

Abstract
Hydrazine-assisted water splitting is a promising strategy for energy-efficient hydrogen production, yet challenges remain in developing effective catalysts that can concurrently catalyze both the hydrogen evolution reaction (HER) and hydrazine oxidation reaction (HzOR) in acidic media. Herein, we report an effective bifunctional catalyst consisting of Rh clusters anchored on Co3O4 branched nanosheets (Rh-Co3O4 BNSs) synthesized via an innovative arginine-induced strategy. The Rh-Co3O4 BNSs exhibit unique Rh–O–Co interfacial sites that facilitate charge redistribution between Rh clusters and the Co3O4 substrate, thereby optimizing their valence electronic structures. When the current density reaches 10 mA cm−2, the Rh-Co3O4 BNSs require working potentials of only 32 mV for the HER and 0.26 V for the HzOR, far surpassing commercial Pt/C. Furthermore, the Rh-Co3O4 BNSs can work efficiently for hydrazine-assisted water electrolysis with a low voltage of 0.34 V at 10 mA cm−2 and excellent stability. Theoretical calculations reveal that the optimized valence electronic structure within interfacial Rh–O–Co sites not only reduces the adsorption energy barrier of Co3O4 for H* in the HER; but also optimizes the hydrazine adsorption in the HzOR and lowers the free energy change in the potential-determining step, where the facilitated dehydrogenation is observed in in situ Raman spectra. This work provides a viable approach for designing efficient bifunctional catalysts for future hydrazine-assisted hydrogen production.
Asymmetric Rh–O–Co bridge site are built to effectively catalyze both the hydrogen evolution reaction and hydrazine oxidation reaction for energy-efficient hydrazine-assisted hydrogen production.
Introduction
Hydrogen (H2) is regarded as a promising clean energy carrier to replace conventional fossil fuels due to its high energy density and sustainability.1–4 Consequently, substantial research efforts have been focused on the production of high-purity hydrogen through electrochemical overall water splitting (OWS), owing to its environmental friendliness and zero carbon emission in the cathodic hydrogen evolution reaction (HER).5–7 However, the sluggish kinetics and high theoretical potential of the anodic oxygen evolution reaction (OER, 1.23 V vs. RHE) require a cell voltage exceeding 1.5 V to drive the OWS process, which hinders the large-scale application.8,9 To address this obstacle, several more favorable small-molecule oxidation reactions (e.g., tetrahydroisoquinoline oxidation,10,11 urea oxidation,12,13 and hydrazine oxidation14,15) have been proposed as alternatives to the OER. Among these, an overall hydrazine splitting (OHzS) system, which integrates the hydrazine oxidation reaction (HzOR) with a low thermodynamic voltage (−0.23 V vs. RHE, pH = 0) and HER, offers an advantageous strategy for green hydrogen production.16,17 Within such a system, the generation of N2 as the sole byproduct from the HzOR mitigates the risk associated with explosive H2/O2 mixtures produced by conventional OWS.18
In terms of the HzOR, the multi-step reaction pathway in acidic media includes the adsorption of N2Hx intermediates, successive intermediate dehydrogenation, and final nitrogen desorption (N2H5+ ⇌ N2 + 5H+ + 4e−). However, since the catalytic process of the HzOR involves the coupling of 2p orbitals of N-containing intermediates and d orbitals of metal sites, the interaction strength of p–d coupling largely inhibits reactant transformation, which may result in a high activation energy barrier.19 Moreover, the challenge of activating the N–H bond also limits the kinetics of the dehydrogenation process. At present, pioneering studies in the HzOR converge on the use of 3d transition metals, particularly the modulation for Co-based materials.20,21 For example, Xia et al. demonstrated that tuning the electronic structure of ordered Co3Ta leads to exceptional electrocatalytic activity for the HzOR, where the Co–Ta bridge acts as the most active site.22 Wang et al. revealed that the Ni(Co)-Ru-P interfacial sites on Ru1-NiCoP could effectively lower the vacant d-band center of NiCoP, which strengthens *N2H2 adsorption and decreases the reaction energy barrier, thereby significantly improving HzOR activity.23 These findings indicate that the formation of distinctive bridges between Co-based materials and other transition metals has the potential to optimize the d-band center of the Co site, thus enhancing its adsorption capacity for nitrogen-containing intermediates.24,25 Furthermore, the selection of suitable coupling species to accelerate the dehydrogenation of continuous intermediates in the HzOR while reducing the free energy of hydrogen adsorption (ΔGH*) in the HER is of paramount importance for the development of efficient HzOR/HER bifunctional catalysts. According to Trassati's volcano diagram, the Rh is located near the apex, suggesting that Rh has a desirable ΔGH*, and is promising to serve as the active site for the HER on coupling with Co-based materials.26,27 As a result, the introduction of Rh can not only modulate the d-band center of Co-based materials, but is also promising to accelerate the dehydrogenation reaction kinetics by serving as an active site for N–H bond activation, thereby enhancing intermediate adsorption during the HzOR and lowering the reaction energy barriers. Nevertheless, further investigation is needed to elucidate the mode of interaction between Co-based hosts and Rh species, and to determine whether this interaction can achieve the predicted bifunctional activity and stability under acidic conditions.
Herein, we present a novel arginine-induced synthesis approach for the fabrication of Co3O4 branched nanosheets uniformly anchored with Rh clusters (Rh-Co3O4 BNSs). Using this catalyst as a model, we systematically investigated the role of the interaction mode between Co3O4 and Rh in modulating the catalytic activities of both the HzOR and HER under acidic conditions. X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS) analyses demonstrated that the Rh–O–Co asymmetric unit induces charge redistribution, effectively modulating the electronic states of both Rh and Co. Electrochemical tests demonstrate that Rh-Co3O4 BNSs exhibit exceptional bifunctional activity and stability for the HER and HzOR under highly acidic conditions. Specifically, the Rh-Co3O4 BNSs require working potentials of only 32 mV for the HER and 0.26 V for the HzOR to achieve 10 mA cm−2, far surpassing the performance of commercial Pt/C. When applied in a hydrazine-assisted water electrolysis system, the Rh-Co3O4 BNSs achieve a cell voltage of 0.34 V at 10 mA cm−2 and maintain excellent stability over 20 h. Density functional theory (DFT) calculations elucidate that the coupling between Rh and Co3O4 leads to electronic redistribution through the asymmetric Rh–O–Co bridge, which not only reduces the adsorption energy barrier of Co3O4 for H* in the HER, but also optimizes the hydrazine adsorption of N-containing intermediates in the HzOR. With the construction of a Rh–O–Co bridge, the potential-determining step of 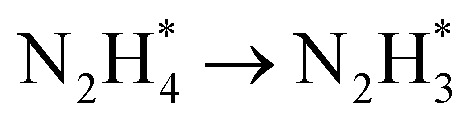 is shifted to
is shifted to 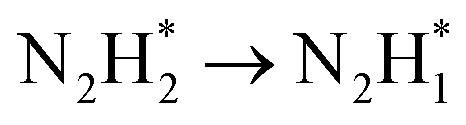 , which facilitated dehydrogenation with a lower free energy change. In situ Raman spectroscopy during the HzOR process confirms that the introduction of Rh into Co3O4 BNSs enhances the adsorption of *N2H4 and promotes the low-potential driving of the rapid dehydrogenation process. This work presents a promising strategy for the development of highly efficient bifunctional catalysts aimed at advancing hydrazine-assisted hydrogen production under acidic conditions.
