [14,15]. Thus, it is a great need to develop a multi-target treatment of hepatic steatosis by anti-inflammation and reversing the lipid metabolism disorder.
Reactive oxygen species (ROS) act as the second messenger to concurrently regulate the inflammation and lipid metabolism disorder [[16], [17], [18]]. For instance, the strategies on quenching excessive ROS could contribute to inhibit M1-type macrophages mediated inflammation [19]. Further, the hepatocellular lipid transport ability would be also boosted by decreasing excessive ROS in hepatocytes [20]. Moreover, the mitochondria in both macrophages and hepatocytes are the most important location for generation of ROS [21,22]. Therefore, mitochondria are considered as effective targets for anti-inflammation and reversing the lipid metabolism disorder in hepatic steatosis [[23], [24], [25]]. Due to the negative potential from −180 mV to −200 mV on the matrix surface of the inner mitochondrial membrane [26], cationic molecule-modified drug carriers can achieve mitochondrial targeting through electrostatic interaction [27]. At the same time, positively charged nanocarriers could also cause serious aggregation and be quickly eliminated during the body circulation [28]. In response to this contradiction, the pH-controlled charge reversal strategy from negative charge to positive charge initiated by the inflammatory environment (pH ∼ 6.5) or cellular lysosomes (pH ∼ 4.6) has been proven to be an effective way to promote drug target delivery [29].
In this study, we tailor a charge reversal fullerene based nano-assembly targeting on the mitochondria of both macrophages and hepatocytes, inhibits inflammation and promotes lipid transport for reversing hepatic steatos (Scheme 1). This nano-assembly FPPD comprises a free radical scavenging dicarboxy fullerene poly(ethylene glycol) molecule (FP), electropositive polyetherimide (PEI), poly(lactic-co-glycolic acid) (PLGA, offered the hydrophobic chains for self-assembly with FP), and charge-shielded dimethylmaleic anhydride (DMA). The charge reversal nano-assembly would emerge negative charge under normal physiological environment, while appear positive charge in an acidic environment, including the inflammatory environment or cellular lysosomes. The positive charge nano-assembly could further target the cellular mitochondria both in pro-inflammatory macrophages and steatosis hepatocytes. Subsequently, the anti-oxidative FPPD markedly inhibits inflammation and lipid accumulation by reducing excessive ROS and improving mitochondrial function in macrophages and hepatocytes. Of note, we demonstrate that FPPD nano-assembly has a superior anti-hepatic steatosis effect by migrating inflammation and boosting lipid transport in obesity mice. Together, our charge reversal nano-assembly reduces macrophages inflammation and promotes lipid metabolism that provides an exciting potential for hepatic steatosis treatment.
Scheme 1.

Schematic illustration of the charge reversal FPPD preparation and the mechanism diagram of FPPD on migrating hepatic steatosis. The FPPD nano-assemblys is comprised of electropositive PEI, PLGA, charge-shielded DMA and free radical scavenging FP. The obtained FPPD nano-assembly owns a charge reversal ability that switches to a positive charge in an acidic environment and targets the electronegative mitochondria both in pro-inflammatory macrophages and steatosis hepatocytes, resulting in reducing inflammation and lipid accumulation by quenching excessive ROS and improving mitochondrial function.
2. Materials and methods
2.1. Materials
Solid C60 was made by ourselves. Polyetherimide (PEI), poly (lactic-co-glycolic acid) (PLGA), dicyclohexylcarbodiimide (DCC), NaOH, NaCl, tert-butanol, dimethylmaleic anhydride (DMA), 4-dimethylaminopyridine (DMAP), hydrochloric acid, benzyl glycine hydrochloride, oleic acid (OA), O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate (HBTU), Cy5.5, sodium bicarbonate, ethyl acetate (EtOAc), dichloromethane (CH2Cl2), methanol (MeOH), trifluoroacetic acid (TFA), N,N-Diisopropylethylamine (DIPEA) and sodium cyano borohydride (NaBH3CN) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). The antibodies of immunofluorescence (F4/80 and CD11c) and Western blot (WB) (NLRP3, iNOS, CD86, ARG-1Plin2, MTTP, and ApoB 100) were purchased from Abcam (Cambridge, UK) and Thermo Fisher Scientific Inc. (Waltham, USA).
2.2. Preparation of C60-(COOH)2 (DCF)
DCF was synthesized according to the route shown in Fig. S1. 14.61 g 1,3 acetone dicarboxylic acid, 22.24 g tert-butanol, 1.00 g DMAP and 100 mL dry methylene chloride were dropped to a 250 mL round-bottomed flask. Stir the solution in an ice bath while 22.79 g of DIPEA was added to the system. After 10 min at 0 °C, stir the mixture for 3 h at room temperature. Filter out the dicyclohexyl urea, and the solution was washed by 0.5 N diluted hydrochloric acid (25 mL) and saturated sodium bicarbonate solution (25 mL). Filter out the precipitates in the process, and concentrate the organic phase by a rotary evaporator. The remaining mixture was purified by silica column chromatography to obtain compound 1.
Dissolve 0.5 g of compound 1 in MeOH, add 395 mg benzyl glycine hydrochloride and 90 mg NaBH3CN, stir the solution for 48 h. The solution was extracted with CH2Cl2, and a rotary evaporator was used to remove the solvent to obtain a colorless oily substance. 0.7 g of the oil was dissolved in EtOAc (105 mL) and MeOH (25 mL). Next, drop Pd/C (10 %, 120 mg) into the solution and stirred the solution at hydrogen atmosphere for 24 h. At the end of the reaction, the reaction solution was filtered and precipitated with n-hexane to obtain compound 2.
Drop 300 mg of compound 2 and 45 mg of paraformaldehyde to 330 mL C60 saturated toluene solution, stirred the solution at 110 °C for 1 h. Remove most of the solvent, and the remaining mixture was purified by silica column chromatography to obtain compound 3.
250 mg of compound 3 and 10 mL of TFA were added to CHCl3 (30 mL) solvent. The solution was stirred for 18 h. Remove the solvent and DCF was obtained after drying.
2.3. Preparation of FP
FP is synthesized by amidation of one DCF molecule with two mPEG2000-NH2 molecules. Briefly, DCF (50 mg), mPEG2000-NH2 (300 mg), HBTU and DIPEA were dissolved in dimethyl formamide (DMF), and then stirred overnight. The product was precipitated with ether and then re-dissolved in water. The solution was dialyzed with dialysis bag (MWCO3500) for 48 h, and FP was obtained by freeze-drying.
