

Abstract
Background
Genome diagnostics is considered gold standard diagnostics for epidermolysis bullosa (EB), a phenotypically and genetically heterogeneous group of rare disorders characterized by blistering and wounding of mucocutaneous tissues. EB is caused by pathogenic variants in genes encoding proteins of the dermo‐epidermal junction. Accurate genetic diagnosis of EB is crucial for prognostication, counselling and precision‐medicine. Genome diagnostics for EB started in 1991 with the introduction of Sanger sequencing (SS), analysing one gene at a time. In 2013, SS was superseded by next‐generation sequencing (NGS), that allow for high‐throughput sequencing of multiple genes in parallel. Several studies have shown a beneficial role for NGS in EB diagnostics, but its true benefit has not been quantified.
Objectives
To determine the benefit of NGS in EB by systematically evaluating the performance of different genome diagnostics used over time based on robust data from the Dutch EB Registry.
Methods
The diagnostic performances of SS and NGS were systematically evaluated in a retrospective observational study including all index cases with a clinical diagnosis of EB in whom genome diagnostics was performed between 01 January 1994 and 01 January 2022 (n = 308), registered at the Dutch EB Expertise Centre.
Results
Over time, a genetic diagnosis was made in 289/308 (94%) EB cases. The diagnostic yield increased from 89% (SS) to 95% (NGS). Most importantly, NGS significantly reduced diagnostic turnaround time (39 days vs. 211 days, p < 0.001). The likelihood of detecting variants of uncertain significance and additional findings increased from 5% and 1% (SS) to 22% and 13% (NGS) respectively.
Conclusions
Our study quantifies the benefit of NGS‐based methods and demonstrate they have had a major impact on EB diagnostics through an increased diagnostic yield and a dramatically decreased turnaround time (39 days). Although our diagnostic yield is high (95%), further improvement of genome diagnostics is urgently needed to provide a genetic diagnosis in all EB patients.
INTRODUCTION
Epidermolysis bullosa (EB) comprises a heterogeneous group of rare genetic disorders featuring mechanical fragility, blistering and wounding of mucocutaneous tissues. 1 The prevalence of EB in the Netherlands has recently been reported to be 22.4 per million people. 2 Four major types of EB are recognized, depending on the level of skin blistering: EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler EB (KEB). The major EB types are further subdivided into >30 EB subtypes based on clinical features and the specific gene variant(s) involved. 1 To date, 16 genes have been found to underlie the different EB subtypes, all encoding structural proteins of the dermo‐epidermal junction. 1 It is essential to know the disease‐causing gene variants in EB for (early) prognostication of the disease course, genetic counselling and future precision‐medicine approaches. 3 Although microscopic analysis on skin biopsies can provide important clues to the diagnosis, genome diagnostics is currently considered the gold standard method in EB diagnostics.
Over the past three decades, genome diagnostics for EB has witnessed many changes. Sanger sequencing (SS)‐based methods (‘first‐generation sequencing’) for EB were introduced in the early 90s and implemented in diagnostics at the Dutch EB Expertise Centre (EB Centre) in 1994. 4 , 5 SS is based on PCR amplification of specific protein‐coding regions in genomic DNA, followed by Sanger sequencing of the amplicons. 3 , 6 In EB, SS was usually initiated after identification of a candidate gene by immunofluorescence microscopy and transmission electron microscopy studies on skin biopsies. However, these genetic test results do not always reveal a candidate gene, leaving a number of EB cases genetically unsolved, and the many amplicons and genes that had to be sequenced frequently led to diagnostic delays.
Overcoming these shortcomings, next‐generation sequencing (NGS) for EB was introduced in 2013. 7 NGS refers to a collection of methods that share the ability to sequence multiple genes in parallel. In March 2014, targeted panel sequencing (TPS) was implemented in our EB Centre. TPS captures the protein‐coding regions of genes of interest from genomic DNA, and these regions are subsequently amplified and sequenced in parallel. In 2017, TPS was replaced by whole‐exome sequencing (WES), 4 years after its first reported use for genodermatoses. 7 WES captures and sequences most of the protein‐coding regions of the genome, with analysis then bioinformatically restricted to the regions of interest. If no causative variants are found in the initial analysis, WES offers the opportunity to scrutinize other regions of the exome by altering the bioinformatic data analysis strategy.
