

Abstract
A monocationic dicopper(I,I) nitrite complex [Cu2(μ-κ1:κ1-O2N)DPFN][NTf2] (2) (DPFN = 2,7-bis(fluoro-di(2-pyridyl)methyl)-1,8-naphthyridine, NTf2– = N(SO2CF3)2–), was synthesized by treatment of a dicopper acetonitrile complex, [Cu2(μ-MeCN)DPFN][NTf2]2 (1), with tetrabutylammonium nitrite ([nBu4N][NO2]). DFT calculations indicate that 2 is one of three linkage isomers that are close in energy and presumably accessible in solution. Reaction of the μ-κ1:κ1-O2N complex with p-TolSH produces nitrous acid (HONO) and the corresponding dicopper thiolate species via an acid–base exchange reaction. Notably, treatment of 2 with HNTf2 results in N–O bond cleavage in the putative, HONO-ligated complex to form the more thermodynamically favorable nitrosyl-bridged dicopper complex [Cu2(μ-NO)(μ-OH)DPFN][NTf2]2 (4). This scission can be reversed via deprotonation of the hydroxy ligand with KOtBu. X-ray diffraction studies confirmed the solid-state molecular structures of 2 and 4. DFT calculations were used to construct a reaction coordinate diagram detailing formation of the μ-NO complex and to describe its electronic structure. The nitrosyl ligand in 4 is chemically labile, as demonstrated by its ready displacement in reactions with CO or NO2–.
Introduction
The reduction of nitrite (NO2–) to nitric oxide (NO) is a key denitrification step in the nitrogen cycle.1,2 The bacterial enzyme copper-containing nitrite reductase (CuNiR) catalyzes this proton-coupled one-electron reaction through the cooperation of type-1 and type-2 Cu sites separated by a Cys-His bridge.3−5 Crystallographic studies indicate that in its resting state, the active site (the type-2 Cu ion) is ligated by three histidine residues and a water molecule in a tetrahedral fashion.6,7 The type-1 Cu ion is bound by a (His)2-Cys-Met donor set, consistent with its role as an electron transfer site (Scheme 1a).8 The postulated mechanism involves initial displacement of water and binding of a chelating nitrito (O-bound) ligand to the oxidized type-2 Cu site. Protonation by a nearby amino acid residue triggers electron transfer from the type-1 to the type-2 Cu site and subsequent nitrite N–O bond cleavage to produce NO and a Cu(II)–OH species (Scheme 1b).9,10 Moreover, the physiological pH determines the efficiency and direction of the reaction: a pH of 6 gives rise to an activity maximum while more basic conditions promote the oxidation of NO back to NO2–.11,12
Scheme 1. (a) CuNiR Active Site with Nitrite Bound; (b) Postulated Mechanism for Nitrite Reduction by CuNiR.

Significant effort has been focused toward the development of molecular copper nitrite complexes that serve as structural and reactive mimics of CuNiR.13 Such studies are also expected to aid in the design of catalysts for the removal of nitrogen oxide pollutants from the environment.2 These functional models usually contain a nitrite-bound copper(II) or copper(I) center, which performs the reduction in the presence of a proton source such as acetic acid.14−17 Monometallic copper complexes are often the focus of these studies, given that enzymatic catalysis only occurs at the mononuclear, type-2 Cu site. However, the nitrite ligand’s ability to engage in several coordination modes and chelate more than one metal center suggests the possibility of multiple activation pathways.18 Uyeda and Peters have shown that bimetallic Co/Mg sites successfully reduce nitrite, with cobalt acting as the reductant and magnesium as a Lewis acid that aids in activating the N–O bond toward cleavage.19 Along these lines, viable catalyst design strategies might involve multiple copper centers that cooperate in substrate activation and/or multielectron and multiproton transfer.
The few reported dicopper nitrite complexes have been primarily investigated for their structural and magnetic properties, rather than for their ability to mediate NO2– reduction.18,20−26 Recently, Zhang and co-workers reported the activation of nitrite at a dicopper core to yield a novel [Cu2(μ-NO)(μ-O)Py4DMB]2+ (Py4DMB = 1,2-bis(di(pyridine-2-yl)methoxy)benzene) complex that engages in hydrogen atom abstraction (HAA) and C–H bond hydroxylation.27 However, the putative dicopper nitrite intermediate remains undefined, and only the final [Cu2(μ-O)(μ-NO)]2+ product of nitrite activation was observed.
A dicopper platform that permits stable nitrite coordination and selective reduction should contribute to a more comprehensive understanding of the factors controlling N–O bond activation. To this end, this laboratory has employed complexes of the rigid dinucleating ligand 2,7-bis(fluoro-di(2-pyridyl)methyl)-1,8-naphthyridine (DPFN), which supports dicopper complexes that exhibit bimetallic cooperativity and possess unusually high electrophilicity.28,29 Herein, we describe a rare dicopper(I,I) nitrite complex that undergoes reversible proton-assisted N–O bond cleavage at the μ-κ1:κ1-NO2– ligand, to produce a unique [Cu2(μ-NO)(μ-OH)DPFN][NTf2]2 complex.
Results and Discussion
The acetonitrile ligand of [Cu2(μ-MeCN)DPFN][NTf2]2 (1) is readily displaced by nitrite upon reaction with [nBu4N][NO2] in THF solution, resulting in a color change from orange to dark red. The diamagnetic product [Cu2(μ-κ1:κ1-O2N)DPFN][NTf2] (2; Scheme 2 and Figure 1), was isolated as an analytically pure brown powder in 98% yield by precipitation from the reaction solution upon addition of diethyl ether. The infrared spectrum of 2 contains a sharp band at 1406 cm–1 that shifts to 1378 cm–1 with 15N labeling at the μ-κ1:κ1-NO2– ligand (Δν(NO2)14N–15N = 28 cm–1, Figure S24). The 15N NMR spectrum of the enriched sample possesses a single resonance at 556.5 ppm, which is modestly shifted with respect to the resonance of NaNO2 in water (δ = 609.5 vs liquid NH3).
Scheme 2. Synthesis of [Cu2(μ-κ1:κ1-O2N)DPFN][NTf2] (2).

Figure 1.