2.4. Preparation of PPD
Poly (lactic-co-glycolic acid) (PLGA, 11 μmoL), polyetherimide (PEI, 34 μmoL), HBTU (56 μmoL) and DIPEA (56 μmoL) were added to 1 mL DMF. The mixture was stirred overnight and precipitated with ether to obtain PLGA-PEI polymer. PLGA-PEI (35 μmoL) and dimethylmaleic anhydride (DMA, 42 μmoL) were dissolved in 10 mL dimethylsulfoxide (DMSO), then 1 mL triethylamine and 1 mL pyridine were added, and the mixture was stirred overnight under the protection of nitrogen atmosphere at room temperature. After the reaction, five times the volume of deionized water was added to the system, and PLGA-PEI-DMA (PPD) polymer was obtained after dialysis and freeze-drying.
2.5. Preparation of FPPD
FP (64 mg) and PPD (80 mg) were dissolved in 1 mL of DMSO and slowly added to 6 mL of deionized water under agitation at a rate of 50 μL/min. The mixture was stirred at room temperature for 24 h and then dialysed in deionized water for 48 h. The solution was freeze-dried and dissolved in deionized water to obtain FPPD aqueous solution.
2.6. Characterization of FP and FPPD
The surface functional groups of FP, PPD and FPPD samples were determined by Fourier transform infrared spectroscopy (FT-IR, Nicolet iN10 MX, Thermo Fisher Scientific, USA). The hydrated particle size and Zeta potential of the FPPD and FP solutions were determined by a dynamic light scattering (DLS, NanoZS ZEN3600, Malvern Instruments, Britain). The particle size and morphology of the FPPD were observed by transmission electron microscopy (TEM, JEM-1011, JEOL, Japan). The samples were filtered by 220 nm aqueous filter membrane and then placed into special cuvets for particle size and potential testing. The apparent morphology of the sample was determined by atomic force microscope (AFM, Bruker, Multimode 8, Germany). The diluted FPPD solution was added to the mica sheet and measured by atomic force microscope. Finally, the FP and FPPD samples were dissolved in physiological media such as PBS buffer, serum and cell medium, respectively, and their stability in different media was tested.
2.7. In vitro hydroxyl radical's elimination
20 μL H2O2 (100 mM) and 40 μL of DMPO (100 mM) were mixed with 20 μL deionized water, 20 μL FP solution (50 μM), and 20 μL FPPD solution (50 μM), respectively. First, shine a 500-W UV light on the mixture for 4 min, and then the electron paramagnetic resonance (EPR) spectra of DMPO-OH were recorded in the dark with an EPR spectrometer (FA-200, JEOL, Japan).
2.8. Cell culture and OA induced lipid accumulation cell model
The human hepatocytes L02 cells and RAW264.7 macrophages were purchased from the Institute of Basic Medicine, Chinese Academy of Medical Sciences (Beijing, China). L02 cells were cultured in RPMI 1640 medium supplemented with 10 % (W/V) FBS, streptomycin (1 μg/mL) and penicillin (1 μg/mL) in 5 % CO2 in an incubator at 37 °C. RAW264.7 cells were cultured in DMEM medium supplemented with 10 % (W/V) FBS in 5 % CO2 in an incubator at 37 °C.
0.2 mmol OA was dissolved in 2.5 mL phosphate buffered saline solution (PBS) containing 10 % (W/V) fatty acid-free bovine serum albumin (BSA). The OA reserve solution was stored at −20 °C. L02 cells were incubated with 0–2.0 mM OA for 24 h. The culture-medium was removed, the cells were washed twice by PBS and treated with Hoechst 33342 (excitation wavelength: 346 nm; Emission wavelength: 461 nm) and Nile red (excitation wavelength: 530 nm; Emission wavelength: 576 nm) for 30 min. After that, the cells were washed twice and the fluorescent signal was detected using an Infinite M1000 Pro (Swiss Tecan). In addition, FPPD (0.2 mM) was incubated with OA (1.0 mM)-induced L02 cells for 24 h, and the cells were detected by the same method.
2.9. Colocalization detection of FPPD and organelles
RAW264.7 cells or L02 cells were incubated with Cy5.5 labeled FPPD for 12 h. After that, the cells were stained with organelle specific fluorescent dyes according to the instructions. These organelle dyes include cell nucleus fluorescent dye (Hoechst 33342), lysosome fluorescent dye (LysoTracker Green DND-26), endoplasmic reticulum fluorescent dye (ER-Tracker Green), and mitochondrial fluorescent dye (MitoTracker Green FM).
2.10. WB tests
Cell samples: Appropriate amount of cells were digested and transferred to 1.5 mL eppendorf (EP) tubes, washed twice with PBS. Then 300 μL radioimmunoprecipitation assay buffer (RIPA, Beyotime, China) containing phosphatase and protease inhibitors was added, and the EP tubes was incubated on ice for 40 min. After that, centrifuge the tubes at 12,000 g and 4 °C for 10 min, and take the supernatant into a new tube.
Tissue samples: Put 50 mg of liver tissue into 1.5 mL EP tubes, washed with tris buffered saline (TBS), and 300 μL RIPA containing phosphatase and protease inhibitors was added. The tissue was homogenized and crushed by ultrasonic cell crusher. After incubating on ice for 1 h, centrifuge the tubes at 12,000 g and 4 °C for 10 min, and take the supernatant into a new tube.
WB detection: The protein concentration of the samples was quantified by a BCA protein assay kit (Beyotime, China). Proteins were separated by polyacrylamide gel electrophoresis. The protein was mixed with loading buffer and heated at 95 °C for 5 min. Each sample was loaded with 20 μg of total protein per small gel well. The gel was immersed in electrophoresis buffer. Then, we ran the gel and stopped when the dye reached the bottom of the gel. Next, we transferred the protein from the gel to a polyvinylidene difluoride membrane (Millipore, USA) by wet transfer. After transfer, we blocked the membrane with 5 % BSA for 1 h at 4 °C under stirring. We then incubated with the primary antibody overnight at 4 °C at the default dilution. The next day, we washed the membranes using TBST buffer solution and incubated them with secondary antibodies for 1 h at 4 °C at the default dilution. After another wash, proteins were visualized by electrochemiluminescence (Milliboll, USA) and imaging systems (Tanon 4200, Tanon, China). Gray level analysis was performed on gel image System version 4.00 (Tanon, China).
2.11. Observation of tissue and subcellular organelles
After co incubating L02 cells with FPPD (0.2 mM) for 3 h, the cells were collected, and fixed with 2.5 % glutaraldehyde solution. The fixed L02 cells were immersed in 1 % osmium tetroxide for 2 h, then dehydrated with a series of ethanol solutions with increasing concentrations, and embedded in SPI-Pon812 resin. The slices were made into ultra-thin sections with a thickness of 70 nm, stained with uranyl acetate and lead citrate, and observed by transmission electron microscope.
