

Abstract
Chromosomal instability (CIN) generates micronuclei—aberrant extranuclear structures that catalyze the acquisition of complex chromosomal rearrangements present in cancer. Micronuclei are characterized by persistent DNA damage and catastrophic nuclear envelope collapse, which exposes DNA to the cytoplasm. We found that the autophagic receptor p62/SQSTM1 modulates micronuclear stability, influencing chromosome fragmentation and rearrangements. Mechanistically, proximity of micronuclei to mitochondria led to oxidation-driven homo-oligomerization of p62, limiting endosomal sorting complex required for transport (ESCRT)–dependent micronuclear envelope repair by triggering autophagic degradation. We also found that p62 levels correlate with increased chromothripsis across human cancer cell lines and with increased CIN in colorectal tumors. Thus, p62 acts as a regulator of micronuclei and may serve as a prognostic marker for tumors with high CIN.
INTRODUCTION:
A main feature of cancer cells is their high frequency of chromosome segregation errors, a condition known as chromosomal instability (CIN), which is associated with poor prognosis and chemoresistance. CIN leads to the formation of micronuclei—abnormal extranuclear bodies widely found in cancer cells.
The nuclear envelopes of micronuclei are often fragile and compromised, which causes irreparable ruptures and structural collapse. This exposes micronuclear DNA to the cytosol, resulting in DNA damage and extensive chromosomal rearrangements, which fuels genomic instability and cancer progression. In addition, rupture of the micronuclear membrane activates the cGAS-STING [cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS)–stimulator of interferon genes (STING)] pathway, triggering inflammatory responses that promote tumor invasion and metastasis.
RATIONALE:
Although the consequences of loss of micronuclei compartmentalization are well studied, the events leading to micronuclear envelope rupture and the mechanisms involved in their repair are less clear. Given the importance of those mechanisms in maintaining micronuclei integrity, we aimed to identify proteins and pathways that specifically modulate these events. We isolated micronuclei and primary nuclei, analyzed their protein composition using mass spectrometry, and identified p62/SQSTM1—a key autophagic pathway component—as specifically enriched in micronuclei compared with primary nuclei.
RESULTS:
We found that p62 localizes to micronuclei and is crucial for maintaining their integrity. The localization of p62 to micronuclei relied on its oxidation-driven homo-oligomerization, which was induced by reactive oxygen species (ROS) released by mitochondria proximal to micronuclei. We used electron tomography and quantitative imaging to analyze the proximity of mitochondria to micronuclei and found that p62-positive micronuclei were closer to mitochondria compared with those without p62. We tested how mitochondrial ROS affected p62 localization and found that reducing ROS decreased p62-positive micronuclei and prevented their rupture, whereas increasing ROS led to more p62-positive and ruptured micronuclei in both normal and cancer cells. Homo-oligomerization of p62 induced by ROS influenced micronuclear integrity by inhibiting the repair activity of the endosomal sorting complex required for transport–III (ESCRT-III), which is involved in envelope resealing. This control was achieved through the p62-mediated autophagic degradation of ESCRT components. The p62-dependent regulation of micronuclear integrity affected several features associated with micronuclei, such as chromosomal rearrangements and cGAS-STING–mediated inflammation. Furthermore, p62 levels correlated with chromothripsis status in cancer cells and tumor samples and were strongly linked to poor prognosis in colorectal cancer with high CIN.
CONCLUSION:
In this work, we identified a critical mechanism by which p62 regulates micronuclear stability by influencing envelope repair. Given p62’s frequent deregulation in tumors and its control over micronuclear envelope integrity, our findings suggest that p62-mediated effects on micronuclear repair activities could influence cancer development and progression.
Graphical Abstract
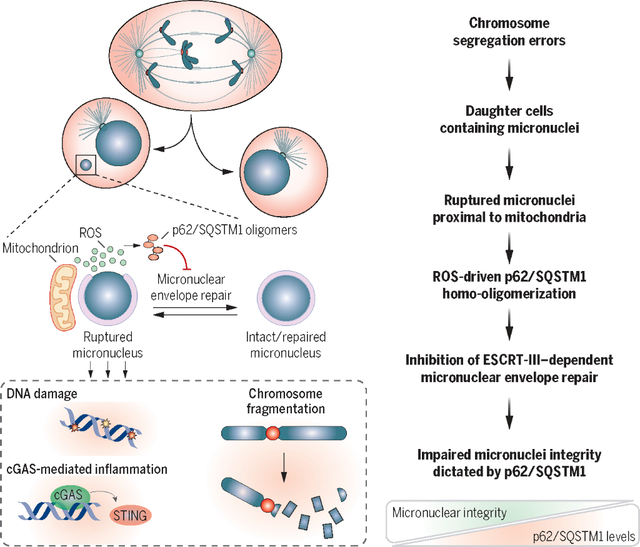
p62/SQSTM1 drives micronuclei collapse and complex chromosome rearrangements by restraining micronuclear envelope repair. Micronuclei-mitochondria proximity promotes ROS-mediated homo-oligomerization of p62/SQSTM1 through cysteine oxidation. ROS-induced p62 homo-oligomerization affects micronuclear integrity by inhibiting components of ESCRT-III repair through their autophagic degradation. This affects chromosomal rearrangements and cGAS-STING inflammation. High p62 levels correlate with chromothripsis and poor prognosis in colorectal cancer with high CIN.
Chromosomal instability (CIN) is a hallmark of cancer associated with poor prognosis and chemotherapeutic resistance (1–7). A main consequence of CIN is the generation of micronuclei (8–10)—aberrant extranuclear bodies that are a major feature of cancer cells and act as a hub for complex chromosomal rearrangements often found in tumors (11–14). Micronuclei are characterized by defective nuclear membranes and are responsible for events that can lead to mutagenesis and cancer development, including chromothripsis (11, 12, 15–17). Micronuclear envelopes exhibit reduced functionality and improper composition (18–21), acquiring unrepaired ruptures that eventually lead to irreversible collapse of the whole micronuclear structure (18, 22, 23). Disruption of the micronuclear envelope and the subsequent loss of compartmentalization result in impaired micronuclear functions (18–20, 24) and expose micronuclear DNA to the cytosol (18, 25–28). Because of this, micronuclear DNA acquires epigenetic abnormalities (18, 29, 30), persistent DNA damage, and fragmentation (16, 17, 31) as well as extensive chromosomal rearrangements that trigger genomic instability, thus fueling cancer genome evolution (9, 11, 15, 16, 32). Upon micronuclear membrane rupture, DNA is recognized by the cytosolic DNA–sensing pathway mediated by cGAS-STING [cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS)–stimulator of interferon genes (STING)], eliciting inflammatory responses that promote tumor invasion and metastasis (25, 26, 33). In contrast to the extensive characterization of the pathophysiological consequences of loss of micronuclei compartmentalization, little is known about the events that lead to micronuclear envelope rupture and controlling their repair. Previous work has demonstrated that micronuclei undergo rupture owing to defective nuclear envelope assembly (18–21) and subsequent endoplasmic reticulum (ER) invasion (18, 27). Micronuclei integrity is maintained, at least partially, by the endosomal sorting complex required for transport–III (ESCRT-III), which mediates envelope resealing (22, 23, 34, 35). ESCRT-III activity must be tightly controlled because aberrant ESCRT-III accumulation leads to micronuclear collapse (22, 23), whereas its inhibition might lead to increased rupture as a consequence of impaired repair. Given the critical role of micronuclear envelope rupture and repair in maintaining micronuclei integrity, we sought to identify proteins and pathways modulating those events and selectively operating in micronuclei and not in primary nuclei.
The autophagic receptor p62/SQSTM1 localizes to micronuclei
To identify cellular mechanisms involved in micronuclear recognition, we took advantage of recently described methods for micronuclei isolation (15, 36) and applied an unbiased approach for their characterization. We induced micronuclei formation in HEK293T cells by inhibiting the spindle assembly checkpoint (SAC) using the Mps1 inhibitor (Mps1i) reversine for 48 hours (37–39). Micronuclei and primary nuclei were then isolated, and their protein composition was analyzed through mass spectrometry (Fig. 1A). The analysis revealed that the two compartments present a similar protein composition, although changes in protein abundance could be detected between them (fig. S1A). By performing gene ontology (GO) enrichment analysis, we identified the “ubiquitin protein ligase binding” category to be among the top 10 molecular functions that were more enriched in micronuclei compared with primary nuclei (Fig. 1B and fig. S1A). Because micronuclei are characterized by defective and damaged components, we reasoned that ubiquitin ligases might be targeting those structures. Indeed, p62/SQSTM1—a key component of the autophagy pathway recognizing ubiquitinated structures (40)—was among the significantly up-regulated proteins, which suggests its possible role in recognizing and targeting micronuclear proteins. We next generated micronuclei in untransformed immortalized retinal epithelial cells (hTERT-RPE1) by Mps1i treatment (Fig. 1C), a method that led to the generation of a cell population in which ~50% of cells had at least one micronucleus. We confirmed the localization of p62 on micronuclear structures (Fig. 1D and fig. S1B) and observed a higher enrichment of p62 compared with primary nuclei (fig. S1, B to D). p62 localization to micronuclei was further validated in both untransformed and cancer cells obtained from different tissues of origin (Fig. 1E). To exclude the possibility of a non-specific effect of Mps1i treatment, we selected three colorectal cancer (CRC) cell lines characterized by CIN (thus prone to generate micronuclei). We confirmed the presence of p62 on ~50% of spontaneously generated micronuclei (Fig. 1F). To gain topological details on the interaction between p62 and micronuclei, we generated micronuclei by Mps1i treatment in hTERT-RPE1 and used (i) super-resolution microscopy (Fig. 1,G to I), (ii) correlative light electron microscopy (CLEM) coupled with immunogold staining (Fig. 1J), and (iii) quantitative confocal microscopy (fig. S1E) and found that p62 localized within micronuclear cavities (Fig. 1, G to J, and fig. S1E). Next, we wanted to analyze the kinetics of p62 recruitment to micronuclei and performed live-cell imaging of hTERT-RPE1 cells stably expressing p62–green fluorescent protein (GFP) in which micronuclei were generated by Mps1i (fig. S1, F and G). In 30% of p62-positive micronuclei, p62 was recruited immediately after their generation (i.e., p62 was present in the first frame after cell division in which the micronucleus can be visualized), whereas 70% of micronuclei recruited p62 in the next few hours thereafter (Fig. 1K, fig. S1G, and movie S1). In most of the cases, p62 was recruited before micronuclear collapse [as visualized by the loss of circularity of the micronucleus using H2B signal (18); see fig. S1, H to J] (Fig. 1L). Finally, we analyzed the kinetics of p62 binding to micronuclei using fluorescence recovery after photobleaching (FRAP). We found that p62 kinetics of recovery partially depended on a mobile fraction of p62, possibly caused by the protein diffusing from the cytoplasm, and mainly on an immobile fraction. This finding was suggestive of p62 being able to bind to micronuclear complexes and/or components that limit its diffusion on and from those structures (fig. S1, K to M). Thus, our data indicate that p62 localizes to micronuclei in both untransformed and cancer cells.
Fig. 1. The autophagic receptor p62 recognizes micronuclear structures.
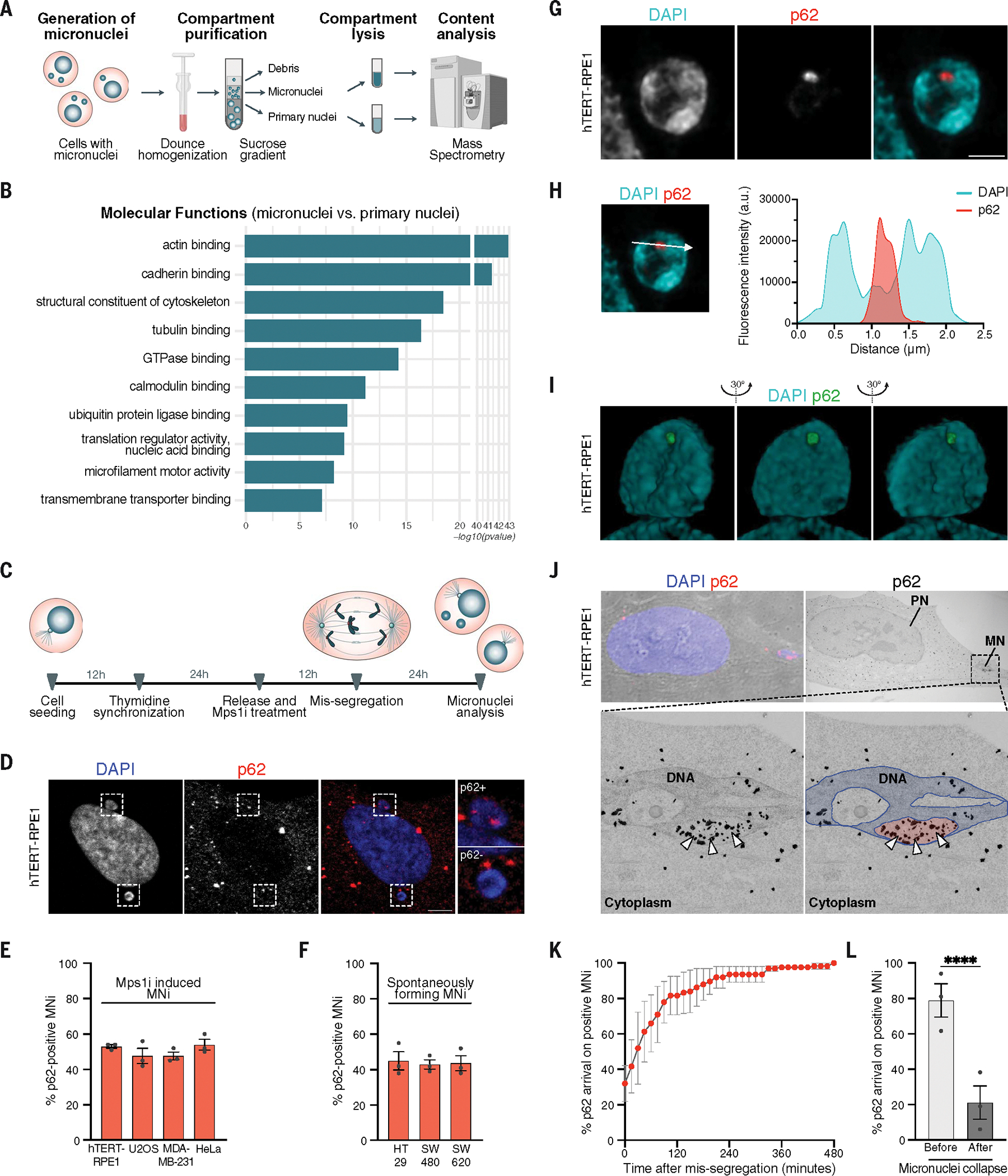
(A) Experimental workflow for the analysis of micronuclei (MNi) and primary nuclei (PNi) in HEK293T cells. (B) Top 10 enriched terms among up-regulated proteins by comparing MNi and PNi proteomes (enrichment analysis cutoff: FDR 0.05). (C) Experimental setup for the generation and analysis of MNi. (D) Representative confocal images of a cell harboring p62-positive (p62+, top) and p62-negative (p62−, bottom) MNi. Scale bar, 5 μm. (E and F) Quantification of p62+ MNi generated with Mps1i (E) or spontaneously forming (F). N ≥ 100 MNi; three biological replicates indicated by data points. Data are means ± SEMs. (G) Representative super-resolution images of a p62+ MN. Scale bar, 1 μm. N = 8 MNi; two biological replicates. (H) Line scan graph of DAPI (micronuclear DNA) and p62 fluorescence intensities (a.u., arbitrary units), respective to a single Z stack of the MN represented in (G). The arrow indicates the directionality of the x axis of the graph. Line scan is representative of 8 MNi analyzed (two biological replicates). (I) Representative super-resolution 3D visualization of a p62+ MN. N = 8 MNi; two biological replicates. (J) (Top) CLEM representative images of a p62+ MN: confocal image of brightfield coupled with DAPI and p62 staining (left) and electron microscopy (EM) image after immunogold labeling of p62 (right). Scale bar, 5 μm. (Bottom) Magnified EM image showing the micronuclear DNA and p62 visualized as black dots (indicated by white arrowheads) within a micronuclear cavity (left), then recolored to highlight micronuclear DNA (blue) and the micronuclear cavity (red). N = 10 MNi; two biological replicates. (K and L) Quantification of cumulative recruitment of p62 to MNi during its formation (K) and with respect to its collapse (L) in H2B-RFP/p62-GFP hTERT-RPE1 cells. Three biological replicates are indicated by data points. Data are means ± SEMs. Chi-squared test, P < 0.0001.