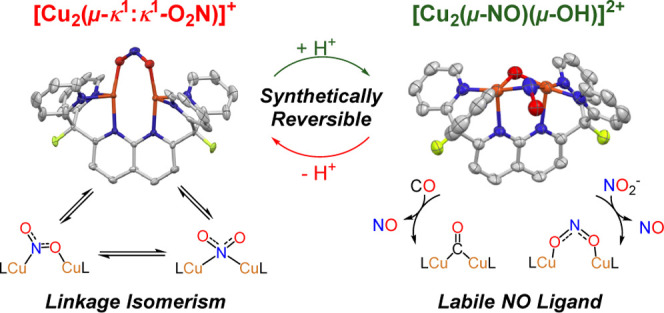
Solid-state structure of 2 as determined by single-crystal X-ray diffraction. The NTf2– counterion and all hydrogens are omitted for clarity. Thermal ellipsoids are set at the 50% probability level.
The solid-state molecular structure of 2, determined by single crystal X-ray diffraction, revealed that the copper(I) ions exist in an unusual, distorted seesaw geometry (τ4 ≈ 0.65, τ4′ ≈ 0.44)30,31 with an average Cu–Cu distance of 2.55(3) Å. The nitrite bonding metrics in the three cationic complexes within the asymmetric unit cell exhibit statistically significant variations, likely resulting from molecular motion. Nevertheless, the symmetrically O,O-bound nitrito ligand (Figure 1) displays surprisingly short Cu–O contacts (Cu–O(avg) = 1.96(2) Å), indicating strong bonding interactions. The enhanced bonding interactions may also result from the reduced ring strain inherent to a μ-κ1:κ1 binding mode as compared to a nitrite ligand chelating a single copper. The N–O bond distances (N–O(avg) = 1.23(11) Å) are comparable to those found in the few mononuclear copper(I) nitrito complexes that have been reported.32−37 To our knowledge, 2 is the only dicopper(I,I) μ-κ1:κ1-nitrite complex to be structurally characterized. In accordance with hard–soft acid–base (HSAB) theory, the nitrito binding mode is most common in cupric systems.38,39 Consequently, mononuclear copper(I) nitrito complexes are often unstable.40,41 The few that have been reported are almost exclusively stabilized by soft phosphine ligands, with only one possessing a bidentate N-donor ligand.32−37 In contrast, complex 2 is stable indefinitely in the solid state and in solution. A possible explanation for the enhanced stability of 2 is that bonding to two copper(I) ions results in a coordinatively nonlabile nitrito ligand.
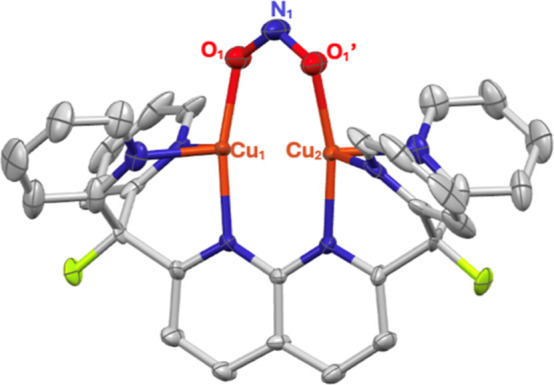
While multinuclear NMR spectroscopy and X-ray crystallography indicate a symmetrical μ-κ1:κ1-nitrite binding mode, DFT computations (B3LYP/def2-TZVPP/CPCM-THF) suggest that the ligand can engage in several coordination modes akin to those of previously reported mononuclear copper nitrite complexes.42−44 Two closed-shell singlet linkage isomers were found to be close in free energy to the O,O μ-κ1:κ1 model complex (2*). The model linkage isomer closest in energy (+0.5 kcal mol–1) to the observed ground state features a μ-(η1-N:η1-O) (2*′) binding mode for the bridging nitrite (Figure 2a), reminiscent of the nitrite-bridged cobalt–magnesium complex reported by Uyeda and Peters.19 The other linkage isomer, with a nitro (N-bound) binding mode (2*″), is only 1.6 kcal mol–1 higher in free energy than the ground state (Figure 2b). Calculations indicate that higher solvent dielectric constants lessen the energy gap between the isomers (Figure S34). A small energetic difference between linkage isomers is not unprecedented for copper(I) nitrite complexes,41−45 and in this case the gap is likely minimized by the presence of a second Lewis acidic center that aids in accommodating different binding modes. These computational results indicate that all isomers are energetically accessible for reaction chemistry.
Figure 2.
DFT calculated molecular structures of closed-shell singlet linkage isomers of [Cu2(μ-κ1:κ1-O2N)DPFN]+ (2*). The [Cu2(μ-η1:η1-NO2)DPFN]+ (2*′, a) and the [Cu2(μ-η1-NO2)DPFN]+ (2*″, b) linkage isomers are 0.5 and 1.6 kcal mol–1 higher in free energy than the ground state, respectively. All hydrogens are omitted for clarity.
To ascertain its capability for proton-coupled nitrite reduction, complex 2 was treated with acids of varying strength. Thiols were of particular interest given their ubiquity in metabolic processes and the reported protonation of a nonheme dinitro complex to produce NO.46−48 Treatment of 2 with one equiv of p-TolSH in THF solution resulted in a rapid solution color change from dark red to orange-red. The 1H NMR spectrum of the crude mixture indicated consumption of the nitrite complex and appearance of new resonances for [Cu2(μ-STol)DPFN][NTf2] (3; Scheme 3). The product was isolated after workup as an analytically pure beige powder in high yield (97%).
Scheme 3. Synthesis of [Cu2(μ-STol)DPFN][NTf2] (3).

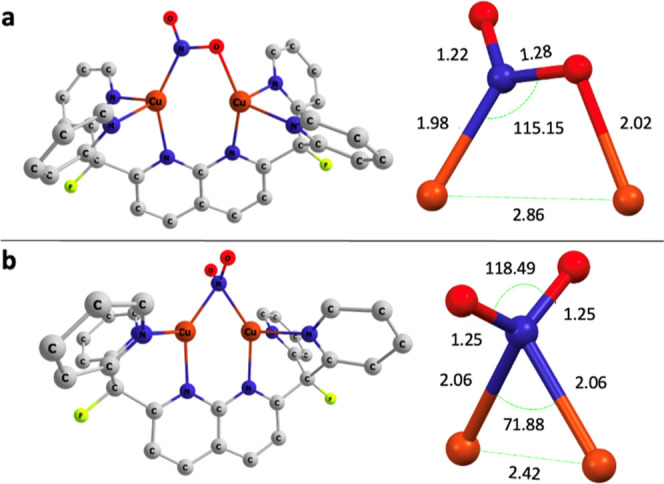
Complex 3 is likely formed by an acid–base exchange akin to the reactivity previously observed for a tris-pyrazolyl zinc nitrite complex reported by Warren and co-workers.49 Although the expected nitrous acid byproduct was not directly observed, exposure of cobalt(II) 5,10,15,20-tetraphenylporphyrin (CoTPP) to the gaseous product released from the reaction produced the {CoNO}8 product (61%) and water, as determined by 1H NMR spectroscopy (Figure S11). The formation of these byproducts in the reaction vessel is likely a result of nitrous acid decomposition after expulsion from the dicopper core.13 The solid-state molecular structure of 3 displays a thiolate ligand bridging the dicopper unit with the tolyl substituent canted away from the plane containing the naphthyridine backbone (Figure 3).