2.12. Cellular mitochondrial function
The L02 cells were incubated with 1.0 mM OA, 0.2 mM FP or 0.2 mM FPPD for 24 h. Then, removing the medium, the cells were washed twice with PBS and treated with Hoechst 33342 and JC-1 kit (Beyotime, China). The samples were then imaged with a confocal laser scanning microscope (FV1000-IX81, Olympus, Japan). Fluorescence intensity analysis was performed by Image-Pro Plus 6.0 (Media Cybernetics, USA).
The NAD+/NADH kit and malondialdehyde (MDA) kit were purchased from Beyotime (Shanghai, China), the mitochondrial ROS specific fluorescence probe, and the activity for the mitochondrial complex I kit was purchased from Solarbio (Beijing, China). The L02 cells were incubated with 1.0 mM OA, 0.2 mM FP or 0.2 mM FPPD for 24 h. The cells were then collected and washed with PBS and analyzed according to the manufacturer's instructions.
2.13. In vivo ROS elimination
The L02 cells were incubated with 1.0 mM OA, 0.2 mM FP or 0.2 mM FPPD for 24 h. After that, we collected the cells and stained them with DCF diacetate (DCF-DA) and Nile Red for 30 min. Then, the cells were washed twice with PBS buffers. laser scanning microscope (FV1000- IX81, Olympus, Japan) and enzymoleter (Bio-RAD, iMark, USA) were used for detection.
2.14. In vivo fluorescence imaging
Cy5.5 labeled FPPD was obtained through self-assembly of Cy5.5, FP, and PPD at room temperature. Simply, Cy5.5, FP (64 mg) and PPD (80 mg) were dissolved in 1 mL DMSO, and the solution was slowly added to 6 mL deionized water at the rate of 50 μL/min under stirring. The mixture was stirred at room temperature for 24 h and then re-dissolved in deionized water after dialysis and freeze-drying.
The OB mice (ob/ob mice, are mice with homozygous mutations in the Leptin gene) were randomly divided into 3 groups (n = 4) and injected with normal saline, free Cy5.5 (100 μg/kg), and FPPD-Cy5.5 (The fluorescence intensity is the same as free CY5.5) once, respectively. The heart, liver, spleen, lung and kidney were harvested 30 h after injection. Another group of mice were anesthetized 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 3 d, 5 d, and 7 d after injection, and the biological distribution of FPPD-Cy5.5 (excitation wavelength: 675 nm; emission wavelength: 720 nm) was observed.
2.15. Animal models and treatments
Male obese mice (5 weeks of age) and C57BL/6J lean littermates (5 weeks of age) were purchased from Beijing Huafukang Biotechnology Co., LTD. (Beijing, China). All mice were reared in a temperature-controlled environment with dark and light cycles for 12 h. In addition, all mice were fed standard normal food purchased from Beijing Huafukang Biotechnology Co., LTD. (Beijing, China) and sterile water. All experimental programs involving live animals are reviewed and approved by the Animal Ethics Committee of the Institute of Chemistry, Chinese Academy of Sciences. The different concentrations of FPPD were adjusted to appropriate osmotic pressure by sodium chloride before treatment.
After feeding for 1 week, these OB mice were randomly divided into 3 groups. (1) Control group (C57): normal saline (200 μL) was injected intraperitoneally every day; (2) Model group: normal saline (200 μL) was injected intraperitoneally every day; (3) FP group: daily intraperitoneal injection of FP (0.01 mmol/kg); (4) FPPD group: daily intraperitoneal injection of FPPD (0.01 mmol/kg). The weight and feed weight of each group were recorded each 3 days. After 18 days of treatment, all mice were euthanized, and the blood, liver, adipose tissue, heart, spleen, lungs, and kidneys were taken. Blood was collected and centrifuged (80 rpm, 4 min) to collect serum. Liver and adipose tissue (perirenal, subcutaneous, and gonad) were weighed, rapidly frozen using liquid nitrogen, and stored at −80 °C. The liver, heart, spleen, lung and kidney of each mouse were fixed with 5 % paraformaldehyde, and part of the liver was fixed in 4 % glutaraldehyde for TEM observation.
2.16. Histopathology examination
H&E staining: Liver, heart, spleen, lung, and kidney tissues were dehydrated through successive alcohol gradients and embedded in paraffin blocks. The embedded tissue was cut into 5 μm thick slices, which were dewaxed, hydrated, stained, dehydrated and sealed by standard methods.
Oil red O staining: Fixed liver tissue was treated overnight with 15 % and 30 % sucrose solution. We then embedded the liver tissue into OCT (optimal cutting temperature compound) and sliced the embedded tissue into 6–8 μm slices using a cryotome. After that, the slices were washed, dyed, distinguished and sealed according to standard methods.
Oil red O staining and H&E staining were scanned with a scanner (NanoZoomer-SQ, Hamamatsu, Japan). Oil red O staining was performed by Image-Pro Plus 6.0 (Media Cybernetics, USA) for optical density analysis.
2.17. Biochemical analysis
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and triglyceride (TG) were detected by an automatic biochemical analyzer (TBA-2000FR, Toshiba, Japan) and corresponding assay kits (Nanjing Jiancheng Bioengineering Institute, China). The MDA was measured by an MDA assay kit (Nanjing Jiancheng Bioengineering Institute, China).
2.18. Routine blood examination
20 μL blood samples were obtained from each mouse for hematological analysis using an automated hematology analyzer (BC5000, Mindray, China).
2.19. Statistics analysis
Data were provided as means ± SD. Comparisons between two or several groups were analyzed using unpaired Student's t-test or one-way analysis of variance (ANOVA). The differences were considered significant for ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
3. Results and discussion
3.1. Preparation and characterization of FPPD
The synthesis and assembly of FPPD are shown in Fig. 1a. Firstly, we synthesized an electroneutral dicarboxy fullerene poly(ethylene glycol) molecule (FP) [30], and an acid-responsive polymer poly-(L-lactide-co-glycolide)-poly-etherimide-dimethylmaleic anhydride (PPD). Specifically, the FP molecule was obtained by covalently conjugating the mono-addition dicarboxy fullerene (DCF) derivative with PEG2000-NH2. Then, the PPD was prepared by connecting the polyetherimide (PEI) with poly (lactic-co-glycolic acid) (PLGA) [31]. Among them, the electropositive PEI was used to target the cellular mitochondria, and the PLGA offered the hydrophobic chains for self-assembly with FP. Before assembly, the excessive electropositive amino groups in PEI was shielded by dimethylmaleic anhydride (DMA) [32], which prevent the positive charge nano-assembly to adsorb on the cellular membrane. Subsequently, by self-assembling FP and FPPD, we obtained a charge reversal FPPD. Under normal physiological environment, DMA molecules modified in the outermost layer of FPPD will incubate the negative charge of nanoparticles. While in a slightly acidic environment, the amide bond between DMA and PEI will be broken, thus FP falls off from the nano-assembly. At this time, PEI in the outermost layer of FPP will incubate the positive charge of the nano-assembly. Thus, FPPD acquires the characteristic of charge reversal.