p62 targets ubiquitinated micronuclei but does not mediate their degradation
In the canonical autophagy pathway, autophagic receptors—such as p62—selectively bind their targets on the basis of the target’s ubiquitination (41, 42). In agreement with this idea and with the mass spectrometry results pointing at increased abundance of ubiquitin-related proteins in micronuclei (Fig. 1B and fig. S1A), we found an enrichment of ubiquitination signals on micronuclei compared with primary nuclei (Fig. 2, A and B, and fig. S2A). The majority of p62-positive micronuclei showed ubiquitin signal, consisting of both poly-Lys K63 and poly-Lys K48 (Fig. 2, A to H, and fig. S2, A to C). In agreement with the abundance of ubiquitinated proteins on micronuclei, we also visualized the presence of other autophagic receptors (42) recruited to these structures—namely NBR1 (fig. S2, D to F), TAX1BP1 (fig. S2, G to I), and NDP52 (fig. S2, J to L)—whose signals largely overlapped with p62 on micronuclei (fig. S2M). Furthermore, super-resolution microscopy and quantitative confocal microscopy confirmed p62 and ubiquitin colocalization on micronuclei (Fig. 2, I to K), which prompted us to test whether p62 recognizes ubiquitinated structures associated with micronuclei. To this aim, we first used an inhibitor of ubiquitin-activating enzyme [E1 inhibitor (E1i) TAK243] (43) for 1 hour to reduce the level of ubiquitination in the cells without affecting p62 protein levels (fig. S2N). This treatment strongly decreased ubiquitination levels on micronuclei (Fig. 2, L and M), preventing p62 localization (Fig. 2, L and N). Next, we set out to map the p62 domain responsible for its binding to micronuclei. We monitored the distribution of a battery of GFP-tagged p62 deletion mutants lacking one particular domain at a time (Fig. 2O and fig. S2O). After verifying that the levels of micronuclear ubiquitination did not change on expression of the different constructs (Fig. 2P and fig. S2P), we analyzed p62-GFP–deleted constructs recruitment to micronuclei (fig. S2P). The UBA domain (ubiquitin-associated domain, responsible for ubiquitin binding) of p62 was essential for its recruitment to ubiquitinated micronuclei, whereas all of the other p62 mutants behaved similarly to p62 wild-type (p62-WT) (Fig. 2Q and fig. S2P). Thus, p62 recruitment to micronuclei depends on micronuclear ubiquitination. In the canonical autophagy pathway, the recognition of ubiquitinated structures by p62 initiates a series of events culminating in their degradation by the lysosome (42). Accordingly, we asked whether ubiquitination of micronuclei might lead to their whole degradation, as previously proposed (44). We thus first examined whether the downstream components of the autophagy pathway were recruited to micronuclei and found that the autophagosomal marker LC3 was present only in a fraction of micronuclei positive for p62 (fig. S3, A to C) or for the other autophagic receptors (fig. S3B), and its recruitment depended on p62 (fig. S3C). Although we observed the recruitment of LC3 (fig. S3, A, E, and F) and other autophagic components, namely FIP200, WIPI, and DFCP1 (fig. S3, D to F), almost no lysosomal markers were visualized on micronuclei in our experiments (fig. S3, G and H). To definitively address whether micronuclei are degraded through lysosomes, we modulated the autophagy pathway at different stages by inhibiting or stimulating its activity (40) while quantifying the number of micronuclei. We used untransformed (hTERT-RPE1) and cancer (MDA-MB-231) cells and monitored micronuclei generated by either Mps1i treatment or spontaneously forming. We exposed cells to both short (6-hour) and long (24-hour) treatments aimed to either increase (starvation) or inhibit (SAR405, Baf-A1, and chloroquine) autophagic degradation. We did not observe changes in micronuclei number upon any of these treatments (fig. S3, I to K). As an orthogonal approach for autophagy inhibition, we also depleted the essential autophagy component ATG7 (fig. S3L) and, similarly, did not find differences in the number of micronuclei (fig. S3M). Thus, p62 recognizes ubiquitinated structures on micronuclei, but there is no evidence of autophagic turnover of micronuclei in this setting.
Fig. 2. Molecular characterization of p62 binding to micronuclei.
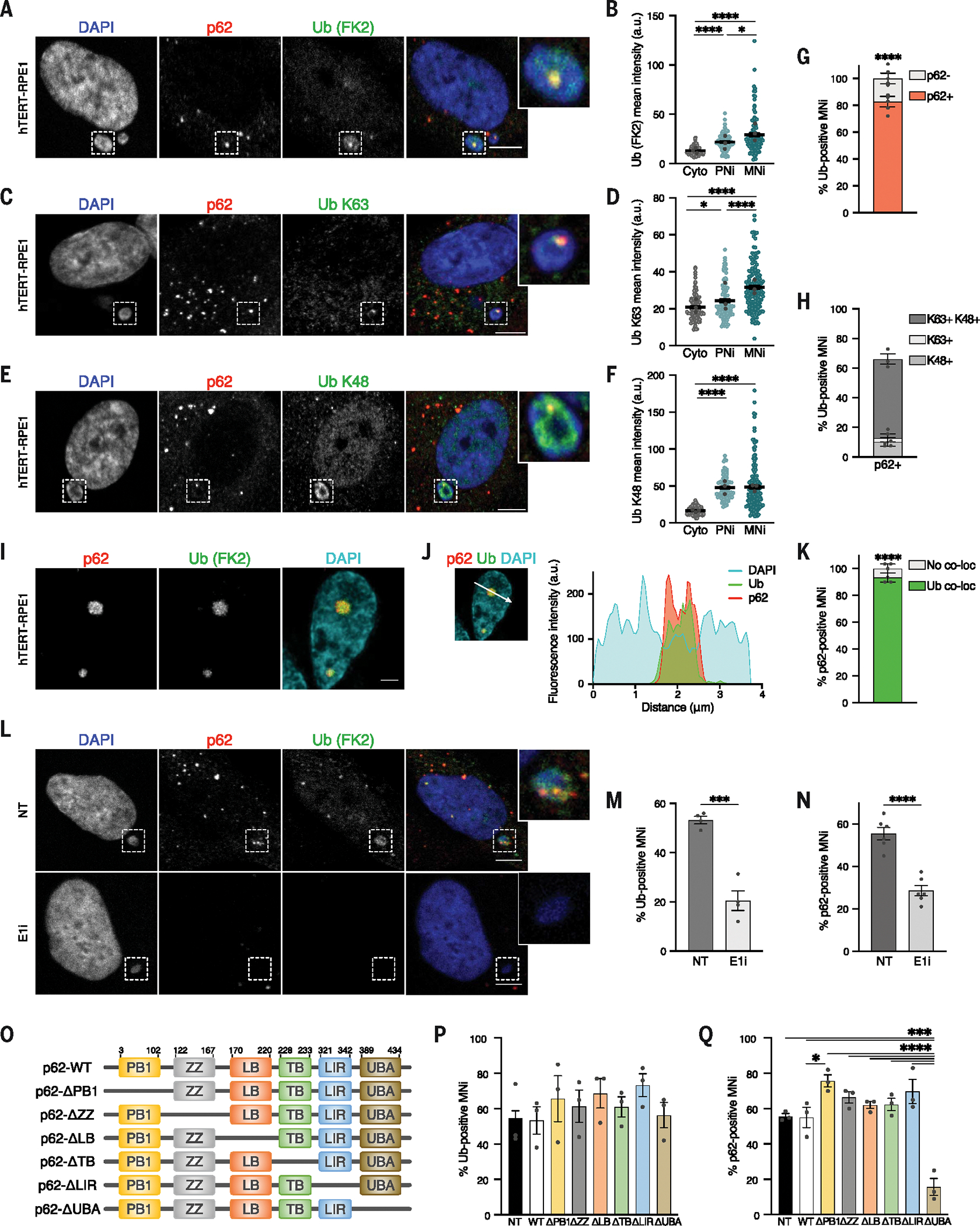
(A to F) Representative images [(A), (C), and (E)] and their quantifications [(B), (D), and (F)] of p62+ and pan-ubiquitin–positive (Ub) (A), Ub poly-Lys K63–positive (C), and Ub poly-Lys K48–positive (E) MNi. Scale bars, 5 μm. Three biological replicates; colored data points indicate the mean of a biological replicate. Data are means ± SEMs. Mann-Whitney test: (B) cytoplasm (Cyto) versus PNi and Cyto versus MNi, P < 0.0001, and PNi versus MNi, P = 0.0327; (D) Cyto versus MNi and PNi versus MNi, P < 0.0001, and Cyto versus PNi P = 0.0323; (F) Cyto versus PNi and Cyto versus MNi, P < 0.0001. (G and H) Quantification of p62− and p62+ MNi within pan-Ub (FK2)–positive ones (G) (N ≥ 170 MNi, five biological replicates; Chi-squared test, P < 0.0001) or of Ub poly-Lys K63–positive and poly-Lys K48–positive MNi among the p62+ ones (H) (N ≥ 100 MNi analyzed, three biological replicates). Replicates are indicated by data points. Data are means ± SEMs. (I and J) Super-resolution (I) and line scan graph (J) of p62 and pan-Ub (FK2) colocalization to a MN. Scale bar, 1 μm. N = 6 MNi analyzed; two biological replicates. (K) Quantification of colocalization of pan-Ub with p62 within p62+ MNi. N ≥ 100 MNi; three biological replicates indicated by data points. Data are means ± SEMs. Chi-squared test, P < 0.0001. (L to N) Representative images (L) and quantification [(M) and (N)] of p62 and pan-Ub signals of MNi in untreated (NT; DMSO) or E1i-treated (bottom) hTERT-RPE1. Scale bars, 5 μm. N ≥ 140 MNi, from at least four biological replicates indicated by data points. Data are means ± SEMs. Unpaired Student’s t test: (M) P = 0.0003; (N) P < 0.0001. (O) Domain organization of p62-deleted constructs. (P and Q) Quantification of pan-Ub–positive (P) and p62+ (Q) MNi in hTERT-RPE1 expressing the indicated constructs [non-transfected (NT)]. N ≥ 100 micronuclei; three biological replicates indicated by data points. Data are means ± SEMs. One-way analysis of variance (ANOVA) and Tukey’s multiple comparison test: NT versus ΔUBA, P = 0.0001; WT versus ΔPB1, P = 0.0491; WT versus ΔUBA, P = 0.0001; ΔPB1, ΔZZ, ΔLB, ΔT, ΔLIR versus ΔUBA, P < 0.0001.
p62 levels modulate micronuclear integrity and collapse
We next sought to understand the consequences of p62 recruitment to micronuclei. High-resolution imaging revealed p62 localization within micronuclear cavities (Fig. 1, G to J, and fig. S1E); thus, we reasoned that micronuclear membranes might be ruptured within these invaginations. Using staining for LSD1—a histone demethylase whose nuclear staining indicated intact micronuclear envelope (18)—we observed that p62 localization strongly correlated with ruptured micronuclei, with 70% of p62-positive micronuclei displaying loss of integrity (Fig. 3, A and B). This localization was specific to ruptures in micronuclei because p62 was not found at rupture sites of the primary nuclei (Fig. 3, C and D, and fig. S4, A to C) (45). These data further suggest a preferential binding of p62 to micronuclei, in agreement with mass spectrometry, fractionation, and imaging experiments (Fig. 1, A to D, and fig. S1, A to D). To gain information about the status of micronuclear membranes in p62-positive micronuclei, we used electron tomography. By comparing p62-positive and p62-negative micronuclei selected by CLEM, we confirmed that the presence of p62 on micronuclei correlated with more extensively damaged and ruptured membranes (Fig. 3E). p62-negative micronuclei had ruptures in the outer nuclear membrane and an intact inner membrane, whereas p62-positive micronuclei presented with an almost absent outer membrane and had holes in their inner layer (Fig. 3E).
Fig. 3. p62-positive micronuclei have lost their envelope integrity.
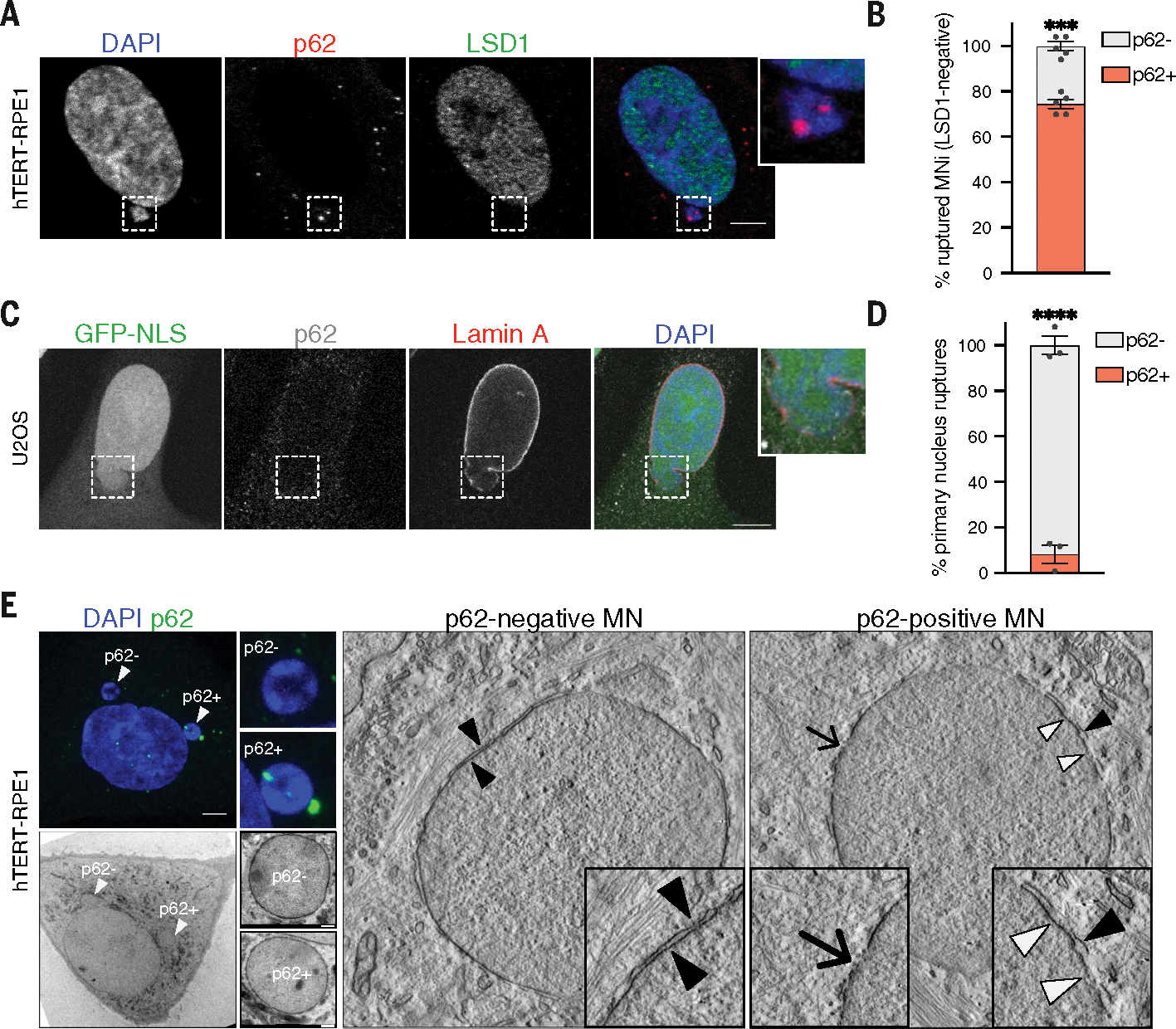
(A and B) Representative image of a cell harboring a p62+ ruptured MN (LSD1−) in hTERT-RPE1 cells (A) and quantification of p62− and p62+ MNi within the ruptured ones (B). Scale bar, 5 μm. N ≥ 170 MNi; five biological replicates indicated by data points. Data are means ± SEMs. Chi-squared test, P = 0.0005. (C and D) Representative image of a p62− herniation relative to a ruptured PN in fixed shRNA–lamin B1 NLS-GFP U2OS cells treated with hydroxyurea (C) and quantification of p62− and p62+ herniations (D). Scale bar, 10 μm. N ≥ 100 MNi analyzed; three biological replicates indicated by data points. Data are means ± SEMs. Chi-squared test, P < 0.0001. (E) (Left) Representative images of a hTERT-RPE1 cell harboring p62− and p62+ MNi selected for CLEM analysis: confocal image of DAPI and p62 staining (top) and EM image (bottom). Scale bar, 5 μm. (Right) Electron tomography representative images of p62− and p62+ MNi. In p62-negative micronucleus, black arrowheads point at the intact double layer of the nuclear envelope (NE). In p62-positive micronucleus, black arrowhead indicates a double layer of NE, white arrowheads point at the absent outer membrane of NE, and the arrow points at the point of rupture of NE. Images are representative of seven micronuclei analyzed from two biological replicates. Confocal images scale bar, 5 μm, EM images scale bar, 500 nm.