, which facilitated dehydrogenation with a lower free energy change. In situ Raman spectroscopy during the HzOR process confirms that the introduction of Rh into Co3O4 BNSs enhances the adsorption of *N2H4 and promotes the low-potential driving of the rapid dehydrogenation process. This work presents a promising strategy for the development of highly efficient bifunctional catalysts aimed at advancing hydrazine-assisted hydrogen production under acidic conditions.
Results and discussion
Synthesis and characterization
The synthesis of Rh clusters anchored on Co3O4 branched nanosheets (Rh-Co3O4 BNSs) is schematically illustrated in Fig. 1a. First, the Co3O4 BNSs were obtained by a facile hydrothermal treatment of arginine and Co2+ salt in a mixed ethanol and water solution. Then, the as-prepared Co3O4 BNSs were transferred to ethylene glycol containing RhCl3 aqueous solution for the reflux treatment to obtain the Rh-Co3O4 BNSs. Arginine has both unique groups of guanidino and carboxyl, which makes it easy to bind with Co2+ cations through the coordination interaction, thereby allowing the synthesis of Co3O4 BNSs to be kinetically controlled.28 The coordination between arginine and Co2+ cations is confirmed by using the ultraviolet and visible spectroscopy (UV-vis) spectra (Fig. 1b), where the absorption peak of Co2+ aqueous solution shows a significant blue shift after the introduction of arginine. The Fourier transform infrared (FTIR) spectrum of arginine-Co(NO3)2 shows a distinct multi-peak overlapping phenomenon between –OH and –NH2 vibrational peaks compared with that of arginine, indicating the coordination between Co2+ and arginine. The crystal structures of Co3O4 BNSs and Rh-Co3O4 BNSs were characterized by X-ray diffraction (XRD) analysis. All the diffraction peaks of both samples are in good agreement with the standard pattern of the spinal Co3O4 phase (JCPDS no. 42-1467), and no Rh-related peaks were detected in Rh-Co3O4 BNSs (Fig. 1c), indicating a high Rh dispersion with small particle sizes.24,29 The electron paramagnetic resonance (EPR) spectra show that both samples possess strong EPR signals at g = 2.003 (Fig. 1d), which are assigned to oxygen vacancies.30,31 Note that the density of oxygen vacancies of Rh-Co3O4 BNSs increase significantly compared with that of Co3O4 BNSs, which indicates a strong interaction between Rh sites and the Co3O4 support.32 The vacancies increase the disorder of the crystal, as verified by Raman spectra, where the peaks of Rh-Co3O4 BNSs show a significant redshift compared to those of Co3O4 BNSs (Fig. S2†), especially the A1g peak located at 678 cm−1.
Fig. 1. (a) Schematic illustration of the preparation of Rh-Co3O4 BNSs; (b) UV-vis absorption spectra of Co(NO3)2, arginine and arginine-Co(NO3)2 aqueous solution; (c) XRD patterns of Co3O4 BNSs and Rh-Co3O4 BNSs; (d) EPR spectra of Co3O4 BNSs and Rh-Co3O4 BNSs.

Transmission electron microscopy (TEM) characterization shows that the Co3O4 BNSs present a branched nanosheet structure with a lateral width of about 150 nm (Fig. 2a and S3†), which is further verified by scanning electron microscopy (SEM) images (Fig. S4†). The atomic force microscopy (AFM) image reveals a uniform height distribution of Co3O4 BNSs with an average thickness of about 2.40 nm (Fig. S5†). The formation of the branched nanosheet structure is strongly dependent on the appropriate concentration of arginine. Without arginine or with an excess amount, the Co3O4 substrate exhibits a non-uniform nanosheet morphology lacking any branches (Fig. S6†). Furthermore, the substrate prepared in water exhibits a nano-needle structure (Fig. S7†), indicating that ethanol is also conducive to the formation of nanosheets. After the deposition of Rh species, the Rh-Co3O4 BNSs still retain the structure and morphology integrity of the branched nanosheet (Fig. 2b and S8†). The magnified TEM image shows that the Rh nanoparticles are uniformly dispersed on the surface of Co3O4 BNSs with an ultrasmall particle size of about 1.60 nm (Fig. 2c). To further clarify the atomic structure of Rh species on the Co3O4 substrate, aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM) was carried out. As shown in Fig. 2d and e, many bright speckles appear on the surface of Co3O4 BNSs, which are considered to be Rh cluster sites. From the high-resolution HAADF-STEM image (Fig. 2f), the lattice spacings of 0.219 nm and 0.214 nm correspond to the (111) and (400) planes of Rh and Co3O4, respectively. The energy dispersive X-ray (EDX) element mapping images of Rh-Co3O4 BNSs show that the Rh signals are uniformly distributed on Co3O4 BNSs without obvious segregation (Fig. 2g and S9†). The mass loading of Rh on Rh-Co3O4 BNSs is about 15.1 wt%, determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES). By varying the addition of RhCl3, other Rh-Co3O4 BNSs with different Rh loadings of 7.9 wt% and 31.7 wt% could be obtained, which exhibit a similar crystal structure and morphology to that of Rh-Co3O4 BNSs (Fig. S10 and S11†). The pure Rh sample was also synthesized for comparison (Fig. S12†).
Fig. 2. (a) TEM image of Co3O4 BNSs; (b) and (c) TEM images of Rh-Co3O4 BNSs with different resolutions, the inset of (c) shows the corresponding particle-size distribution; (d)–(f) AC-HAADF-STEM image of Rh-Co3O4 BNSs; (g) EDX element mapping images of Rh-Co3O4 BNSs.