Earlier studies demonstrated the benefit of NGS for elucidating genetically unsolved EB cases via the discovery of sequence variants in new EB genes, and these studies showed a higher diagnostic yield (DY) and shorter turnaround time (TAT). 8 , 9 , 10 , 11 , 12 , 13 , 14 NGS‐based methods are thus generally perceived as hugely beneficial in current EB diagnostics. However, no studies have been performed to thoroughly assess the true power of NGS. In this study, we aimed to unveil the diagnostic power of NGS‐based methods for EB by systematically analysing its performance compared to first‐generation methods.
MATERIALS AND METHODS
Design and study population
In this retrospective observational study, we included all index patients with a certain clinical diagnosis of EB (n = 308) in whom EB genome diagnostics was performed between 01 January 1994 and 01 January 2022 at the Dutch EB Centre (source: Dutch EB Registry). The study was conducted in accordance with the Declaration of Helsinki and approved by the UMCG Medical Ethical Committee (2023/386).
We also included patients with suprabasal EBS because these phenotypes were part of the EB classification until 2020; they are currently classified among the EB‐related skin fragility disorders. 1 , 15 To explore the WES EB gene‐panel's ability to detect pathogenic variants in patients with a less certain clinical diagnosis of EB, or with disorders with overlapping features, we also investigated these test results.
Genome diagnostics
To understand the diagnostic value of NGS, we compared the performances of the evolving genome diagnostic methods over the years. For this study, we recognized two different genome diagnostic methods: SS‐based methods, including pre‐screening methods, and NGS‐based methods, specifically TPS and WES. From 1994 to 2014, SS was validated for each newly identified EB gene. In the early years, DNA materials were sent to foreign expertise centres for SS gene analysis.
As several genodermatoses exhibit overlapping clinical features with EB, our NGS EB gene panels have always included more than the current 16 EB genes. In 2014, our first TPS EB gene panel was validated by applying it to 20 EB cases with known pathogenic variants, and all but one large intragenic deletion was detected (data not shown). The contents of the EB gene panels are revisited twice annually and updated based on new genetic findings in the literature (Table S1).
Definitions
We used DY and TAT as primary indicators of diagnostic performance. DY was calculated as the percentage of cases in whom (likely) pathogenic gene variant(s) that explain the EB phenotype were found. TAT is the duration of a genetic analysis (time period between start and end dates). In case of multiple consecutive analyses, the cumulative TAT (cTAT) was used (time periods between analyses were excluded).
Variant classification
Gene variants were classified (retrospectively for SS) by the clinical laboratory geneticist according to the ACMG‐AMP variant classification guideline as: benign (B), likely benign (LB), variant of uncertain significance (VUS), likely pathogenic (LP) or pathogenic (P). 16 LP/P variants in genes that did not explain the EB phenotype were classified as additional findings (AF). 17 To investigate the extent of AF and VUSs, we compared the occurrence of both between NGS and SS.
Statistical analysis
Statistical analyses were performed using SPSS (v.28.0; SPSS Inc., Chicago, IL, USA). Diagnostic characteristics were calculated using descriptive statistics (median [IQR]) and compared using a chi‐square (X 2) test or Mann–Whitney U test. Missing values were excluded from relevant calculations.
RESULTS
Genetic diagnosis
Over the past three decades, technical improvements and discovery of new EB genes led to a genetic diagnosis in increasing numbers of EB cases (Figure 1), with 94% (289/308) of clinically diagnosed EB cases ultimately receiving a confirming genetic diagnosis.
FIGURE 1.