Because defects in nuclear membrane assembly and composition have been suggested as potential mechanisms of micronuclear rupture (18–21), we investigated the localization of p62 relative to components of the nuclear envelope. Among the nuclear markers analyzed, including lamin A, emerin, and nuclear pore complex proteins (NPCs), we observed a strong correlation of p62 localization with the inner nuclear membrane component lamin B receptor (LBR) (Fig. 4, A to C, and fig. S4D). In agreement with the fact that LBR has previously been proposed to be enriched on collapsed micronuclei (18), we observed that collapsed micronuclei lacked lamin B and displayed persistent epigenetic dysregulation (Fig. 4D). We confirmed micronuclear collapse and loss of compartmentalization using CLEM (Fig. 4E). In addition, we correlated p62 localization to gene densities of the chromosomes enclosed within micronuclei to test whether p62 recruitment might be influenced by chromosome identity. For this, we used probes for human specific arm (HSA) of chromosomes 17 and 19—as gene-dense chromosomes—and HSA 18—as gene-poor chromosome (21)—and did not observe differences in p62 recruitment (fig. S4, E and F). We also checked p62 localization with respect to micronuclear size and found its recruitment to be strongly anticorrelated with micronuclear area (fig. S4G). We observed higher p62 in micronuclei with smaller areas, in agreement with the fact that those micronuclei are more prone to collapse (21, 22). Together, these data reveal a strong correlation between p62 localization and micronuclear membrane rupture and collapse. Because p62 localization to micronuclei mainly occurs before their collapse (Fig. 1L), we looked for a causal relationship between p62 and micronuclear collapse. To assess this, we first down-regulated p62 by small interfering RNA (siRNA) or deleted it using CRISPR-Cas9 in untransformed and cancer cell lines (fig. S5, A to D) and observed that loss of p62 significantly increased the fraction of intact micronuclei (Fig. 4, F and G, and fig. S5, E and F). Similarly, p62 overexpression in untransformed and cancer cells exacerbated the loss of micronuclear integrity (Fig. 4,H and I, and fig. S5G). Because nuclear envelope rupture can result in the collapse of the whole micronuclear structure, p62 down-regulation or deletion decreased the extent of micronuclear collapse (Fig. 4J and fig. S5, H to J). In agreement with the idea that p62 levels modulate micronuclear integrity, its overexpression increased micronuclear collapse in untransformed and cancer cell lines (Fig. 4K and fig. S5, K and L). Next, we examined which p62 domain (Fig. 2O) was responsible for modulation of micronuclear integrity. Limiting p62 recruitment to micronuclei through depletion of the UBA domain prevented micronuclear rupture and collapse (fig. S5, M, N, Q, and R). In agreement with this, inhibition of ubiquitination through E1i treatment (Fig. 2, L to N) led to a similar phenotype (fig. S5, O and P). Moreover, p62 modulated the integrity of micronuclei and not primary nuclei (fig. S5, Q and R), in agreement with its preferential recruitment to the micronuclear structures (Fig. 3, C and D; fig. S1, A to D; and fig. S4, A and B). p62 modulation of micronuclear integrity partially depended on the LIR (LC3-interacting) domain (fig. S5, M and N). This observation supports a scenario in which p62 modulates the degradation of specific micronuclear components necessary for micronuclear stability through the canonical autophagy pathway. Finally, we checked whether p62 contributes to micronuclear integrity loss in breast cancer cell lines spontaneously forming micronuclei and harboring different levels of p62 (fig. S5S). p62 levels correlated with micronuclear rupture, with high p62 levels corresponding to elevated rupture and loss of integrity (fig. S5T). Thus, p62 acts as regulator of micronuclear membrane integrity and collapse in both untransformed and cancer cells.
Fig. 4. p62 levels regulate micronuclear integrity.
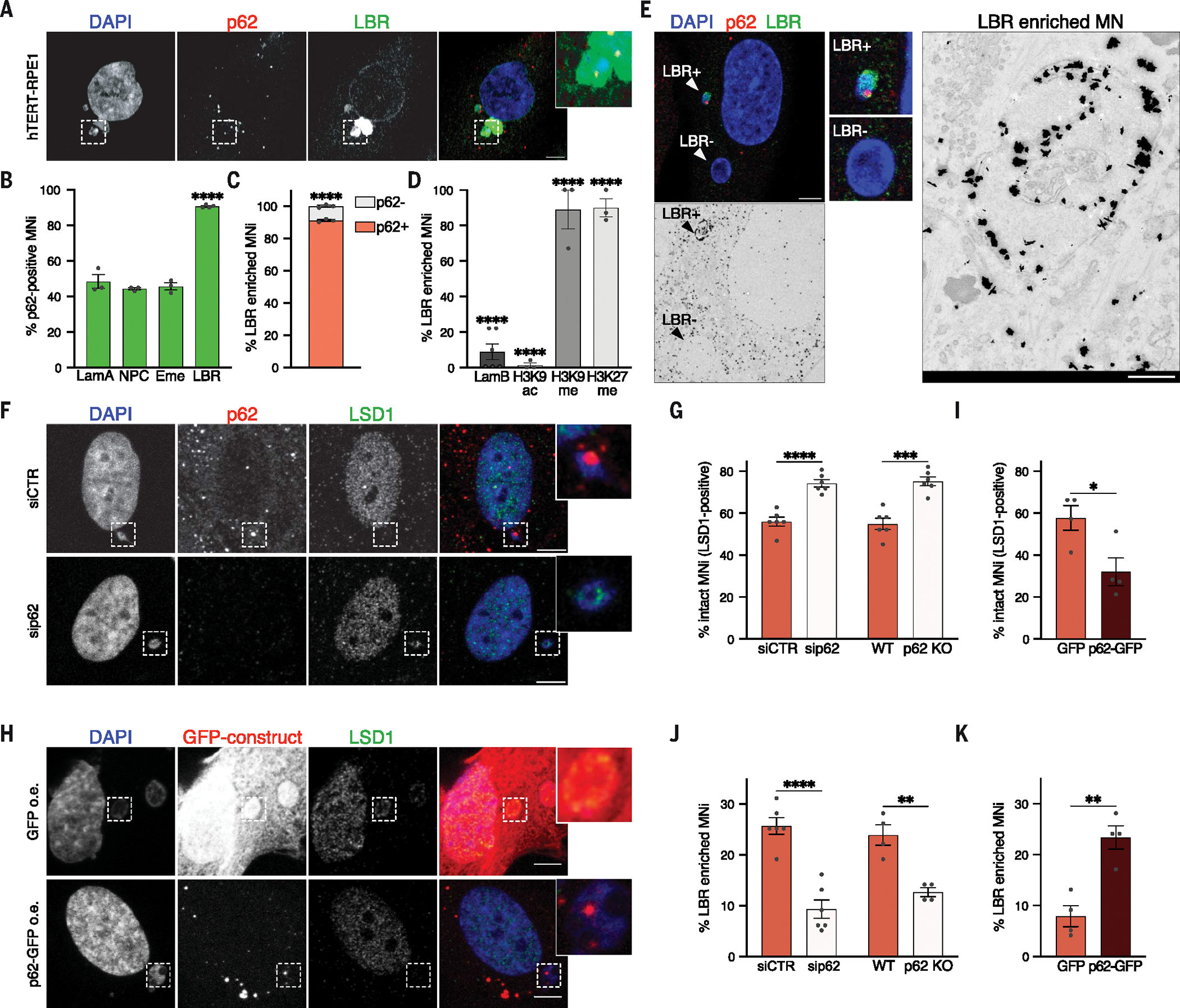
(A) Representative images of a cell with a p62+ LBR-enriched MN. Scale bar, 5 μm. (B) Quantification of p62+ MNi within lamin A–positive (LamA), NPC-positive, emerin-positive (Eme), and LBR-positive ones. N ≥ 100 MNi; at least three biological replicates (N = 3, 3, 3, 4). Chi-squared test: LBR, P < 0.0001. (C) Quantification of p62− and p62+ MNi within LBR-enriched ones in hTERT-RPE1 cells. N ≥ 100 MNi; three biological replicates. Chi-squared test, P < 0.0001. (D) Quantification of LBR-enriched MNi within lamin B– (LamB), H3K9ac-, H3K9me3-, and H3K27me2me3-positive ones in hTERT-RPE1 cells. N ≥ 100 MNi; at least three biological replicates (N = 6, 3, 3, 3). Chi-squared test, all P < 0.0001. (E) (Left) Representative images of a hTERT-RPE1 cell harboring an LBR-enriched (LBR+), p62+ MN and an LBR−, p62− MN selected for CLEM. (Top) Confocal image of DAPI, LBR, and p62. (Bottom) EM image after immunogold labeling of LBR (black dots), magnified on the right. N = 6 MNi; two biological replicates. Scale bars: confocal, 5 μm; EM, 500 nm. (F and G) Representative images of ruptured (LSD1−) and intact (LSD1+) MNi upon ± sip62 (F) and quantification of intact MNi in hTERT-RPE1 ± sip62 or ± p62 KO (G). Scale bar, 5 μm. N ≥ 200 MNi; six biological replicates. Unpaired t test: siRNA, P < 0.0001; KO, P = 0.0001. (H and I) Representative images of an intact (LSD1+) and a ruptured (LSD1−) MN (H) and quantification of intact MNi (I) upon overexpression (o.e.) of GFP and p62-GFP in hTERT-RPE1 cells. Scale bar, 5 μm. N ≥ 100 MNi; four biological replicates. Unpaired t test, P = 0.0270. (J and K) Quantification of collapsed MNi (LBR-enriched) in hTERT-RPE1 cells ± sip62 or ± p62 KO (J) or ± p62 o.e. (K). N ≥ 100 MNi, from at least four biological replicates (N = 6, 4, 4). Unpaired t test: siRNA, P < 0.0001; KO, P = 0.0022; o.e., P = 0.0024. In (B), (C), (D), (G), (I), (J), and (K), each data point indicates a biological replicate. Data are means ± SEMs.
Micronuclei-mitochondria proximity leads to oxidation-driven homo-oligomerization of p62 and autophagic degradation of ESCRT components
Next, we sought to elucidate the molecular determinants of p62-dependent modulation of micronuclear integrity. To this aim, we analyzed, using mass spectrometry, the proximity-proteome of p62 in micronuclei isolated from cells stably expressing APEX2-p62 (or APEX2, as negative control) (Fig. 5A and fig. S6A) (46). This analysis was particularly insightful in defining the functional interaction of p62 with micronuclei, elucidating both its target and the mechanism responsible for its localization. The most enriched cellular components included the ESCRT machinery and mitochondrial components (Fig. 5B and fig. S6B). Given that the ESCRT machinery plays a role in repairing nuclear envelope ruptures through ER recruitment in primary nuclei (34, 35), we reasoned that p62 at micronuclei might hamper their nuclear envelope repair process by limiting ESCRT machinery functioning. To test this, we evaluated the localization of ESCRT components to micronuclei upon p62 deletion in untransformed and cancer cells. We observed an increase in the ESCRT components CHMP7, CHMP4B, and CHMP2A localization to micronuclei upon loss of p62 (Fig. 5, C to F, and fig. S6, C to G). This modulation occurred through autophagic regulation of ESCRT proteins. Preventing autophagosome formation through SAR405 or blocking lysosome-mediated autophagosome degradation through Baf-A1 or chloroquine treatment increased micronuclear localization of CHMP7 and CHMP4B (Fig. 5, G and H, and fig. S6, H and I). These findings suggest that p62 negatively regulates the ESCRT proteins’ localization and activity. We further confirmed the direct involvement of p62 in autophagic degradation by inhibiting lysosomal activity in either p62 WT or knockout (KO) cells harboring micronuclei and measuring the percentage of CHMP7 signal within lysosomes. The percentage of CHMP7 signal within lysosomes decreased by ~30% upon p62 depletion (from 17 to 12%), confirming that p62 directly participates in the autophagy-mediated degradation of CHMP7 (fig. S6J).
Fig. 5. p62 controls peri-micronuclear autophagic degradation of ESCRT components.
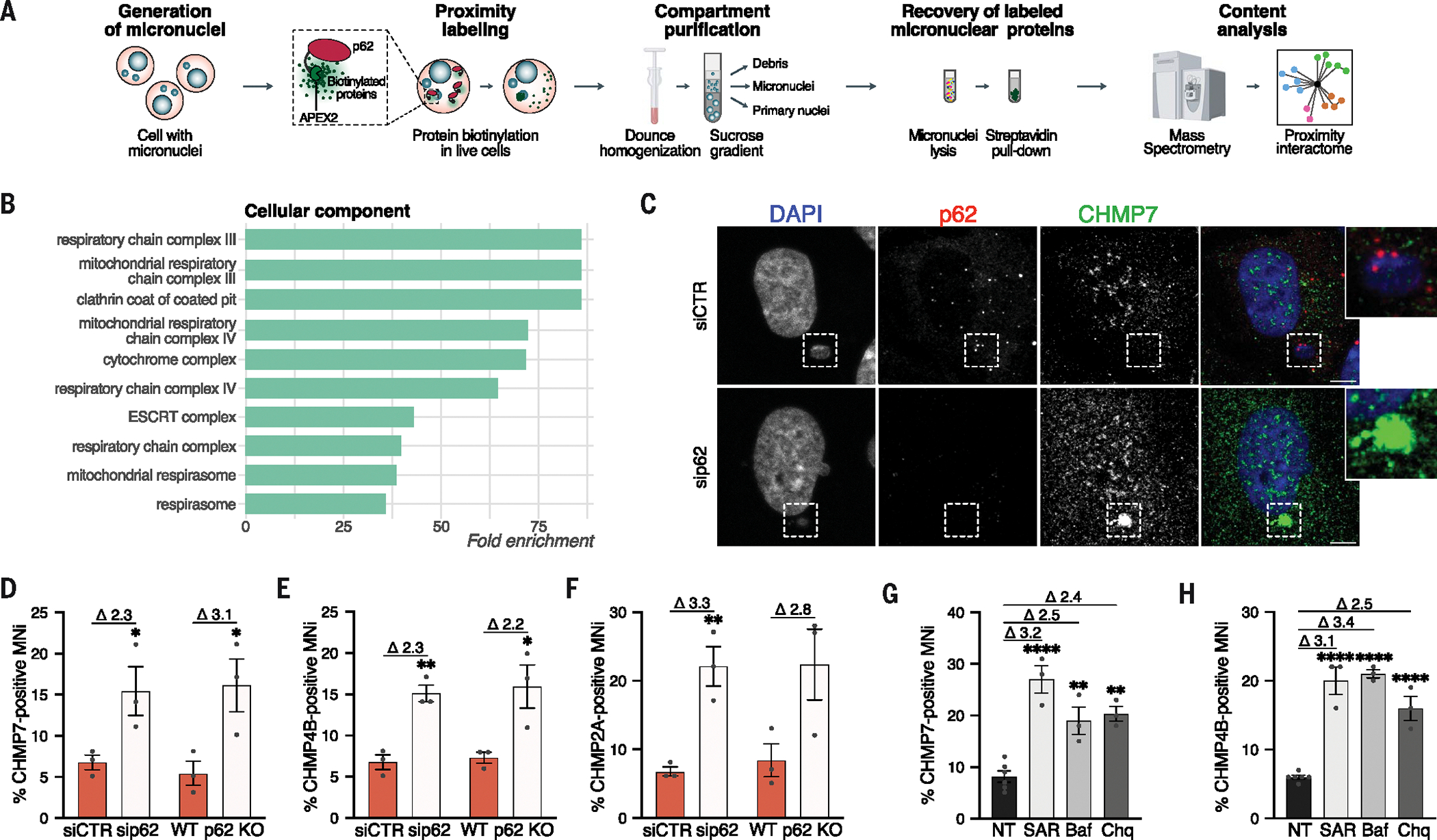
(A) Experimental workflow for the mass spectrometry analysis of p62 proximity-proteome of MNi in HEK293T cells. (B) Top 10 enriched GO cellular component terms in the up-regulated proteins from p62 proximity-proteome of MNi (enrichment analysis cutoff: FDR 0.05). (C and D) Representative images showing a CHMP7− p62+ MN upon siCTR and a CHMP7+ MN upon sip62 (C) and quantification of CHMP7+ MNi (D) in hTERT-RPE1 cells upon ± sip62 or ± p62 KO. Scale bars, 5 μm. Unpaired t test: siRNA, P = 0.0486; KO, P = 0.0390. (E) Quantification of CHMP4B+ MNi in hTERT-RPE1 cells upon ± sip62 or ± p62 KO. Unpaired t test: siRNA, P = 0.0033; KO, P = 0.0337. (F) Quantification of CHMP2A+ MNi in hTERT-RPE1 cells upon ± sip62 or ± p62 KO. Unpaired t test: siRNA, P = 0.0066. (G and H) Quantification of CHMP7+ (G) or CHMP4B+ (H) MNi in hTERT-RPE1 cells upon treatment with SAR405 (SAR) or Baf-A1 (Baf) or chloroquine (Chq) or not treated (NT). Ordinary one-way ANOVA test followed by Tukey’s multiple comparison test (all versus NT). In (G): NT versus SAR405, P < 0.0001; NT versus Baf-A1, P = 0.0028; NT versus chloroquine, P = 0.0012. In (H): all P < 0.0001. In (D) to (H), fold changes upon normalization to relative controls are shown above the graphs. N ≥ 100 MNi; at least three biological replicates indicated by data points. Data are means ± SEMs.
Next, we wanted to investigate the molecular mechanism of p62 localization and activity on micronuclei. Notably, among the proteins enriched in the p62 proximity-proteome on micronuclei, we found several mitochondrial components (Fig. 5B). We confirmed the proximity of mitochondria to micronuclei using electron tomography (Fig. 6, A and B, and movie S2) and quantitative DeepSIM [structured illumination microscopy (SIM)] imaging (Fig. 6C). This analysis also revealed that p62-positive micronuclear cavities were more proximal to mitochondria compared with those lacking p62 (Fig. 6, B and C). We then tested whether reactive oxygen species (ROS) released by mitochondria contribute to p62 localization and activity. Treatment with the ROS scavenger N-acetyl cysteine (NAC) decreased the percentage of p62-positive micronuclei and prevented micronuclear rupture (Fig. 6D and fig. S6K), whereas increasing ROS levels with H2O2 treatment led to an increase in p62-positive micronuclei and ruptured micronuclei (Fig. 6E and fig. S6L) in both untransformed and cancer cells. We hypothesized that these effects might be explained by ROS-driven cysteine oxidation of p62 because p62 can be oxidized upon oxidative damage (fig. S6M), which leads to enhanced autophagic activity (47). After micronuclei fractionation, we found that p62 undergoes large levels of oxidation in the micronuclear fraction, resulting in its homo-oligomerization (Fig. 7A).
Fig. 6. p62 localizes to micronuclear cavities proximal to mitochondria.