X-ray photoelectron spectroscopy (XPS) was performed to understand the surface chemical and electronic states of Rh-Co3O4 BNSs. The survey XPS spectrum shows the existence of Rh, Co and O elements on the surface of Rh-Co3O4 BNSs (Fig. S13†). For the high-resolution Rh 3d spectrum (Fig. 3a), the intense peaks observed at 306.9 and 311.7 eV are attributed to metallic Rh0 species, while the peaks centered at 308.0/313.2 and 309.7/314.7 eV respectively correspond to Rh+ and Rh3+.33 The geometric areas of the Rh+ and Rh3+ peaks obviously increase when compared with pure Rh, indicating that the Rh species within Rh-Co3O4 BNSs has a higher oxidation state. The Co 2p spectrum of Rh-Co3O4 BNSs (Fig. 3b) corresponds to spin–orbit splitting into 2p1/2 (781.0) and 2p3/2 (796.2 eV), where the peaks at 781.0 and 797.2 eV are assigned to Co3+ species and the peaks at 784.4 and 801.6 eV correspond to Co2+ species.34,35 It is worth noting that the binding energies of Rh 3d and Co 2p spectra in Rh-Co3O4 BNSs exhibit a positive shift of about 0.23 eV and 0.65 eV compared to those in pure Rh and Co3O4 BNSs, respectively. This shift indicates a strong interaction between Rh and the Co3O4 support, leading to the charge redistribution. In the high-resolution O 1s spectra (Fig. S14†), the peaks of O1 at 530.4 eV and O3 at 532.6 eV are attributed to the metal–oxygen bond and surface-adsorbed water molecule, while the O2 peak at 531.3 eV is caused by oxygen vacancies.36,37 Compared to Co3O4 BNSs, the O2 peak area in Rh-Co3O4 BNSs shows a significant increase, suggesting an increase in oxygen vacancies of Rh-Co3O4 BNSs. X-ray absorption spectroscopy (XAS) was conducted with the aim of distinguishing the electronic structure and the coordination environment of Rh and Co in Rh-Co3O4 BNSs. As shown by the normalized Co K-edge X-ray absorption near-edge structure (XANES), the Rh-Co3O4 BNSs exhibit a lower energy compared with the Co3O4 reference (Fig. 3c), indicating a slight decrease in the average valence state of Co species in Co3O4 after the introduction of Rh. The Co K-edge Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) of Rh-Co3O4 BNSs is similar to that of Co3O4 (Fig. 3d), with three distinct peaks identified as Co–O, octahedral Co–Co and tetrahedral Co–Co bonds, respectively.38,39Fig. 3e depicts the normalized XANES spectra for the Rh K-edge of Rh-Co3O4 BNSs and standard samples. The Rh K-edge absorption edge for Rh-Co3O4 BNSs is located between that of Rh foil and Rh2O3, with a greater proximity to the line of Rh. The Rh valence of Rh-Co3O4 BNSs was quantitatively measured by using the adsorption energy (E0) to be +0.51 (Fig. S15†). This fact proves that the coupling of Rh and Co3O4 leads to the transfer of electrons from the Rh sites to the Co sites via the bridged O atoms. Furthermore, the presence of Ov as Lewis acids, while exhibiting a weaker metal-electron withdrawing effect than O atoms, also facilitates electron donation by Rh.40,41 The information derived from Rh EXAFS shows that the Rh-Co3O4 BNS sample exhibits a strong Rh–Rh peak and a more pronounced weak Rh–O peak relative to the Rh foil (Fig. 3f), implying the existence of Rh–O–Co bridging sites between the Rh clusters and the Co3O4 BNSs.42,43 Consistent results were observed from the wavelet transform (WT) analyses of the Rh-Co3O4 BNS spectrum. The oscillation of the WT spectrum of Rh-Co3O4 BNSs is similar to that of pristine Co3O4, suggesting their similar spinel structure (Fig. 3g). In addition, the Rh–O scattering of Rh-Co3O4 BNSs (R = 1.61 Å, k = 5.65 Å−1) are quite different from those of Rh–O scattering in Rh2O3 (R = 1.55 Å, k = 7.40 Å−1), which further demonstrates the presence of asymmetric Rh–O–Co bridging sites rather than pure Rh–O bridging sites in the Rh-Co3O4 BNSs (Fig. 3h).
Fig. 3. (a) Rh 3d XPS spectra of Rh-Co3O4 BNSs and pure Rh; (b) Co 2p XPS spectra of Rh-Co3O4 BNSs and Co3O4 BNSs; (c) the normalized Co K-edge XANES spectra; (d) Fourier-transforms of K3-weight Co K-edge EXAFS spectra; (e) the normalized Rh K-edge XANES spectra; (f) Fourier-transforms of the K3-weight Rh K-edge EXAFS spectra; (g) wavelet transforms for the K3-weight Co K-edge EXAFS signals; (h) wavelet transforms for the K3-weight Rh K-edge EXAFS signals.

Electrocatalytic performance towards the HER
The HER performance of Rh-Co3O4 BNSs was evaluated in 0.5 M H2SO4 electrolyte using a typical three-electrode setup, where Co3O4 BNSs, pure Rh, and commercial Pt/C catalysts were also measured under the same conditions for comparison. Fig. 4a shows the HER polarization curves of the catalysts. The Rh-Co3O4 BNSs require remarkably low overpotentials of 32 mV and 56 mV to achieve current densities of 10 mA cm−2 (E50) and 50 mA cm−2 (E50), respectively (Fig. 4a). These values are superior to those of pure Rh (E10 = 53 mV and E50 = 90 mV), Co3O4 BNSs (E10 = 244 mV and E50 = 491 mV), and are even comparable to those of commercial Pt/C (E10 = 38 mV and E50 = 76 mV). The reaction kinetics of the HER process on all catalysts was revealed by the Tafel slope (Fig. S16†). As depicted in Fig. 4b, the Rh-Co3O4 BNSs exhibit a sharply decreased Tafel slope (27.0 mV dec−1) relative to those of Pt/C (48.6 mV dec−1), pure Rh (42.3 mV dec−1) and Co3O4 BNSs (285 mV dec−1), implying the fastest HER kinetics at the Rh-Co3O4 BNS surface. The Tafel slope of Rh-Co3O4 BNSs is close to 29.9 mV dec−1, illustrating that the Rh-Co3O4 BNSs catalyze the HER by a Volmer–Tafel mechanism.44 The low overpotentials and small Tafel slope of Rh-Co3O4 BNSs are also significantly lower than those of the Rh7.9 wt%-Co3O4 BNSs and Rh31.7 wt%-Co3O4 BNSs (Fig. 4c and S17†), as well as most of the previously reported typical catalysts (data confirmed by using the DigCat database: https://www.digcat.org/; illustrated in Fig. 4d and Table S1†). The above results suggest that the synergistic effect of Rh and Co3O4 components facilitates the HER process. To better understand the synergistic effect of Rh and Co3O4 components, electrochemical impedance spectroscopy (EIS) was carried out and the electrochemical double-layer capacitance (Cdl) were investigated. As shown in EIS Nyquist plots (Fig. 4e), the Rh-Co3O4 BNSs present the lowest electron transfer resistance (Rct) relative to pure Rh and Co3O4 BNSs, indicating that the interaction between Rh and Co3O4 enhances the charge transfer efficiency of Rh-Co3O4 BNSs. The Cdl was calculated by using cyclic voltammetry (CV) measurements at different scan rates in the non-faradaic region (Fig. S18†). The Rh-Co3O4 BNSs exhibit a high Cdl of about 10.32 mF cm−2 (Fig. 4f), which is higher than those of Pt/C (8.49 mF cm−2), pure Rh (5.85 mF cm−2), and Co3O4 BNSs (3.24 mF cm−2). Given the linear relationship between the Cdl and the electrochemical surface area (ECSA), Rh-Co3O4 BNSs exhibit a larger ECSA and expose more active sites, thus increasing HER activity. To evaluate the stability of Rh-Co3O4 BNSs, the accelerated durability test (ADT) was performed by 2000 CV cycles in the voltage window from −0.08 to −0.02 V (vs. RHE) with a scan rate of 100 mV s−1. As shown in Fig. 4g, the overpotential at 10 mA cm−2 in the HER polarization curve of Rh-Co3O4 BNSs after 2000 cycles only increases by 3 mV compared with the original one, which is smaller than that of Pt/C (13 mV), pure Rh (5 mV), and Co3O4 BNSs (65 mV). The chronopotentiometry measurement was further applied to evaluate the long-term durability of Rh-Co3O4 BNSs at a current density of at 10 mA cm−2 (Fig. 4h). In comparison to the other three catalysts, the superior alkaline HER activity is well retained on Rh-Co3O4 BNSs after continuous hydrogen production for more than 20 h. After chronopotentiometry, the Rh-Co3O4 BNSs still retain the branched nanosheet structure and uniform height distribution of Rh particles on the Co3O4 surface (Fig. S19†). The XPS spectra of recovered Rh-Co3O4 BNSs show a slight decrease in the proportion of Rh in the oxidized state with no significant change for Co species (Fig. S20†). In addition, the Rh-Co3O4 BNSs exhibit a high faradaic efficiency (FE) of about 100% during the HER, indicating the high HER selectivity of Rh-Co3O4 BNSs (Fig. 4i and S21†).