Figure 3.
Solid-state structure of 3 as determined by single-crystal X-ray diffraction. The NTf2– counterion and all hydrogens are omitted for clarity. Thermal ellipsoids are set at the 50% probability level.
Since the reaction of 2 with a thiol results in displacement of the presumed nitrous acid product formed by protonation, a Brønsted acid with a weakly coordinating conjugate base was employed to favor HONO retention at the dicopper core. Treatment of 2 with HNTf2 in thawing THF at −196 °C, followed by warming to 23 °C, resulted in a color change from dark red to dark green, corresponding to the formation of [Cu2(μ-NO)(μ-OH)DPFN][NTf2]2 (4; Figures 4 and 5). Addition of pentane to a saturated THF solution of the compound enabled its isolation as an analytically pure dark green powder in high yield (92%). The diamagnetic complex exhibits a 1H NMR spectrum reflecting a loss of symmetry evident by the presence of 11 resonances that together integrate to 21 protons. The 19F{1H} NMR spectrum reveals a shift of the ligand resonance from −172.90 ppm in 2 to −174.41 ppm (with respect to the resonance of benzene-f6 in THF) and the presence of two NTf2– counterions per formula unit (Figure S9). Infrared spectroscopy confirms the presence of two new bands belonging to 4, a broad band at 3547 cm–1, assigned as an O–H stretch, and another weak band at 1555 cm–1. The latter band shifts to 1522 cm–1 with 15N labeling (Δν(NO)14N–15N = 33 cm–1, Figure S28) and is assigned as an N–O stretch. This low νN–O value is consistent with those reported in the literature for a NO– ligand bridging an oxidized dicopper core.27,50,51
Figure 4.

Stoichiometric cycle illustrating the synthesis of [Cu2(μ-NO)(μ-OH)DPFN][NTf2]2 (4) and its deprotonation to yield [Cu2(μ-κ1:κ1-O2N)DPFN][NTf2] (2) via a dicopper(II,II) μ-NO, μ-O intermediate that undergoes N–O bond formation. The DFT-calculated geometry of [Cu2(μ-NO)(μ-O)DPFN]+ is also shown with hydrogens omitted for clarity. *Yield determined by integration of the 1H NMR spectrum against an internal standard.
Figure 5.
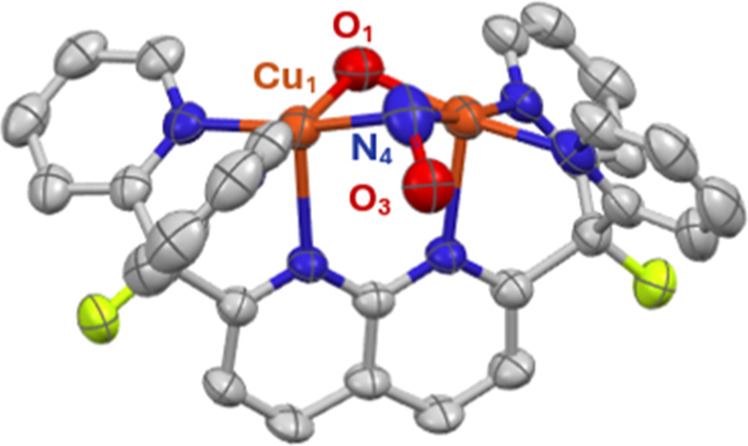
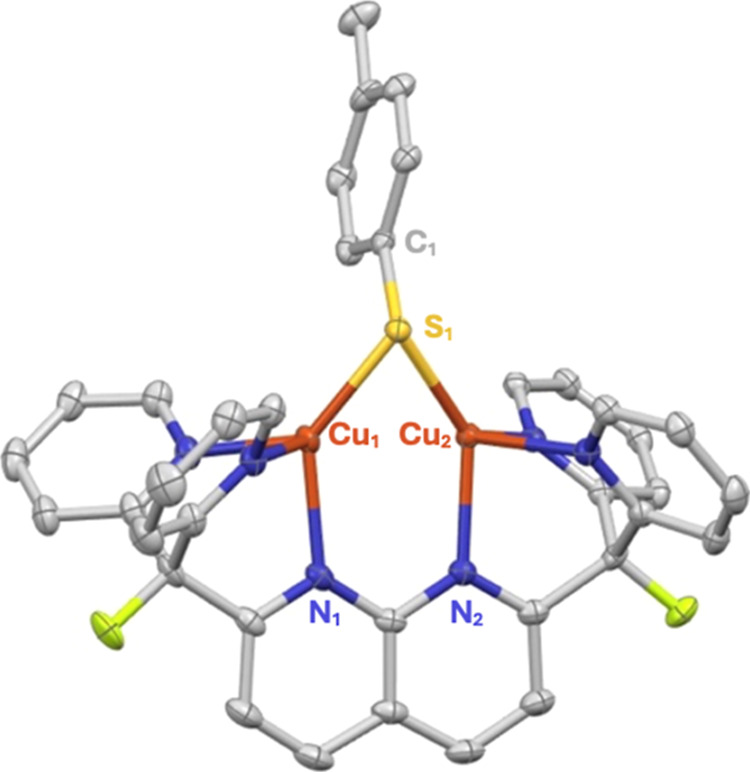
Solid-state structure of 4 as determined by single-crystal X-ray diffraction. The NTf2– counterions and hydrogens are omitted for clarity. Thermal ellipsoids are set at the 50% probability level.