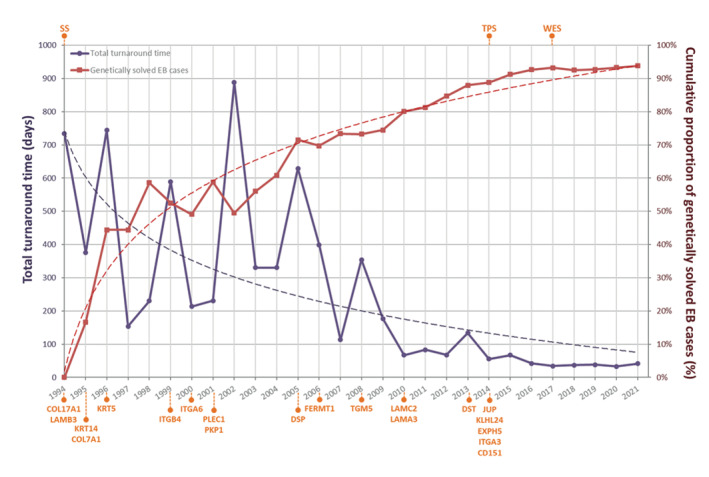
Evolution of genome diagnostics at the Dutch EB Expertise Centre from 1994 to 2022. Figure shows the cumulative yield and turnaround time of genome diagnostics for epidermolysis bullosa (EB). Turnaround time decreased from over 700 days in 1994 to 36 days in 2022, while the cumulative proportion of genetically solved cases with a clinical diagnosis of EB increased to 94% (289/308). X‐axis: Years indicate 31 December of the respective years. Abbreviations at top indicate when different diagnostic methods were implemented. Gene names at bottom indicate when these genes were implemented in SS or NGS diagnostics. Dashed lines indicate logarithmic trend‐lines following both data sets. EB, epidermolysis bullosa; SS, Sanger sequencing; TPS, targeted panel sequencing; WES, whole‐exome sequencing.
Diagnostic yield
We first compared the DY of the different diagnostic methods as a primary performance indicator. From February 1994 to mid‐2014, 240 cases with a certain clinical diagnosis of EB were analysed with SS‐based methods (SS group). SS led to a genetic diagnosis in 89% (214/240) of these EB cases, leaving 11% (26/240) genetically unsolved and 42% (11/26) of these unsolved EB cases were later solved with NGS (Figure 2; Table S2). In more than half (117/214) of the SS group, screening of the first candidate EB gene resulted in a genetic diagnosis. In the remaining 45% (97/214), multiple EB genes needed to be analysed to detect the pathogenic variant(s) (median 2.0, IQR 1.0).
FIGURE 2.
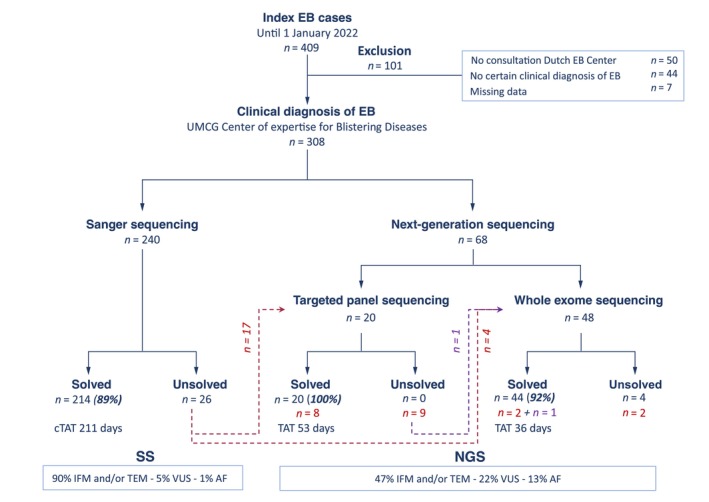
Overview of genome diagnostics for epidermolysis bullosa from 1994 to 2022. Flow chart showing the diagnostic yield and turnaround times for SS‐ and NGS‐based methods in cases with a certain clinical diagnosis of epidermolysis bullosa (EB) at the Dutch EB Expertise Centre during the study period 1994–2022 (n = 308). Red dashed lines and red numbers indicate patients genetically unsolved with SS who were later re‐analysed using TPS or WES. Purple dashed line and purple numbers indicates one patient who was genetically unsolved with SS and TPS who was re‐analysed with WES. (c) TAT, (cumulative) turnaround time; EB, epidermolysis bullosa; AF, additional finding(s); IFM, immunofluorescence microscopy; TEM, transmission electron microscopy; VUS, variant(s) of uncertain significance.