Fig. 1.

Preparation and characterization of FPPD. (a) The schematic diagram of the synthesis of FPPD. (b) FT-IR of FP, PPD and FPPD. (c) AFM image of the FPPD. Scale bar, 800 nm. (d) TEM image of the FPPD. Scale bar, 100 nm. (e) Hydrodynamic size distribution of FPPD (pH = 7.4). (f) Hydrodynamic size distribution of FPPD after surface charge inversion (pH = 6.0). (g) The schematic diagram of the surface charge inversion of FPPD. (h) Surface charges of FP, PPD and FPPD at pH = 7.4 (black), pH = 6.0 (red), and pH = 4.6 (blue). (i) Optical images of FPPD in water, PBS, FBS, and DMEM before (left) and after (right) centrifugation at 10,000 rpm for 10 min. (j) ESR spectra of •OH captured by DMPO after deionized water (Blank), FP and FPPD treatment. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001.
The molecular structures of FP, PPD and FPPD were characterized by Fourier transform infrared spectroscopy (FT-IR). As shown in Fig. 1b, PPD owned characteristic tensile vibration peaks of N(O)-H at 3400 cm−1 and C=O at 1750 cm−1. The characteristic peaks at 2900-2800 cm−1, 1600-1500 cm−1, 1410 cm−1 and 1200-1000 cm−1 in FP, assigned to C-H, C =C, C-C, and C-O stretching bands, respectively. While FPPD owned infrared signatures of both FP and PPD, proving that FPPD is assembled from FP and PPD. To accurately observe the morphology and size of FPPD, the nano-assembly were characterized using atomic force microscopy (AFM) and transmission electron microscopy (TEM) (Fig. 1c and d). AFM results showed that the average height and diameter of FPPD are ca. 15 nm and ca. 95 nm, respectively. TEM image showed that the FPPD were spherical, and the size was consistent with that of AFM. Further, the size distributions and zeta potentials of FP and FPPD were measured by dynamic light scattering (DLS). The average hydrodynamic diameter of the FP and FPPD are 183.4 ± 4.6 nm and 172.2 ± 1.9 nm (Fig. 1e and Fig. S2). A fter being treated under pH = 6.0 buffer solution, the average hydrodynamic diameter of the FPPD is 171.4 ± 2.7 nm (Fig. 1f). In deionized water, the surface charges of FP, PPD and FPPD are −0.05 mV, −8.79 mV and −1.34 mV, respectively. After being treated under pH = 6.0 or pH = 4.6 buffer solutions, the surface charge of FP, PPD and FPPD became −0.21 mV or −0.14 mV, 16.8 mV or 29 mV, and 12.2 mV or 16.4 mV, respectively. These indicated that the obtained FPPD nano-assembly was endowed the charge reversal ability in a weak acidic environment (Fig. 1g and h). In addition, the FPPD nano-assembly were also highly stable in water, phosphate buffered brine (PBS), fetal bovine serum (FBS), and Dulbecco's Modified Eagle Medium (DMEM), with no significant aggregation even after 10 min centrifuge at 10,000 rpm (Fig. 1i).
3.2. ROS scavenging performance of FPPD
Hydroxyl radical (•OH) is considered to be the most powerful ROS induced by lipid accumulation in hepatic steatosis. Here, we examined the ability of FPPD to eliminate •OH in vitro by electron spin resonance (ESR). H2O2 produces •OH when exposed to ultraviolet light (UV). These •OH are captured by 5, 5-dimethyl-1-pyrroline N-oxide (DMPO). Our tests showed that over 95 % •OH was eliminated by 50 μM FPPD. It also revealed that FP showed similar •OH elimination with the of FPPD (Fig. 1j), which demonstrated that the fullerene in FPPD nano-assembly had a highly efficient ROS capacity.
3.3. FPPD suppressed inflammation by quenching excess ROS in macrophages
In order to investigate the mitochondrial targeting ability of charge-reversed FPPD in macrophages, we inoculated RAW264.7 cells (mouse mononuclear macrophage leukemia cells) with Cy5.5 labeled FPPD for 12 h. After that, we stained the RAW264.7 cells with organelle specific fluorescent dyes including cell nucleus fluorescent dye (Hoechst 33342), lysosome fluorescent dye (LysoTracker Green DND-26), endoplasmic reticulum fluorescent dye (ER-Tracker Green), and mitochondrial fluorescent dye (MitoTracker Green FM). The confocal microscopy was used to observe the co-localization of FPPD with these organelles in RAW264.7 cells (Fig. 2a). We found that FPPD barely entered into the nucleus and the lysosome. Whereas, the FPPD could specifically distribute into endoplasmic reticulum as well as mitochondria, especially the latter. It showed that the co-localization curve of FPPD-Cy5.5 was highly consistent with that of mitochondrial fluorescent dyes (Fig. 2b), indicating that FPPD could target into mitochondria of the macrophages. Next, we tested the cytotoxicity and cytoprotection of FPPD in RAW264.7 cells by the cell counting kit-8 (CCK-8). Our results showed that FPPD had no significant toxicity towards RAW264.7 cells within the concentration range of 200 μM (Fig. S3). Then RAW264.7 cells were oxidatively damaged with media containing hydrogen peroxide (H2O2), and cultured with FPPD. The cell viability was further measured using CCK-8. The results showed that the cell viability was notably restored after FPPD administration with a concentration dependent manner (Fig. S4). In addition, we determined the anti-inflammation of FPPD. The RAW264.7 cells were treated with lipopolysaccharide (LPS), incubated with FPPD for 24 h, and then Western blot (WB) was used to detect the representative markers of M1-type and M2-type macrophages (Fig. 2c). Of note, we found that the protein expression of the markers of M1-type macrophages including inducible nitric-oxide synthase (iNOS) and CD86 were highly reduced by FPPD treatment. Further, our results also exhibited that the protein expression of Arginase 1 (ARG-1, the marker of M2-type macrophages) was markedly enhanced by FPPD compared with the controls. However, the FP showed weak regulatory effects on inflammasome and macrophage markers. Further, we detected the effects of FP or FPPD on the levels of ROS in LPS treated RAW264.7 cells. We found that the intracellular 2′,7′-dichlorofluorescein (DCF) fluorescent intensity was significantly reduced after FP or FPPD treatment (Figs. S5 and S6). These results indicated that FPPD could specifically distribute into mitochondria and remove excessive ROS to prevent oxidative injury and relieve inflammation in RAW264.7 cells.