The obtained solid state molecular structure for this compound is a mixture (30:70) of 4 and the previously reported [Cu2(μ-OH)2DPFN][NTf2]2 (5), likely resulting from unavoidable exposure of single crystals of 4 to air during the diffraction experiment.53 Further collection attempts yielded X-ray diffraction data with similar mixtures. While the structural data illustrates the NO binding mode and the lack of NO/OH ligand disorder across the plane containing the naphthyridine backbone, the structural metrics are less informative as they represent a superposition of both species. Nonetheless, both copper atoms have a slightly distorted square pyramidal geometry (τ5 ≈ 0.04)52 and are 2.7569(3) Å apart. This distance is much larger than that of 2, which suggests that the nitrite functions to bring the copper ions closer together. The hydroxy ligand symmetrically bridges the dicopper core (Cu–O = 1.9047(11) Å) in a manner similar to that of other hydroxy-bridged dicopper complexes supported by DPFN (1.89–1.96 Å).53,54 The nitrosyl N–O bond distance (N–O = 1.3816(14) Å) is longer than those of the other reported dicopper μ-nitrosyl complexes, with the Zhang group’s [Cu2(μ-NO)(μ-OMe)Py4DMB]2+ complex being the closest at 1.32(5) Å. The Cu–Nμ bond distance [1.9601(16) Å] in the complex is the smallest reported for such a moiety, but remains consistent with structural data of the other nitrosyl-bridged dicopper complexes.27,50,51,55
Computational modeling shows that the ground state of 4 is best described as a dicopper(II,II) open-shell singlet. The copper(II) ions show no coupling between each other, but instead engage in antiferromagnetic interactions with the nitrosyl ligand. This is in good agreement with the diamagnetic character reflected in sharp NMR resonances of the complex. Analysis of its Löwdin spin population shows ca. +0.4 unpaired e– on each of the copper atoms. The remaining spin density is mostly located on the nitrosyl ligand with approximately −0.6 unpaired e– on each of its atoms. The oxygen atom on the hydroxy ligand minimally contributes to the antiferromagnetic behavior with a spin density of +0.1 e–. Together, the computational and spectroscopic results indicate that complex 4 is formally a dicopper(II,II) complex possessing one OH–, one NO–, and two NTf2– counterions completing the charge balance.
DFT computations (B3LYP/def2-TZVPP/CPCM-THF) were also employed to gain insight into the mechanism of protonation and nitrite cleavage from the model complex [Cu2(μ-κ1:κ1-O2N)DPFN]+ (2*) to yield [Cu2(μ-NO)(μ-OH)DPFN]2+ (4*). The calculations suggest that protonation initially occurs at one of the oxygen atoms of the μ-κ1:κ1 bridging nitrite, which requires 8.4 kcal mol– of free energy and forms a transient [Cu2(μ-κ1:κ1-HONO)DPFN]2+ complex, intermediate A, analogous to those spectroscopically observed during nitrite reduction by functional mimics of the CuNiR.56 The following step involves an exergonic linkage isomerism of the HONO ligand to intermediate B with a nitro binding mode (μ-NO2H). This is in good agreement with calculations showing that 2*″ possesses a more negative free energy of protonation compared to the other two linkage isomers of the nitrite complex (−2.46 kcal mol–1 for 2*″ vs 8.43 kcal mol–1 for 2*, Figure S35). Intermediate B then undergoes an oxidative addition of the N–OH bond at the dicopper core to yield 4*. Cleavage of the N–O bond of intermediate B is accompanied by a moderate release of energy (11.1 kcal mol–1) which drives the process forward (Figure 6). Another possible reaction pathway involves cleaving one N–O bond in μ-κ1:κ1-NO2– prior to protonation. However, to form a [Cu2(μ-NO)(μ-O)DPFN]+ species from complex 2*, the system must overcome a significant energetic barrier of 45 kcal mol–1 (Figure S36). Similar calculations conducted on the CuNiR active site have attributed the large energetic cost to the destabilizing charge that develops on the O2– atom as the N–O bond of the nitrite ligand is being cleaved.9 Thus, reminiscent of the CuNiR mechanism, the protonation-induced N–O cleavage pathway is more accessible.
Figure 6.
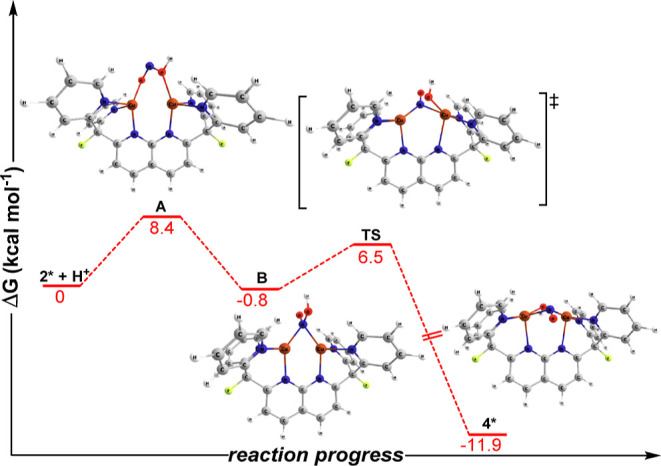
Reaction coordinate diagram for the formation of 4* derived by DFT at the B3LYP/def2-TZVPP/CPCM-THF level of theory.
Nitrosyl complex 4 bears an unusual level of thermal stability given that, to our knowledge, only one other reported dicopper nitrosyl-bridged compound is stable at ambient temperature.51 Under an inert atmosphere, complex 4 is stable for several days in THF solution or over months in the solid state without signs of decomposition. Interestingly, DFT analysis reveals that there is a 18.9 kcal mol–1 barrier to the thermoneutral dissociation of NO from 4 (ΔG of −0.25 kcal mol–1, Figure S38). Treatment of 4 with stoichiometric amounts of CoTPP resulted in immediate formation of the {CoNO}8 species (97%) along with [Cu3(DPFN)2][NTf2]3 (17%) and [Cu2(μ-OH)2DPFN][NTf2]2 (37%) (5), as determined by 1H and 19F{1H} NMR spectroscopy. Both copper complexes have been isolated and fully characterized in this laboratory, and the tricationic helical complex is known to result from loss of the bridging ligand at the dicopper core.53,57 This indicates a dissociative loss of the nitrosyl ligand from the dicopper center, followed by reaction with CoTPP to yield {CoNO}8. Nonetheless, a bimolecular reaction between both metal complexes resulting in NO transfer cannot be completely ruled out. A proposed pathway involves an initial trapping of NO by Co(TPP), followed by disproportionation of the resulting [CuICuII(μ-OH)]2+ core to form the stable complexes [CuII2(μ-OH)2]2+ and [Cu3(DPFN)2][NTf2]3 (Scheme 4).
Scheme 4. Reactivity of [Cu2(μ-NO)(μ-OH)DPFN][NTf2]2 (4) with CoTPP, CO, and [nBu4N][NO2].