Since the introduction of NGS at our EB Centre in March 2014, NGS has been performed in 90 EB cases, and 68 received NGS as the first tier of diagnostics (Figure 2). In 22 other EB cases, NGS was performed after SS failed to provide a genetic diagnosis.
TPS was performed as initial tier of diagnostics in 20 EB cases, leading to a genetic diagnosis in all. TPS was further performed in 17 unsolved EB cases from the SS group and revealed the pathogenic variant(s) in eight cases (47%). Interestingly, in 5/8 of these EB cases, the genetic diagnosis matched a different EB type than clinically suspected (atypical presentation). In another 2/8 EB cases, the variants resided in genes analysed with SS but apparently missed for technical reasons. In the one remaining case, the variants resided in EXPH5, a gene that was only discovered after the SS era.
From May 2017 until January 2022, 48 EB cases were analysed using WES as initial tier, which solved the diagnosis in 92% (44/48) of EB cases (Figure 2). Given the similarities in capturing between TPS and WES, the higher DY for the TPS cases is most likely coincidental and due to the small number analysed (n = 20). WES was further performed in five unsolved EB cases from the SS group, enabling a genetic diagnosis in three (60%). In one EB case, the pathogenic variants were located in EXPH5. In another, an intronic splice variant in COL7A1 was detected that had been missed by SS. In a third unsolved SS case, WES uncovered a pathogenic variant in KLHL24 that had also been missed by TPS because KLHL24 was not part of the first TPS‐panel design (Table S1). 18 Four unsolved SS cases were not available for follow‐up screening by NGS methods. Therefore, the total DY in our cohort of EB cases who received state‐of‐the‐art diagnostics was 95% (289/304).
Turnaround time
To measure the effect of NGS in accelerating genome diagnostics, we compared the (cumulative) TAT between the different diagnostic methods as a primary indicator of diagnostic performance. The median cTAT for all genetically solved EB cases (n = 214) in the SS group was 211 days (IQR 464), although this was already declining during the SS era (Figure 1). As screening of the first candidate gene led to a genetic diagnosis in more than half of these EB cases (117/214), the TAT was ‘only’ 133 days for this group (IQR 350). However, in the remaining 45% (97/214), multiple genes needed to be sequenced, which prolonged the cTAT to 348 days (IQR 484). For TPS as initial tier diagnostics, we calculated a median TAT of 53 days (IQR 46). With the arrival of WES, TAT is now 36 days in genetically solved EB cases (IQR 16). Therefore, NGS decreased TAT more than five times (39 days, p < 0.001).
Additional findings
In 13/240 (5%) EB cases in the SS group, a single VUS was found (Table S3), and in 4/13 EB cases (31%), this VUS was later reclassified as (L)P based on additional genetic studies. In only one of the remaining nine cases was the causative variant not identified, leaving it open whether the identified VUS is the disease‐causing variant. In the other 8/9 EB cases, the VUS was found in addition to causative gene variants, and the question remains whether these VUSs play a disease‐modifying role. Furthermore, AF were found in only 3/240 (1%) EB cases in the SS group (Table S3).
VUSs were found in 20/90 (22%) EB cases in the NGS‐group, including 27 VUSs in total (≥2 VUSs in five cases) (Table S3). This proportion is more than four times higher than during the SS era. Of these EB cases, 16/20 (80%) were genetically solved. In 4/16, the VUS was found in one of the candidate EB genes, and two were later reclassified as (L)P (considered solved). Two other VUSs were located in the same EB gene as the pathogenic variant, but it remains open whether these two variants contribute to the EB phenotype. The VUS was found in another EB gene in 7/16, in both an EB gene and an EB‐related gene in another two and in an EB‐related gene in the remaining three. It cannot be ruled out that these VUSs play a disease‐modifying role. In the remaining 4/20 EB cases in whom VUSs were found, NGS detected no pathogenic variant(s). The question is whether these VUSs explain the EB phenotype, or are clinically unrelated. Finally, AF considered to be unrelated to the EB‐phenotype were found in 12/90 (13%) NGS EB cases (Table S3).