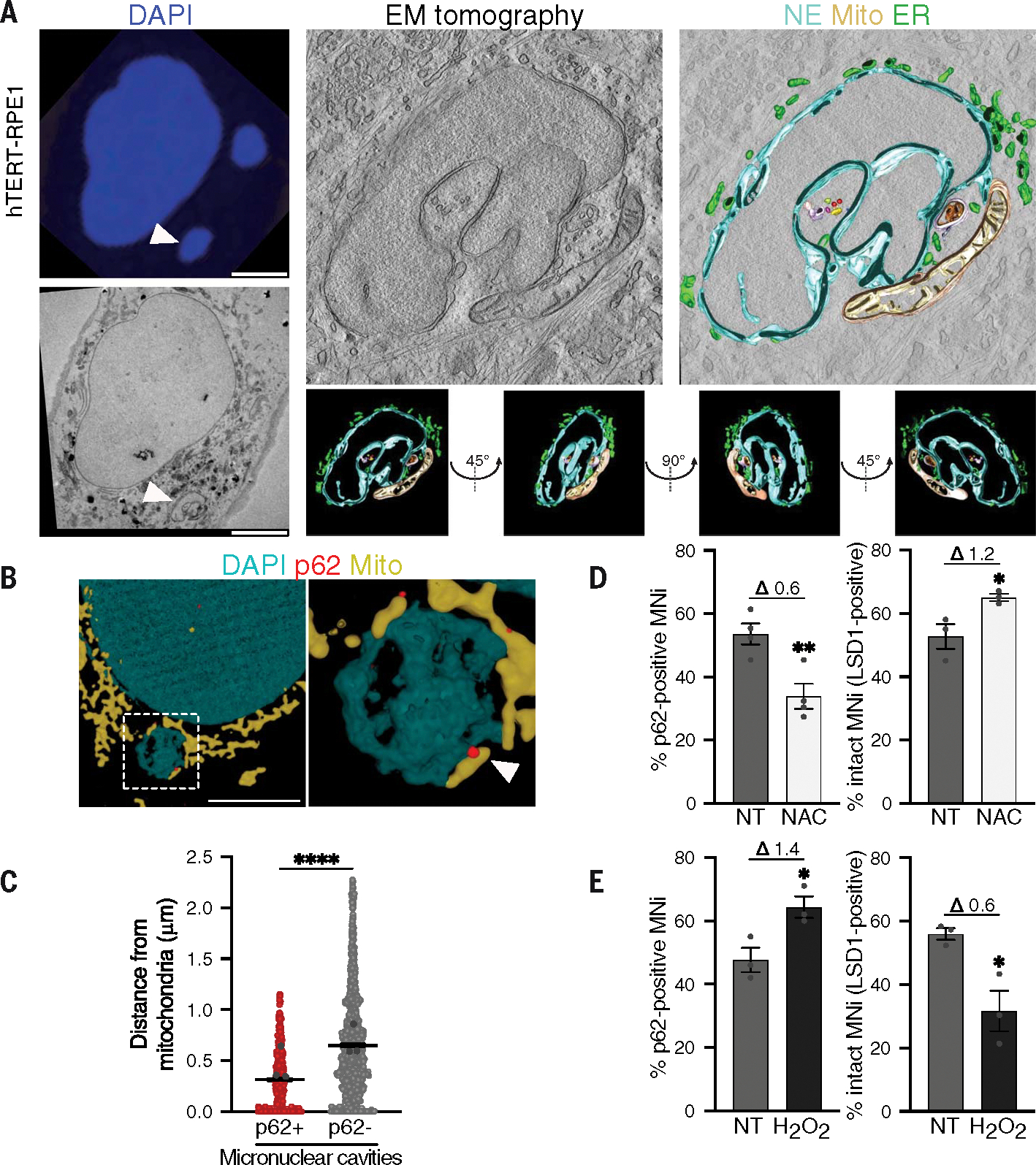
(A) (Left) Representative CLEM analysis: confocal (top) and EM images (bottom). (Right) Representative electron tomography analysis of mitochondria proximity to the MN and tomography reconstruction (z-slices, gray; mitochondria, yellow; NE, cyan; ER, green). In total, 131 images were acquired (from −65° to +65°, acquisition every 1°) with the reconstructed tomogram encompassing a total of 200-nm depth; z-slices are shown from different directions (bottom). N = 8 MNi; two biological replicates. Scale bars, 5 μm. (B) Representative images of DeepSIM reconstruction of a p62+ MN showing mitochondria (visualized by mito-tracker) in proximity in hTERT-RPE1. Scale bar, 5 μm. (C) Quantification of the distance between mitochondria and p62+ or p62− cavities of MNi in hTERT-RPE1. Three biological replicates; each colored data point indicates the mean of a biological replicate. Data are means ± SEMs. Two-sided Mann-Whitney test, P < 0.0001. (D) Quantification of p62+ MNi and intact (LSD1+) MNi in hTERT-RPE1 cells ± NAC treatment (NT, not treated), labeled with DAPI, p62, and LSD1. Unpaired t test: p62-pos, P = 0.0088; LSD1-pos, P = 0.0395. (E) Quantification of p62+ MNi and intact (LSD1+) MNi in hTERT-RPE1 cells ± H2O2 treatment (NT, not treated). Unpaired t test: p62-pos, P = 0.0312; LSD1-pos, P = 0.0216. In (D) and (E), fold changes upon normalization to the relative controls are displayed above the graphs. N ≥ 100 MNi; three biological replicates. Each data point indicates a biological replicate. Data are means ± SEMs.
Fig. 7. Micronuclei-mitochondria proximity leads to oxidation-driven homo-oligomerization of p62.
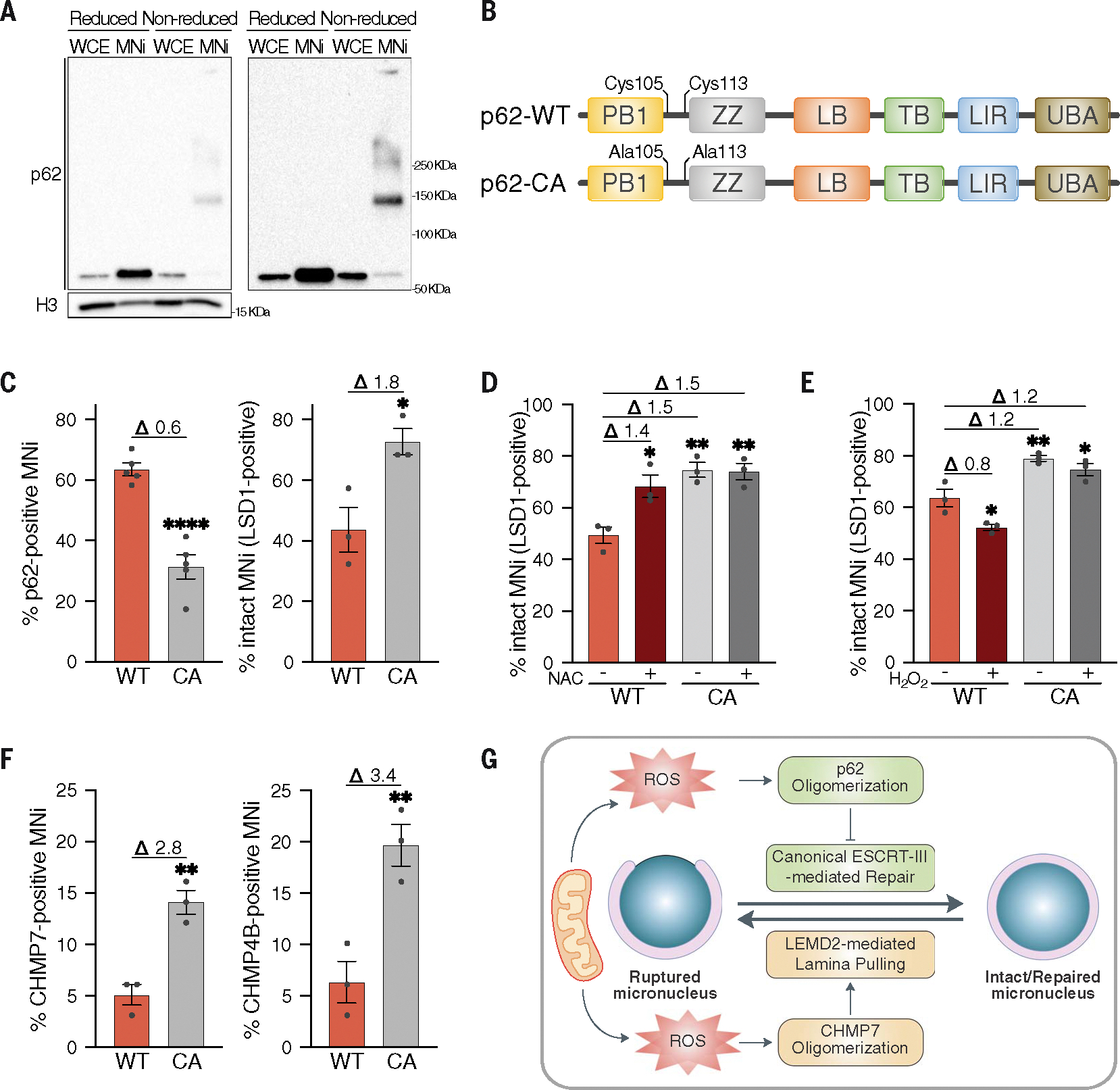
(A) Reduced and nonreduced Western blot analysis of p62 homo-oligomerization in whole cell extracts (WCEs) and MNi isolated from HEK293T cells, showing low (left) and high (right) p62 exposure. H3 used as loading control. Four biological replicates. (B) Schematic representation of p62-CA mutant showing protein domains and mutated residues. (C) Quantification of p62+ MNi and intact (LSD1+) MNi in hTERT-RPE1 p62 KO stably expressing FLAG-tagged p62-WT or p62-CA mutant. Unpaired t test: p62+, P < 0.0001; LSD1+, P = 0.0270. (D) Quantification of intact (LSD1+) MNi in hTERT-RPE1 p62 KO stably expressing FLAG-tagged p62-WT or p62-CA mutant upon ± NAC treatment. Ordinary one-way ANOVA test followed by Tukey’s multiple comparison test (all versus WT NT): WT NT versus WT NAC, P = 0.0119; WT NT versus CA NT, P = 0.0022; WT NT versus CA NAC, P = 0.0026. (E) Quantification of intact (LSD1+) MNi in hTERT-RPE1 p62 KO stably expressing FLAG-tagged p62-WT or p62-CA mutant upon ± H2O2 treatment, labeled with DAPI, FLAG, and LSD1. Ordinary one-way ANOVA test followed by Tukey’s multiple comparison test (all versus WT NT): WT NT versus WT NAC, P = 0.0195; WT NT versus CA NT, P = 0.0036; WT NT versus CA NAC, P = 0.0226. (F) Quantification of CHMP7+ and CHMP4B+ MNi in hTERT-RPE1 p62 KO stably expressing FLAG-tagged p62-WT or p62-CA mutant. Unpaired t test: CHMP7+, P = 0.0041; CHMP4B+, P = 0.0097. (G) Schematic model showing p62 and ROS functioning in modulating micronuclear integrity via inhibiting ESCRT-III–mediated repair activity. See text for more details. In (C) to (F), fold changes upon normalization to the relative controls are displayed above the graphs. N ≥ 100 MNi; at least three biological replicates indicated by data points. Data are means ± SEMs.
To interrogate the function of such oxidation on p62 behavior mechanistically and to better delineate the functional interplay between p62 and mitochondria-derived oxidative damage on micronuclei, we used a p62 oxidation-resistant mutant in which the two cysteines (Cys105 and Cys113) crucial for its oxidation are mutated to alanine (p62-CA) (Fig. 7B and fig. S6, N and O) (47). Cells expressing p62-CA showed a reduction in p62 localization to micronuclei and an increase in intact micronuclei (Fig. 7C and fig. S6P), which was not further affected by ROS scavenging or H2O2 treatment (Fig. 7, D and E). In agreement with the idea that ROS dictate p62 localization and functioning by inducing its oxidation, cells expressing p62-CA showed an increased percentage of CHMP7- and CHMP4B-positive micronuclei (Fig. 7F). Thus, oxidation-driven homo-oligomerization of p62 is crucial for its autophagic activity, which negatively regulates the canonical ESCRT-mediated micronuclear repair. A companion Research Article (48) found that, besides its canonical function in micronuclear envelope repair, the ESCRT component CHMP7 plays a pivotal role in initiating micronuclear envelope rupture in an ESCRT-III–independent manner (48). This suggests that CHMP7 is involved in micronuclei stability through its canonical ESCRT-mediated repair activity—which is negatively affected by p62— and by being directly involved in micronuclear envelope rupture in an ESCRT-III–independent manner. This prompted us to test whether deletion of CHMP7 could rescue p62-mediated loss of micronuclear envelope integrity. Micronuclei rupture mediated by p62 overexpression was completely abolished by deletion of CHMP7 or its receptor LEMD2 (fig. S6Q). Also, p62 overexpression was unable to alter micronuclear envelope integrity under conditions in which CHMP7-driven rupture and p62 homo-oligomerization were prevented by ROS scavenging (fig. S6R). Thus, we suggest that ROS triggered by the proximity of mitochondria to micronuclei leads to homo-oligomerization of p62, which in turn inhibits ESCRT-III–mediated repair through autophagic degradation of CHMP7 (Fig. 7G). Furthermore, mitochondria-driven ROS negatively influence the integrity of the micronuclear envelope by triggering ESCRT-independent CHMP7 micronuclear rupture (48). Thus, by promoting a p62-dependent inhibition of repair and a CHMP7-driven rupture, mitochondria-derived oxidative damage leads to micronuclei collapse (Fig. 7G).
p62-mediated loss of micronuclear integrity drives catastrophic events
We next investigated the consequences of p62-dependent micronuclei rupture on cell physiology and genome stability. First, because micronuclear DNA gets exposed through ruptures and recognized by the cytosolic DNA cGAS-STING sensing pathway (25, 26, 33), we examined cGAS localization to micronuclei and the activation of inflammatory response in breast cancer cells MDA-MB-231 upon modulation of p62 levels. Depletion of p62 prevented cGAS recruitment to micronuclei (Fig. 8, A and B, and fig. S7A), in agreement with the finding that a smaller fraction of micronuclei was ruptured and there was a decrease in exposed micronuclear DNA (Fig. 4, F to I, and fig. S5, E to J). Also, we observed reduced nuclear RelB translocation (Fig. 8C and fig. S7B) and decreased expression of cGAS-STING target genes (Fig. 8D), revealing an overall dampened activation of the downstream inflammatory response. Down-regulation of inflammatory gene expression was specifically due to a decreased cGAS localization to micronuclei upon p62 KO. The down-regulation of those genes obtained after cGAS depletion was not further enhanced in p62 KO cells (fig. S7, C to F). This observation underscores the relationship between p62’s role in maintaining micronuclei integrity and the downstream cGAS activation, and it indicates the involvement of p62 and cGAS in the same regulatory axis.
Fig. 8. p62 drives micronuclear catastrophe.