*Yield determined by integration of the 19F NMR spectrum against an internal standard.
To further probe the lability of the nitrosyl ligand, 4 was exposed to excess carbon monoxide, leading to a gradual color change from dark green to light blue over the course of 30 min (Figure S18). The 19F NMR spectrum of the crude mixture shows the formation of [Cu2(μ-CO)DPFN][NTf2]2 and 5 in 35 and 41% yield, respectively (Scheme 4).54 These results suggest that, while CO can competently displace NO, the proposed [CuICuII(μ-OH)(μ-CO)DPFN]2+ species is susceptible to disproportionation. Interestingly, CO is not the only ligand that promotes this type of disproportionation. Treatment of 4 with half an equivalent of [nBu4N][NO2] results in a darkening of the solution to dark red in under 5 min. Multinuclear NMR spectroscopic analysis reveals the formation of 2 (31%), 5 (33%), and the salt metathesis product [nBu4N][NTf2] (95%) (Scheme 4). This reaction also likely proceeds through a mixed-valent intermediate [CuICuII(μ-OH)(μ-κ1:κ1-NO2)DPFN]+, which could further react to ultimately yield 2. Not only do these results establish novel reactivity pathways for a μ-nitrosyl, μ-hydroxy dicopper core, they are also of potential relevance to current interest in nitric oxide releasing systems.58
Given the observed stability of Cu2(μ-κ1:κ1-O2N)DPFN][NTf2], it was hypothesized that deprotonation of the hydroxy ligand of 4 would result in N–O bond formation and reductive elimination of NO2–. Indeed, addition of a stoichiometric amount of KOtBu to a THF solution of 4 gave an instant color change to dark red along with a noticeable exotherm evidenced by warming of the reaction flask. The 1H and 19F NMR spectra of the crude reaction mixture confirmed successful deprotonation of the hydroxy ligand of 4 to yield 2 (72%), tert-butanol (98%), and potassium bistriflimide (Figure 4). Computational modeling (B3LYP/def2-SVP/CPCM-THF) of a likely initial product of deprotonation [Cu2(μ-NO)(μ-O)DPFN][NTf2], suggests a similar electronic structure to 4. The optimized structure of this intermediate shares similar Cu–Cu (2.756 Å) and Cu(II)–O (1.859 Å) distances to those observed in [Cu2(μ-NO)(μ-O)Py4DMB]2+. The Cu–Nμ distances (2.104 Å), however, are longer than that of [Cu2(μ-NO)(μ-O)Py4DMB]2+(Cu(II)–Nμ = 2.03(1) Å) and those in the nitrosyl-bridged dicopper complex (Figure 4). This intermediate is significantly destabilized relative to nitrite-bridged dicopper complex 2 by 29.6 kcal mol–1 and overcomes a 13.9 kcal mol–1 barrier to N–O bond formation (Figure S37).
Conclusions
Mononuclear copper coordination compounds have been extensively studied in the context of biomimetic nitrite reduction. Meanwhile, nitrite bond activation at dicopper cores has only recently garnered attention, despite the valuable information these systems can offer. These results demonstrate that two copper(I) centers in proximity can work in concert to stabilize what historically has been an unstable linkage for mononuclear complexes. Moreover, this cooperativity allows the nitrito ligand to be reversibly cleaved by a proton, a process permitted by nitrosyl retention at the dicopper core. To our knowledge, this is the first instance of a stoichiometric cycle reminiscent of the CuNiR’s pH-dependent, bidirectional catalysis occurring on a dicopper platform. Computational and spectroscopic methods were used to describe a rare antiferromagnetically coupled dicopper(II,II) nitrosyl hydroxy complex that possesses unusual thermal stability. The lability of the nitrosyl ligand provides a new avenue for the formation and study of other reactive bimetallic species and possesses relevance toward the study of NO-releasing systems.
Acknowledgments
This work was primarily funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, under Contract no. DE-AC02-05CH11231 (to TDT.) and DE-FG02-03ER15387 (to TRC.). The LBNL Catalysis Laboratory provided HR-ESI-MS instrumentation. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE 1752814 (to JMF). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. We thank Dr. Nick Settineri and the UC Berkeley CheXray crystallographic facility under grant no. S10-RR027172. We thank Drs. Hasan Celik, Raynald Giovine, and the Pines Magnetic Resonance Center’s Core NMR Facility (PMRC Core) for spectroscopic assistance. The instrument used in this work was in part supported by NIH S10OD024998. We also thank Drs. Tianchang Liu, Marcel Bamberg, Laurent Severy, and Rex C. Handford for useful conversations and advice.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c14642.