Fig. 2.

Mitochondria-targeted FPPD suppressed macrophage inflammation by quenching excess ROS. (a) Co-localization images of FPPD and organelles in RAW264.7 cells. Scale bars, 50 μm (left) and 5 μm (right). (b) Co-localization curves of FPPD and organelles in RAW264.7 cells. (c) The protein expression of iNOS, CD86 and ARG-1 detected by WB. (d) Schematic diagram of the co-culture system for L02 cells treated with macrophages. (e) Confocal images of ROS and lipids in L02 cells treated with macrophages. Scale bars, 50 μm. (f) Relative lipid contents in L02 cells treated with macrophages. ∗, p < 0.05; ∗∗, p < 0.01.
To investigate whether the anti-inflammatory FPPD could further regulate lipid accumulation in steatosis hepatocytes, we established an oleic acid (OA) accumulation model of L02 cells (human hepatocytes). Firstly, to determine the optimal OA concentration, L02 cells were incubated with 0.5, 1.0, 1.5 and 2.0 mM OA for 24 h, then the proportion of viable cells, the lipid content, and the level of ROS of L02 cells were detected using Hoechst 33342, and Nile red, DCFH dyes, respectively. The results showed that when OA concentration was 1.0 mM, the levels of lipids in L02 cells were relatively high, and the number of living cells did not change significantly (Figs. S7 and S8). Thus, we chose the OA concentration as 1.0 mM for the following experiment.
Then we treated RAW264.7 cells with LPS for 24 h, replaced fresh media and incubated for another 24 h. The obtained culture medium of M1 macrophage was used to incubate with L02 cells with OA accumulation, and the lipid content of L02 cells was detected using confocal microscopy (Fig. 2d). The experiment results showed that the fluorescence signal of Nile red and DCF in L02 cells were remarkably reduced in L02 cells incubated with the culture medium of M1 macrophage pre-treated FPPD (Fig. 2e). Quantitative results showed that the Nile red signal in L02 cells after FPPD treatment was reduced by about 25 % (Fig. 2f). Together, these showed that mitochondria-targeted FPPD could inhibit the secretion of pro-inflammatory factors from the RAW264.7 cells, which further resulting in reducing the lipid content and ROS level of OA accumulation L02 cells.
3.4. FPPD inhibited lipid accumulation and reduced excess ROS in steatosis hepatocytes
We further detected the cellular distribution of FPPD in hepatocytes. Specifically, the L02 hepatic cells were incubated with Cy5.5-labeled FPPD and organelle fluorescent dyes. The co-localization of FPPD with organelles were observed by confocal microscopy. We also found that the fluorescence signal of FPPD was highly consistent with that of mitochondrial fluorescent dyes, with a highly consistent co-localization curve, indicating that FPPD could enter into the hepatocytes and effectively target the hepatocellular mitochondria (Fig. 3a and b). Next, we evaluated the effect of FPPD on lipid accumulation and ROS level in L02 hepatocytes in a lipid accumulation L02 cell model using confocal microscopy. Our results revealed that and the content of lipid dyed by Nile red the level of ROS dyed by DCF probe were observably boosted in the model group, which were notably reduced after FPPD treatment (Fig. 3c). In addition, to quantify investigate the lipid and ROS, the microplate reader was used to detect the fluorescent signals of Nile red and DCF (Fig. 3d and e). It showed that the fluorescence intensity of both DCF and Nile red were significantly reduced after FPPD treatment, which were superior than that of FP treatment. All of these results indicated that FPPD could specially target the mitochondria and reduce excess ROS as well as inhibit lipid accumulation in the steatosis hepatocytes.
Fig. 3.

FPPD inhibited lipid accumulation and reduced excess ROS in steatosis hepatocytes. (a) Co-localization images of FPPD and organelles in L02 cells. Scale bars, 50 μm (left) and 5 μm (right). (b) Co-localization curves of FPPD and organelles in L02 cells. (c) Confocal fluorescence images of Nile red and DCF in L02 cell treated by FPPD. Scale bars, 50 μm. (d) Relative lipid contents in L02 cells detected by microplate reader. L: low concentration; M: medium concentration; H: high concentration. (e) Relative ROS contents in L02 cells detected by microplate reader. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗∗, p < 0.0001.
3.5. FPPD improved the mitochondrial function in steatosis hepatocytes
To investigate the effect of FPPD on mitochondria in steastosis hepatocytes, we firstly used a mitochondrial ROS specific fluorescence probe to detect the regulation ability of FPPD on mitochondrial ROS in OA and palmitic acid (PA) treated L02 cells (Fig. 4a). Our results showed that the fluorescence intensity in L02 cells was highly increased after incubation with OA and PA. Of note, the levels of mitochondrial ROS were reduced almost to the normal level after FPPD treatment, which exhibited better regulation effect on mitochondrial ROS than FD. In addition, the quantitative mitochondrial ROS tests measured by the microplate reader were quite consistent with the above confocal results (Fig. 4b). Next, we measured the levels of MDA (a typical biomarker of lipid peroxidation) in hepatic steatosis L02 cells after FPPD treatment. Compared with the control group without OA administration, the level of MDA was remarkably increased in L02 cells treated with OA (Fig. 4c). Whereas, FPPD could significantly reverse the abnormity of MDA in steatosis L02 cells, better than FP.
Fig. 4.

FPPD improved the mitochondrial function in steatosis hepatic cells. (a) Confocal microscopy images of mitochondrial ROS. Scale bars, 50 μm. (b) The levels of mitochondrial ROS detected by microplate reader. (c) The contents of MDA in L02 cells. (d) Confocal microscopy images of JC-1 fluorescence probe. Scale bars, 50 μm. (e) The ratio of JC-1 fluorescence intensity (green/red). (f) The activity of mitochondrial complex I. (g) The contents of NAD+&NADH, NAD+ and NADH. (h) TEM images of L02 cells. Scale bars, 2 μm (top) and 1 μm (bottom). ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
Subsequently, we tested a range of indicators to evaluate the mitochondrial function in L02 cells induced by OA, including the mitochondrial membrane potential (MMP), the mitochondrial complex activity, and the contents of NAD+ & NADH. The stability of MMP is a prerequisite for intracellular ion transport, apoptosis regulation, ATP production, and other mitochondrial functions. Normally, the inner membrane potential of mitochondria is higher and the outer membrane potential is lower. The decrease of MMP indicates mitochondrial abnormalities and early apoptosis. We tested MMP in L02 cells using the JC-1 kit, and the reduction of MMP was represented by higher green color and lower red fluorescence. It revealed that a higher green/red ratio appeared in OA treated model group than that in the untreated control group (Fig. 4d and e). While, both of FP and FPPD could reduce the green/red ratio, particularly for FPPD. We also tested the mitochondrial complex I activity in OA-induced L02 cells. It showed that the activity of mitochondrial complex I was enhanced in model group, which was reduced by FP and FPPD treatment, especially the latter (Fig. 4f). Then, we analyzed the amounts of oxidation state NAD+ and NADH, which is produced during the cellular respiration and is involved in the mitochondria respiratory chain. Our results showed that the contents of NAD+&NADH, NAD+ and NADH were observably reduced after FPPD treatment compared with the model and FP treated groups (Fig. 4g). Moreover, the micro-structure of mitochondria in hepatocytes was observed by cellular TEM. The images showed that the abnormal hepatic mitochondria in the OA treated model group with membrane rupture, cristosis, matrix loss, and internal vacuoles (Fig. 4h). On the contrary, the mitochondrial structure integrity of FPPD treated group was more notably promoted than that in the FP treated group. These suggested that FPPD could remarkably improve the mitochondrial function by reducing mitochondrial ROS, lipid peroxidation, MMP, the mitochondrial complex I activity, NAD+ and NADH, inducing to normalize the lipid accumulation in OA induced steatosis hepatic cells.