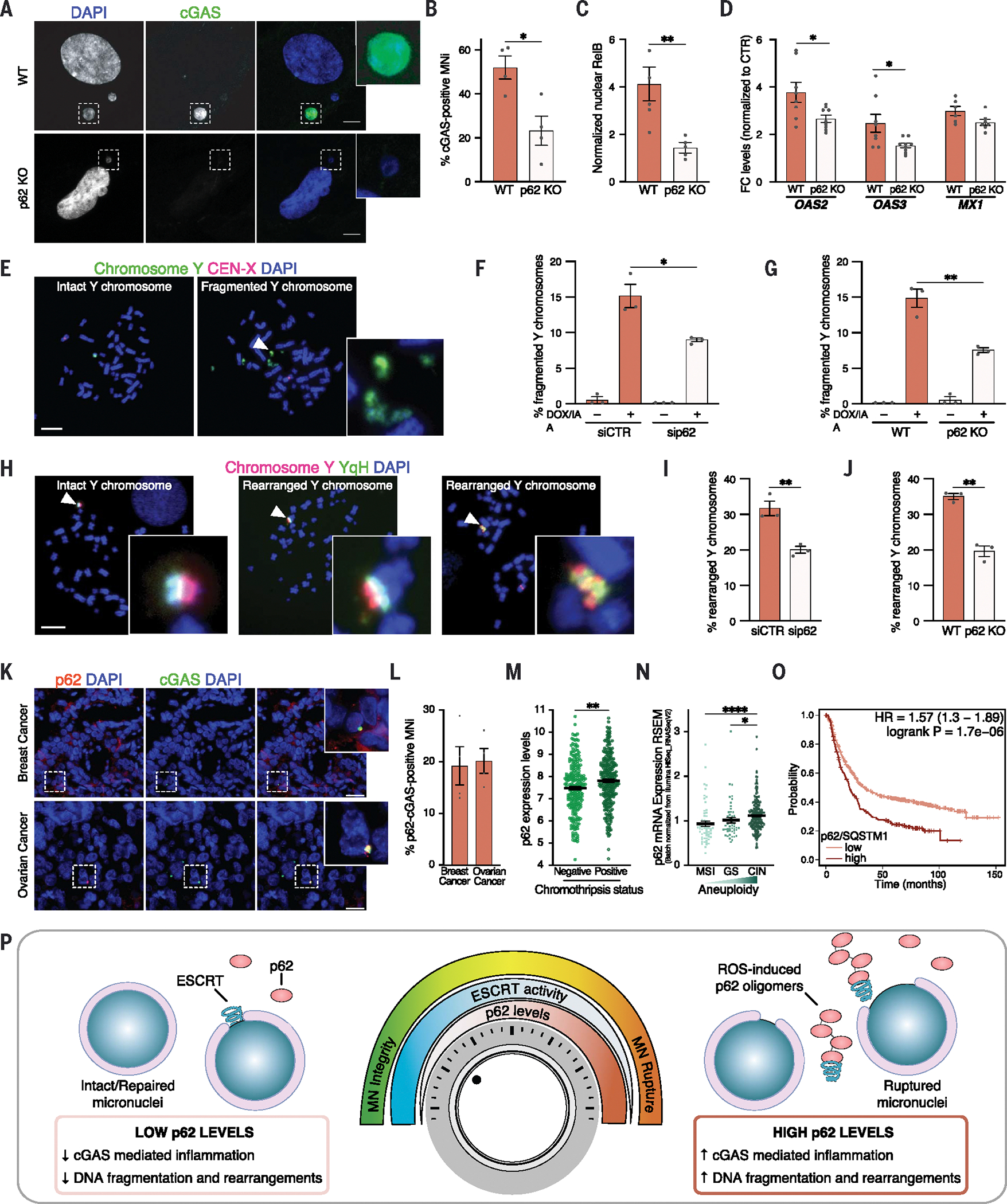
(A and B) Representative images (A) and quantification (B) of cGAS+ MNi in WT and p62 KO cells in MDA-MB-231 cells. Scale bars, 5 μm. N ≥ 140 MNi; four biological replicates. Unpaired t test, P = 0.0145. (C) Quantification of RelB translocation into PN in MDA-MB-231 cells ± p62 KO. N ≥ 200 cells; three biological replicates. Unpaired t test, P = 0.0072. (D) Quantification of OAS2, OAS3, and MX1 levels in MDA-MB-231 cells ± p62 KO, normalized to the respective controls (DMSO). GAPDH used as loading control. N = 8, 9, 8, 9, 6, 7. Unpaired t test: OAS2, P = 0.0197; OAS3, P = 0.0228. (E to J) Representative metaphase spreads of intact, fragmented (E) and rearranged (H) Y chromosomes in DLD-1 cells after 3 days of DOX/IAA treatment and G418 selection labeled with DAPI and with FISH probes targeting the Y chromosome and X centromere (E) or the euchromatic (red) and heterochromatic (YqH, green) regions of the Y chromosome (H). Quantification of fragmented [(F) and (G)] and rearranged [(I) and (J)] Y chromosomes in ± sip62 [(F) and (I)] or ± p62KO [(G) and (J)]. Scale bars, 10 μm. N ≥ 120 metaphases; three biological replicates. Unpaired t test: (F), P = 0.0203; (G), P = 0.0053; (I), P = 0.0067; (J), P = 0.0080. (K and L) Representative images of breast and ovarian tumor tissues harboring p62+ cGAS+ MNi, labeled with DAPI, p62, and cGAS (K) and quantification of p62+-cGAS+ double-positive MNi (L). Scale bars, 100 μm. Four case studies for each tumor are indicated by data points. Data are means ± SEMs. (M) p62 mRNA levels [log2(TPM +1)] of 517 cancer cell lines. Data are means ± SEMs. Two-sided Mann-Whitney test, P = 0.001. (N) p62 mRNA levels in colorectal adenocarcinoma (COAD) classified by subtype. Data are means ± SEMs. Kruskal-Wallis test followed by Dunn’s multiple comparison test: MSI versus CIN, P < 0.0001; GS versus CIN, P = 0.0312. (O) Kaplan-Meier plot of gastric tumors stratified for p62 expression. Hazard ratio (HR) = 1.57; P = 1.7 × 10−6. (P) Schematic model illustrating p62 as a rheostat in regulating micronuclear integrity. See text for more details. In (B), (C), (D), (F), (G), (I), and (J), each data point indicates a biological replicate. Data are means ± SEMs.
One of the major consequences of the loss of micronuclear integrity is the generation of complex chromosomal rearrangements, also known as chromothripsis (2, 9, 11). Given the role of p62 in micronuclei integrity, we wanted to determine whether p62 promotes chromothripsis. We used the established Y centromere–selective inactivation strategy to assess chromosome fragmentation and rearrangements upon p62 loss in an inducible and tractable micronucleus model (12, 15). Down-regulation or deletion of p62 in DLD-1 cells (fig. S7G) was sufficient to suppress fragmentation (Fig. 8, E to G) and rearrangements of the micronuclear chromosome (Fig. 8, H to J). Catastrophic chromosomal rearrangements—such as chromothripsis—have been shown to be strong mutagenic drivers and to be frequent in tumors (12, 14). Given the role of p62 in chromosome fragmentation and rearrangements (Fig. 8, E to J), we wanted to test whether p62 localizes to micronuclei in tumor samples. We investigated this in chromosomally unstable breast and ovarian tumor tissues and found p62 to be recruited to micronuclei and to colocalize with cGAS on these structures (Fig. 8, K and L). Furthermore, because p62 levels can modulate the frequency of chromosomal rearrangements (Fig. 8, E to J), we analyzed p62 expression and chromothripsis status in 517 human cancer cell lines and found that high p62 levels were associated with chromothripsis occurrence (Fig. 8M).
Micronuclei, and the resulting CIN and chromothripsis, are potent drivers of metastasis and poor prognosis (13, 14, 33). Thus, we wanted to check whether and how p62 levels correlate with CIN and with prognosis in cancer. For this, we first analyzed the levels of p62 in colon adenocarcinoma—a tumor with defined molecular subtypes that are affected to a different degree by copy number changes—and found p62 levels to positively correlate with the degree of aneuploidy and CIN [highest aneuploidy in CIN subtype and lowest in microsatellite instability (MSI)] (Fig. 8N). Furthermore, higher p62 mRNA expression levels were strongly associated with shortened overall survival in gastric cancer (Fig. 8O). Thus, p62 controls several micronuclei-associated features, including chromosome fragmentation and rearrangement. Furthermore, p62 mRNA levels are positively correlated with the presence of chromothripsis in cancer cells and might be used as a prognostic marker for tumors with high CIN (49–53).
Discussion
Micronuclei have historically been recognized as simple by-products of chromosome mis-segregation and a prominent feature of CIN tumors (1, 2). In recent years, a large body of evidence has pointed to a causal role of these dysfunctional structures in mutagenic processes and cancer development (11–15). Nuclear envelope rupture and collapse, together with DNA damage and rearrangements occurring within micronuclei, contribute to chromothripsis and metastasis, further inducing CIN and driving tumorigenesis (11, 12, 15, 16, 18, 33).
Given the catastrophic consequences of micronuclear rupture and collapse, many recent studies have focused on whether and how specific micronuclear features, such as defective nuclear envelope composition and chromosome identity (18–21), contribute to the loss of integrity observed in micronuclei. However, it remains poorly understood whether cellular machineries might modulate micronuclear integrity independently of the intrinsic characteristic of the micronucleus. In this work, we identify and characterize a modulator of micronuclear repair, the autophagic receptor p62. Using a combination of approaches—including mass spectrometry of purified micronuclei, super-resolution microscopy, and CLEM—we show that p62 localizes to micronuclei, and its levels correlate with micronuclear rupture and collapse, acting as a rheostat in modulating micronuclear integrity (Fig. 8P). Mechanistically, we show that proximity of micronuclei to mitochondria promotes ROS-mediated homo-oligomerization of p62 through cysteine oxidation. Although p62 oligomerization can also be observed in the cytoplasm (47), it is particularly high in the micronuclear fraction. We speculate that the proximity of micronuclei to mitochondria, along with the extensive invaginated surface area provided by micronuclear cavities, increases the local concentration of ROS, thus enhancing p62 oxidation. Homo-oligomerized p62 negatively affects the activity of ESCRT-III components by targeting them for autophagic degradation, thus limiting their ability to repair the ruptured micronuclear envelope. Our data, which indicate that p62 specifically localizes to micronuclei rather than primary nuclei upon rupture, might also explain why ESCRT-III activity has greater effectiveness in primary nuclei compared with micronuclei (22, 23). This difference is likely because the repair process is properly functioning in the former, whereas it is inhibited by p62 in the latter.
Previous studies have demonstrated that uncontrolled activity of the ESCRT-III machinery on micronuclei can lead to micronuclear collapse (22, 23, 54). Consistent with this, we hypothesize that ESCRT-III functionality must be tightly regulated and maintained at physiological levels. Our data support the idea that p62 plays a pivotal role in this regulation, as evidenced by p62-mediated disruption of micronuclear integrity resulting from inhibition of ESCRT-III activity. However, if the activity and accumulation of ESCRT-III components on micronuclei exceed the control exerted by p62, it could lead to catastrophic consequences (22, 23). Notably, recent findings by Di Bona et al. (48) underscore the essential role of the ESCRT machinery in maintaining micronuclear envelope integrity, revealing that ROS-induced accumulation of CHMP7 on micronuclei drives micronuclear rupture. Hence, the proximity of micronuclei to mitochondria negatively affects micronuclear envelope integrity owing to elevated ROS levels, which in turn alter the biological properties of peri-micronuclear p62 and CHMP7. Although oxidation of the former inhibits the repair process, oxidative damage of the latter triggers the rupture of micronuclei, providing a potential explanation for the irreversible catastrophic collapse of the micronuclear envelope. Our data reveal a correlation between p62 levels and micronuclear DNA fragmentation, chromosome rearrangements, and the occurrence of chromothripsis across hundreds of cancer cell lines. Thus, p62-mediated regulation of micronuclear stability might further trigger CIN, as suggested by the correlation between p62 levels and aneuploidy in high-CIN colon adenocarcinoma. Given the fact that p62 levels are highly deregulated in tumors, the effects of p62-mediated micronuclear integrity modulation—including chromosome rearrangements and cGAS-mediated inflammation—might affect cancer development and metastasis formation in CIN-high tumors. This prediction fits our survival analysis in gastric tumors stratified by their p62 levels, in which we observed an unfavorable prognosis in tumors characterized by high levels of p62. Future studies will expand these observations and hold the promise to address the potential role of p62 as a prognostic factor in CIN-high tumors.
Materials and methods
Cell culture conditions
BT-549, Cal-51, HEK293T, HEK293T cells expressing APEX2 or APEX2-p62 (all generated in house), HeLa, HT-29, hTERT-RPE1 WT, hTERT-RPE1 p62 KO (generated in house), hTERT-RPE1 cells stably expressing H2B-RFP and p62-GFP (generated in house), hTERT-RPE1 p62 KO cells stably expressing p62-WT or p62-CA (all generated in house), MCF-7, MDA-MB-231 WT, MDA-MB-231 p62 KO (generated in house), MDA-MB-361, U2OS WT, and U2OS p62 KO (generated in house) cell lines were cultured in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% penicillin/streptomycin. SW480 and SW620 were cultured in Leibovitz L15 Medium supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. MDA-MB-468 were cultured in 1:1 DMEM and DMEM F12 supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. All cell lines were tested free of mycoplasma contamination using Myco Alert (Lonza) according to manufacturer’s instructions. All cell lines were cultured at 37°C in 5% CO2.
For immunofluorescence (IF)–fluorescence in situ hybridization (FISH) experiments, hTERT-RPE1 cells were grown in DMEM/F12 supplemented with 10% FBS, 1% penicillin/streptomycin, and 0.01 mg/ml hygromycin at 37°C in 5% CO2.
For evaluation of nuclear herniations and micronuclear rupture, U2OS cells expressing 3xGFP-NLS (GFP-NLS) and shRNA–lamin B1 were grown in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 0.01 mg/ml hygromycin at 37°C in 5% CO2.
For FISH experiments, DLD-1 cells were cultured in DMEM supplemented with 10% tetracyclin-free FBS and 100 U/ml penicillin-streptomycin at 37°C in 5% CO2 and were routinely tested for mycoplasma.
Cell synchronization and treatments
To analyze p62 recruitment and localization to micronuclei, HeLa, hTERT-RPE1, MDA-MB-231, and U2OS cells were synchronized at the G1/S boundary with thymidine (Sigma Aldrich; 5 mM in hTERT-RPE1, MDA-MB-231, U2OS cells or 2,5 mM in HeLa cells), washed three times in 1X phosphate-buffered saline (PBS) and pulsed with the Mps1 inhibitor (Mps1i) reversine (500 nM, Cayman Chemical), then cells were fixed 36 hours later (i.e., 24 hours after mis-segregation). To analyze p62 effects on micronuclei, HeLa, hTERT-RPE1, MDA-MB-231, and U2OS cells were treated with the Mps1 inhibitor reversine (500 nM, Cayman Chemical) for 24 hours before fixation. To evaluate micronuclei-mediated inflammation effects, MDA-MB-231 cells were treated with the Mps1 inhibitor reversine (500 nM, Cayman Chemical) or vehicle control [dimethyl sulfoxide (DMSO)] for 24 hours, washed three times in 1XPBS, then fixed or harvested 60 hours later.
To investigate micronuclear ubiquitination, cells were treated with E1 inhibitor (E1i) TAK243 (25 μM, Selleck Chemicals) or vehicle control (DMSO) for 1 hour before fixation.
To investigate micronuclear removal via autophagy, cells were treated with either SAR405 (1 μM), Bafilomycin A1 (100 nM), chloroquine (50 μM), or vehicle control (DMSO) or starved with Hank’s balanced salt solution (HBSS 1X, Gibco) for 6 hours or 24 hours before fixation.
To investigate autophagic involvement in ESCRT-III activity on micronuclei, cells were treated with either SAR405 (1 μM) for 12 hours, Bafilomycin A1 (100 nM) for 6 hours, chloroquine (50 μM) for 12 hours, or vehicle control (DMSO) before fixation.
To investigate ROS and p62 interplay in hTERT-RPE1 and MDA-MB-231 cell lines, cells were treated with either NAC (10 mM) for 12 hours, H2O2 300 μM for 6 hours, or vehicle control (H2O) before fixation.
To investigate autophagic removal of CHMP7, cells were treated with Bafilomycin A1 (100 nM) for 6 hours, before fixation.
To investigate p62 and ROS interplay in modulating micronuclear integrity in HeLa cells, both the conditional KO and the expression of p62 were induced with 1 mg/ml doxycycline for 72 hours. Then, cells were treated with either H2O2 300 μM (or the control vehicle, H2O) for 4 hours, or with NAC 5 mM (or the control vehicle, H2O) for 24 hours.
For FISH experiments, doxycycline (DOX) and auxin (indole-3-acetic acid, IAA) (Sigma-Aldrich) were dissolved in cell culture-grade water and used at 1 μg/ml and 500 μM, respectively.
KO cell lines generation
p62 KO U2OS cell line was generated by CRISPR-Cas9 with a double transfection of SQSTM1 CRISPR-Cas9 KO (Santa Cruz no. sc-400099) and SQSTM1 HDR plasmids (Santa Cruz no. sc-400099-HDR) using Lipofectamine 3000 according to manufacturer’s instructions (Life Technologies), followed by selection with puromycin (1 μg/ml).
p62 KO hTERT-RPE1, MDA-MB-231, and DLD-1 cell lines were generated by CRISPR-Cas9 using the following three sequences of single-guide RNAs (sgRNAs): sgRNA no. 1 GACTTGTGTAGCGTCTGCGA, sgRNA no. 2 TCAGGAGGCGCCCCGCAACA, sgRNA no. 3 TAGTGCGCCTGGAAGCCGCC (Sigma-Aldrich). sgRNAs were cloned in a PX458 vector (GFP-tagged) to generate hTERT-RPE1 and MDA-MB-231 KO cell lines and in Lenti-Cas9-gRNA-TagBFP2 vector to generate DLD-1 KO cell line.
Plasmid expression
p62-GFP– and p62-GFP–deleted constructs were generated from HA-p62 (Addgene no. 28027) and cloned in a pEFGP-C1 vector by VectorBuilder. For micronuclei analysis, hTERT-RPE1 WT and p62 KO and HeLa cells were transfected with Lipofectamine 3000 according to manufacturer’s instructions (Thermo Fisher) and analyzed 48 hours later. To generate hTERT-RPE1 H2B-RFP cells stably expressing p62-GFP, cells were selected using G418 (800 μg/ml) and positive-cells were sorted using FACSJazz flow cytometer (BD). DFCP1 construct was previously described. For micronuclei analysis, hTERT-RPE1 cells were transfected with Lipofectamine 3000 according to manufacturer’s instructions (Thermo Fisher) and analyzed 48 hours later.
APEX2 and APEX2-p62 constructs were cloned in a pLV-FLAG vector by VectorBuilder. HEK293T cells were transfected using calcium/phosphate precipitation with lentiviral constructs pLV-APEX2 and pLV-APEX2-p62 and after a double cycle of infection, HEK293T cells were selected with puromycin (1 μg/ml).
FLAG-p62 (p62-WT) and FLAG-C105A, C113A-p62 (p62-CA) constructs were previously described and were received from V. Korolchuk (Newcastle University, UK). HEK293T cells were transfected using calcium/phosphate precipitation with lentiviral constructs pLENTI6/V5-DEST FLAG-p62 and pLENTI6/V5-DEST FLAG-C105A, C113A-p62 and after a double cycle of infection, hTERT-RPE1 cells were selected with blasticidin (5 μg/ml).