Pseudogenes
A well‐known challenge in genome diagnostics is the risk of mistaking sequence variants in non‐functional homologous sequences (pseudogenes) for variants in the functional genes, especially KRT14. 19 , 20 , 21 In the SS era, this issue was covered by designing specific primers to only amplify the functional genes. Our question was thus whether NGS‐based methods are sensitive enough to discriminate between functional genes and their pseudogenes. In all our KRT14 cases (n = 38), it was possible to distinguish between variants in KRT14 and its pseudogenes based on other variants specific for KRT14 or its pseudogenes detected in cis or trans with the variant of interest (Figure S1). This highlights that the sequence depth of NGS methods is sufficiently high and the data analysis pipeline sufficiently robust to correctly interpret sequence variants in the presence of pseudogenes.
Mutation detection rate
Finally, we investigated the mutation detection rate (percentage of tests requested in which causative mutations were detected) and diagnostic accuracy (percentage of positive test results precisely matching the request reasons). Here, we took the perspective of the Genome Diagnostics laboratory and compared all test results to the request reasons given on the request forms (Figure S2). From the implementation of WES‐based EB diagnostics in October 2017 up until January 2022, our laboratory received 74 internal requests. In 50/74 internal requests, pathogenic variants were found that explained the EB phenotypes (68% mutation detection rate), and the results matched the request reasons exactly in 46 (92% diagnostic accuracy). However, when looking more closely at these results, the mutation detection rate and diagnostic accuracy were much higher in cases with a clinically certain EB diagnosis (88% [46/52 requests] and 93% [43/46] respectively) than in cases with a less certain clinical diagnosis (18% [4/22] and 75% [3/4] respectively).
DISCUSSION
In this study, we aimed to unveil the diagnostic power of NGS‐based genome diagnostic methods for EB by systematically analysing their performance in a well‐characterized Dutch cohort of patients with a certain clinical diagnosis of EB. Over the past three decades, a genetic diagnosis could be made in more and more EB cases in an ever shorter period of time (Figure 1). Our most important finding is that the strength of NGS lies not only in its higher DY, but also in its significantly reduced TAT (39 days NGS vs. 226 days SS) (Figure 2).
Although cTAT was already decreasing during the SS era due to significant technical improvements, it declined considerably further with the arrival of NGS (Figure 1). This is undoubtedly because of the high‐throughput sequencing of multiple genes in parallel, which does not depend on the identification of a candidate gene using microscopic analyses on skin biopsies. Although analysis of the first candidate gene led to identification of the causative variant(s) in 55% of the SS group, multiple EB genes had to be sequenced in 45%, resulting in long diagnostic delays (cTAT 348 days), something we urgently intend to prevent because early prognostication is extremely important in EB diagnostics.
NGS further increased DY in our Dutch EB cohort to 95%. DY was already high during the 20‐year SS era (89%), but it is important to keep in mind that genome diagnostics were largely directed by the results of microscopic analyses on skin biopsies during that time. During the SS era, our EB Centre also saw relatively more patients with severe EB phenotypes, which makes it easier to make an accurate clinical diagnosis and detect the pathogenic variant(s). 2 An effect of phenotypic severity on DY is also suggested by the higher DY for DEB we found for SS (94%) compared to NGS (88%), which correlates to the fact that more dominant DEB cases were seen in recent years. We speculate that the dominant‐negative COL7A1 variants underlying dominant DEB are more frequently located in intronic sequences influencing splicing and therefore not detected by the current WES strategy (Table S2). Interestingly, the relatively low percentage of EB cases in the NGS group in whom microscopic analyses on skin biopsies were performed (47%) did not negatively influence the DY. In our opinion, this justifies a genetics‐first approach, which will become more feasible in routine diagnostic practice with increasing speed of genome diagnostics.
Additionally, NGS has proven effective when no candidate gene was available, as seen by NGS genetically solving 11 EB cases not solved by SS (Figure 2). In five of these EB cases, the pathogenic variant(s) were in different EB genes than initially suspected, strengthening the added value of an NGS‐based approach. In another three EB cases, NGS detected pathogenic variants in EB genes that were sequenced but missed with SS, probably due to insufficient coverage of intronic regions. In our opinion, the major benefit in TAT and the ability to detect gene variants that match EB (sub)types other than that clinically suspected, without diagnostic delays, makes massive parallel sequencing approaches like NGS the gold standard in EB diagnostics.