3.6. Bio-distribution study of FPPD in OB mice
We used the in vivo fluorescence imaging to study the bio-distribution of FPPD in mice with hepatic steatosis. The ob/ob obesity (OB) mice were intraperitoneally injected with normal saline, Cy5.5-labeled FPPD (Cy5.5-FPPD) and free Cy5.5 and imaged using the IVIS live fluorescence imaging at 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 3 d, 5 d, and 7 d (Fig. 5a). A group of mice were sampled 30 h after administration to detect the fluorescence intensity in the organs (Fig. 5b). The results showed that Cy5.5-FPPD exhibited higher fluorescence intensity and longer retention time than free Cy5.5 in the liver, secondly in the kidney tissue, indicating that FPPD could be effectively targeted into mouse liver.
Fig. 5.

Bio-distribution study of FPPD in OB mice. (a) Fluorescence imaging in vivo of OB mice (Mice were injected with saline, free Cy5.5 and Cy5.5-FPPD from left to right). (b) Fluorescence imaging of mouse major organs ex vivo. ∗∗, p < 0.01.
3.7. FPPD ameliorated hepatic steatosis in obesity mice
To investigate the effect of FPPD on ameliorating hepatic steatosis in vivo, we selected OB mice as animal models for hepatic steatosis treatment studies. We set up (1) C57 mice as the control group: normal saline (200 μL) was intraperitoneally injected daily; (2) hepatic steatosis model group: normal saline (200 μL) was injected into OB mice daily; (3) FP treated group: FP (0.01 mmol/kg) was injected into OB mice daily; (4) FPPD treated group: FPPD was injected into OB mice daily (0.01 mmol/kg) (Fig. 6a). The body weights and food intakes of both C57 and OB mice were recorded each 3 days during the 18 days treatment (Fig. 6b). It showed that the average body weight of C57 mice and OB mice treated with normal saline increased by about 2 g and 8 g at the end of treatment, weight gain was reduced in OB mice treated with FP and FPPD and the weight loss was more pronounced in the FPPD group than in the FP group (Fig. 6c). In addition, FPPD treatment significantly reduced the ratio of liver weight to body weight and the adipose tissue (including subcutaneous fat, perirenal fat, and gonadal fat) weight to body weight in OB mice (Fig. 6d and e). The food intake of OB mice also decreased after FPPD treatment (Fig. 6f). Importantly, we found that the liver of OB mice was significantly enlarged and white, while the appearance of the liver of mice treated with FPPD was well improved, with small size and no abnormal color, which was close to the normal liver of normal C57 mice and better than FP treatment (Fig. 6g). Of note, H&E staining sections of liver in OB mice showed extensive lipid droplets, hepatocyte vacuoles, and hepatocyte damage. These typical pathological changes of hepatic steatosis were improved and reversed after treatment with FPPD (Fig. 6h). In oil red O staining of liver sections, neutral lipid content in liver was reduced over 80 % after treatment with FPPD, respectively, compared with the model group (Fig. 6i and j). For common indicators of hepatic steatosis, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), two major biomarkers of liver function, returned to normal levels after FPPD treatment (Fig. 6k and l), and total serum TG was reduced after 18 days of treatment (Fig. 6m). In addition, we measured MDA levels in serum and liver of mice. Compared with C57 control group, the levels of MDA in the serum and liver were increased in model group, which were decreased in different levels after treatment with FP and FPPD (Fig. 6n and o), and FPPD had a better therapeutic effect. Then, H&E staining of major organs showed no significant damage to the heart, spleen, lung, and kidney in the FPPD treatment group (Fig. S9). Additionally, the main blood routine indicators of mice had no significant change after FPPD treatment (Fig. S10), indicating that FPPD exhibited high security. In conclusion, our results confirmed that FPPD had a superior anti-hepatic steatosis effect in OB mice.
Fig. 6.

Therapeutic effect of FPPD on hepatic steatosis in OB mice. (a) The schematic diagram of normal saline, FP and FPPD treatment (n = 8 in each group). (b) Changes in body weight of mice in each group during treatment. (c) Body weight gain of each group after 18 days of treatment. (d) Liver coefficients (liver weight/body weight) in each group after 18 days of treatment. (e) Adipose coefficient (adipose weight/body weight) of each group after 18 days of treatment. (f) Food intake increment of each group during treatment. (g) Photographs of acquired livers taken after 18 days of treatment. (h) H&E staining section images of liver of mice in each group after treatment. Scale bars, 250 μm (top) and 100 μm (bottom). (i) Oil red staining section images of liver of mice in each group after treatment. Scale bars, 250 μm (top) and 100 μm (bottom). (j) Quantitative determination of oil red staining of liver of mice in each group after treatment. (k) Serum ALT content in each group after 18 days of treatment. (l) Serum AST content of mice in each group after 18 days of treatment. (m) Serum TG content of each group after 18 days of treatment. (n) Serum MDA content in mice. (o) Hepatic MDA content in mice. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001.