To investigate p62 and ROS interplay in modulating micronuclear integrity, p62 overexpression was obtained through transfection of 500 ng of HA-p62 plasmid (Addgene no. 28027) using the reverse transfection protocol with Lipofectamine 2000 according to manufacturer’s instructions (Thermo Fisher) on HeLa cells bearing the conditional KO for the indicated proteins [as described in (48)].
RNA interference
hTERT-RPE1 and MDA-MB-231 cells were transfected with ATG7 (Dharmacon no. L-020112-00-0005), cGAS (Dharmacon no. L-015607-02-0005), p62/SQSTM1 (Dharmacon no. L-010230-00-0010), or nontargeting (Dharmacon no. D-001810-10-20) smartPool siRNAs at a final concentration of 20 nM by using Lipofectamine RNAiMax transfection reagent (Thermo Fisher) according to the manufacturer’s instructions. Cells were analyzed 72 hours after scGAS and sip62 and 48 hours after siATG7.
IF
Cells were plated onto coverslips coated with 5 μg/ml fibronectin (Sigma-Aldrich). Cells were washed in 1XPBS and fixed with 4% paraformaldehyde (in 1XPBS, Santa Cruz) for 15 min at room temperature (RT). Cells were permeabilized using 1XPBS-0,5% Triton X-100 for 10 min at RT and blocked in 5% bovine serum albumin (BSA) in 1XPBS for 30 min at RT. Then, cells were incubated with primary antibodies diluted in the same buffer for 90 min at RT: CHMP2A (Proteintech no. 10477-1-AP), CHMP4B (Proteintech no. 13683-1-AP), CHMP7 (Proteintech no. 16424-1-AP), cGAS (Cell Signaling no. D1D3G), emerin (Proteintech no. 10351-1-AP), FIP200 (Invitrogen no. PA5-28563), FLAG (Sigma-Aldrich no. F1865), H3K9ac (Active Motif no. 39585), H3K9me3 (Abcam no. ab8898), H3K27me2me3 (Active Motif no. 39536), lamin A (Abcam no. ab8980), lamin B1 (Abcam no. ab16048), LAMP2 (Santa Cruz no. 18822), LBR (Abcam no. ab32535), LBR (Sigma-Aldrich no. SAB1400151), LC3 (Cell Signaling no. L7543), LSD1 (Cell Signaling no. 2139), NBR1 (Cell Signaling no. 9891), NDP52 (Abcam no. ab68588), NPCs mAb414 (Abcam no. ab24609), p62 (Santa Cruz no. 28359), p62 (Enzo no. BML-PW9860-0100), RelA (Santa Cruz no. 8008), RelB (Abcam no. ab180127), TAX1BP1 (Sigma-Aldrich no. HPA024432), Ub FK2 (Enzo no. BML-PW8810), Ub poly-Lys K48 (Millipore no. 05-1307), Ub poly-Lys K63 (Millipore no. 05-1308), Ub poly-Lys K63 (Genentech), WIPI (Abcam no. ab105459). After three washes with 1XPBS, coverslips were incubated with secondary antibodies [for confocal microscopy, Alexa-fluor 488–, Alexa-fluor 555–, Alexa-fluor 647–labeled anti-rabbit or anti-mouse or anti-human (Jackson ImmunoResearch); for super-resolution microscopy, ATTO 594– and ATTO 647–labeled anti-rabbit or anti-mouse secondary antibodies (AttoTech)] and 4′,6-diamidino-2-phenylindole (DAPI) diluted in the same buffer for 45 min at RT. After three washes with 1XPBS, and one wash with ddH2O, cells were mounted using Mowiol (Sigma-Aldrich).
For micronuclei analyses, confocal images were acquired by using Leica DMi8 (inverted) SP8 confocal microscope, controlled by Leica confocal software. 63x/1.4 oil-immersion objective was used. Images were acquired as a tile scan of Z-sections per image and a pixel size of 90 nm and a Z-step size of 0.3 μm. To evaluate micronuclei-mediated inflammation effects, confocal images were acquired by using Leica DMi8 (inverted) SP8 confocal microscope, controlled by Leica confocal software. 40x/1.3 oil-immersion objective was used. Images were acquired as a tile scan of Z-sections per image and a pixel size of 90 nm and a Z-step size of 0.5 μm. To count micronuclei, wide-field images were acquired by using Leica DM6 B wide-field fluorescence microscope, controlled by Leica confocal software. 40x/0.95 dry objective was used. Super-resolution images were acquired using Leica CLSM TCS SP8 STED, controlled by Leica confocal software. 100x/1.4 oil-immersion objective was used. Images were acquired as a tile scan of Z-sections per image and a pixel size of 11.27 nm and a Z-step size of 0.07 μm. To investigate p62 and ROS interplay in modulating micronuclear integrity in HeLa cells, wide-field images were acquired by using Zeiss LSM880 microscope equipped of a 63x objective, using 10% illumination intensity of an EPI-fluorescence lamp [as described in (48)]. FIJI software was used for image processing of confocal and wide-field microscope images. Super-resolution images were deconvoluted using Huygens Professional Software, then Las X software was used for three-dimensional (3D) reconstruction.
IF analysis
Protein localization to micronuclei (e.g., p62, ubiquitin, autophagic components, ESCRT-III proteins), micronuclear integrity (LSD1 and LBR), and micronuclei-associated features (as NE components) were analyzed by selecting the best Z-stack for micronucleus visualization (using DAPI) and then manually scoring the signal on this specific Z-stack. Proteins localizing as puncta were scored as positive if a signal was observed within the micronucleus (visualized by DAPI). Diffuse nuclear proteins were scored as positive if the signal was observed within the whole micronucleus (visualized by DAPI). LBR was scored as enriched if the signal was higher compared with the one of the primary nuclei and observed within the whole micronucleus (visualized by DAPI).
Evaluation of protein levels within cellular compartments
To evaluate levels of proteins within primary nuclei and micronuclei, DAPI masking was used to identify nuclear objects and manually revised, then primary nuclei and micronuclei were categorized based on their size. Cytoplasm region was manually drawn based on the signal of cytoplasmic proteins (as ubiquitin), and by excluding nuclear objects. Finally, the mean intensities of the specific regions of interest (ROIs) were measured in the specific channel of interest.
Evaluation of micronuclear geometric features
DAPI masking was used to identify nuclear objects and manually revised, then micronuclei were categorized based on their size. The mean intensities of specific proteins, area, and circularity score were then measured in the identified ROIs.
Evaluation of protein colocalization with p62 on micronuclei
Upon identification of a p62-positive micronucleus, a line (thickness = 1 pixel) was drawn from the outside to the inside of the micronucleus, encompassing the p62 signal. Then the line scan plots of p62 and of the other protein/s were analyzed. By manually reviewing the plots, the peaks of the proteins were identified as coordinates of the maximum value identified. Colocalization was scored as follows: distance between the 2 peaks < 90 nm (1 pixel) = colocalization, distance between the 2 peaks > 90 nm (1 pixel) = no colocalization.
Evaluation of p62 localization within micronuclear cavities
Upon identification of a p62-positive micronucleus, a line (thickness = 2 pixels) was drawn from the outside to the inside of the micronucleus, encompassing the p62 signal. By manually reviewing the line scan, the peak of p62 was identified as coordinates of the maximum value and width of the peak: “p62-positive region” was identified as the region representing the p62 peak (width 3 or 4 pixels) and “p62-negative region” was identified as a region of the same width (3 or 4 pixels) in close proximity to the end of the peak (1 or 2 pixels of distance). In the DAPI channel, the mean intensities of the p62-positive and p62-negative regions were measured as area under the curve. Finally, a ratio of the p62-positive and p62-negative region was calculated. In p62-negative micronuclei, a line (thickness = 2 pixels) was randomly drawn from the outside to the inside of the micronucleus, then the coordinates of the peak and the different ROIs were randomly selected, and the mean intensities and the ratio were calculated as above mentioned. Ratios (R) were scored as follows: R ≥ 1 region of the peak enclosed within DNA, thus no cavity; R < 1 region of the peak excluded from DNA, thus enclosed within a cavity.
DeepSIM imaging analysis
Cells were plated onto coverslips coated with 5 μg/ml fibronectin (Sigma-Aldrich). Mitochondria were stained by adding MitoTracker Red CMXRos (M7512) 1:10,000 in the cultured medium for 40 min at 37°C in 5% CO2. Then, cells were washed with fresh medium and incubated for 5 min at 37°C in 5% CO2. Finally, cells were fixed with 4% paraformaldehyde (in 1XPBS, Santa Cruz) for 15 min at RT and stained as mentioned above. Cells were imaged by the DeepSIM super-resolution module (CrestOptics S.p.A.) mounted on an Eclipse Ti2 fluorescence microscope (Nikon Europe B.V.) equipped with solid-state lasers (Lumencor Celesta light engine), a sCMOS camera (Kinetix, Teledyne Photometrics) and a 100x/1.49 NA oil immersion objective lens (Nikon Europe B.V.). The standard structured illumination mask (CrestOptics S.p.A.) was used and 17 images per channel per plane were acquired. Overall, four channels and 21 Z planes spaced of 0.15 μm were acquired. The super-resolution reconstructed images were analyzed with Arivis4D scientific image analysis software (Carl Zeiss Microscopy Software Center Rostock GmbH). The DAPI channel was duplicated to segment both micronuclei and cavities using Watershed method and a customized trained machine learning algorithm, respectively. p62 signal was used to segment p62 particles through a Blob Finder method while MitoTracker signal was used to segment mitochondria through an Intensity Threshold method. Segmented micronucleus objects were manually revised. Then, the distance between the cavities’ edges and the nearest p62 particles’ edges in 3D space was measured to categorize the cavities into p62-positive and p62-negative classes by using a 200-nm threshold (p62-positive, distance ≤ 200 nm; p62-negative, distance > 200 nm). Subsequently, the distance between the cavities’ edges and the closest mitochondria in the 3D space was measured.
Lysosome content analysis
Cells were imaged with spinning-disk X-Ligth V3 module (CrestOptics S.p.A.) mounted on an Eclipse Ti2 fluorescence microscope (Nikon Europe B.V.) equipped with solid-state lasers (Lumencor Celesta light engine), a sCMOS camera (Kinetix, Teledyne Photometrics) and a 100x/1.49 NA oil immersion objective lens (Nikon Europe B.V.). Overall, three channels and 25 Z planes spaced 0.2 μm were acquired. Images were deconvolved using Blind algorithm method (30 iterations) within the deconvolution module of NIS-elements software (Nikon Europe B.V.). The deconvoluted images were analyzed with Arivis4D scientific image analysis software (Carl Zeiss Microscopy Software Center Rostock GmbH). DAPI, LAMP2, and CHMP7 channels were used to segment nuclei, lysosomes, and CHMP7 particles, respectively. Nuclei and lysosomes were segmented using Intensity Threshold method while CHMP7 particles using Blob Finder method. Nuclei of cells without micronuclei were manually excluded. For each cell, CHMP7 particles and lysosomes with a distance ≤15 μm from the nucleus were considered for the analysis. Subsequently, the distance of CHMP7 particles’ center from the lysosome border was measured, CHMP7 particles with distances ≤0 were considered inside lysosomes. For each field of view, the percentage of CHMP7 particles inside lysosomes was calculated and plotted as a single point.
IF and DNA FISH
hTERT-RPE1 cells were grown on poly-L-lysine–coated coverslips and arrested in G1 by addition of 1 uM PD-0332991 isethionate (Cdk4/6i; Sigma-Aldrich) for 24 hours. Cells were released by washing three times in 1XPBS before incubation in 100 nM BAY-1217389 (Mps1i; Fisher) for an additional 24 hours to yield a cell population enriched in G1/S cells. Coverslips were fixed in 100% methanol at −20°C for 10 min. Coverslips were blocked in 3% BSA + 0.1% − 0.4% Triton X-100 + 0.02% sodium azide in 1XPBS for 30 min, then incubated with primary antibodies: CREST (Antibodies Incorporated no. 15-234), p62 (Santa Cruz no. 28359) for 30 min at RT. Coverslips were washed three times in 1XPBS and incubated in the following secondary antibodies for 30 min at RT: Alexa Fluor 488–conjugated goat anti–mouse 1:500 (Thermo Fisher no. A-11029), Alexa Fluor 647–conjugated goat anti-human 1:1000 (Thermo Fisher no. A-21445). Coverslips were refixed for 5 min with 4% paraformaldehyde (Electron Microscopy Services) in 1XPBS. Coverslips were washed twice with 2x SSC for 5 min, then permeabilized with 0.2 M HCl + 0.7% Triton X-100 for 12 min at RT. Coverslips were denatured in 50% formamide 2x SSC for 1 hour, then inverted onto 3.5 μL of Spectrum Orange XCE or XCP probe (MetaSystems) and sealed with rubber cement. Probes and targets were codenatured at 75°C for 2 min and hybridized 2 hours (HSA 17, HSA 18) or overnight (HSA 19) at 37°C in a humidified chamber. Coverslips were washed once in preheated 1x SSC buffer at 74°C for 5 min then twice in 2x SSC + 0.05% Tween-20 for 2 min. Coverslips were incubated in DAPI (1 μg/ml in 1XPBS; Roche) for 5 min and mounted in Vectashield (VectorLabs). Confocal image stacks were acquired with a Leica DMi8 laser scanning confocal microscope using the Leica Application Suite (LAS X) software and a Leica ACS APO 40x/1.15 Oil CS, or ACS APO 63x/1.30 Oil CS objective. Images were acquired as a tile scan of 10 z-sections per image and a pixel size between 60 and 80 nm and a z-step size of 0.5 μm. Sum intensity projections were overlaid without merging at edges and presence (higher than background) or absence of p62 was recorded for each micronucleus. Single chromosome micronucleus contained a single CREST focus and a single FISH focus of the chromosome of interest. Micronuclei calls were validated using the full z-stack.
Evaluation of p62 localization after primary nuclear rupture
U2OS cells expressing 3xGFP-NLS and shRNAs against lamin B1 were arrested for 24 hours with 2 mM hydroxyurea (Sigma-Aldrich) and then fixed in 4% paraformaldehyde in 1XPBS for 5 min. Cells were blocked with 3% BSA + 0.4% Tx100 for 30 min at RT, followed by incubations with primary antibodies for 30 min at RT: p62 (Santa Cruz no. 28359) and lamin A (Sigma-Aldrich no. L1293), then with secondary antibodies for 30 min at RT (Alexa 568– and Alexa 647–labeled anti-rabbit or anti-mouse; Thermo Fisher). Confocal images were acquired with a Leica DMi8 laser scanning confocal microscope using the Leica Application Suite (LAS X) software and with the Leica ACS APO 40x/1.15 Oil CS objective. Postacquisition, images were cropped and levels adjusted through Adobe Photoshop to make use of the entire histogram spectrum. In addition, a gamma correction was performed on the GFP-NLS channel to highlight the cytoplasmic GFP-NLS signal. For quantification, rupture sites were defined as the location of highest nuclear GFP-NLS loss (rupturing nuclei) or as a lamin A focus [ruptured nuclei (55)]. Any overlap between p62 signal and a lamin A focus or herniated chromatin in a rupturing nucleus was scored as positive.
FISH of Y chromosome
Cells were treated with 100 ng/ml colcemid (KaryoMAX, Thermo Fisher) for 4 hours before harvesting by mitotic shake-off. Cells were resuspended in 75 mM KCl hypotonic solution for 6 min at 37°C, then pelleted and resuspended in freshly prepared 3:1 methanolglacial acetic acid and dropped onto slides. DNA FISH probes (MetaSystems) were applied to metaphase spreads and sealed with a coverslip using rubber cement. Slides were codenatured on a heat block at 75°C for 2 min and then hybridized at 37°C in a humidified chamber overnight. The next day, coverslips were removed, and the slides were washed with 0.4X SSC at 72°C for 2 min and rinsed with 2X SSC with 0.05% Tween-20 at RT for 30 s. After washing, slides were counterstained with DAPI, air dried, and mounted in ProLong Gold antifade mounting solution. DNA FISH images were captured on a DeltaVision Ultra (GE Healthcare) microscope system equipped with 4.2 MPx sCMOS detector, at 60x magnification (PlanSApo, 1.4 NA) and a Z-sectioning of 5/10 × 0.2-μm z-section was performed. Y chromosome fragmentation and rearrangements were scored as previously described (15, 56). Y chromosome–positive metaphase spreads were manually scored for fragmentation based on three criteria: (i) Y chromosome paint signal must colocalize with DAPI-positive fragments, (ii) each fragmentation event must generate at least three distinct Y chromosome fragments, and (iii) at least one acentric Y fragment must be generated. Chromosomal rearrangement frequencies were scored as described in (12); Y chromosome–positive metaphases were imaged and analyzed for structural chromosomal abnormalities when compared with the parental DLD-1 karyotype.
Live-cell imaging
Cells were plated in 12-well plates with glass bottom (MatTek), previously coated with 5 μg/ml fibronectin (Sigma-Aldrich). Cells were treated as above-mentioned, Mps1i reversine was added in fresh medium without phenol red (Euroclone). Imaging was performed using a Nikon Eclipse Ti inverted using a 40x/0.95 dry objective controlled by NIS software. No binning was applied. For the entire observation period, cells were kept in an incubated microscope stage at 37°C and 5% CO2. Cells were filmed for 24 hours and images acquired every 15 min. For each frame, red fluorescent protein (RFP) and GFP were imaged, and three Z-stacks were acquired in a 10-μm range. 3×3 fields of view with a 2% overlap were acquired for each well. For the analysis, single Z-stacks were chosen and large images comprising all fields of view (3×3) were generated using the FIJI software Stitching Plugin. Then, images were processed and analyzed using FIJI software.
