

Abstract

Chiral cyclopentadienyl (CpX) metal complexes are frequently used in asymmetric catalysis by virtue of their high reactivity and selectivity. Planar-chiral-only rhodium and iridium cyclopentadienyl complexes are particularly promising due to unrestricted chemical space for CpX ligand design while retaining structural simplicity. However, they are currently still niche because of a lack of efficient synthetic strategies that avoid lengthy chiral auxiliary routes or chiral preparatory HPLC resolution of the complexes. To streamline access to such planar-chiral-only CpX-metal complexes, we designed a straightforward, highly enantiospecific, point-to-planar chirality transfer complexation via facially selective concerted-metalation-deprotonation between metal-carboxylate precursor [M(olefin)2OAc]2 and a chiral cyclopentadiene. This entirely avoids the typical stereoablative complexation of an achiral cyclopentadienyl anion that detrimentally yields a racemate. Exploiting the described enantiospecific complexation protocol and a simple divergent synthetic route to suitable chiral cyclopentadienes, we generated a structurally diverse library of new planar chiral Cp-Rh(I), Cp-Ir(I), Cp-Rh(III), and Cp-Ir(III) complexes. Moreover, the enantiospecific complexation step can be concatenated with a preceding Au-catalyzed cyclization in an efficient one-pot process that likely involves an elaborate point-to-axial-to-point-to-planar chirality transfer. Guided by computational selectivity predictions, the structure of a CpX-Rh complex in our library was tuned to optimize reactivity and selectivity in the asymmetric C–H functionalization of a benzamide with various challenging alkenes. With an optimized CpX-Rh complex in hand, we showcased its excellent catalytic performance and high selectivity for refractory alkene substrates that reacted in poor selectivity with previous CpX-Rh catalysts.
Introduction
Chiral cyclopentadienyl (CpX) ligands have been used as a stereochemical element in transition-metal-based asymmetric catalysis to great success, delivering high performance with respect to both reactivity and selectivity.1 For example, CpX complexes of Group 9 metals (Co, Rh, and Ir) have enabled challenging enantioselective C–H functionalization reactions to be carried out.2 Other attractive characteristics of CpX-metal complexes include low molecular weight, simplicity, rigidity, and a largely unexplored chemical space. Despite this, fundamental problems exist in their preparation on a general level.
Traditionally, CpX-metal complexes are formed by deprotonation of a cyclopentadiene (CpH) and subsequent complexation (i.e., transmetalation) of a metal precursor.3,4 The geometry of the intermediate Cp anion falls into one of three possible categories: (I) homotopic, (II) diastereotopic, and (III) enantiotopic (Scheme 1). By far the most common geometry type seen in literature is Type I, in which a corresponding CpH typically contains a C2-symmetric (chiral) backbone.5−11 The metal complex formed from a Type I anion is always enantiomerically pure, as complexation to either face of the Cp anion is identical. This design avoids any need for chiral separation postcomplexation. However, Type I CpHs with different chiral backbones do not share a common synthetic intermediate, and therefore, traversal of chemical space can be tedious. Additionally, CpX-metal complexes with C2-symmetric backbones are not structurally optimal as only one half of the backbone (facing the metal) significantly affects the relevant chiral environment. A non-C2-symmetric cyclopentadiene with one or more stereogenic centers in its backbone will generate a Type II Cp anion.12 Complexation in this case leads to two possible diastereomers. Good control of facial selectivity in the complexation of Type II anions is rare and restricted to very specific examples. For instance, Loginov describes one diastereoselective complexation of RhCl3 with an indene containing an α-pinene-derived backbone.13 Wang reports a diastereoselective complexation of [Rh(COD)Cl]2 with a planar chiral indene containing a [2.2]benzoindenophane backbone.14 However, other examples of Type II anion complexation in literature are not selective,15 indicating the highly restrictive nature of such CpH backbone design. The Type III Cp anion is prochiral and, upon addition of a binding metal, delivers a complex that is planar chiral without other sources of chirality (planar-chiral-only). Assuming that the typical transmetalation process takes place, there is no facial selectivity in their complexation, and the resulting product is a racemate. Nevertheless, CpHs that lead to Type III anions have a large design space as they do not need to be chiral themselves and do not require a structurally elaborate backbone.
Scheme 1. Overview of Three Different Cp-metal Complexation Outcomes Depending on Cp Anion Geometry.

To our knowledge, all literature reports of synthesized planar-chiral-only CpX-metal complexes (Type III) describe the need for some variation of chiral separation of the complexes themselves.16,17 There exist enterprising methods to, at the very least, avoid the use of chiral preparatory HPLC. Perekalin has previously used a chiral resolution strategy that involved addition of a chiral amine or amino acid to a racemic CpX-Rh(III) complex, followed by diastereomeric separation and finally removal of the auxiliary.18,19 A clear drawback to this approach is the number of extra operational steps. More recently, Wang20 reported a unique protocol using a tailored [Rh(chiral diene)OAc]2 complex. Direct complexation with achiral CpH led to a diastereomeric mixture that was separable by flash column chromatography. While an improvement over existing methods, this strategy still required effort in synthesizing the chiral diene auxiliary, and the facial selectivity of the complexation ranged from good to very poor, depending on the substitution pattern of the CpH.