3.8. FPPD inhibited inflammation and promoted lipid transport in OB mice
We then used immunofluorescence to test the effect of FPPD on the phenotype of macrophages in the mouse liver. Specifically, we stained frozen sections of mouse liver tissue with F4/80 and CD11c fluorescent antibodies. F4/80 fluorescent antibodies with red fluorescence labeled macrophages, and the M1-type macrophages were jointly labeled by F4/80 and CD11c. From the immunofluorescence images, we found that the number of M1 inflammatory macrophages in the liver of mice treated with FPPD nano-assembly is significantly reduced compared to the untreated group (Fig. 7a). In addition, we also detected the levels of inflammatory factors in serum of mice and culture medium supernatant of M1 macrophages after FPPD treatment, and the results showed that the levels of IFN-γ and IL-6 in serum and cell supernatant were significantly decreased, which was consistent with the results of immunofluorescence (Fig. S11). Subsequently, we observed the microstructure of hepatocyte mitochondria in liver tissue by cellular TEM. The results showed that FPPD repaired cellular mitochondrial abnormalities in liver tissue and improved mitochondrial structural integrity, which was consistent with the results of previous cell experiments (Fig. 7b). In order to further clarify the molecular biological mechanism of FPPD in the treatment of liver steatosis, we used WB to detect the expression levels of key proteins related to lipid catabolism and transport (Fig. 7c). We found that Plin2 protein, which is closely related to the intracellular storage of lipids, was significantly increased in the liver of mice in model group and decreased after treatment with FPPD. The down-regulation of Plin2 in FPPD group indicated that the accumulation of lipids in hepatocytes was decreased after treatment with FPPD. Microsomal triglyceride transfer protein (MTTP), a transfer protein in microsomes on the endoplasmic reticulum, plays an important role in lipid metabolism, which is involved in the transport and synthesis of lipids. Apolipoprotein B 100 (ApoB 100), is mainly used to transport triglyceride. Compared with control group, the levels of MTTP and lipid transporters ApoB 100 protein in liver of model group were down-regulated, and the expressions of MTTP and ApoB 100 proteins in liver of FPPD group were increased. Notably, FPPD exhibits higher changes in the above proteins than FP. In the cell experiment, we found that after FPPD incubation, the level of triglyceride in the cells decreased, while the level of triglyceride in the medium increased, which confirmed the outward transport of triglyceride (Fig. S12). These results indicated that FPPD reduced macrophage inflammation and promoted the outward transfer of liver lipids, contributing to reverse hepatic steatosis.
Fig. 7.

FPPD inhibited inflammation and promoted lipid transport in OB mice. (a) Immunofluorescence straining with antibodies against F4/80 and CD11c in liver tissues. Scale bars, 100 μm. (b) TEM images of liver mitochondria after FPPD treatment. Scale bar, 2 μm (left) and 1 μm (right). (c) The protein expressions of Plin2, MTTP, and ApoB 100 in mouse liver detected by WB. ∗, p < 0.05; ∗∗, p < 0.01.
4. Conclusion
In this study, we explore a charge reversal and antioxidative nano-assembly that inhibiting inflammation and promoting lipid transport for hepatic stestosis treatment in obesity mice. We demonstrate that the charge reversal nano-assembly appeares a positive charge under an acid condition resulting in targeting the cellular mitochondria of macrophages and hepatocytes. By quenching excessive ROS, the antioxidative nano-assembly significantly reduces the over-activated macrophage inflammation and boosts lipid transport in vitro. Importantly, this nano-assembly achieves an efficient treatment effect on hepatic stestosis in obesity mice. Our strategy provides a highly effective means to mitigate hepatic stestosis by resolving inflammation and improving lipid metabolism disorder.
CRediT authorship contribution statement
Haoyu Wang: Writing – original draft, Methodology, Investigation, Data curation, Conceptualization. Sheng'e Su: Methodology, Investigation, Data curation. Xin An: Validation, Methodology, Investigation, Data curation. Yuan Xu: Validation, Methodology, Investigation. Jiacheng Sun: Validation, Formal analysis, Data curation. Mingming Zhen: Writing – review & editing, Writing – original draft, Supervision, Formal analysis, Data curation, Conceptualization. Chunru Wang: Writing – review & editing, Resources, Project administration, Funding acquisition, Conceptualization. Chunli Bai: Supervision, Resources, Funding acquisition, Conceptualization.
Ethics approval and consent to participate
All mice were fed standard normal food purchased from Beijing Huafukang Biotechnology Co., LTD. (Beijing, China) and sterile water. All experimental programs involving live animals are reviewed and approved by the Animal Ethics Committee of the Institute of Chemistry, Chinese Academy of Sciences (ICCAS2022090101). All mice were purchased from Beijing Huafukang Biotechnology Co., LTD. (Beijing, China). All efforts were made to minimize the suffering of animals.
Declaration of competing interest
All the authors declare that there is no conflict of interest in this manuscript.
Acknowledgment
This work was supported by the National Key R&D Program of China (Grant No. 2022YFA1205900), the National Natural Science Foundation of China (52172055) and the Key Research Program of the Chinese Academy of Sciences (QYZDJ-SSW-SLH01). Mingming Zhen particularly thanks the Youth Innovation Promotion Association of CAS (2022036).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2024.11.023.
Contributor Information
Mingming Zhen, Email: zhenmm@iccas.ac.cn.
Chunru Wang, Email: crwang@iccas.ac.cn.
Chunli Bai, Email: clbai@cas.cn.
Appendix A. Supplementary data
The following is/are the supplementary data to this article.