FRAP
Cells were plated on 3-mm dishes with glass bottom (MatTek) coated with 5 μg/ml fibronectin (Sigma-Aldrich). Cells were treated as above-mentioned, and after Mps1i reversine wash out, fresh medium without phenol red (Euroclone) was added on the cells. Cells were imaged using Leica SP8 confocal microscope with a magnification objective of 63x, equipped with an incubation chamber at 37°C and 5% CO2. The experiment was performed using FRAP Mode of Leica LasX Software. Three images were acquired prebleach, then GFP was bleached using laser line 488 at 30% for 30 ms, postbleach images were acquired every 300 ms up to 10 s PB (postbleaching), every 1 s up to 1 min PB, every 10 s up to 5 min PB. For the entire observation period, cells were kept in an incubated microscope stage at 37°C and 5% CO2. For the analysis, the ROI containing the GFP signal within the bleached area (B), the ROI containing the GFP signal inside the cell outside the bleached area (NB), the ROI containing the background outside the cells (BG) were determined using FIJI software. The mean intensity (I) of the ROIs was measured in all the frames acquired. GFP intensity values (I measured in a.u., arbitrary units) of the ROI (B) was calculated as follow: (IB-IBG)/(INB-IBG). Using GraphPad Prism software, the exponential curve of recovery was generated from the mean intensity values and frame times. One-phase association was used to analyze nonlinear fitting curve, and calculate plateau, tau, and half-time. FIJI software was used for image processing.
CLEM and immunogold labeling of p62
Cells seeded at low confluency on 3-mm dishes with glass bottom (MatTek) and previously imaged at the confocal microscope were fixed for1 hour at RT in 4% paraformaldehyde electron microscopy (EM)–grade in 0.2 M HEPES buffer. After three washes in 1XPBS, cells were incubated 10 minutes with 50 mM glycine, permeabilized for 10 min with 0.25% saponin, 0.1% BSA in 1XPBS, and blocked 1 hour in blocking buffer (5% goat serum, 0.2% bovine serum albumin, 0.1% saponin, 50 mM NH4Cl, 20 mM PO4 buffer, 150 mM NaCl). Cells were then incubated with anti-p62 (Santa Cruz no. 28359) primary antibody for 1 hour at RT, washed (0.1% BSA, 0.1% saponin in 1XPBS) and incubated for1 hour at RT with secondary antibodies conjugated with nanogold (Nanoprobes). Samples were then fixed with 1% glutaraldehyde for 30 min and nanogold was enlarged with a gold enhancement solution (Nanoprobes) according to manufacturer’s instructions. Cells were then postfixed in 1% osmium tetroxide, 1,5% potassium ferricyanide in 100 mM sodium cacodylate buffer for 1 hour on ice. After rinsing in sodium cacodylate buffer, the samples were washed with distilled water for five times and stained with 0.5% uranyl acetate in distilled water for an overnight incubation at 4°C keeping them shielded from light. Lastly, the specimens were once again rinsed five times in distilled water, gradually dehydrated using increasing concentrations of ethanol, and embedded in Epon before curing at 60°C for 48 hours. Once embedding was complete, the cells of interest imaged by confocal microscopy were identified using the reference coordinate system present on the MaTtek chamber and sectioned with an ultramicrotome (UC7, Leica microsystem). Ultrathin sections (70 nm) were collected, stained with uranyl acetate and Sato’s lead solutions, and observed with a TALOS L120C Transmission Electron Microscope (Thermo Fisher) while images were acquired with a CETA 4×4k CMOS camera (Thermo Fisher). FIJI software with the BigWarp plugin was used for image processing and alignment.
CLEM and immunogold labeling of LBR
Cells seeded at low confluency on 3-mm dishes with glass bottom (MatTek) and previously imaged at the confocal microscope were fixed with a mixture of 4% paraformaldehyde and 0.05% glutaraldehyde (EMS) in 0.15 M HEPES (pH 7.3) for 5 min at RT and then replaced with 4% paraformaldehyde in 0.15 M HEPES (pH 7.3) for 30 min. Afterward, the cells were washed three times in 1XPBS and incubated with blocking solution for 30 min at RT. Then, cells were incubated with the primary antibody (anti-LBR, Abcam no. ab32535) diluted in blocking solution overnight at 4°C. On the following day, the cells were washed three times with 1XPBS and incubated with goat anti-rabbit Fab’ fragments coupled to 1.4-nm gold particles (diluted in blocking solution 1:100) for 2 hours and washed six times with 1XPBS. Meanwhile, the activated Gold Enhance TM-EM was prepared according to the instructions and 250 μl were added into each sample well. The reaction was monitored by a conventional light microscope and was stopped after 5 to 10 min when the cells had turned “dark enough” by washing several times with 1XPBS. Then cells were fixed with of 4% paraformaldehyde and 2.5% glutaraldehyde (EMS) mixture in 0.2 M sodium cacodylate pH 7.2 for 2 hours at RT, followed by six washes in 0.2 sodium cacodylate pH 7.2 at RT. Then, cells were incubated in 1:1 mixture of 2% osmium tetra oxide and 3% potassium ferrocyanide for 1 hour at RT followed by six times rinsing in cacodylate buffer. The samples were sequentially treated with 0.3% thiocarbohydrazide in 0.2 M cacodylate buffer for 10 min and 1% OsO4 in 0.2 M cacodylate buffer (pH 6.9) for 30 min. Then, samples were rinsed with 0.1 M sodium cacodylate (pH 6.9) buffer until all traces of the yellow osmium fixative have been removed. The samples were subsequently subjected to dehydration in ethanol and embedded in epoxy resin at RT and polymerized for at least 72 hours in a 60°C oven. Embedded samples were then sectioned with diamond knife (Diatome) using Leica EM UC7 ultramicrotome. Sections were analyzed with a Tecnai20 High Voltage EM (Thermo Fisher) operating at 200 kV. Images were then processed with FIJI software.
CLEM and tomography
Cells were plated on 3-mm dishes with Grid glass bottom (MatTek) coated with 5 μg/ml fibronectin (Sigma-Aldrich), fixed with paraformaldehyde 4% + 0.05% glutaraldehyde in 0.15 M HEPES (pH 7.2 to 7.4) for 5 min at RT and then three times with paraformaldehyde 4% in 0.15 M HEPES (pH 7.2 to 7.4) for 10 min at RT. IF staining was performed as described above using a blocking and permeabilization solution of 5% BSA + 1% saponin (also used to dilute antibodies). Images were acquired using Leica SP8 confocal microscope (20 to 63x magnification objectives) and processed with FIJI software. Then, cells were washed three times in 1XPBS, incubated with blocking solution for 30 min at RT and with primary antibodies diluted in blocking solution overnight at 4°C. After three washes with 1XPBS, cells were incubated with goat anti-Rb or -Ms Fab’ fragments coupled to 1.4-nm gold particles (diluted in blocking solution 1:100) for 2 hours, washed six times with 1XPBS and finally activated Gold Enhance TM-EM solution (prepared as manufacturer’s instructions) was added. The reaction was monitored using a light microscope and stopped after 5 to 10 min (when cells had turned “dark enough”) by washing several times with 1XPBS. Then, cells were fixed with a 4% paraformaldehyde and 2.5% glutaraldehyde (EMS, USA) mixture in 0.2 M sodium cacodylate pH 7.2 for 2 hours at RT, washed six times with 0.2 sodium cacodylate pH 7.2 at RT, incubated in 1:1 mixture of 2% osmium tetra oxide and 3% potassium ferrocyanide for 1 hour at RT, rinsed six times in cacodylate buffer, treated with 0.3% thiocarbohydrazide in 0.2 M cacodylate buffer for 10 min, 1% OsO4 in 0.2 M cacodylate buffer (pH 6.9) for 30 min and rinsed with 0.1 M sodium cacodylate (pH 6.9) buffer until all traces of the yellow osmium fixative have been removed. Samples were subsequently subjected to dehydration in ethanol, embedded in epoxy resin at RT and polymerized for at least 72 hours at 60°C. Embedded samples were then sectioned with diamond knife (Diatome) using Leica EM UC7 ultramicrotome (cutting 70-nm serial thin sections and 200-nm serial semithick sections) and collected onto 1% Formvar films adhered to slot grids labeled with fiduciary 10-nm gold. Sections were analyzed with a Tecnai20 High Voltage EM (Thermo Fisher) at 200 kV at a magnification of 9600 to 25,000x.
Quantitative immunolocalization analyses
Human breast and ovarian cancer tissue samples for immunolocalization analyses were selected from the archives of the Tumor Immunology Laboratory of the University of Palermo. Samples were collected and handled according to the Helsinki Declaration, and the study was approved by the University of Palermo Ethical Review Board (approval no. 04/2023).
4-μm-thick sections of human tissues were deparaffinized, rehydrated, and unmasked using Novocastra Epitope Retrieval Solutions at pH 9 (Leica Novocastra) in a thermostatic bath at 98°C for 30 min. Subsequently, the sections were brought to RT and washed in 1XPBS. After neutralization of the endogenous peroxidase with 3% H2O2 and Fc blocking by 0.4% casein in 1XPBS (Leica Novocastra), double-marker IF was carried out by incubation with the primary antibodies cGAS (1:100, overnight at 4°C; Cell Signaling no. D1D3G) and p62 (1:100, 90 min at RT; Abnova no. H00008878-M01). The binding of the primary antibodies to their respective antigenic substrates was revealed by made-specific secondary antibodies conjugated with Alexa-488 (Life Technologies, 1:250) and Alexa-568 (Life Technologies, 1:300) fluorochromes. The slides were counterstained with DAPI.
The slides were analyzed under a Zeiss Axioscope A1 microscope equipped with four fluorescence channels widefield IF. Microphotographs were collected using a Zeiss Axiocam 503 Color digital camera with the Zen 2.0 Software (Zeiss). Quantitative analyses of IF stainings were performed by calculating the percentage of cGAS+ and cGAS+/p62+ micronuclei in 10 nonoverlapping fields at a medium magnification (x200) using HALO image analysis software (v3.2.1851.229, Indica Labs).
RNA extraction, reverse transcription polymerase chain reaction (RT-PCR), and quantitative PCR (qPCR)
RNA was extracted from cells using RNeasy Plus Mini Kit (QIAGEN), according to manufacturer’s protocol. 500 ng of RNA from each sample was reverse-transcribed using OneScript Plus cDNA Synthesis Kit (abm) according to the manufacturer’s instructions. mRNA expression was performed by real-time quantitative PCR reactions using Fast SYBR Green reaction mix (Thermo Fisher) and achieved on an Applied Biosystems 7500 Fast Real-time PCR system. The relative expression level was calculated with the 2[DDCt] method and expressed as a “fold change”: Normalization of data was performed on house-keeping gene (GAPDH) expression and compared with the respective controls. Primers used for profiling the mRNA expression levels of genes are as follows: GAPDH Fw: 5-CAACTACATGGTTTACATG-3, Rv: 5-GCCAGTGGACTCCACGAC-3; OAS2 Fw: 5-GAGCCAGTTGCAGAAAACCAG-3, Rv: 5-GCATTGTCGGCACTTTCCAA-3; OAS3 Fw: 5-GAAGCCCAGGCCTATCATCC-3, Rv: 5-TCATCCAGTAGGACCGCTGA-3; MX1 Fw: 5-TGGCATAACCAGAGTGGCTG-3, Rv: 5-CCACATTACTGGGGACCACC-3.
Western blot analysis
Cells were lysed in 1XRIPA lysis buffer (Cell Signaling) with the addition of protease inhibitor cocktail (Millipore), phosphatase inhibitor (Roche) and then sonicated. Protein lysates were centrifuged at maximum speed for 15 min and resolved on SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels. The following primary antibodies were used: FLAG (Sigma Aldrich no. 3165), GAPDH (Santa Cruz no. 32233), GFP (Roche no. 11814460001), H3 (Cell Signaling no. 4499), p62 (Santa Cruz no. 28359), Ub FK2 (Enzo no. BML-PW8810), Ub K48 (Millipore no. 05-1307), Ub K63 (Millipore no. 05-1308), and vinculin (Sigma Aldrich no. V9131). Blots were imaged using Chemidoc XRS and quantified using ImageLab Software.
Reducing and nonreducing Western blot analysis
Cells were lysed in 1XRIPA lysis buffer [150 mM NaCl, 1% NP40, 0.5% NaDoC, 0.1% SDS, 50 mM Tris pH 7.4, supplemented with protease inhibitor cocktail (Millipore), phosphatase inhibitor (Roche)] added with 50 mM N-ethylmaleimide, which interacts with reduced cysteines and prevents new disulfide bond formation. Then, cell lysates were centrifuged at 4°C at maximum speed for 10 minutes. Samples were prepared by boiling in SDS-loading buffer at 100°C for 5 min in the presence or absence of 2.5% β-ME (β-mercaptoethanol, Sigma). Proteins were resolved on SDS-PAGE gels. The following primary antibodies were used: H3 (Cell Signaling no. 4499) and p62 (Santa Cruz no. 28359). Blots were imaged using Chemidoc XRS and quantified using ImageLab Software.
Micronuclei and primary nuclei purification
The protocol was adapted from previously described methods (15, 35). HEK293T cells were seeded and treated with Mps1i for 48 hours at 37°C under 5% CO2. After 48 hours, cells (at least 500,000,000 per condition for each biological replicate) were harvested using trypsin and washed twice in DMEM without serum. Washed cells were resuspended in prewarmed (37°C) DMEM without serum supplemented with cytochalasin B (Cayman) at 10 μg/ml at a concentration of 107 cells/ml DMEM and incubated at 37°C for 30 min. Cells were centrifuged at 300 g for 5 min and the cell pellet was resuspended in cold lysis buffer [10 mM Tris-HCl, 2 mM Mg-acetate, 3 mM CaCl2, 0.32 M sucrose, 0.1 mM EDTA, 0.1% (v/v) NP-40, pH 8.5] freshly complemented (with 1 mM DTT, 0.15 mM spermine, 0.75 mM spermidine, 10 μg/ml cytochalasin B and protease inhibitors) at a concentration of 2 × 107 cells/ml lysis buffer. Resuspended cells were then dounce homogenized by 10 strokes with a loose-fitting pestle (Wheaton). Cell lysates were mixed with an equal volume of ice-cold 1.8 M sucrose buffer (10 mM Tris-HCl, 1.8 M sucrose, 5 mM Mg-acetate, 0.1 mM EDTA, pH 8.0) freshly complemented (with 1 mM DTT, 0.3% BSA, 0.15 mM spermine, 0.75 mM spermidine) before use. Then, 10 ml of the mixture (1:1 lysate and 1.8 M sucrose buffer) was slowly laid on top of a sucrose cushion freshly prepared (20 ml 1.8 M sucrose buffer as above at the bottom and 15 ml of 1.4 M sucrose buffer on the top layer) in a 50-ml Falcon tube. Sucrose gradients were centrifuged at 944 g for 20 min at 4°C. Generally, fractions were collected as follows: upper 3 ml typically contains debris and was discarded; next 6 ml contains micronuclei and was collected; next 6 ml contains a mixed population of micronuclei and primary nuclei and was discarded; next 10 ml contains primary nuclei and was collected. Then, to-be-used fractions were diluted fivefold with 1XPBS and centrifuged at 944 g for 20 min at 4°C. Finally, the pellets were washed once with 1XPBS and stored at −80°C.
Proximity labeling and micronuclei purification
Proximity biotinylation assay was performed on HEK293T cells stably expressing APEX2 and APEX2-p62 before micronuclei isolation and biotinylated protein pull-down, in accordance to previously published protocols (46). HEK293T cells were seeded and treated with Mps1i for 48 hours at 37°C under 5% CO2. After 48 hours, biotin-phenol labeling was initiated by adding to the cell media 500 μM biotin-phenol (Iris-Biotech), incubated at 37°C under 5% CO2 for 30 min. Afterward,1 mM H2O2 (Sigma) was added to the cells and the plates were gently agitated for 1 min at RT. The reaction was then quenched by washing three times with a freshly prepared quencher solution (5 mM Trolox, 10 mM sodium ascorbate, 10 mM sodium azide). Then, cells (at least 500,000,000 per condition for each biological replicate) were harvested using trypsin and washed twice in DMEM without serum. Washed cells were resuspended in prewarmed (37°C) DMEM without serum supplemented with cytochalasin B (Cayman) at 10 μg/ml at a concentration of 107 cells/ml DMEM and incubated at 37°C for 30 min. Cells were centrifuged at 300 g for 5 min and the cell pellet was resuspended in cold lysis buffer [10 mM Tris-HCl,2 mM Mg-acetate, 3 mM CaCl2, 0.32 M sucrose, 0.1 mM EDTA, 0.1% (v/v) NP-40, pH 8.5] freshly complemented (with 1 mM DTT, 0.15 mM spermine, 0.75 mM spermidine, 10 μg/ml cytochalasin B and protease inhibitors) at a concentration of 2 × 107 cells/ml lysis buffer. Resuspended cells were then dounce homogenized by 10 strokes with a loose-fitting pestle (Wheaton). Cell lysates were mixed with an equal volume of ice-cold 1.8 M sucrose buffer (10 mM Tris-HCl, 1.8 M sucrose, 5 mM Mg-acetate, 0.1 mM EDTA, pH 8.0) freshly complemented (with 1 mM DTT, 0.3% BSA, 0.15 mM spermine, 0.75 mM spermidine) before use. Then, 10 ml of the mixture (1:1 lysate and 1.8 M sucrose buffer) was slowly laid on top of a sucrose cushion freshly prepared (20 ml 1.8 M sucrose buffer as above at the bottom and 15 ml of 1.4 M sucrose buffer on the top layer) in a 50-ml Falcon tube. Sucrose gradients were centrifuged at 944 g for 20 min at 4°C. Generally, fractions were collected as follows: upper 3 ml typically contains debris and was discarded; next 6 ml contains micronuclei and was collected; bottom ml contains primary nuclei and was discarded. Then, fraction of micronuclei was diluted fivefold with 1XPBS and centrifuged at 944 g for 20 min at 4°C. Finally, the pellets were washed once with 1XPBS and stored at −80°C.