Detailed experimental methods, including characterization data, spectra, details of X-ray crystallography, and computational studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Maia L. B.; Moura J. J. G. How Biology Handles Nitrite. Chem. Rev. 2014, 114 (10), 5273–5357. 10.1021/cr400518y. [DOI] [PubMed] [Google Scholar]
- Cioncoloni G.; Roger I.; Wheatley P. S.; Wilson C.; Morris R. E.; Sproules S.; Symes M. D. Proton-Coupled Electron Transfer Enhances the Electrocatalytic Reduction of Nitrite to NO in a Bioinspired Copper Complex. ACS Catal. 2018, 8 (6), 5070–5084. 10.1021/acscatal.8b00361. [DOI] [Google Scholar]
- Godden J. W.; Turley S.; Teller D. C.; Adman E. T.; Liu M. Y.; Payne W. J.; LeGall J. The 2.3 Angstrom X-Ray Structure of Nitrite Reductase from Achromobacter Cycloclastes. Science 1991, 253 (5018), 438–442. 10.1126/science.1862344. [DOI] [PubMed] [Google Scholar]
- Adman E. T.; Godden J. W.; Turley S. The Structure of Copper-Nitrite Reductase from Achromobacter Cycloclastes at Five pH Values, with NO2– Bound and with Type II Copper Depleted. J. Biol. Chem. 1995, 270 (46), 27458–27474. 10.1074/jbc.270.46.27458. [DOI] [PubMed] [Google Scholar]
- Lawton T. J.; Bowen K. E.; Sayavedra-Soto L. A.; Arp D. J.; Rosenzweig A. C. Characterization of a Nitrite Reductase Involved in Nitrifier Denitrification. J. Biol. Chem. 2013, 288 (35), 25575–25583. 10.1074/jbc.M113.484543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonyuk S. V.; Strange R. W.; Sawers G.; Eady R. R.; Hasnain S. S. Atomic Resolution Structures of Resting-State, Substrate- and Product-Complexed Cu-Nitrite Reductase Provide Insight into Catalytic Mechanism. Proc. Natl. Acad. Sci. U.S.A. 2005, 102 (34), 12041–12046. 10.1073/pnas.0504207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocheva E. I.; Rosell F. I.; Mauk A. G.; Murphy M. E. P. Side-On Copper-Nitrosyl Coordination by Nitrite Reductase. Science 2004, 304 (5672), 867–870. 10.1126/science.1095109. [DOI] [PubMed] [Google Scholar]
- Kujime M.; Izumi C.; Tomura M.; Hada M.; Fujii H. Effect of a Tridentate Ligand on the Structure, Electronic Structure, and Reactivity of the Copper(I) Nitrite Complex: Role of the Conserved Three-Histidine Ligand Environment of the Type-2 Copper Site in Copper-Containing Nitrite Reductases. J. Am. Chem. Soc. 2008, 130 (19), 6088–6098. 10.1021/ja075575b. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Dey A.; Sun Y.; Scholes C. P.; Solomon E. I. Spectroscopic and Computational Studies of Nitrite Reductase: Proton Induced Electron Transfer and Backbonding Contributions to Reactivity. J. Am. Chem. Soc. 2009, 131 (1), 277–288. 10.1021/ja806873e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore C. M.; Szymczak N. K. Nitrite Reduction by Copper through Ligand-Mediated Proton and Electron Transfer. Chem. Sci. 2015, 6 (6), 3373–3377. 10.1039/C5SC00720H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Lukoyanov D. A.; Toropov Y. V.; Wu K.; Shapleigh J. P.; Scholes C. P. Catalytic Function and Local Proton Structure at the Type 2 Copper of Nitrite Reductase: The Correlation of Enzymatic pH Dependence, Conserved Residues, and Proton Hyperfine Structure. Biochemistry 2002, 41 (23), 7464–7474. 10.1021/bi0256274. [DOI] [PubMed] [Google Scholar]
- Wijma H. J.; Canters G. W.; De Vries S.; Verbeet M. Ph. Bidirectional Catalysis by Copper-Containing Nitrite Reductase. Biochemistry 2004, 43 (32), 10467–10474. 10.1021/bi0496687. [DOI] [PubMed] [Google Scholar]
- Bouchey C. J.; Tolman W. B. Involvement of a Formally Copper(III) Nitrite Complex in Proton-Coupled Electron Transfer and Nitration of Phenols. Inorg. Chem. 2022, 61 (5), 2662–2668. 10.1021/acs.inorgchem.1c03790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.-L.; Lin Y.-F.; Chuang W.-J.; Kao C.-L.; Narwane M.; Chen H.-Y.; Chiang M. Y.; Hsu S. C. N. Structure and Nitrite Reduction Reactivity Study of Bio-Inspired Copper(I)–Nitro Complexes in Steric and Electronic Considerations of Tridentate Nitrogen Ligands. Dalton Trans. 2018, 47 (15), 5335–5341. 10.1039/C7DT03843G. [DOI] [PubMed] [Google Scholar]
- Hsu S. C. N.; Chang Y.-L.; Chuang W.-J.; Chen H.-Y.; Lin I.-J.; Chiang M. Y.; Kao C.-L.; Chen H.-Y. Copper(I) Nitro Complex with an Anionic [HB(3,5-Me2Pz)3]− Ligand: A Synthetic Model for the Copper Nitrite Reductase Active Site. Inorg. Chem. 2012, 51 (17), 9297–9308. 10.1021/ic300932a. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Dixon N. A.; Merkle A. C.; Zeller M.; Lehnert N.; Papish E. T. Hydrotris(Triazolyl)Borate Complexes as Functional Models for Cu Nitrite Reductase: The Electronic Influence of Distal Nitrogens. Inorg. Chem. 2012, 51 (13), 7004–7006. 10.1021/ic300160c. [DOI] [PubMed] [Google Scholar]
- Maji R. C.; Barman S. K.; Roy S.; Chatterjee S. K.; Bowles F. L.; Olmstead M. M.; Patra A. K. Copper Complexes Relevant to the Catalytic Cycle of Copper Nitrite Reductase: Electrochemical Detection of NO(g) Evolution and Flipping of NO2 Binding Mode upon CuII → CuI Reduction. Inorg. Chem. 2013, 52 (19), 11084–11095. 10.1021/ic401295t. [DOI] [PubMed] [Google Scholar]
- Halfen J. A.; Mahapatra S.; Wilkinson E. C.; Gengenbach A. J.; Young V. G.; Que L.; Tolman W. B. Synthetic Modeling of Nitrite Binding and Activation by Reduced Copper Proteins. Characterization of Copper(I)–Nitrite Complexes That Evolve Nitric Oxide. J. Am. Chem. Soc. 1996, 118 (4), 763–776. 10.1021/ja952691i. [DOI] [Google Scholar]