Fig. 4. (a) HER polarization curves of the catalysts in 0.5 M H2SO4 at a scan rate of 5 mV s−1; (b) Tafel slope and overpotential at 10 mA cm−2 and 50 mA cm−2 of the catalysts; (c) comparison of the Tafel slope and overpotential for Rh-Co3O4 BNSs at different Rh loadings; (d) comparison of the Tafel slope and overpotential of Rh-Co3O4 BNSs and the reported catalysts; (e) EIS Nyquist plots of the catalysts; (f) Cdl values of the catalysts; (g) HER polarization curves of the catalysts before and after 2000 CV cycles; (h) chronopotentiometric curves of the catalysts at 10 mA cm−2; (i) Faraday efficiency of Rh-Co3O4 BNSs.

Electrocatalytic performance towards the HzOR
The electrochemical performance of Rh-Co3O4 BNSs for the HzOR was then evaluated in H2SO4/N2H4 electrolyte. We first determined that the HzOR activity of Rh-Co3O4 BNSs is optimal in 0.5 M H2SO4 + 0.05 M N2H4 electrolyte (Fig. S22†). When the concentration of N2H4 exceeds 0.05 M in H2SO4 solution, hydrazine sulfate precipitation is easily observed.45Fig. 5a shows the HzOR and OER polarization curves of the Rh-Co3O4 BNS electrode in 0.5 M H2SO4 with and without 0.05 M N2H4. It can be seen that the Rh-Co3O4 BNSs require a low working potential of 0.26 V vs. RHE to reach a current density of 10 mA cm−2 for the HzOR, which is 1.39 V less than that of the OER and other reported small-molecule oxidative reactions (Table S2†), indicating that the Rh-Co3O4 BNSs possess excellent HzOR activity and can be utilized as an alternative OER catalyst to reduce the electrical energy consumption. The HzOR activity of Rh-Co3O4 BNSs was then compared with that of Co3O4 BNSs, pure Rh and Pt/C control samples (Fig. 5b). As observed in Fig. 5c, the working potentials at a current density of 10 and 50 mA cm−2 for Rh-Co3O4 BNSs are merely 0.26 V and 0.44 V, which are much lower than that of Co3O4 BNSs and pure Rh, and even exceeds that of commercial Pt/C, suggesting the significantly improved HzOR activity by the synergistic effect of Rh and Co3O4 components. The corresponding Tafel slope value of Rh-Co3O4 BNSs is about 76.0 mV dec−1 (Fig. 5c and S23†), which is considerably smaller than that of Pt/C (168.9 mV dec−1), pure Rh (204.5 mV dec−1), and Co3O4 BNSs (264.5 mV dec−1), proving the faster HzOR kinetics of Rh-Co3O4 BNSs. The interfacial electron transfer kinetics of catalysts during the HzOR was investigated by EIS, where the Rh-Co3O4 BNSs exhibit a smaller charge-transfer resistance than that of pure Rh and Co3O4 BNSs (Fig. S24†). Based on the CV measurement at varying scan rates (ν) shown in Fig. 5d, the peak oxidation current density (Jp) of the HzOR increases in proportion to the square root of ν (ν1/2); while the Ep value increases linearly with log(ν) from 20 to 100 mV s−1 (Fig. S25†), indicating that the HzOR of Rh-Co3O4 BNSs is a diffusion-controlled irreversible process.45 The Rh-Co3O4 BNSs also present excellent electrocatalytic stability for the HzOR, as verified by ADT and chronopotentiometry measurements. The ADT measurement shows that the working potential of Rh-Co3O4 BNSs at 10 mA cm−2 only increases by 3 mV after 2000 CV cycles (Fig. 5e), which is significantly lower than that of other comparative samples. Meanwhile, the chronoamperometric curve at 10 mA cm−2 remains relatively stable over 20 h (Fig. 5f). In addition, the Rh-Co3O4 BNSs show superior stability in the HzOR in comparison to their performance in the OER (Fig. S26†). After the prolonged HzOR process, the Rh clusters on Co3O4 BNSs exhibit a slight tendency to aggregate, while the dispersion remains relatively homogeneous (Fig. S27†). The XPS full spectra of Rh-Co3O4 BNSs after the HzOR show that the Co and Rh species still exist after the long oxidation process (Fig. S28a†). The main peaks of Co 3d and the ratio of Co3+/Co2+ for recovered Rh-Co3O4 BNSs did not change significantly (Fig. S28b†). For the Rh3d XPS spectrum, the peaks of recovered Rh-Co3O4 BNSs shift to higher binding energies compared with those of the original one, accompanied by an increase in the oxidized state (Fig. S28c†). The preferential oxidation of Rh clusters facilitates the HzOR process by avoiding excessive oxidation of Co3+ in Co3O4 to acidic soluble Co4+.46
Fig. 5. (a) The polarization curves of Rh-Co3O4 BNSs in 0.5 M H2SO4 with and without 0.05 M N2H4 at a scan rate of 5 mV s−1; (b) HzOR polarization curves of the catalysts in 0.5 M H2SO4 + 0.05 M N2H4 electrolyte; (c) comparison of the Tafel slope and working potential at 10 mA cm−2 and 50 mA cm−2 recorded from panel (c); (d) CV curves of Rh-Co3O4 BNSs in 0.5 M H2SO4 + 0.05 M N2H4 electrolyte at different scanning rates from 20 to 100 mV s−1, the inset shows the Ipvs. ν1/2 plot; (e) HzOR polarization curves of the catalysts before and after 2000 CV cycles; (f) chronopotentiometry curves of the catalysts for the HzOR recorded at 10 mA cm−2 in 0.5 M H2SO4 + 0.05 M N2H4 electrolyte; (g) schematic illustration of a homemade OhzS electrolyzer; (h) OHzR polarization curves of Rh-Co3O4 BNSs‖Rh-Co3O4 BNSs and Pt/C‖Pt/C; (i) chronopotentiometry curves for OHzR recorded at 10 mA cm−2.