However, in 5% (15/304) of EB cases, NGS still failed to provide a genetic diagnosis (Figure 2). There could be several reasons for this. First, currently unknown EB genes may be involved. However, although new genes may play a role in a small subset of EB cases, the high DY in our study and others makes it unlikely that there are many new EB genes to be discovered. 7 , 11 , 12 , 13 , 14 , 22 , 23 As a major advantage of WES is the possibility to dig further into regions of the exome not included in the initial EB gene‐panel analysis, bioinformatic follow‐up strategies on existing WES data may eventually identify causative variants in novel genes in a subset of unsolved EB cases. 24 Second, pathogenic variants in regulatory sequences and deep intronic regions, as well as copy number variations, are expected to be missed with NGS (standard variant calling currently is ±20 intronic nucleotides). However, newer sequencing methods like whole‐genome sequencing and long‐read sequencing are expected to identify those variants as well. Third, low‐grade mosaic sequence variants could also be missed. 25 , 26 Finally, clinical diagnosis of EB may prove incorrect in a subset of unsolved EB cases, especially for relatively milder phenotypes that may, for instance, be multifactorial or autoimmune. 27 , 28
An issue reported in NGS literature is the challenge of clinical interpretation of VUSs and AF, which requires bioinformatic expertise and reproductive counselling. 13 , 14 Our results confirm that NGS has increased the likelihood of detecting additional sequence variants compared to SS (VUSs 22% vs. 5%; AF 13% vs. 1%) (Table S3). In the absence of pathogenic variants, VUSs are of particular interest, as additional studies found them to be the disease‐causing variant in six EB cases. However, VUSs in other genes raise extra challenges to the diagnostic process. This is also true for AF, for example, carriership of pathogenic variants in other genes, which may require reproductive counselling. The question remains whether these additional sequence variants play a modifying role in the EB phenotype. 20 , 21 As AF were found in 1% of EB cases with SS, one may argue that EB gene‐panels should only contain true candidate genes (Table S1).
CONCLUSION AND FUTURE PERSPECTIVES
This study demonstrates that NGS‐based approaches are powerful techniques in current EB diagnostics, providing genetic diagnoses with high sensitivity and a continuously decreasing TAT which is so crucial for prognostication and personalized therapies. It is likely that this power of NGS can be extended to other heterogeneous genetic disorders.
As the DY of WES is not 100%, there is still need for further improvement of current EB diagnostics. Deep‐phenotyping, developing and implementing more powerful techniques (e.g. [long‐read] whole‐genome sequencing, RNA‐sequencing) and improving bioinformatic data analysis algorithms are all crucial to identify the causative variants in EB patients who are still genetically unsolved.
FUNDING INFORMATION
This study was funded by a clinical research fellowship grant from DEBRA Netherlands to RB and research grants from Stichting Vlinderkind (Dutch Butterfly Child Foundation) to MCB and PCvdA.
CONFLICT OF INTEREST STATEMENT
None declared.
ETHICAL APPROVAL
This study was performed with routinely collected healthcare data for the evaluation of genome diagnostics at the Dutch EB Expertise Centre. The study was conducted in accordance with the Declaration of Helsinki and the Institutional Review Board of the UMCG confirmed that the study did not fall under the scope of the Dutch Medical Scientific Research with People Act (WMO) (2023/386). The objection registry was checked for all included cases.
Supporting information
Data S1
ACKNOWLEDGEMENTS
We thank our patients with EB and their families for their trust and cooperation in the Dutch EB Expertise Centre. We also thank our colleagues from the UMCG Genetics Laboratory for their continuing support in finding the genetic diagnosis in EB patients over the past three decades, further improvement of genome diagnostics and help with this study. Furthermore, we thank late professor Marcel Jonkman for his tremendous contribution to the current knowledge and diagnostics in EB and for founding the Dutch EB Expertise Centre. Finally, we also thank Kate McIntyre, English editor, for her tremendous help with editing this article.