References
- 1.Loomba R., Friedman S.L., Shulman G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184(10):2537–2564. doi: 10.1016/j.cell.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z.M., Zelber-Sagi S., Henry L., Gerber L.H. Lifestyle interventions in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2023;20(11):708–722. doi: 10.1038/s41575-023-00800-4. [DOI] [PubMed] [Google Scholar]
- 3.Dixon J.B., Bhathal P.S., O'Brien P.E. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121(1):91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 4.Woo Baidal J.A., Lavine J.E. The intersection of nonalcoholic fatty liver disease and obesity. Sci. Transl. Med. 2016;8(323) doi: 10.1126/scitranslmed.aad8390. 323rv321-323rv321. [DOI] [PubMed] [Google Scholar]
- 5.Díaz L.A., Arab J.P., Louvet A., Bataller R., Arrese M. The intersection between alcohol-related liver disease and nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2023;20(12):764–783. doi: 10.1038/s41575-023-00822-y. [DOI] [PubMed] [Google Scholar]
- 6.Kuraji R., Sekino S., Kapila Y., Numabe Y. Periodontal disease–related nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an emerging concept of oral-liver axis. Periodontol. 2021;87(1):204–240. doi: 10.1111/prd.12387. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kazankov K., Jørgensen S.M.D., Thomsen K.L., Møller H.J., Vilstrup H., George J., Schuppan D., Grønbæk H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019;16(3):145–159. doi: 10.1038/s41575-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 8.Tacke F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017;66(6):1300–1312. doi: 10.1016/j.jhep.2017.02.026. [DOI] [PubMed] [Google Scholar]
- 9.Sica A., Invernizzi P., Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59(5):2034–2042. doi: 10.1002/hep.26754. [DOI] [PubMed] [Google Scholar]
- 10.Toubal A., Kiaf B., Beaudoin L., Cagninacci L., Rhimi M., Fruchet B., da Silva J., Corbett A.J., Simoni Y., Lantz O., et al. Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat. Commun. 2020;11(1):3755. doi: 10.1038/s41467-020-17307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren L., Sun Y., Lu H., Ye D., Han L., Wang N., Daugherty A., Li F., Wang M., Su F., et al. Pro)renin receptor inhibition reprograms hepatic lipid metabolism and protects mice from diet-induced obesity and hepatosteatosis. Circ. Res. 2018;122(5):730–741. doi: 10.1161/CIRCRESAHA.117.312422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haas J.T., Staels B. Understanding lipid metabolism through hepatic steat-omics. Nat. Rev. Endocrinol. 2019;15(6):321–322. doi: 10.1038/s41574-019-0203-9. [DOI] [PubMed] [Google Scholar]
- 13.Schoeler M., Ellero-Simatos S., Birkner T., Mayneris-Perxachs J., Olsson L., Brolin H., Loeber U., Kraft J.D., Polizzi A., Martí-Navas M., et al. The interplay between dietary fatty acids and gut microbiota influences host metabolism and hepatic steatosis. Nat. Commun. 2023;14(1):5329. doi: 10.1038/s41467-023-41074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bozic M., Guzmán C., Benet M., Sánchez-Campos S., García-Monzón C., Gari E., Gatius S., Valdivielso J.M., Jover R. Hepatocyte vitamin D receptor regulates lipid metabolism and mediates experimental diet-induced steatosis. J. Hepatol. 2016;65(4):748–757. doi: 10.1016/j.jhep.2016.05.031. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L., Tian R., Yao X., Zhang X.J., Zhang P., Huang Y., She Z.G., Li H., Ji Y.X., Cai J. Milk fat globule–epidermal growth factor–factor 8 improves hepatic steatosis and inflammation. Hepatology. 2021;73(2):586–605. doi: 10.1002/hep.31277. [DOI] [PubMed] [Google Scholar]
- 16.Leavy O. Regulating ROS. Nat. Rev. Immunol. 2014;14(6) doi: 10.1038/nri3685. 357-357. [DOI] [PubMed] [Google Scholar]
- 17.El-Kenawi A., Ruffell B. Inflammation, ROS, and mutagenesis. Cancer Cell. 2017;32(6):727–729. doi: 10.1016/j.ccell.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 18.Liu J., Lu W., Shi B., Klein S., Su X. Peroxisomal regulation of redox homeostasis and adipocyte metabolism. Redox Biol. 2019;24 doi: 10.1016/j.redox.2019.101167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn S., Siddiqi M.H., Noh H.-Y., Kim Y.-J., Kim Y.-J., Jin C.-G., Yang D.-C. Anti-inflammatory activity of ginsenosides in LPS-stimulated RAW 264.7 cells. Sci. Bull. 2015;60(8):773–784. [Google Scholar]
- 20.Zhou C., Zhen M., Yu M., Li X., Yu T., Liu J., Jia W., Liu S., Li L., Li J., et al. Gadofullerene inhibits the degradation of apolipoprotein B100 and boosts triglyceride transport for reversing hepatic steatosis. Sci. Adv. 2020;6(37) doi: 10.1126/sciadv.abc1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chinnery P.F., DiMauro S. Mitochondrial hepatopathies. J. Hepatol. 2005;43(2):207–209. doi: 10.1016/j.jhep.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y., Tang B., Long L., Luo P., Xiang W., Li X., Wang H., Jiang Q., Tan X., Luo S., et al. Improvement of obesity-associated disorders by a small-molecule drug targeting mitochondria of adipose tissue macrophages. Nat. Commun. 2021;12(1):102. doi: 10.1038/s41467-020-20315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan X.-x., Xu M.-z., Leung E.L.-H., Jun C., Yuan Z., Liu L. ROS-responsive berberine polymeric micelles effectively suppressed the inflammation of rheumatoid arthritis by targeting mitochondria. Nano-Micro Lett. 2020;12(1):76. doi: 10.1007/s40820-020-0410-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He F., Huang Y., Song Z., Zhou H.J., Zhang H., Perry R.J., Shulman G.I., Min W. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J. Exp. Med. 2020;218(3) doi: 10.1084/jem.20201416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meçe O., Houbaert D., Sassano M.-L., Durré T., Maes H., Schaaf M., More S., Ganne M., García-Caballero M., Borri M., et al. Lipid droplet degradation by autophagy connects mitochondria metabolism to Prox1-driven expression of lymphatic genes and lymphangiogenesis. Nat. Commun. 2022;13(1):2760. doi: 10.1038/s41467-022-30490-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alpert N.M., Pelletier-Galarneau M., Petibon Y., Normandin M.D., El Fakhri G. In vivo quantification of mitochondrial membrane potential. Nature. 2020;583(7815):E17–E18. doi: 10.1038/s41586-020-2366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y., Zhang J., Tu Y., Zhu L. Potential-independent intracellular drug delivery and mitochondrial targeting. ACS Nano. 2022;16(1):1409–1420. doi: 10.1021/acsnano.1c09456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu N., Liu X., Zhong Y., Zhou Z., Piao Y., Miao L., Zhang Q., Tang J., Huang L., Shen Y. Esterase-activated charge-reversal polymer for fibroblast-exempt cancer gene therapy. Adv. Mater. 2016;28(48):10613–10622. doi: 10.1002/adma.201603095. [DOI] [PubMed] [Google Scholar]
- 29.Zhang P., Chen D., Li L., Sun K. Charge reversal nano-systems for tumor therapy. J. Nanobiotechnol. 2022;20(1):31. doi: 10.1186/s12951-021-01221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aroua S., Schweizer W.B., Yamakoshi Y. C60 pyrrolidine bis-carboxylic acid derivative as a versatile precursor for biocompatible fullerenes. Org. Lett. 2014;16(6):1688–1691. doi: 10.1021/ol500363r. [DOI] [PubMed] [Google Scholar]
- 31.Wang S., Zhang J., Wang Y., Chen M. Hyaluronic acid-coated PEI-PLGA nanoparticles mediated co-delivery of doxorubicin and miR-542-3p for triple negative breast cancer therapy. Nanomed.-Nanotechnol. 2016;12(2):411–420. doi: 10.1016/j.nano.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 32.Tang S., Meng Q., Sun H., Su J., Yin Q., Zhang Z., Yu H., Chen L., Gu W., Li Y. Dual pH-sensitive micelles with charge-switch for controlling cellular uptake and drug release to treat metastatic breast cancer. Biomaterials. 2017;114:44–53. doi: 10.1016/j.biomaterials.2016.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.