- Uyeda C.; Peters J. C. Selective Nitrite Reduction at Heterobimetallic CoMg Complexes. J. Am. Chem. Soc. 2013, 135 (32), 12023–12031. 10.1021/ja4053653. [DOI] [PubMed] [Google Scholar]
- Blake A. J.; Hill S. J.; Hubberstey P. Unique Cationic, Neutral and Anionic Copper(II) Nitrite Species in a Single Compound. Chem. Commun. 1998, (15), 1587–1588. 10.1039/a803762k. [DOI] [Google Scholar]
- Escuer A.; El Fallah M. S.; Vicente R.; Sanz N.; Font-Bardia M.; Solans X.; Mautner F. A. Polynuclear and 1-D Derivatives of 1,3-Bis(Dimethylamino)-2-Propanolato Ligand. Structure and Magnetic Characterization. Dalton Trans. 2004, 12, 1867. 10.1039/b402629b. [DOI] [PubMed] [Google Scholar]
- Arnold P. J.; Davies S. C.; Durrant M. C.; Griffiths D. V.; Hughes D. L.; Sharpe P. C. Copper(II) Nitrite Complexes of Tripodal Ligands Derived from 1,1,1-Tris(2-Pyridyl)Methylamine. Inorg. Chim. Acta 2003, 348, 143–149. 10.1016/S0020-1693(02)01487-1. [DOI] [Google Scholar]
- Biswas S.; Karim S.; Zangrando E.; Chandra A. An Effortless Approach to Synthesize Two Structurally Diverse Nano Copper(II) Materials and Assessment of Their Apoptosis-inducing Ability on Lung Cancer Cell Line. Appl. Organomet. Chem. 2022, 36 (5), e6659 10.1002/aoc.6659. [DOI] [Google Scholar]
- Brooks S. H.; Richards C. A.; Carroll P. J.; Gau M. R.; Tomson N. C. Anion Capture at the Open Core of a Geometrically Flexible Dicopper(II,II) Macrocycle Complex. Inorganics 2023, 11 (9), 348. 10.3390/inorganics11090348. [DOI] [Google Scholar]
- Camus A.; Marsich N.; Manotti Lanfredi A. M.; Ugozzoli F.; Massera C. Copper(II)nitrito complexes with 2,2′-dipyridylamine. Crystal structures of the [(acetato)(2,2′-dipyridylamine)(nitrito-O,O′)copper(II)] and [(2,2′-dipyridylamine) (nitrito-O,O′)(μ-nitrito-O)copper(II)]2·2(acetonitrile). Inorg. Chim. Acta 2000, 309 (1–2), 1–9. 10.1016/S0020-1693(00)00213-9. [DOI] [Google Scholar]
- Halfen J. A.; Mahapatra S.; Olmstead M. M.; Tolman W. B. Synthetic Analogs of Nitrite Adducts of Copper Proteins: Characterization and Interconversion of Dicopper(I,I) and -(I,II) Complexes Bridged Only by NO2–. J. Am. Chem. Soc. 1994, 116 (5), 2173–2174. 10.1021/ja00084a079. [DOI] [Google Scholar]
- Tao W.; Bower J. K.; Moore C. E.; Zhang S. Dicopper μ-Oxo, μ-Nitrosyl Complex from the Activation of NO or Nitrite at a Dicopper Center. J. Am. Chem. Soc. 2019, 141 (26), 10159–10164. 10.1021/jacs.9b03635. [DOI] [PubMed] [Google Scholar]
- Ziegler M. S.; Levine D. S.; Lakshmi K. V.; Tilley T. D. Aryl Group Transfer from Tetraarylborato Anions to an Electrophilic Dicopper(I) Center and Mixed-Valence μ-Aryl Dicopper(I,II) Complexes. J. Am. Chem. Soc. 2016, 138 (20), 6484–6491. 10.1021/jacs.6b00802. [DOI] [PubMed] [Google Scholar]
- Desnoyer A. N.; Nicolay A.; Rios P.; Ziegler M. S.; Tilley T. D. Bimetallics in a Nutshell: Complexes Supported by Chelating Naphthyridine-Based Ligands. Acc. Chem. Res. 2020, 53 (9), 1944–1956. 10.1021/acs.accounts.0c00382. [DOI] [PubMed] [Google Scholar]
- Yang L.; Powell D. R.; Houser R. P. Structural Variation in Copper(I) Complexes with Pyridylmethylamide Ligands: Structural Analysis with a New Four-Coordinate Geometry Index τ4. Dalton Trans. 2007, 955–964. 10.1039/B617136B. [DOI] [PubMed] [Google Scholar]
- Okuniewski A.; Rosiak D.; Chojnacki J.; Becker B. Coordination Polymers and Molecular Structures among Complexes of Mercury(II) Halides with Selected 1-Benzoylthioureas. Polyhedron 2015, 90, 47–57. 10.1016/j.poly.2015.01.035. [DOI] [Google Scholar]
- Chen C.-S.; Dai H.-F.; Chen C.-H.; Yeh W.-Y. S. Synthesis, characterization and protonation reaction of copper and palladium complexes bearing nitrite ligands in O,O-bidentate and N-monodentate bonding fashions. Inorg. Chim. Acta 2011, 376 (1), 396–400. 10.1016/j.ica.2011.06.052. [DOI] [Google Scholar]
- Halfen J. A.; Tolman W. B. Nitrito-O,O’Bis(Triphenylphosphine)Copper(I), (PPh3)2Cu(NO2-O,O’). Acta Crystallogr., Sect. C:Cryst. Struct. Commun. 1995, 51 (2), 215–217. 10.1107/S0108270194008991. [DOI] [PubMed] [Google Scholar]
- Chen C.-S.; Yeh W.-Y. Coordination of NO2– Ligand to Cu(I) Ion in an O,O-Bidentate Fashion That Evolves NO Gas upon Protonation: A Model Reaction Relevant to the Denitrification Process. Chem. Commun. 2010, 46 (18), 3098. 10.1039/b927513d. [DOI] [PubMed] [Google Scholar]
- Chuang W.-J.; Lin I.-J.; Chen H.-Y.; Chang Y.-L.; Hsu S. C. N. Characterization of A New Copper(I)–Nitrito Complex That Evolves Nitric Oxide. Inorg. Chem. 2010, 49 (12), 5377–5384. 10.1021/ic100083b. [DOI] [PubMed] [Google Scholar]
- Kalita A.; Kumar P.; Deka R. C.; Mondal B. First Example of a Cu(i)–(η2-O,O)Nitrite Complex Derived from Cu(ii)–Nitrosyl. Chem. Commun. 2012, 48 (9), 1251–1253. 10.1039/C1CC16316G. [DOI] [PubMed] [Google Scholar]
- Chuang W.-J.; Narwane M.; Chen H.-Y.; Kao C.-L.; Huang B.; Hsu K.-M.; Wang Y.-M.; Hsu S. C. N. Nitric Oxide-Release Study of a Bio-Inspired Copper(I)-Nitrito Complex under Chemical and Biological Conditions. Dalton Trans. 2018, 47 (37), 13151–13157. 10.1039/C8DT02281J. [DOI] [PubMed] [Google Scholar]
- Mondal A.; Reddy K. P.; Bertke J. A.; Kundu S. Phenol Reduces Nitrite to NO at Copper(II): Role of a Proton-Responsive Outer Coordination Sphere in Phenol Oxidation. J. Am. Chem. Soc. 2020, 142 (4), 1726–1730. 10.1021/jacs.9b11597. [DOI] [PubMed] [Google Scholar]