Considering the excellent bifunctional performance of Rh-Co3O4 BNSs for both the HER and HzOR, an overall hydrazine splitting (OHzS) electrolyzer was constructed to explore the possibility of hydrazine-assisted hydrogen production, as schematically illustrated in Fig. 5g. The HER polarization curves of Rh-Co3O4 BNSs remain unchanged before and after the introduction of 0.05 M N2H4 (Fig. S29†), indicating a high HER selectivity of Rh-Co3O4 BNSs.1 The OHzS electrolyzer achieves a current density of 10 mA cm−2 at a lower cell voltage of 0.34 V (Fig. S30†), which is 1.38 V lower than that of overall water splitting (OWS), demonstrating the great potential of the Rh-Co3O4 BNS-based OHzS electrolyzer to reduce the energy consumption. In addition, the Rh-Co3O4 BNSs‖Rh-Co3O4 BNS-based OHzS outperforms the Pt/C‖Pt/C-based OHzS (Fig. 5h), and many reported OWS systems (Table S3†). During the chronopotentiometry measurement, the driving current of the OHzR electrolyzer on Rh-Co3O4 BNSs‖Rh-Co3O4 BNSs decays at a slower rate than that of the Pt/C‖Pt/C-based OHzR electrolyzer, further implying the superior stability of Rh-Co3O4 BNSs (Fig. 5i).
Insights into the catalytic mechanism
DFT calculations were conducted to investigate the HER and HzOR mechanisms on Rh-Co3O4 BNSs. As shown in Fig. 6a, the projected density of states (PDOS) of Rh-Co3O4 reveals that the Rh 4d states are closer to the Fermi level (EF), than the Co 3d states below the EF, indicating the main contribution of Rh 4d states for catalytic performance. Furthermore, the d-band center of Co in Rh-Co3O4 BNSs exhibits an increasing trend towards the Fermi energy level, from −2.92 to −2.81 eV, in comparison to that of the PDOS of Co3O4 (Fig. S31†), confirming the effective electronic interaction within the [Rh–O–Co] site. This upshift is considered to favor the adsorption of intermediates during the HER and HzOR process, thereby improving the kinetic performance.20,47 The charge density difference plot of Rh-Co3O4 was further obtained to understand the electronic redistribution (Fig. 6b). It can be observed that Rh atoms show directional charge depletion, while the O atoms and partially Co atoms exhibit charge accumulation, which is consistent with the XANES analysis. The surface Pourbaix diagrams were obtained to confirm the surface state for initiating the reaction process, including oxygen vacancies, *H, and hydroxyls coverages (Fig. 6c and S32†). Although the formation of oxygen vacancies is facilitated at low bias potential, the coverage of 2 monolayer (ML) *H shares the lowest energy below the bias potential of 0 VRHE, where the exposed surface unsaturated lattice oxygen atoms can be covered by *H. Based on such a surface state, the HER process of Rh-Co3O4 BNSs was further discussed. Generally, the free energy of hydrogen adsorption (ΔGH*) is regarded as a descriptor to characterize the HER activity. As shown in Fig. 6d, Co3O4 exhibits a high ΔGH* of 1.67 eV, implying that adsorption of hydrogen atoms is challenging. Conversely, Rh-Co3O4 exhibits a low ΔGH* of −0.68 eV, which is closer to 0 eV, implying the low reaction energy barrier of the HER on the Rh-Co3O4 surface. Fig. 6e describes the stepwise dehydrogenation process of N2H4 with various intermediates on Rh-Co3O4 and the Co3O4 surface. It is revealed that when the HzOR takes place on the Co3O4 surface, the first dehydrogenation step 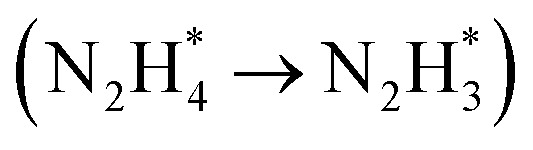 serves as the potential-determining step (PDS) with a change in Gibbs free energy (ΔG) of 0.79 eV. After the introduction of Rh, the PDS on the Rh-Co3O4 surface turns into the third dehydrogenation step
serves as the potential-determining step (PDS) with a change in Gibbs free energy (ΔG) of 0.79 eV. After the introduction of Rh, the PDS on the Rh-Co3O4 surface turns into the third dehydrogenation step 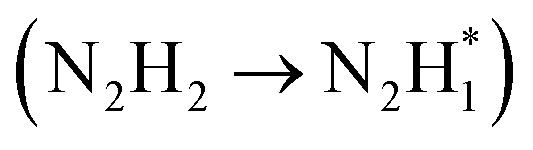 , whose ΔG significantly decreased to 0.38 eV. Therefore, the reaction pathway of Rh-Co3O4 is more favorable than that of Co3O4 in the HzOR process.
, whose ΔG significantly decreased to 0.38 eV. Therefore, the reaction pathway of Rh-Co3O4 is more favorable than that of Co3O4 in the HzOR process.
Fig. 6. (a) PDOS of Rh-Co3O4 (100); (b) charge density difference of Rh-Co3O4 (100) upon the combination of two counterparts (yellow color represents charge accumulation, and blue color represents charge depletion in the space); (c) surface Pourbaix diagram at 25 °C; (d) Gibbs free energy diagrams of Co3O4 and Rh-Co3O4 (100) for the HER; (e) Gibbs free energy diagrams of Co3O4 and Rh-Co3O4 (100) for the HzOR; in situ Raman spectrum of (f) Co3O4 BNSs and (g) Rh-Co3O4 BNSs.

To gain insights into the role of the Rh–O–Co unit in the HzOR mechanism, electrochemical in situ Raman spectroscopy was employed to monitor surface transformations and reaction intermediates of Rh-Co3O4 and Co3O4 BNSs at different potentials in a 0.5 M H2SO4 + 0.05 M N2H4 solution (Fig. 6f and g). The peaks at ∼191 cm−2 and ∼475 cm−2 in in situ Raman spectra correspond to the F2g and Eg modes of spinel Co3O4, respectively.48 An intense peak at ∼976 cm−1 corresponds to the symmetric stretching vibrational mode of SO42−, indicating that the Rh-Co3O4 BNSs and Co3O4 BNSs possess excellent wettability. Compared with Co3O4 BNSs, the Rh-Co3O4 BNSs exhibit increased peak intensities associated with N2H4 (∼1050 cm−1) bond absorption, which suggests that the introduction of Rh into Co3O4 enhances the adsorption of N2H4.49–51 When the working potential of Rh-Co3O4 BNSs increases to 0.20 V vs. RHE, the Co–N bond exhibits a slight weakening, and a scattering peak representing the vibrational mode of Rh–O stretching appeared at 460 cm−2.52 Note that the Raman spectra of Rh-Co3O4 BNSs display a new peak (∼1013 cm−2) between the N2H4 and SO42− peaks corresponding to the *N2H3 intermediate produced after the first dehydrogenation step. However, this peak is unobservable in Raman spectra of Co3O4 BNSs. The in situ Raman results indicate that Rh clusters anchored on Co3O4 BNSs are susceptible to exhibit a high oxidation state during the HzOR, which can serve as an important active site to lower the continuous dehydrogenation energy barrier of Co3O4 BNSs.