Streptavidin pull-down
Following the previously published protocol (46), micronuclei were lysed in homemade 1XRIPA buffer (Tris HCl pH 8 10 mM, NaCl 150 mM, SDS 0,1%, Triton 1%, EDTA 1 mM, Na Deoxycholate 0,1%, PMSF 1 mM, DTT 1 mM, PI 1X) and then sonicated using Bioruptor (Diagenode) at high intensity. Protein lysates were centrifuged at maximum speed for 15 min and quantified with Bradford Assay (Biorad), following manufacturer’s instructions. Streptavidin-coated magnetic beads (Pierce) were washed with RIPA buffer, and 150 μg of each protein lysate samples were then incubated for1 hour at RT with 50 μl of the magnetic bead slurry. Subsequently, the beads were washed twice with 1 ml RIPA lysis buffer, once with 1 ml of 1 M KCl, once with 1 ml of 0.1M Na2CO3, once with1 ml of2M urea in 10 mM Tris-HCl pH 8.0, twice with 1 ml RIPA lysis buffer, and twice with 1 ml 1XPBS. At this point, affinity-purified biotinylated protein samples were processed for further analysis.
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis and raw data processing
In all cases, the acquired mass spectrometry (MS) raw data were analyzed using MaxQuant version 2.0.1.0 integrated with Andromeda search engine (57). False discovery rate (FDR) was set to a maximum of 1% both at peptides and protein level. Carbamidomethylcysteine and methionine oxidation were selected as fixed and variable modifications, respectively. The UniProt Human Fasta database UP000005640 (82678 entries) was specified for the search. The label-free quantification (LFQ) intensity calculation and the match between run (MBR) function were both enabled (57). The “protein groups” MaxQuant output file was analyzed using an R-based pipeline implemented in a ShinyApp web framework available at https://bioserver.ieo.it/shiny/app/qproms. Briefly, false positive hits (reverse hits from the Decoy database) and common contaminant proteins (keratin, desmoplakin, plectin, and actin) were filtered out, and 4 out of 5 valid values data completeness in at least one group was required. After data filtering the remaining missing values were imputed (58). Normalized intensities (LFQ) were log2 transformed, and proteins significantly changing between Micronuclei/Primary Nuclei and APEX-p62/APEX pull-down experiments were compared. To this aim, in both cases, a two-sample Student’s t test was used and the original P value was adjusted for an FDR of 0.05 using the Benjamini–Hochberg truncation. Threshold setting for differential protein expression was minimum set to 1 and minimum P value adjusted set to 0.05. Specific biological process enrichment and their plotting was generated in R Studio using R v4.2.0 and clusterProfiler (59, 60) v4.6.0 passing the weight algorithm and Fisher test to the runTest function. A cut off of 0.05 was applied and top 10 enriched terms were visualized with ggplot v3.4.0.
p62 correlation with chromothripsis status
Segmented copy number data of 1040 human cell lines were generated from single-nucleotide polymorphism (SNP) arrays as described in the Cancer Cell Line Encyclopedia (CCLE) (51, 53) and were downloaded directly from the DepMap (version 22q2; https://depmap.org/portal/) (50). The segmented copy number file went through the following transformations: Segment_mean column was log2 transformed and capped at ≤ |3|. Segment_Length column indicating the length of each segment was added as required by the CTLP detecetor4 input format. The segmented copy number file was then placed into the CTLP scanner (49) for chromothripsis detection with the following parameters:
Copy number status change times ≥ 10, log10 of likelihood ratio ≥ 0, signal distance between adjacent segments (log2 transformed) ≥ 0.3, segment filtration of ≥ 10,000 base pairs (bp) and human reference genome build GRCH37/hg19.
Positive cell lines were determined as those in which at least one chromosome had a score of log10 likelihood ratio ≥ 12, and negative cell lines were determined as those in which all chromosomes had a log10 likelihood ratio ≤ 6. 490 “borderline” cell lines (in between these scores) were omitted from the analysis.
mRNA expression of cell lines was downloaded from DepMap (version 22q2; https://depmap.org/portal/) (50). Values are in log2(TPM+1). 33 cell lines that did not have gene expression data were omitted from the analysis.
p62 correlation with aneuploidy
COAD TCGA mRNA expression data were accessed using the cBioPortal platform (58), and tumors were stratified based on their molecular subtype [CIN, genomically stable (GS), or MSI]. The mRNA expression for each subtype was plotted and compared using a Kruskal-Wallis test followed by Dunn’s multiple comparison test. The subtypes were ordered based on their average aneuploidy levels from most aneuploid (CIN) to least aneuploid (MSI).
Survival analysis
Survival analysis was performed using the gastric cancer dataset of the KM plotter (https://kmplot.com/analysis/) (51). Overall survival was compared across 875 gastric cancer patients stratified into groups of high and low mRNA expression of SQSTM1/p62 (probe set 201471_s_at), using the default parameters.
Quantification and data analysis
Statistical analysis was performed using GraphPad Prism software. Details of the statistical tests were reported in figure legends. Chi-squared test was performed by testing the null hypothesis that the phenotypes analyzed were randomly caused (50%) in a heterogeneous population. The values of statistically significant P values are indicated in the figure legends, the values of not–statistically significant P values are not indicated. Error bars represent SDs or SEMs, as indicated in the figure legends. All experiments were performed in at least two biological replicates.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful to all the members of the Santaguida laboratory for fruitful discussions throughout the project. We thank members of the M. Mapelli laboratory for constructive discussions and G. Natoli (IEO, Milan, Italy) for critical reading of the manuscript. We are grateful to V. Korolchuk (Newcastle University, UK) for sharing p62 constructs. We acknowledge help from Flow Cytometry, Imaging, Mass Spectrometry, and Tissue Culture units at IEO for their technical assistance. We thank the ALEMBIC facility at San Raffaele Scientific Institute for the electron microscopy experiments. S.Sc. is a PhD student within the European School of Molecular Medicine (SEMM).
Funding:
This study was supported by: Italian Association for Cancer Research grant AIRC-MFAG 2018, ID. 21665 (S.Sa.); Italian Association for Cancer Research grant AIRC Bridge Grant 2023, ID. 29228 (S.Sa.); Italian Ministry of Health, Ricerca Finalizzata grant GR-2018-12367077 (S.Sa.); Fondazione Cariplo (S.Sa.); Rita-Levi Montalcini program from MIUR (S.Sa.); Italian Ministry of Health with Ricerca Corrente and 5 × 1000 funds (S.Sa.); Fondazione IEO-Monzino Fellowship (S.M.); Italian Association for Cancer Research AIRC Fellowship 1660 ID 26738-2021 (M.R.I.); National Institutes of Health grant R35GM124766 (A.L.G. and E.M.H.); Rita Allen Foundation Scholars Award (A.L.G. and E.M.H.); European Research Council starting grant 945674 (U.B.-D.); Israel Cancer Research Fund Project Award (U.B.-D.); Israel Science Foundation grant 1805/21 (U.B.-D.); BSF project grant 2019228 (U.B.-D.); Israel Cancer Association grant 20230018 (U.B.-D.); EMBO Young Investigator (U.B.-D.); Worldwide Cancer Research grant 19-0003 (S.P.); American-Italian Cancer Foundation Postdoctoral Research Fellowship (M.D.B.); National Institutes of Health NIH DP5OD026395 (S.F.B.); Foundation of the National Institutes of Health NCI P50CA247749 (S.F.B.); National Institutes of Health R01CA256188-01 (S.F.B.); Congressionally Directed Medical Research Program (S.F.B.); Burroughs Wellcome Fund (S.F.B.); the Josie Robertson Foundation (S.F.B.); and the Mark Foundation for Cancer Research (S.F.B.).
Footnotes
Competing interests: S.F.B. owns equity in, receives compensation from, serves as a consultant to, and serves on the scientific advisory board and board of directors of Volastra Therapeutics. U.B.-D. receives consulting fees from Accent Therapeutics. The authors declare no other competing interests.
Data and materials availability:
Data are available in the main text or the supplementary materials. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE database with the dataset identifier PXD052675.
REFERENCES AND NOTES
- 1.Holland AJ, Cleveland DW, Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10, 478–487 (2009). doi: 10.1038/nrm2718; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakhoum SF, Cantley LC, The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 174, 1347–1360 (2018). doi: 10.1016/j.cell.2018.08.027; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ippolito MR et al. , Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev. Cell 56, 2440–2454.e6 (2021). doi: 10.1016/j.devcel.2021.07.006; [DOI] [PubMed] [Google Scholar]
- 4.Lukow DA et al. , Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 56, 2427–2439.e4 (2021). doi: 10.1016/j.devcel.2021.07.009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trakala M et al. , Clonal selection of stable aneuploidies in progenitor cells drives high-prevalence tumorigenesis. Genes Dev. 35, 1079–1092 (2021). doi: 10.1101/gad.348341.121; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shoshani O et al. , Transient genomic instability drives tumorigenesis through accelerated clonal evolution. Genes Dev. 35, 1093–1108 (2021). doi: 10.1101/gad.348319.121; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garribba L et al. , Short-term molecular consequences of chromosome mis-segregation for genome stability. Nat. Commun. 14, 1353 (2023). doi: 10.1038/s41467-023-37095-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soto M, Raaijmakers JA, Medema RH, Consequences of Genomic Diversification Induced by Segregation Errors. Trends Genet. 35, 279–291 (2019). doi: 10.1016/j.tig.2019.01.003; [DOI] [PubMed] [Google Scholar]
- 9.Ly P, Cleveland DW, Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 27, 917–930 (2017). doi: 10.1016/j.tcb.2017.08.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santaguida S, Amon A, Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 16, 473–485 (2015). doi: 10.1038/nrm4025; [DOI] [PubMed] [Google Scholar]
- 11.Zhang CZ et al. , Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015). doi: 10.1038/nature14493; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ly P et al. , Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 51, 705–715 (2019). doi: 10.1038/s41588-019-0360-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens PJ et al. , Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011). doi: 10.1016/j.cell.2010.11.055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortés-Ciriano I et al. , Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 52, 331–341 (2020). doi: 10.1038/s41588-019-0576-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ly P et al. , Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 19, 68–75 (2017). doi: 10.1038/ncb3450; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crasta K et al. , DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 (2012). doi: 10.1038/nature10802; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH, Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333, 1895–1898 (2011). doi: 10.1126/science.1210214; [DOI] [PubMed] [Google Scholar]
- 18.Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW, Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60 (2013). doi: 10.1016/j.cell.2013.06.007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S et al. , Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561, 551–555 (2018). doi: 10.1038/s41586-018-0534-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffelder DR et al. , Resolution of anaphase bridges in cancer cells. Chromosoma 112, 389–397 (2004). doi: 10.1007/s00412-004-0284-6; [DOI] [PubMed] [Google Scholar]
- 21.Mammel AE, Huang HZ, Gunn AL, Choo E, Hatch EM, Chromosome length and gene density contribute to micronuclear membrane stability. Life Sci. Alliance 5, e202101210 (2022). doi: 10.26508/lsa.202101210; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vietri M et al. , Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 22, 856–867 (2020). doi: 10.1038/s41556-020-0537-5; [DOI] [PubMed] [Google Scholar]
- 23.Willan J et al. , ESCRT-III is necessary for the integrity of the nuclear envelope in micronuclei but is aberrant at ruptured micronuclear envelopes generating damage. Oncogenesis 8, 29 (2019). doi: 10.1038/s41389-019-0136-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soto M, García-Santisteban I, Krenning L, Medema RH, Raaijmakers JA, Chromosomes trapped in micronuclei are liable to segregation errors. J. Cell Sci. 131, jcs214742 (2018). doi: 10.1242/jcs.214742; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackenzie KJ et al. , cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). doi: 10.1038/nature23449; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harding SM et al. , Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017). doi: 10.1038/nature23470; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohr L et al. , ER-directed TREX1 limits cGAS activation at micronuclei. Mol. Cell 81, 724–738.e9 (2021). doi: 10.1016/j.molcel.2020.12.037; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joo YK et al. , ATR promotes clearance of damaged DNA and damaged cells by rupturing micronuclei. Mol. Cell 83, 3642–3658.e4 (2023). doi: 10.1016/j.molcel.2023.09.003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agustinus AS et al. , Epigenetic dysregulation from chromosomal transit in micronuclei. Nature 619, 176–183 (2023). doi: 10.1038/s41586-023-06084-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papathanasiou S et al. , Heritable transcriptional defects from aberrations of nuclear architecture. Nature 619, 184–192 (2023). doi: 10.1038/s41586-023-06157-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terradas M, Martín M, Tusell L, Genescà A, DNA lesions sequestered in micronuclei induce a local defective-damage response. DNA Repair 8, 1225–1234 (2009). doi: 10.1016/j.dnarep.2009.07.004; [DOI] [PubMed] [Google Scholar]
- 32.Lin YF et al. , Mitotic clustering of pulverized chromosomes from micronuclei. Nature 618, 1041–1048 (2023). doi: 10.1038/s41586-023-05974-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakhoum SF et al. , Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018). doi: 10.1038/nature25432; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denais CM et al. , Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358 (2016). doi: 10.1126/science.aad7297; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raab M et al. , ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362 (2016). doi: 10.1126/science.aad7611; [DOI] [PubMed] [Google Scholar]
- 36.Toufektchan E, Maciejowski J, Purification of micronuclei from cultured cells by flow cytometry. STAR Protoc. 2, 100378 (2021). doi: 10.1016/j.xpro.2021.100378; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santaguida S, Tighe A, D’Alise AM, Taylor SS, Musacchio A, Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 190, 73–87 (2010). doi: 10.1083/jcb.201001036; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santaguida S, Vasile E, White E, Amon A, Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 29, 2010–2021 (2015). doi: 10.1101/gad.269118.115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santaguida S et al. , Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 41, 638–651.e5 (2017). doi: 10.1016/j.devcel.2017.05.022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galluzzi L et al. , Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836 (2017). doi: 10.15252/embj.201796697; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pankiv S et al. , p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 (2007). doi: 10.1074/jbc.M702824200; [DOI] [PubMed] [Google Scholar]
- 42.Pohl C, Dikic I, Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822 (2019). doi: 10.1126/science.aax3769; [DOI] [PubMed] [Google Scholar]
- 43.Zhuang J et al. , Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 133, 1572–1584 (2019). doi: 10.1182/blood-2018-06-859686; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rello-Varona S et al. , Autophagic removal of micronuclei. Cell Cycle 11, 170–176 (2012). doi: 10.4161/cc.11.1.18564; [DOI] [PubMed] [Google Scholar]
- 45.Hatch EM, Hetzer MW, Nuclear envelope rupture is induced by actin-based nucleus confinement. J. Cell Biol. 215, 27–36 (2016). doi: 10.1083/jcb.201603053; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam SS et al. , Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015). doi: 10.1038/nmeth.3179; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carroll B et al. , Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 9, 256 (2018). doi: 10.1038/s41467-017-02746-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Bona M et al. , Micronuclear collapse from oxidative damage. Science 385, eadj8691 (2024). doi: 10.1126/science.adj8691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J et al. , CTLPScanner: A web server for chromothripsis-like pattern detection. Nucleic Acids Res. 44, W252–W258 (2016). doi: 10.1093/nar/gkw434; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghandi M et al. , Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). doi: 10.1038/s41586-019-1186-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cancer Cell Line Encyclopedia Consortium, Genomics of Drug Sensitivity in Cancer Consortium, Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–87 (2015). doi: 10.1038/nature15736; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szász AM et al. , Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7, 49322–49333 (2016). doi: 10.18632/oncotarget.10337; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barretina J et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). doi: 10.1038/nature11003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coyne AN et al. , Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci. Transl. Med. 13, eabe1923 (2021). doi: 10.1126/scitranslmed.abe1923; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le Berre M, Aubertin J, Piel M, Fine control of nuclear confinement identifies a threshold deformation leading to lamina rupture and induction of specific genes. Integr. Biol. 4, 1406–1414 (2012). doi: 10.1039/c2ib20056b; [DOI] [PubMed] [Google Scholar]
- 56.Lin Y-F, Hu Q, Guyer A, Fachinetti D, Ly P, “Induction of chromosome-specific micronuclei and chromothripsis by centromere inactivation” in Methods in Cell Biology, vol. 182, Zierhut C, Galluzzi L, Eds. (Academic Press Inc., 2024), pp. 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cox J et al. , Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011). doi: 10.1021/pr101065j; [DOI] [PubMed] [Google Scholar]
- 58.Tyanova S, Temu T, Cox J, The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016). doi: 10.1038/nprot.2016.136; [DOI] [PubMed] [Google Scholar]
- 59.Wu T et al. , clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021). doi: 10.1016/j.xinn.2021.100141; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu G, Wang L-G, Han Y, He Q-Y, clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). doi: 10.1089/omi.2011.0118; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in the main text or the supplementary materials. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE database with the dataset identifier PXD052675.