- Stauffer M.; Sakhaei Z.; Greene C.; Ghosh P.; Bertke J. A.; Warren T. H. Mechanism of O-Atom Transfer from Nitrite: Nitric Oxide Release at Copper(II). Inorg. Chem. 2021, 60 (21), 15968–15974. 10.1021/acs.inorgchem.1c00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chand K.; Chu Y.-C.; Wang T.-W.; Kao C.-L.; Lin Y.-F.; Tsai M.-L.; Hsu S. C. N. Nitric Oxide Generation Study of Unsymmetrical β-Diketiminato Copper(ii) Nitrite Complexes. Dalton Trans. 2022, 51 (9), 3485–3496. 10.1039/D1DT03711K. [DOI] [PubMed] [Google Scholar]
- Lehnert N.; Cornelissen U.; Neese F.; Ono T.; Noguchi Y.; Okamoto K.; Fujisawa K. Synthesis and Spectroscopic Characterization of Copper(II)–Nitrito Complexes with Hydrotris(Pyrazolyl)Borate and Related Coligands. Inorg. Chem. 2007, 46 (10), 3916–3933. 10.1021/ic0619355. [DOI] [PubMed] [Google Scholar]
- Borowski P.; Kutniewska S. E.; Kamiński R.; Krówczyński A.; Schaniel D.; Jarzembska K. N. Exploring Photoswitchable Properties of Two Nitro Nickel(II) Complexes with (N, N, O)-Donor Ligands and Their Copper(II) Analogues. Inorg. Chem. 2022, 61 (17), 6624–6640. 10.1021/acs.inorgchem.2c00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woollard-Shore J. G.; Holland J. P.; Jones M. W.; Dilworth J. R. Nitrite reduction by Copper Complexes. Dalton Trans. 2010, 39 (6), 1576–1585. 10.1039/B913463H. [DOI] [PubMed] [Google Scholar]
- Roger I.; Wilson C.; Senn H. M.; Sproules S.; Symes M. D. An Investigation into the Unusual Linkage Isomerization and Nitrite Reduction Activity of a Novel Tris(2-Pyridyl) Copper Complex. R. Soc. Open Sci. 2017, 4 (8), 170593. 10.1098/rsos.170593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H.; Yamaguchi K.; Sugimoto M.; Suzuki S. CuI and CuII Complexes Containing Nitrite and Tridentate Aromatic Amine Ligand as Models for the Substrate-Binding Type-2 Cu Site of Nitrite Reductase. Eur. J. Inorg. Chem. 2005, 2005 (8), 1435–1441. 10.1002/ejic.200400808. [DOI] [Google Scholar]
- Sanders B. C.; Hassan S. M.; Harrop T. C. NO2– Activation and Reduction to NO by a Nonheme Fe(NO2)2 Complex. J. Am. Chem. Soc. 2014, 136 (29), 10230–10233. 10.1021/ja505236x. [DOI] [PubMed] [Google Scholar]
- Kundu S.; Kim W. Y.; Bertke J. A.; Warren T. H. Copper(II) Activation of Nitrite: Nitrosation of Nucleophiles and Generation of NO by Thiols. J. Am. Chem. Soc. 2017, 139 (3), 1045–1048. 10.1021/jacs.6b11332. [DOI] [PubMed] [Google Scholar]
- Cortese-Krott M. M.; Fernandez B. O.; Kelm M.; Butler A. R.; Feelisch M. On the Chemical Biology of the Nitrite/Sulfide Interaction. Nitric Oxide 2015, 46, 14–24. 10.1016/j.niox.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Cardenas A. J. P.; Abelman R.; Warren T. H. Conversion of Nitrite to Nitric Oxide at Zinc via S-Nitrosothiols. Chem. Commun. 2014, 50 (2), 168–170. 10.1039/C3CC46102E. [DOI] [PubMed] [Google Scholar]
- Tao W.; Moore C. E.; Zhang S. Redox-Neutral S-nitrosation Mediated by a Dicopper Center. Angew. Chem., Int. Ed. 2021, 60 (29), 15980–15987. 10.1002/anie.202102589. [DOI] [PubMed] [Google Scholar]
- Paul P. P.; Tyeklar Z.; Farooq A.; Karlin K. D.; Liu S.; Zubieta J. Isolation and X-Ray Structure of a Dinuclear Copper-Nitrosyl Complex. J. Am. Chem. Soc. 1990, 112 (6), 2430–2432. 10.1021/ja00162a060. [DOI] [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; Van Rijn J.; Verschoor G. C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-Bis(N-Methylbenzimidazol-2′-Yl)-2,6-Dithiaheptane]Copper(II) Perchlorate. J. Chem. Soc., Dalton Trans. 1984, 1349–1356. 10.1039/DT9840001349. [DOI] [Google Scholar]
- Desnoyer A. N.; Nicolay A.; Ziegler M. S.; Torquato N. A.; Tilley T. D. A Dicopper Platform That Stabilizes the Formation of Pentanuclear Coinage Metal Hydride Complexes. Angew. Chem., Int. Ed. 2020, 59 (31), 12769–12773. 10.1002/anie.202004346. [DOI] [PubMed] [Google Scholar]
- Desnoyer A. N.; Nicolay A.; Ziegler M. S.; Lakshmi K. V.; Cundari T. R.; Tilley T. D. A Dicopper Nitrenoid by Oxidation of a CuICuI Core: Synthesis, Electronic Structure, and Reactivity. J. Am. Chem. Soc. 2021, 143 (18), 7135–7143. 10.1021/jacs.1c02235. [DOI] [PubMed] [Google Scholar]
- Tao W.; Yerbulekova A.; Moore C. E.; Shafaat H. S.; Zhang S. Controlling the Direction of S-Nitrosation versus Denitrosation: Reversible Cleavage and Formation of an S–N Bond within a Dicopper Center. J. Am. Chem. Soc. 2022, 144 (7), 2867–2872. 10.1021/jacs.1c12799. [DOI] [PubMed] [Google Scholar]
- Kujime M.; Fujii H. Spectroscopic Characterization of Reaction Intermediates in a Model for Copper Nitrite Reductase. Angew. Chem., Int. Ed. 2006, 45 (7), 1089–1092. 10.1002/anie.200503555. [DOI] [PubMed] [Google Scholar]
- Ziegler M. S.; Torquato N. A.; Levine D. S.; Nicolay A.; Celik H.; Tilley T. D. Dicopper Alkyl Complexes: Synthesis, Structure, and Unexpected Persistence. Organometallics 2018, 37 (16), 2807–2823. 10.1021/acs.organomet.8b00443. [DOI] [Google Scholar]
- Ren H.; Wu J.; Xi C.; Lehnert N.; Major T.; Bartlett R. H.; Meyerhoff M. E. Electrochemically Modulated Nitric Oxide (NO) Releasing Biomedical Devices via Copper(II)-Tri(2-Pyridylmethyl)Amine Mediated Reduction of Nitrite. ACS Appl. Mater. Interfaces 2014, 6 (6), 3779–3783. 10.1021/am406066a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.