Conclusion
In summary, we have developed a highly effective bifunctional catalyst by anchoring Rh on Co3O4 branched nanosheets (Rh-Co3O4 BNSs) synthesized through an innovative arginine-induced strategy. The prepared Rh-Co3O4 BNSs require ultra-low working potentials of only 32 mV for the acidic HER and 0.26 V for the acidic HzOR to achieve current densities of 10 mA cm−2, and significantly outperform commercial Pt/C and other reported catalysts. When used in two-electrode hydrazine-assisted water electrolysis, it delivers a current density of 10 mA cm−2 at a low voltage of 0.34 V, which is 1.38 V lower than that of traditional overall water splitting, and provides excellent stability over 20 h. XAS and XPS analyses demonstrated that charge redistribution between Rh clusters and Co3O4 substrates occurs via distinctive Rh–O–Co sites. This optimizes their valence electronic structures, which has the potential to enhance their catalytic activity and stability. Theoretical calculations further confirmed that the optimized valence electronic structure within the Rh–O–Co sites not only reduced the adsorption energy barrier of Co3O4 for H* in the HER, but also enhanced hydrazine adsorption in the HzOR with an accelerated dehydrogenation process, as indicated by in situ Raman spectroscopy. This work provides new ideas for the construction of bifunctional catalysts with excellent performance and stability for efficient hydrogen production under acidic conditions.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Author contributions
Jinrui Hu and Xuan Wang did the majority of experimental work, including synthesis, characterization and analysis. Yi Zhou, Meihan Liu and Caikang Wang participated in the electrochemistry test and analysis. Meng Li also participated in the data analysis and contributed to the preparation of the manuscript. Heng Liu and Hao Li were responsible for the DFT calculation analysis. Yawen Tang and Gengtao Fu conceived the idea, supervised the experimental work, and analyzed the data. The initial draft of the manuscript was written by Jinrui Hu and later refined by Gengtao Fu and Hao Li. All authors have reviewed and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (22279062 and 22232004), JSPS KAKENHI (JP24K23069) and Ensemble Grants for Early Career Researchers 2024. The authors are grateful for the support from the National and Local Joint Engineering Research Center of Biomedical Functional Materials and a project sponsored by the Priority Academic Program Development of Jiangsu Higher Education Institutions. H. Li acknowledges the Center for Computational Materials Science, Institute for Materials Research, Tohoku University for the use of MASAMUNE-IMR (202312-SCKXX-0203) and the Institute for Solid State Physics (ISSP) at the University of Tokyo for the use of their supercomputers.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07442d
References
- Zhu Y. Zhang J. Qian Q. Li Y. Li Z. Liu Y. Xiao C. Zhang G. Xie Y. Dual Nanoislands on Ni/C Hybrid Nanosheet Activate Superior Hydrazine Oxidation-Assisted High-Efficiency H2 Production. Angew. Chem., Int. Ed. 2021;61:e202113082. doi: 10.1002/anie.202113082. [DOI] [PubMed] [Google Scholar]
- Xue Y. Zhao J. Huang L. Lu Y. R. Malek A. Gao G. Zhuang Z. Wang D. Yavuz C. T. Lu X. Stabilizing ruthenium dioxide with cation-anchored sulfate for durable oxygen evolution in proton-exchange membrane water electrolyzers. Nat. Commun. 2023;14:8093. doi: 10.1038/s41467-023-43977-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Zhu W. Fang J. Xu Z. Xue Y. Pei J. Sui R. Wang X. Zhang X. Zhuang Z. Electrochemical converting ethanol to hydrogen and acetic acid for large scale green hydrogen production. Nano Res. 2023;17:1542–1551. [Google Scholar]
- Wang Y. Ma F. Zhang G. Zhang J. Zhao H. Dong Y. Wang D. Precise synthesis of dual atom sites for electrocatalysis. Nano Res. 2024;17:9397–9427. [Google Scholar]
- Zhang J. Xu L. Yang X. Guo S. Zhang Y. Zhao Y. Wu G. Li G. Amorphous MnRuOx Containing Microcrystalline for Enhanced Acidic Oxygen-Evolution Activity and Stability. Angew. Chem., Int. Ed. 2024;63:e202405641. doi: 10.1002/anie.202405641. [DOI] [PubMed] [Google Scholar]
- Liu Y. Zhang J. Li Y. Qian Q. Li Z. Zhang G. Realizing the Synergy of Interface Engineering and Chemical Substitution for Ni3N Enables its Bifunctionality Toward Hydrazine Oxidation Assisted Energy-Saving Hydrogen Production. Adv. Funct. Mater. 2021;31:2103673. [Google Scholar]
- Zhu Y. Wu X. Wu Z. Wang X. Wang X. Wang C. Zhu X. Li M. Sun D. Li H. Tang Y. Fu G. Europium Oxide Evoked Multisite Synergism to Facilitate Water Dissociation for Alkaline Hydrogen Evolution. Adv. Funct. Mater. 2024:2409324. [Google Scholar]
- Rong C. Wang S. Shen X. Jia C. Sun Q. Zhang Q. Zhao C. Defect-balanced active and stable Co3O4−x for proton exchange membrane water electrolysis at ampere-level current density. Energy Environ. Sci. 2024;17:4196–4204. [Google Scholar]
- Li Z. Wu X. Jiang X. Shen B. Teng Z. Sun D. Fu G. Tang Y. Surface carbon layer controllable Ni3Fe particles confined in hierarchical N-doped carbon framework boosting oxygen evolution reaction. Adv. Powder Mater. 2022;1:100020. [Google Scholar]
- Huang C. Huang Y. Liu C. Yu Y. Zhang B. Integrating Hydrogen Production with Aqueous Selective Semi-Dehydrogenation of Tetrahydroisoquinolines over a Ni2P Bifunctional Electrode. Angew. Chem., Int. Ed. 2019;58:12014–12017. doi: 10.1002/anie.201903327. [DOI] [PubMed] [Google Scholar]