In general, as illustrated with the various caveats of the Type I–III Cp anions, the synthesis of enantiopure planar chiral CpX-metal complexes remains challenging. This makes the development of a globally applicable facially selective complexation strategy a high priority endeavor. However, this could only be made possible by escaping the paradigm of deprotonation and complexation occurring in separate steps. Carboxylate ligands in metal complexes can facilitate C–H activation of appropriate substrates via a concerted metalation-deprotonation (CMD) mechanism. This has not only been long exploited in C–H functionalization reactions21 but also more recently to prepare derivative complexes under mild conditions.22,23 One such complexation protocol, which uses either [Rh(COD)OAc]2 or [Ir(COD)OAc]2, circumvents the need for any additional base. We reasoned, using the same or similar conditions, that an appropriate CpH with a core tertiary carbon stereocenter could undergo metal complexation preferentially on the same face as its tertiary hydrogen through a CMD pathway (Scheme 2). The process would in effect be a full point-to-planar chirality transfer and, unlike the examples in Scheme 1, not involve formal deprotonation of a CpH.
Scheme 2. Enantiospecific Complexation of CpHs Containing a Core Tertiary Carbon Stereocenter via Point-To-Planar Chirality Transfer.

We herein report a very successful proof of this concept, establishing a general method for the enantiospecific complexation of chiral CpHs with [M(olefin)2OAc]2 precursors. We also demonstrate that several of the new CpX-Rh complexes prepared by this approach set new performance benchmarks in asymmetric C–H functionalization reactions.
Results and Discussion
Using compound 1a, an enantioenriched CpH that importantly contained a single tertiary carbon stereocenter in its core, we first applied typical complexation methods to establish a baseline for chirality transfer, or to confirm its absence (Scheme 3). The reaction between 1a and [Rh(COD)Cl]2 in the presence of thallium ethoxide5 delivered Rh(I) complex 2a in 77% yield as a fully racemic mixture. When nBuLi11 was used as the base, 2a was formed, again with full stereochemical ablation. Clearly, in both of these cases, complexation occurs from the generated achiral Cp anion with no facial selectivity. The microwave-assisted reaction of 1a with RhCl3 afforded [CpXRhCl2]23a in 93% yield.24 Chiral SFC analysis of the trimethyl phosphite derivative of 3a confirmed that a virtually racemic product was formed. Finally, a complexation attempt using [Ir(COD)Cl]2 and aqueous HCl gave the analogous Ir(III) complex 4a in 84% yield.25 Again, trimethylphosphite derivatization and chiral SFC analysis were carried out to indicate this complexation proceeded with only negligible enantiospecificity. Therefore, none of these traditional methods allow any significant degree of chirality transfer of CpH to a planar chiral CpX-metal complex.
Scheme 3. Application of Typical Complexation Protocols on Enantioenriched CpH 1a.

We then screened various reaction conditions for the complexation of 1a and different rhodium and iridium precursors (Table 1). A 1:1 toluene/methanol mixture was optimal in balancing between dissolution of reactants and deaggregation of the [M]2 dimer. After 3 h of stirring at ambient temperature, the desired chiral CpX-Rh(I) complex 2a was cleanly formed and isolated in 83% yield. Pleasingly, the enantiomeric excess of 2a was 90%, corresponding to >99% enantiospecificity (es) and a full transfer of point chirality of 1a to planar chirality of 2a. Using alternative complexes [Rh(COD)OMe]2 and [Rh(COD)OH]2 also yielded 2a in full enantiospecificity, albeit with somewhat lower yields (entries 2 and 3). No complexation was observed at all when using [Rh(COD)Cl]2 (entry 4), providing evidence that CMD facilitated by an anionic oxygen-containing ligand was indeed occurring with the other precursors. [Rh(COD)Cl]2 could instead be converted to the [Rh(COD)OAc]2 in situ with KOAc (entry 5), and these conditions delivered 2a but in both lower yield and enantioselectivity compared to entry 1. This outcome can be explained by a possible competing pathway involving coordination of the metal to the “wrong” face of CpH and then CMD with an external acetate. The reaction of 1a with iridium analogue [Ir(COD)OAc]2 proceeded more slowly than that for rhodium (entry 6), requiring elevated temperature to deliver 5a in 73% yield (entry 7) but also with complete enantiospecificity. Complexation of 1a with [Ir(C2H4)2OAc]2 generated in situ from [Ir(COE)2Cl]2, KOAc, and ethylene gas (entry 8) delivered the bis–ethylene complex 5b in 86% yield, again with full chirality transfer.26
Table 1. Optimization of the Enantiospecific Complexation.

| entry | [M] | T (°C) | % yield | % es |
|---|---|---|---|---|
| 1 | [Rh(COD)OAc]2 | 23 | 83 | >99 |
| 2 | [Rh(COD)OMe]2 | 23 | 63 | >99 |
| 3 | [Rh(COD)OH]2 | 23 | 70 | >99 |
| 4 | [Rh(COD)Cl]2 | 23 | 0 | n.d |
| 5a | [Rh(COD)Cl]2 | 23 | 59 | 79 |
| 6 | [Ir(COD)OAc]2 | 23 | <5 | n.d |
| 7 | [Ir(COD)OAc]2 | 70 | 73 | >99 |
| 8b | [Ir(COE)2Cl]2 | 60 | 86 | >99 |
Isolated yields shown. Enantiospecificity calculated as ee of complex ÷ ee of CpH. with 1.2 eq. KOAc.