Baardman R, Lemmink HH, Yenamandra VK, Commandeur‐Jan SZ, Viel M, Kooi KA, et al. Evolution of genome diagnostics in epidermolysis bullosa: Unveiling the power of next‐generation sequencing. J Eur Acad Dermatol Venereol. 2025;39:154–160. 10.1111/jdv.19938
Linked article: A. Klausegger et al. J Eur Acad Dermatol Venereol 2025;39:19–20. https://doi.org/10.1111/jdv.20433.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner‐Tuderman L, Diem A, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614–627. [DOI] [PubMed] [Google Scholar]
- 2. Baardman R, Yenamandra VK, Duipmans JC, Pasmooij AMG, Jonkman MF, van den Akker PC, et al. Novel insights into the epidemiology of epidermolysis bullosa (EB) from the Dutch EB registry: EB more common than previously assumed? J Eur Acad Dermatol Venereol. 2021;35:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Has C, Liu L, Bolling MC, Charlesworth AV, El Hachem M, Escámez MJD, et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol. 2020;182:574–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van den Akker PC, Hettema W, Meijer R, Jonkman MF, Hofstra RMW, Scheffer H. Design and validation of a conformation‐sensitive capillary electrophoresis system for mutation identification of the COL7A1 gene with automated peak comparison. Genet Test Mol Biomarkers. 2009;13:589–597. [DOI] [PubMed] [Google Scholar]
- 5. Coulombe PA, Hutton ME, Letai A, Hebert A, Paller AS, Fuchs E. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: genetic and functional analyses. Cell. 1991;66:1301–1311. [DOI] [PubMed] [Google Scholar]
- 6. Harvey N, Youssefian L, Saeidian AH, Vahidnezhad H, Uitto J. Pathomechanisms of epidermolysis bullosa: beyond structural proteins. Matrix Biol. 2022;110:91–105. [DOI] [PubMed] [Google Scholar]
- 7. Takeichi T, Nanda A, Liu L, Salam A, Campbell P, Fong K, et al. Impact of next generation sequencing on diagnostics in a genetic skin disease clinic. Exp Dermatol. 2013;22:825–831. [DOI] [PubMed] [Google Scholar]
- 8. Lin Z, Li S, Feng C, Yang S, Wang H, Ma D, et al. Stabilizing mutations of KLHL24 ubiquitin ligase cause loss of keratin 14 and human skin fragility. Nat Genet. 2016;48:1508–1516. [DOI] [PubMed] [Google Scholar]
- 9. McGrath JA, Stone KL, Begum R, Simpson MA, Dopping‐Hepenstal PJ, Liu L, et al. Germline mutation in EXPH5 implicates the Rab27B effector protein Slac2‐b in inherited skin fragility. Am J Hum Genet. 2012;91:1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vahidnezhad H, Youssefian L, Saeidian AH, Mahmoudi H, Touati A, Abiri M, et al. Recessive mutation in tetraspanin CD151 causes kindler syndrome‐like epidermolysis bullosa with multi‐systemic manifestations including nephropathy. Matrix Biol. 2018;66:22–33. [DOI] [PubMed] [Google Scholar]
- 11. Takeichi T, Liu L, Fong K, McMillan JR, Salam A, Campbell P, et al. Whole‐exome sequencing improves mutation detection in a diagnostic epidermolysis bullosa laboratory. Br J Dermatol. 2015;172:94–100. [DOI] [PubMed] [Google Scholar]
- 12. Tenedini E, Artuso L, Bernardis I, Artusi V, De Rosa L, Contin R, et al. Amplicon‐based next‐generation sequencing: an effective approach for the molecular diagnosis of epidermolysis bullosa. Br J Dermatol. 2015;173:731–738. [DOI] [PubMed] [Google Scholar]
- 13. Has C, Kusel J, Reimer A, Hoffmann J, Schauer F, Zimmer A, et al. The position of targeted next‐generation sequencing in epidermolysis bullosa diagnosis. Acta Derm Venereol. 2018;98:437–440. [DOI] [PubMed] [Google Scholar]