- Zhou Z. Pan X. Sun L. Xie Y. Zheng J. Li L. Zhao G. Boosting Hydrogen Production via Selective Two-electron Mild Electrochemical Oxidation of Tetrahydroisoquinolines Completely to Dihydroisoquinolines. Angew. Chem., Int. Ed. 2023;62:e202216347. doi: 10.1002/anie.202216347. [DOI] [PubMed] [Google Scholar]
- Zhong M. Xu M. Ren S. Li W. Wang C. Gao M. Lu X. Modulating the electronic structure of Ni(OH)2 by coupling with low-content Pt for boosting the urea oxidation reaction enables significantly promoted energy-saving hydrogen production. Energy Environ. Sci. 2024;17:1984–1996. [Google Scholar]
- Xu X. Ullah H. Humayun M. Li L. Zhang X. Bououdina M. Debecker D. P. Huo K. Wang D. Wang C. Fluorinated Ni-O-C Heterogeneous Catalyst for Efficient Urea-Assisted Hydrogen Production. Adv. Funct. Mater. 2023;33:2303986. [Google Scholar]
- Qian Q. Zhang J. Li J. Li Y. Jin X. Zhu Y. Liu Y. Li Z. El-Harairy A. Xiao C. Zhang G. Xie Y. Artificial Heterointerfaces Achieve Delicate Reaction Kinetics towards Hydrogen Evolution and Hydrazine Oxidation Catalysis. Angew. Chem., Int. Ed. 2021;60:5984–5993. doi: 10.1002/anie.202014362. [DOI] [PubMed] [Google Scholar]
- Senthil R. A. Jung S. Min A. Kumar A. Moon C. J. Singh M. Choi M. Y. Revealing the Impact of Pulsed Laser-Produced Single-Pd Nanoparticles on a Bimetallic NiCo2O4 Electrocatalyst for Energy-Saving Hydrogen Production via Hybrid Water Electrolysis. ACS Catal. 2024;14:3320–3335. [Google Scholar]
- Burshtein T. Y. Yasman Y. Muñoz-Moene L. Zagal J. H. Eisenberg D. Hydrazine Oxidation Electrocatalysis. ACS Catal. 2024;14:2264–2283. [Google Scholar]
- Wang T. Cao X. Jiao L. Progress in Hydrogen Production Coupled with Electrochemical Oxidation of Small Molecules. Angew. Chem., Int. Ed. 2022;61:e202213328. doi: 10.1002/anie.202213328. [DOI] [PubMed] [Google Scholar]
- Li Y. Niu S. Liu P. Pan R. Zhang H. Ahmad N. Shi Y. Liang X. Cheng M. Chen S. Du J. Hu M. Wang D. Chen W. Li Y. Ruthenium Nanoclusters and Single Atoms on alpha-MoC/N-Doped Carbon Achieves Low-Input/Input-Free Hydrogen Evolution via Decoupled/Coupled Hydrazine Oxidation. Angew. Chem., Int. Ed. 2024;63:e202316755. doi: 10.1002/anie.202316755. [DOI] [PubMed] [Google Scholar]
- Zhu Y. Chen Y. Feng Y. Meng X. Xia J. Zhang G. Constructing Ru-O-TM Bridge in NiFe-LDH Enables High Current Hydrazine-assisted H2 Production. Adv. Mater. 2024;36:2401694. doi: 10.1002/adma.202401694. [DOI] [PubMed] [Google Scholar]
- Liu Y. Zhang J. Li Y. Qian Q. Li Z. Zhu Y. Zhang G. Manipulating dehydrogenation kinetics through dual-doping Co3N electrode enables highly efficient hydrazine oxidation assisting self-powered H2 production. Nat. Commun. 2020;11:1853. doi: 10.1038/s41467-020-15563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y. Wang L. Ren J. T. Tian W. W. Sun M. L. Yuan Z. Y. Heteroatom-Induced Accelerated Kinetics on Nickel Selenide for Highly Efficient Hydrazine-Assisted Water Splitting and Zn-Hydrazine Battery. Nano-Micro Lett. 2023;15:155. doi: 10.1007/s40820-023-01128-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G. An L. Li B. Zuo Y. Song J. Ning F. Jiang N. Cheng X. Zhang Y. Xia D. Atomically ordered non-precious Co3Ta intermetallic nanoparticles as high-performance catalysts for hydrazine electrooxidation. Nat. Commun. 2019;10:4514. doi: 10.1038/s41467-019-12509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. Chao T. Li Y. Liu P. Zhao T. Yu G. Chen C. Liang X. Jin H. Niu S. Chen W. Wang D. Li Y. Cooperative Ni(Co)-Ru-P Sites Activate Dehydrogenation for Hydrazine Oxidation Assisting Self-powered H2 Production. Angew. Chem., Int. Ed. 2023;62:e202308800. doi: 10.1002/anie.202308800. [DOI] [PubMed] [Google Scholar]
- Liu G. Nie T. Wang H. Shen T. Sun X. Bai S. Zheng L. Song Y.-F. Size Sensitivity of Supported Palladium Species on Layered Double Hydroxides for the Electro-oxidation Dehydrogenation of Hydrazine: From Nanoparticles to Nanoclusters and Single Atoms. ACS Catal. 2022;12:10711–10717. [Google Scholar]
- Feng G. Pan Y. Su D. Xia D. Constructing Fully-Active and Ultra-Active Sites in High-Entropy Alloy Nanoclusters for Hydrazine Oxidation-Assisted Electrolytic Hydrogen Production. Adv. Mater. 2024;36:e2309715. doi: 10.1002/adma.202309715. [DOI] [PubMed] [Google Scholar]
- Fan Z. Liao F. Ji Y. Liu Y. Huang H. Wang D. Yin K. Yang H. Ma M. Zhu W. Wang M. Kang Z. Li Y. Shao M. Hu Z. Shao Q. Coupling of nanocrystal hexagonal array and two-dimensional metastable substrate boosts H2-production. Nat. Commun. 2022;13:5828. doi: 10.1038/s41467-022-33512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H. Wang Q. Zhang Z. Yan H. M. Zhao H. Yang H. B. Liu B. Li J. Wang Y. G. Engineering Single-Atom Electrocatalysts for Enhancing Kinetics of Acidic Volmer Reaction. J. Am. Chem. Soc. 2023;145:13038–13047. doi: 10.1021/jacs.2c13418. [DOI] [PubMed] [Google Scholar]
- Fu G. Liu H. You N. Wu J. Sun D. Xu L. Tang Y. Chen Y. The dendritic platinum-copper bimetallic nanoassemblies with tunable composition and structure: arginine driven self-assembly and their enhanced electrocatalytic activity. Nano Res. 2016;9:755–765. [Google Scholar]
- Lin C. Li J.-L. Li X. Yang S. Luo W. Zhang Y. Kim S.-H. Kim D.-H. Shinde S. S. Li Y.-F. Liu Z.-P. Jiang Z. Lee J.-H. In situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 2021;4:1012–1023. [Google Scholar]
- Wang X. Wang J. Wang P. Li L. Zhang X. Sun D. Li Y. Tang Y. Wang Y. Fu G. Engineering 3d-2p-4f Gradient Orbital Coupling to Enhance Electrocatalytic Oxygen Reduction. Adv. Mater. 2022;34:e2206540. doi: 10.1002/adma.202206540. [DOI] [PubMed] [Google Scholar]