With 2.0 eq. KOAc under ethylene atmosphere for 16 h. n.d. = not determined.
Encouraged by these initial results, we then sought to build a library of chiral CpHs containing a single core stereogenic tertiary carbon and demonstrate that the enantiospecific complexation method is applicable to all of these compounds. A moderately sized collection of these CpHs was obtained (several on gram scale) via a short divergent sequence, as described in Scheme 4, starting from a variety of chiral propargylic alcohols (6). Compounds 6 all contained a cyclohexenyl group, and some contained a 3,3-dimethyl or 6,6-dimethyl substitution pattern on the cyclohexene ring. 6 was prepared either by the Sonogashira coupling of a cyclohexenyl triflate with an appropriate alkynol, or by Carreira alkynylation of aldehydes.27 Esterification of 6 with a variety of acyl chlorides yielded 7, from which a gold-catalyzed cyclization protocol was carried out according to Zhang.28 Depending on the substrate and the conditions of the cyclization reaction, either cyclopentadienyl ester 8 (one of two isomers) or cyclopentenone 9 was obtained as the major product, generally with a high degree of chirality transfer. Replacing tBuBrettPhosAuCl with PPh3AuCl and replacing NaBARF with AgSbF6 led to a higher reaction rate, which was necessary for full conversion in some cases. When wet solvent was used, 9 would be preferentially formed over 8, presumably by some hydrolytic process taking place. Arylation of 9 subsequently yielded a group of aryl-substituted chiral CpHs, including the first example 1a. Acyloxy-substituted CpHs such as 8 are previously completely unexplored candidates for metal complexation. Overall, a chemical space for 8 and 1 was traversed in three dimensions: (a) presence of a gem-dimethyl group on the cyclohexane, (b) the alkyl substituent on the Cp, and (c) the acyloxy/aryl group.
Scheme 4. Enantioselective Synthesis of Chiral CpHs.

Isolated yields over multiple steps from 6 are shown.
Alkynylation and acylation were performed in one pot.
AgSbF6 instead of NaBARF.
PPh3AuCl instead of tBuBrettPhosAuCl.
10 mol % of additive.
Acylation reaction time of 18 h and cyclization time of 6 h.
Wet DCM as the solvent.
During workup of ArLi addition, 1.5 equiv of 2-NsCl was added to induce dehydration of the direct addition intermediate.
50% probability thermal ellipsoids. Hydrogen atoms omitted for clarity, except for on the stereocenter.
We found that all of the synthesized CpHs (8 and 1) were amenable to enantiospecific complexation, delivering a range of unique planar-chiral-only Cp-Rh(I) and Cp-Ir(I) complexes in generally high yield and high enantiopurity (Scheme 5). There was virtually complete point-to-planar transfer of chirality over nearly the entire chemical space of CpHs. The presence of the acyloxy group on the Cp ring was well tolerated despite initial concerns with electrophilicity. All the Rh(I) and Ir(I) complexes generated were stable under ambient conditions and could be purified by typical chromatographic methods without precaution to moisture or air.
Scheme 5. Planar Chiral CpX-Rh(I) and CpX-Ir(I) Complexes Formed by Enantiospecific Complexation of Chiral CpHs, and Subsequent CpX-Rh(III) and CpX-Ir(III) Complexes.

Conditions A: [Rh(COD)OAc]2 (0.51 equiv) in 1:1 PhMe/MeOH at 23 °C for 3 h. Conditions B: [Ir(COD)OAc]2 (0.51 equiv) in 1:1 PhMe/MeOH at 70 °C for 3 h. Conditions C: [Ir(COE)2Cl]2 (0.60 equiv), KOAc (2.0 equiv) in 1:1 PhMe/MeOH under C2H4 atmosphere at 60 °C for 16 h. Conditions D: Used crude CpH, [Ir(COD)OAc]2 (0.51 equiv) in 1:1 PhMe/MeOH at 40 °C for 16 h. Isolated yields of the last step shown, unless stated otherwise. Enantiospecificity calculated as ee of complex ÷ ee of CpH, determined from chiral HPLC analysis unless indicated otherwise.
ee of complex was determined by chiral HPLC or SFC analysis of the [CpXMCl2(P(OMe)3)] derivative.
Reaction temperature of 0 °C and time of 30 min.
Reaction time: 16 h.
Isolated yield over two steps, enantiospecificity determined from ee of crude CpH.
Synthesized from 5b.
Isolated yield over two steps from 2m.
50% probability thermal ellipsoids. Hydrogen atoms omitted for clarity.
We also demonstrated that various Rh(I) and Ir(I) complexes (2 and 5) could be readily oxidized into the catalytically active Rh(III) and Ir(III) halide complexes (3 and 4) by treatment with sulfuryl chloride, bromine, or iodine.29−31 Several of the M(III) chlorides, in particular, were also converted into the [CpXMCl2(P(OMe)3)] derivatives (10 and 11).32 M(III) complexes with an aryl substituent on the Cp ring were all completely stable under ambient conditions. The Rh(III) chloride derivative of 2m, containing an acyloxy group, proved to be a transient species (likely susceptible to hydrolysis), but immediate conversion to the trimethylphosphite derivative yielded stable complex 10c.