- 14. Lucky AW, Dagaonkar N, Lammers K, Husami A, Kissell D, Zhang K. A comprehensive next‐generation sequencing assay for the diagnosis of epidermolysis bullosa. Pediatr Dermatol. 2018;35:188–197. [DOI] [PubMed] [Google Scholar]
- 15. Fine JD, Bruckner‐Tuderman L, Eady RAJ, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70:1103–1126. [DOI] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blackburn HL, Schroeder B, Turner C, Shriver CD, Ellsworth DL, Ellsworth RE. Management of Incidental Findings in the era of next‐generation sequencing. Curr Genomics. 2015;16:159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yenamandra VK, van den Akker PC, Lemmink HH, Jan SZ, Vermeer M, van den Berg MP, et al. Cardiomyopathy in patients with epidermolysis bullosa simplex with mutations in KLHL24. Br J Dermatol. 2018;179:1181–1183. [DOI] [PubMed] [Google Scholar]
- 19. Hut PH, Vlies P, Jonkman MF, Verlind E, Shimizu H, Buys CH, et al. Exempting homologous pseudogene sequences from polymerase chain reaction amplification allows genomic keratin 14 hotspot mutation analysis. J Invest Dermatol. 2000;114:616–619. [DOI] [PubMed] [Google Scholar]
- 20. Glász‐Bóna A, Medvecz M, Sajó R, Lepesi‐Benko R, Tulassay Z, Katona M, et al. Easy method for keratin 14 gene amplification to exclude pseudogene sequences: new keratin 5 and 14 mutations in epidermolysis bullosa simplex. J Invest Dermatol. 2009;129:229–231. [DOI] [PubMed] [Google Scholar]
- 21. Oldak M, Kowalewski C, Maksym RB, Woźniak K, Pollak A, Podgórska M, et al. Novel keratin 14 hotspot mutation in Dowling‐Meara type of epidermolysis bullosa simplex: strategy to avoid KRT14 pseudogene amplification by a simple approach. J Dermatol Sci. 2010;57:69–70. [DOI] [PubMed] [Google Scholar]
- 22. Mariath LM, Kiszewski AE, Frantz JA, Siebert M, Matte U, Schuler‐Faccini L. Gene panel for the diagnosis of epidermolysis bullosa: proposal for a viable and efficient approach. An Bras Dermatol. 2021;96:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen F, Huang L, Li C, Zhang J, Yang W, Zhang B, et al. Next‐generation sequencing through multigene panel testing for the diagnosis of hereditary epidermolysis bullosa in Chinese population. Clin Genet. 2020;98:179–184. [DOI] [PubMed] [Google Scholar]
- 24. Bolling MC, Jan SZ, Pasmooij AMG, Lemmink HH, Franke LH, Yenamandra VK, et al. Generalized ichthyotic peeling skin syndrome due to FLG2 mutations. J Invest Dermatol. 2018;138:1881–1884. [DOI] [PubMed] [Google Scholar]
- 25. Chen F, Deng D, Pan C, Yao Z, Gu Y, Li M. Detection and characterization of low‐level mosaicism among clinically unaffected parents of ‘sporadic’ epidermolysis bullosa simplex cases. Br J Dermatol. 2022;187:441–443. [DOI] [PubMed] [Google Scholar]
- 26. Natsuga K, Furuta Y, Takashima S, Nohara T, Kosumi H, Mai Y, et al. Detection of revertant mosaicism in epidermolysis bullosa through Cas9‐targeted long‐read sequencing. Hum Mutat. 2022;43:529–536. [DOI] [PubMed] [Google Scholar]
- 27. Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Sotoudeh S, Jazayeri A, et al. Next generation sequencing identifies double homozygous mutations in two distinct genes (EXPH5 and COL17A1) in a patient with concomitant simplex and junctional epidermolysis bullosa. Hum Mutat. 2018;39:1349–1354. [DOI] [PubMed] [Google Scholar]
- 28. Gong L, Liu C, Li Y, Xu X. Whole exome sequencing identified two point mutations of COL7A1 and FLG in a Chinese family with dystrophic epidermolysis bullous pruriginosa and ichthyosis vulgaris. J Dermatol. 2019;46:158–160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.