- Ning S. Li M. Wang X. Zhang D. Zhang B. Wang C. Sun D. Tang Y. Li H. Sun K. Fu G. Importing Antibonding-Orbital Occupancy through Pd-O-Gd Bridge Promotes Electrocatalytic Oxygen Reduction. Angew. Chem., Int. Ed. 2023;62:e202314565. doi: 10.1002/anie.202314565. [DOI] [PubMed] [Google Scholar]
- Zhou L. Shao Y. Yin F. Li J. Kang F. Lv R. Stabilizing non-iridium active sites by non-stoichiometric oxide for acidic water oxidation at high current density. Nat. Commun. 2023;14:7644. doi: 10.1038/s41467-023-43466-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y. Yang X. Liu X. Tong X. Yang N. Coupling Methanol Oxidation with Hydrogen Evolution on Bifunctional Co-Doped Rh Electrocatalyst for Efficient Hydrogen Generation. Adv. Funct. Mater. 2022;33:2209134. [Google Scholar]
- Hu J. Li Z. Zhao D. Han Z. Wu X. Zhai J. Liu Z. Tang Y. Fu G. l-Lysine-induced green synthesis of CoS/Co3O4 nanoframes for efficient electrocatalytic oxygen evolution. Green Chem. 2023;25:7309–7317. [Google Scholar]
- Li M. Wang X. Liu K. Sun H. Sun D. Huang K. Tang Y. Xing W. Li H. Fu G. Reinforcing Co–O Covalency via Ce(4f)–O(2p)–Co(3d) Gradient Orbital Coupling for High-Efficiency Oxygen Evolution. Adv. Mater. 2023;35:2302462. doi: 10.1002/adma.202302462. [DOI] [PubMed] [Google Scholar]
- Li M. Wang X. Zhang D. Huang Y. Shen Y. Pan F. Lin J. Yan W. Sun D. Huang K. Tang Y. Lee J.-M. Li H. Fu G. Atomic rare earths activate direct O-O coupling in manganese oxide towards electrocatalytic oxygen evolution. Nano Energy. 2024;128:109868. [Google Scholar]
- Huang Y. Li M. Pan F. Zhu Z. Sun H. Tang Y. Fu G. Plasma-induced Mo-doped Co3O4 with enriched oxygen vacancies for electrocatalytic oxygen evolution in water splitting. Carbon Energy. 2023;5:e279. [Google Scholar]
- Zhu Y. Wang J. Koketsu T. Kroschel M. Chen J.-M. Hsu S.-Y. Henkelman G. Hu Z. Strasser P. Ma J. Iridium single atoms incorporated in Co3O4 efficiently catalyze the oxygen evolution in acidic conditions. Nat. Commun. 2022;13:7754. doi: 10.1038/s41467-022-35426-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. Liu T. Dong C. L. Huang Y. C. Li Y. Chen J. Zou Y. Wang S. Tuning the Selective Adsorption Site of Biomass on Co3O4 by Ir Single Atoms for Electrosynthesis. Adv. Mater. 2021;33:e2007056. doi: 10.1002/adma.202007056. [DOI] [PubMed] [Google Scholar]
- Huang X. Lee C. Li Y. Xu J. Liu D. Acid-Treated RuO2/Co3O4 Nanostructures for Acidic Oxygen Evolution Reaction Electrocatalysis. ACS Appl. Nano Mater. 2024;7:9244–9251. [Google Scholar]
- Chen Y. Liu Y. Zhai W. Liu H. Sakthivel T. Guo S. Dai Z. Metastabilizing the Ruthenium Clusters by Interfacial Oxygen Vacancies for Boosted Water Splitting Electrocatalysis. Adv. Energy Mater. 2024;14:2400059. [Google Scholar]
- Wu J. Fan J. Zhao X. Wang Y. Wang D. Liu H. Gu L. Zhang Q. Zheng L. Singh D. J. Cui X. Zheng W. Atomically Dispersed MoOx on Rhodium Metallene Boosts Electrocatalyzed Alkaline Hydrogen Evolution. Angew. Chem., Int. Ed. 2022;61:5389. doi: 10.1002/anie.202207512. [DOI] [PubMed] [Google Scholar]
- Zhang B. Zhu C. Wu Z. Stavitski E. Lui Y. H. Kim T.-H. Liu H. Huang L. Luan X. Zhou L. Jiang K. Huang W. Hu S. Wang H. Francisco J. S. Integrating Rh Species with NiFe-Layered Double Hydroxide for Overall Water Splitting. Nano Lett. 2019;20:136–144. doi: 10.1021/acs.nanolett.9b03460. [DOI] [PubMed] [Google Scholar]
- Gu J. Li L. Xie Y. Chen B. Tian F. Wang Y. Zhong J. Shen J. Lu J. Turing structuring with multiple nanotwins to engineer efficient and stable catalysts for hydrogen evolution reaction. Nat. Commun. 2023;14:5389. doi: 10.1038/s41467-023-40972-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. Jia N. Wu X.-R. Li F.-M. Chen P. Jin P.-J. Yin S. Chen Y. Rhodium phosphide ultrathin nanosheets for hydrazine oxidation boosted electrochemical water splitting. Appl. Catal., B. 2020;270:118880. [Google Scholar]
- Zhu W. Yao F. Cheng K. Zhao M. Yang C.-J. Dong C.-L. Hong Q. Jiang Q. Wang Z. Liang H. Direct Dioxygen Radical Coupling Driven by Octahedral Ruthenium–Oxygen–Cobalt Collaborative Coordination for Acidic Oxygen Evolution Reaction. J. Am. Chem. Soc. 2023;145:17995–18006. doi: 10.1021/jacs.3c05556. [DOI] [PubMed] [Google Scholar]
- Zhai Y. Jin C. Xia Q. Han W. Wu J. Zhao X. Zhang X. Atomically Confined Ru Sites in Octahedral Co3O4 for High-Efficiency Hydrazine Oxidation. Adv. Funct. Mater. 2023;34:2311063. [Google Scholar]
- Qiao L. Liu D. Zhu A. Feng J. Zhou P. Liu C. Ng K. W. Pan H. Nickel-facilitated in situ surface reconstruction on spinel Co3O4 for enhanced electrochemical nitrate reduction to ammonia. Appl. Catal., B. 2024;340:123219. [Google Scholar]
- Meng Q. Hou Y. Yang F. Cao C. Zou Z. Luo J. Zhou W. Tong Z. Chen S. Zhou S. Wang J. Deng S. Modulation of surface properties on cobalt phosphide for high-performance ambient ammonia electrosynthesis. Appl. Catal., B. 2022;303:120874. [Google Scholar]
- Zheng W. Liu M. Lee L. Y. S. Electrochemical Instability of Metal–Organic Frameworks: In Situ Spectroelectrochemical Investigation of the Real Active Sites. ACS Catal. 2019;10:81–92. [Google Scholar]
- Meng G. Chang Z. Zhu L. Chen C. Chen Y. Tian H. Luo W. Sun W. Cui X. Shi J. Adsorption Site Regulations of [W-O]-Doped CoP Boosting the Hydrazine Oxidation-Coupled Hydrogen Evolution at Elevated Current Density. Nano-Micro Lett. 2023;15:212. doi: 10.1007/s40820-023-01185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Wang X. Wang Y. Shi Z. Yang Y. Zhao T. Jiang Z. Liu C. Xing W. Ge J. The decisive role of adsorbed OH* in low-potential CO electro-oxidation on single-atom catalytic sites. Carbon Energy. 2023;5:e310. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†