2m and 2n, bearing 4-methoxybenzoyloxy and 4-phenylbenzoyloxy groups, respectively, became important complexes in later stages of the study as they proved to be highly competent in asymmetric C–H functionalization. However, their corresponding yields following the method in Scheme 5 were disappointingly low due to the difficulty in isolating the sensitive electron-rich CpHs 8j and 8k. Logically, we then sought to develop a protocol that combined both the preceding gold-catalyzed cyclization and enantiospecific complexation steps in a single pot. This proved to be highly successful, allowing complexes 2m and 2n to be furnished in much improved yields and with almost no decrease in enantiopurity compared to the stepwise approach (Scheme 6). This method constitutes, according to accepted mechanisms,28 an incredible point-to-axial-to-point-to-planar chirality transfer in a single pot process. In a scaled-up synthesis of 2n, the desired complex was obtained in 60% yield over two steps in 89% ee and was then recrystallized to full enantiopurity. Because 2m was not crystalline, its norbornadiene analogue (2p) was prepared and could then be recrystallized to afford the enantioenriched complex.
Scheme 6. Point-to-Axial-to-Point-to-Planar Chirality Transfer in a One-Pot Synthesis of CpX -Rh(I) Complexes 2m, 2n, and 2p.

Our next major endeavor was to show the catalytic potential of this new library of CpX-metal complexes. We evaluated their performance in the C–H functionalization of benzamide derivative 12 with various alkenes as this is a well-accepted benchmark transformation (Scheme 7).5 There exist numerous examples in literature in which reaction with cyclic alkenes (e.g., norbornene) and electronically activated alkenes (e.g., styrene) give high enantioselectivity,.5,13,14,18,20,33−35 However, simple unbiased terminal alkenes are far more challenging substrates, and regio- and enantioselectivity remain unsolved for their Rh(III) catalysis. We previously reported the C–H functionalization of N-chlorobenzamide with 1-hexene or 1-octene using a CpX-Co catalyst, giving the 3-substituted dihydroisoquinolones exclusively with high enantioselectivity.35 In contrast, poor enantioselectivity has always been observed for the analogous Rh-catalyzed reaction in which the 4-alkyl-substituted product is primarily formed.13,18−20,25 We first attempted the C–H functionalization of 12 with styrene (a, R = Ph) and 1-hexene (b, R = Bu) using our first generation [CpXRh(COD)] complex 2d under standard conditions that formed the catalytically active Rh(III) species in situ (Scheme 7).5 The reaction with styrene led to the formation of only the 3-substituted regioisomer 14a along with side product isoindolinone 15a. For 1-hexene, only the regioisomers 13b/14b were produced, in strong favor of the 4-substituted 13b. While typically enantioselectivity is judged from the ee of the product(s), to make fair comparisons, we used an adjusted enantiomeric excess of the product (ee*) that accounted for the less than full enantiopurity of the catalyst used. Using complex 2d, across both reactions, good to moderate enantioselectivity was already observed (83% for styrene and 55% for 1-hexene). We then proposed to further improve catalyst performance by modifying 2d to incorporate a gem-dimethyl group on the cyclohexyl backbone (analogous to a tert-butyl group) at one of two positions (2g and 2j).
Scheme 7. Steric Trend of Rh Complexes 2d, 2g, and 2j in the C–H Functionalization of Benzamide 12 with Styrene and 1-hexene, with Steric Maps and Computational Predictions.

Conversion determined by isolation of 12. Isolated yields are shown. Adjusted enantiomeric excess ee* was calculated as ee of major product ÷ ee of complex, determined from chiral HPLC analysis. Steric maps generated using SambVca 2.1 (bondi radii scaled by 1.17, sphere radius 3.5 Å, mesh spacing 0.1 Å).47 Computed free energies were determined at the PBE0-D3(BJ)/def2-TZVP//PBE0-D3(BJ)/def2-SVP level (see Supporting Information Section 6 for more detail).
Prior to experimental work on
these derivative complexes, computational
predictions aimed at identifying the lowest energy transition states
for the regio-/enantiodetermining step were undertaken for the C–H
functionalization of 12 with styrene or 1-hexene, leading
to dihydroisoquinolones (13 and 14) using
complexes 2d, 2g, and 2j. To
accomplish this, we employed a computational workflow based on previous
work that capably reproduced experimental enantioselectivites of C2-symmetric Cp derivatives in the rhodium-catalyzed
C–H bond functionalization of hydroxamic acid derivatives.36,37 Beginning from eight preconstructed TS structural templates associated
with formation of possible regio- and enantiomers (two each, leading
to the 3R, 3S, 4R, and 4S products, see Supporting Information Figure S1) we used the SCINE Molassembler library38,39 to generate 3200 total conformers (400 for each of the possible
pathways) with full stereoisomer control for each catalyst/substrate
combination. Generated conformers that lead to chemically reasonable
structures (i.e., those without overlapping chemical moieties) when
projected back to three-dimensional coordinates from their graphs38,39 were then subjected to a series of constrained optimizations, at
the PM740 and PBE041,42-D3(BJ)43,44/def2-SVP45 levels,
followed by full TS optimizations at the PBE0-D3(BJ)/def2-SVP level
and single point energies at the PBE0-D3(BJ)/def2-TZVP level (see Supporting Information Section 6 for full details).
The optimized conformer ensembles associated with each pathway (i.e.,
3DR, 3DS, 4DR, 4DS, etc.) were then analyzed using marc,46 a conformer analysis program that uses k-means
clustering, in order to identify the ten most representative conformers
for each of the aforementioned pathways. Those representative conformers
having free energies within 4.0 kcal/mol from lowest energy species
of the respective pathway were retained and used to determine effective
ΔG values through Boltzmann weighting at 296
K. These ΔG values were subsequently used to
determine regio-/enantioselectivity ratios from the theoretical kinetic
constants: 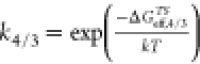 as rr4/3 =
100 ×
as rr4/3 =
100 × 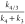 and kS/R =
and kS/R = 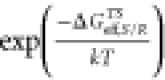 as erS/R = 100 ×
as erS/R = 100 ×  , respectively.
, respectively.
Across the three complexes, regioselectivity in the reaction with styrene was always predicted to be overwhelmingly in favor of the 3-substituted product 14a (ΔGTSeff,4/3 > 4.50 kcal/mol for all ligands). For 1-hexene, the 4-substituted regioisomer 13b was predicted to be the major isomer (ΔGTSeff,4/3 = 1.26 to 3.64 kcal/mol). Furthermore, the computational results (see the Supporting Information for a comprehensive comparison of experimental and computational selectivity data) suggested that, for both styrene and 1-hexene, a steric trend exists where placement of the gem-dimethyl group adjacent to the Cy group (2j) had a strongly positive effect on enantioselectivity for the major isomer compared to 2d, while a negative effect is associated with placing the gem-dimethyl group adjacent to the OBz group (2g). With this insight in hand, analogues 2g and 2j were synthesized and tested. The relevant steric maps of 2d, 2g, and 2j were also obtained from their respective X-ray structures. For complex 2g, the steric bulk of the gem-dimethyl group was directly opposite that of the Cy group, which seems to largely nullify the steric bias. For complex 2j, the gem-dimethyl group proximal to the Cy group appears to enhance the overall steric bias. From this primitive analysis, 2j was expected to induce the highest enantioselectivity of the three. As predicted from the computations, in the reaction with styrene, the steric modification of complex 2d did not affect its already excellent regioselectivity. Meanwhile, for 1-hexene, the observed regioselectivities for 2d, 2g, and 2j were relatively consistent with predictions. At the same time, the steric environment of 2j was predicted to induce superior enantioselectivity relative to 2d and 2g. Supported by prior rationale, complex 2j indeed outperformed 2d with 96% enantioselectivity in the reaction with styrene and 74% for 1-hexene. Catalytic reactivity was also improved over 2d with 76% yield for styrene and 96% yield for 1-hexene.
Other [CpXRh(COD)] complexes in our library were then screened in the same C–H functionalization reactions of 12 with styrene and 1-hexene (Scheme 8). When examining the effect of gem-dimethyl group steric modification in the series 2f/2h/2o and 2a/2c, trends similar to those of 2d/2g/2j for enantioselectivity were observed. However, none outperformed existing 2j. Thus, we next investigated modifying the electron density of the benzoyl group of 2j to determine stereoelectronic effects on the outcome of the reaction for the same two alkene substrates. A series of analogues 2k (para-nitro), 2l (para-chloro) and 2m (para-methoxy) were prepared and subjected to the same catalytic screening. Interestingly, for both the reactions with styrene and 1-hexene, conversion and yield significantly increased as the electron density of the benzoyl group increased, with only slight differences in regio- and enantioselectivity. This is consistent with our assumption that introducing the distal para-substituent does not influence the steric environment already elucidated in 2j. To justify the observed stereoelectronic trend with reactivity, we reason that for more electron-dense Cp ligands the Cp–Rh bond is less labile, and therefore, the active Cp-Rh(III) species is more long-lived and effectively leads to increased catalytic turnover.
Scheme 8. Screening of Other Rh Complexes in the C–H Functionalization of Benzamide 12 with Styrene and 1-Hexene.

Conversion determined by isolation of 12. Isolated yields shown. Adjusted enantiomeric excess ee* calculated as ee of major product ÷ ee of complex, determined from chiral HPLC analysis.
Complex 2n was then designed to combine ideal steric (gem-dimethyl substitution next to Cy), electronic (electron-rich para-phenyl substituent), and physical (easily crystallized to full enantiopurity) properties (Scheme 9). The methodology described in Scheme 4 and Scheme 6 allowed for the rapid synthesis of enantiopure 2n. When subjected to the same C–H functionalization benchmarks as before, the fast catalytic turnover of 2n could be exploited with a lower reaction temperature. In the reaction between 12 and styrene, using 2n under optimized conditions, 14a was formed in 95% yield with virtually full enantioselectivity (>99.5:0.5 er). Using the same reaction conditions with 1-hexene as the substrate, the major regioisomer 13b was formed in 78% yield and 93:7 er. To date, this is by a margin the highest enantioselectivity seen for any existing CpX-Rh complex using terminal unbiased alkenes. Finally, two other highly challenging substrates—allylbenzene (c, R = Bn) and allyl alcohol (d, R = CH2OH)—for which little to no selectivity was previously observed,18 were tested. Again, the high catalytic performance and selectivity of 2n was demonstrated for both substrates, delivering their respective dihydroisoquinolones in good yield and with 95:5 er of both major regioisomers.
Scheme 9. High Selectivity in the C–H Functionalization of Benzamide 12 with Various Alkenes Using Optimized Rh Catalyst 2n.

Conversion determined by isolation of 12. Isolated yields shown. Enantiomeric ratios were determined from chiral HPLC analysis.
Conclusions
In summary, we have reported a novel complexation strategy for efficient access to planar-chiral-only cyclopentadienyl Rh(I) and Ir(I) complexes, avoiding tedious chiral chromatographic separation of the complexes or resolution with chiral auxiliaries. A structurally diverse Cp ligand library (including a unique acyloxy substitution pattern) was assembled, characterized, and evaluated in benchmark transformations. The most salient achievements on different axes are
A first-of-its-kind, straightforward method for the fully enantiospecific complexation of Rh and Ir precursors with chiral cyclopentadienes. This features a point-to-planar chirality transfer via facially selective concerted-metalation-deprotonation between a metal-carboxylate [M(olefin)2OAc]2 and chiral cyclopentadiene, circumventing the typical stereoablative complexation of an achiral cyclopentadienyl anion.
An unprecedented point-to-axial-to-point-to-planar chirality transfer demonstrated by a highly efficient one-pot synthesis of enantiopure CpX-Rh complexes from a chiral propargylic ester precursor, telescoping an Au-catalyzed cyclization and subsequent Rh(I) complexation.
The application of sophisticated computational selectivity predictions, delivering ligand design blueprints that could subsequently be tuned in practice for their synthesizability and stability.
Optimized planar chiral CpX-Rh complexes that display best-in-class enantioselectivities for benchmark asymmetric C–H functionalizations of aryl hydroxamates with terminal aliphatic alkenes.
These accomplishments can ultimately be used to access further novel families of planar chiral group 9 metal complexes and to address yet unsolved challenges in enantioselective catalysis.
Acknowledgments
This work is supported by the EPFL and the NCCR Catalysis. This publication was created as part of NCCR Catalysis (grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation. Prof Markus Reiher (ETH-Zürich) and his group members are acknowledged for assistance with Molassembler.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c13279.
Author Contributions
∥ Y.S.Y. and A.L. contributed equally. The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Mas-Roselló J.; Herraiz A.; Audic B.; Laverny A.; Cramer N. Chiral cyclopentadienyl ligands: design, syntheses and applications in asymmetric catalysis. Angew. Chem., Int. Ed. 2021, 60, 13198–13244. 10.1002/anie.202008166. [DOI] [PubMed] [Google Scholar]
- Newton C.; Kossler D.; Cramer N. Asymmetric catalysis powered by chiral cyclopentadienyl ligands. J. Am. Chem. Soc. 2016, 138, 3935–3941. 10.1021/jacs.5b12964. [DOI] [PubMed] [Google Scholar]
- Scholz H. J.; Werner H.; Basische metalle L. V. I. Synthese von Li2[(C5Me4)2CH2] und ringverbrückter rhodium-zweikernkomplexe mit dem (C5Me4)2CH2-dianion als brückenliganden. J. Organomet. Chem. 1986, 303, C8–C12. 10.1002/ange.19830950426. [DOI] [Google Scholar]
- Enders M.; Kohl G.; Pritzkow H. Rhodium-Carbonyl Complexes with a Quinolyl Functionalized Cp-Ligand: Synthesis and Photochemical Activation. J. Organomet. Chem. 2004, 689, 3024–3030. 10.1016/j.jorganchem.2004.06.046. [DOI] [Google Scholar]
- Ye B.; Cramer N. Chiral cyclopentadienyl ligands as stereocontrolling element in asymmetric C-H functionalization. Science 2012, 338, 504–506. 10.1126/science.1226938. [DOI] [PubMed] [Google Scholar]
- Ye B.; Cramer N. A Tunable Class of Chiral Cp Ligands for Enantioselective Rhodium(III)-Catalyzed C–H Allylations of Benzamides. J. Am. Chem. Soc. 2013, 135, 636–639. 10.1021/ja311956k. [DOI] [PubMed] [Google Scholar]
- Wang S.; Park S. H.; Cramer N. A Readily Accessible Class of Chiral Cp Ligands and Their Application in RuII-Catalyzed Enantioselective Syntheses of Dihydrobenzoindoles. Angew. Chem., Int. Ed. 2018, 57, 5459–5462. 10.1002/anie.201802244. [DOI] [PubMed] [Google Scholar]
- Li G.; Yan X.; Jiang J.; Liang H.; Zhou C.; Wang J. Chiral Bicyclo[2.2.2]octane-Fused CpRh Complexes: Synthesis and Potential Use in Asymmetric C–H Activation. Angew. Chem., Int. Ed. 2020, 59, 22436–22440. 10.1002/anie.202010489. [DOI] [PubMed] [Google Scholar]
- Liang H.; Vasamsetty L.; Li T.; Jiang J.; Pang X.; Wang J. A New Class of C2-Symmetric Chiral Cyclopentadienyl Ligand Derived from Ferrocene Scaffold: Design, Synthesis and Application. Chem.—Eur. J. 2020, 26, 14546–14550. 10.1002/chem.202001814. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Cui W.-J.; Zheng C.; You S.-L. Synthesis and Application of Chiral Spiro Cp Ligands in Rhodium-Catalyzed Asymmetric Oxidative Coupling of Biaryl Compounds with Alkenes. J. Am. Chem. Soc. 2016, 138, 5242–5245. 10.1021/jacs.6b02302. [DOI] [PubMed] [Google Scholar]
- Pan C.; Yin S.; Wang S.; Gu Q.; You S. Oxygen-Linked Cyclopentadienyl Rhodium(III) Complexes-Catalyzed Asymmetric C–H Arylation of Benzo[h]Quinolines with 1-Diazonaphthoquinones. Angew. Chem., Int. Ed. 2021, 60, 15510–15516. 10.1002/anie.202103638. [DOI] [PubMed] [Google Scholar]
- Jia Z.-J.; Merten C.; Gontla R.; Daniliuc C.; Antonchick A.; Waldmann H. General enantioselective C-H activation with efficiently tunable cyclopentadienyl ligands. Angew. Chem., Int. Ed. 2017, 56, 2429–2434. 10.1002/anie.201611981. [DOI] [PubMed] [Google Scholar]
- Kharitonov V. B.; Podyacheva E.; Chusov D.; Nelyubina Y. V.; Muratov D. V.; Loginov D. A. Planar Chiral Rhodium Complex Based on the Tetrahydrofluorenyl Core for Enantioselective Catalysis. Org. Lett. 2023, 25, 8906–8911. 10.1021/acs.orglett.3c03726. [DOI] [PubMed] [Google Scholar]
- Guo W.; Jiang J.; Wang J. [2.2]Benzoindenophane-Based Chiral Indenyl Ligands: Design, Synthesis, and Applications in Asymmetric C–H Activation. Angew. Chem., Int. Ed. 2024, 63, e202400279 10.1002/anie.202400279. [DOI] [PubMed] [Google Scholar]
- Yan X.; Jiang J.; Wang J. A class of readily tunable planar-chiral cyclopentadienyl rhodium(III) catalysts for asymmetric C–H activation. Angew. Chem., Int. Ed. 2022, 61, e202201522 10.1002/anie.202201522. [DOI] [PubMed] [Google Scholar]
- Farr C.; Kazerouni A.; Park B.; Poff C.; Won J.; Sharp K.; Baik M.-H.; Blakey S. Designing a planar chiral rhodium indenyl catalyst for region- and enantioselective allylic C–H amidation. J. Am. Chem. Soc. 2020, 32, 13996–14004. 10.1021/jacs.0c07305. [DOI] [PubMed] [Google Scholar]
- Gross P.; Im H.; Laws D. III; Park B.; Baik M.-H.; Blakey S. B. Enantioselective Aziridination of Unactivated Terminal Alkenes Using a Planar Chiral Rh(III) Indenyl Catalyst. J. Am. Chem. Soc. 2024, 146, 1447–1454. 10.1021/jacs.3c10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonova E.; Ankudinov N.; Mikhaylov A.; Chusov D.; Nelyubina Y.; Perekalin D. A planar-chiral rhodium(III) catalyst with a sterically demanding cyclopentadienyl ligand and its application in the enantioselective synthesis of dihydroisoquinolones. Angew. Chem., Int. Ed. 2018, 57, 7714–7718. 10.1002/anie.201801703. [DOI] [PubMed] [Google Scholar]
- Kolos A.; Nelyubina Y.; Sundararaju B.; Perekalin D. Synthesis of overloaded cyclopentadienyl rhodium(III) complexes via cyclotetramerization of tert-butylacetylene. Organometallics 2021, 40, 3712–3719. 10.1021/acs.organomet.1c00403. [DOI] [Google Scholar]
- Zhang C.; Jiang J.; Huang X.; Wang J. Planar-Chiral Cyclopentadienyl Rhodium Catalysts: Design Concept, Chiral Resolution Strategy, and Applications. ACS Catal. 2023, 13, 10468–10473. 10.1021/acscatal.3c02865. [DOI] [Google Scholar]
- Gandon V.; Hoarau C. Concerted vs Nonconcerted Metalation–Deprotonation in Orthogonal Direct C–H Arylation of Heterocycles with Halides: A Computational Study. J. Org. Chem. 2021, 86, 1769–1778. 10.1021/acs.joc.0c02604. [DOI] [PubMed] [Google Scholar]
- Audic B.; Wodrich M. D.; Cramer N. Mild complexation protocol for chiral CpX Rh and Ir complexes suitable for in situ catalysis. Chem. Sci. 2019, 10, 781–787. 10.1039/c8sc04385j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pototskiy R. A.; Kolos A. V.; Nelyubina Y. V.; Perekalin D. S. Rhodium Catalysts with a Chiral Cyclopentadienyl Ligand Derived from Natural R-Myrtenal. Eur. J. Org. Chem. 2020, 2020, 6019–6025. 10.1002/ejoc.202001029. [DOI] [Google Scholar]
- Brauns M.; Cramer N. Efficient kinetic resolution of sulfur-stereogenic sulfoximes exploiting CpXRhIII-catalyzed C-H functionalization. Angew. Chem., Int. Ed. 2019, 58, 8902–8906. 10.1002/anie.201904543. [DOI] [PubMed] [Google Scholar]
- Amouri H.; Gruselle M.; Jaouen G. bis[Dichloro(η-pentamethylcyclopentadienyl)rhodium(III) and – iridium(III)] from bis[Chloro(1, 5-cyclooctadiene)rhodium(I) and – iridium(I)] Oxidation, and Formation of 1, 5-Cyclooctadiene(η-pentamethylcyclopentadienyl)rhodium(I). Synth. React. Inorg. Met.Org. Chem. 1994, 24, 395–400. 10.1016/S0022-328X(98)00939-5. [DOI] [Google Scholar]
- Wozniak Ł.; Cramer N. Atropo-enantioselective oxidation-enabled iridium(III)-catalyzed C–H arylations with boronic esters. Angew. Chem., Int. Ed. 2021, 60, 18532–18536. 10.1002/anie.202106403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz D.; Fässler R.; Carreira E. Facile enantioselective synthesis of propargylic alcohols by direct addition of terminal alkynes to aldehydes. J. Am. Chem. Soc. 2000, 122, 1806–1807. 10.1021/ja993838z. [DOI] [Google Scholar]
- Zhao K.; Hsu Y.-C.; Yang Z.; Liu R.-S.; Zhang L. Gold-catalyzed synthesis of chiral cyclopentadienyl esters via chirality transfer. Org. Lett. 2020, 22, 6500–6504. 10.1021/acs.orglett.0c02293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fast C. D.; Schley N. D. Light-Promoted Transfer of an Iridium Hydride in Alkyl Ether Cleavage. Organometallics 2021, 40, 3291–3297. 10.1021/acs.organomet.1c00391. [DOI] [Google Scholar]
- Trifonova E. A.; Ankudinov N. M.; Kozlov M. V.; Sharipov M. Y.; Nelyubina Y. V.; Perekalin D. S. Rhodium(III) Complex with a Bulky Cyclopentadienyl Ligand as a Catalyst for Regioselective Synthesis of Dihydroisoquinolones through C–H Activation of Arylhydroxamic Acids. Chem.—Eur. J. 2018, 24, 16570–16575. 10.1002/chem.201804050. [DOI] [PubMed] [Google Scholar]
- Lin W.; Li W.; Lu D.; Su F.; Wen T.-B.; Zhang H.-J. Dual Effects of Cyclopentadienyl Ligands on Rh(III)-Catalyzed Dehydrogenative Arylation of Electron-Rich Alkenes. ACS Catal. 2018, 8, 8070–8076. 10.1021/acscatal.8b01753. [DOI] [Google Scholar]
- Tellers D. M.; Yung C. M.; Arndtsen B. A.; Adamson D. R.; Bergman R. G. Electronic and Medium Effects on the Rate of Arene C–H Bond Activation by Cationic Ir(III) Complexes. J. Am. Chem. Soc. 2002, 124, 1400–1410. 10.1021/ja011809u. [DOI] [PubMed] [Google Scholar]
- Hyster T. K.; Knörr L.; Ward T. R.; Rovis T. Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C–H Activation. Science 2012, 338, 500–503. 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W.-J.; Wu Z.-J.; Gu Q.; You S.-L. Divergent Synthesis of Tunable Cyclopentadienyl Ligands and Their Application in Rh-Catalyzed Enantioselective Synthesis of Isoindolinone. J. Am. Chem. Soc. 2020, 142, 7379–7385. 10.1021/jacs.0c02813. [DOI] [PubMed] [Google Scholar]
- Ozols K.; Jang Y.-S.; Cramer N. Chiral Cyclopentadienyl Cobalt(III) Complexes Enable Highly Enantioselective 3d-Metal-Catalyzed C–H Functionalizations. J. Am. Chem. Soc. 2019, 141, 5675–5680. 10.1021/jacs.9b02569. [DOI] [PubMed] [Google Scholar]
- Laplaza R.; Sobez J.-G.; Wodrich M. D.; Reiher M.; Corminboeuf C. The (Not So) Simple Prediction of Enantioselectivity – A Pipeline for High-Fidelity Computations. Chem. Sci. 2022, 13, 6858–6864. 10.1039/D2SC01714H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodrich M. D.; Laplaza R.; Cramer N.; Reiher M.; Corminboeuf C. Toward in silico Catalyst Optimization. Chimia 2023, 77, 139–143. 10.2533/chimia.2023.139. [DOI] [PubMed] [Google Scholar]
- Sobez J.-G.; Reiher M. MOLASSEMBLER: Molecular Graph Construction, Modification, and Conformer Generation for Inorganic and Organic Molecules. J. Chem. Inf. Model. 2020, 60, 3884–3900. 10.1021/acs.jcim.0c00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensberg M.; Grimmel S.; Sobez J.-G.; Steiner M.; Unsleber J. P.; Rieher M.. qcscine/Molassembler: Release 3.0.0, 2024, https://zenodo.org/records/13372940 (accessed September 22, 2024).
- Stewart J. J. P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Barone V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0Model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Laplaza R.; Wodrich M. D.; Corminboeuf C. Overcoming the Pitfalls of Computing Reaction Selectivity from Ensembles of Transition States. J. Phys. Chem. Lett. 2024, 15, 7363–7370. 10.1021/acs.jpclett.4c01657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falivene L.; Cao Z.; Petta A.; Serra L.; Poater A.; Oliva R.; Scarano V.; Cavallo L. Towards the Online Computer-Aided Design of Catalytic Pockets. Nat. Chem. 2019, 11, 872–879. 10.1038/s41557-019-0319-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.