

Summary
Hemophilia is caused by the deficiency of clotting factors due to a single genetic abnormality. Replacement therapies have evolved from plasma-derived to recombinant coagulation factor concentrates but continue to have certain limitations. Monoclonal antibodies are clinical prophylactic treatment options unaffected by inhibitors and have better compliance than coagulation factor concentrates for patients with hemophilia. Gene therapy is a breakthrough in hemophilia treatment, as it drives the hepatic expression of factor VIII or factor IX and requires only a single administration to enable long-term replacement treatment in adult patients. Furthermore, biopharmaceutical products that target new pathways unaffected by inhibitors, including tissue factor pathway inhibitors, activated protein C, and antithrombin, as well as pharmaceutical technology advances to reduce dosing frequency, have demonstrated promising clinical results. This review provides a comprehensive overview of these biopharmaceutical products and explores the future of hemophilia treatment.
Subject areas: Biopharmaceuticals, Health sciences, Pharmacology
Graphical abstract
Biopharmaceuticals; Health sciences; Pharmacology
Introduction
Hemophilia A (HA) and B (HB) are X-linked recessive congenital bleeding disorders caused by mutations in the genes responsible for producing coagulation factor VIII (FVIII) or factor IX (FIX), leading to clotting factor deficiencies in the blood plasma. The severity and frequency of bleeding in patients with hemophilia (PwH) depend on the levels of these coagulation factors. Traditional treatment for hemophilia involves intravenous supplementation with virus-inactivated plasma or recombinant coagulation factor concentrates (CFCs), which have established applications in episodic treatment, routine prophylaxis, and perioperative management.1
Clotting factor replacement therapies require long-term and frequent intravenous injections to maintain FVIII or FIX at a minimum trough level of 1–3%, which aids in reducing the risk of spontaneous bleeding to an acceptable level and improving the quality of life for PwH.2 During bleeding or the perioperative period, PwH requires additional CFC injections to achieve the desired peak in factor activity.3 However, the currently available clotting factor products impose a major treatment burden, with frequent intravenous administration risking damage to the skin and blood vessels, potentially leading to phlebitis.4
The use of CFCs may lead to the development of neutralizing antibodies, known as inhibitors, which can hinder the activity of coagulation factors.5 PwH with persistent inhibitors require prompt immune tolerance induction (ITI) or bypassing agents to treat bleeding.1,6 However, ITI requires a considerable initial investment and is not always effective. For patients with HB (PwHB) with inhibitors, ITI also carries the risk of allergic reactions and irreversible nephrotic syndrome.6 Compared with CFCs, bypassing agents are more expensive and associated with a high risk of inducing thrombosis.2
Given these limitations, gene therapies, and non-factor drugs, including monoclonal antibodies, small interfering RNAs (siRNAs), and recombinases, have emerged as less burdensome preventive treatment options. With advancements in gene delivery systems, gene therapy products for hemophilia treatment have been approved for marketing and are regulated as biological products.7 Clinical data indicate that a single intravenous injection can achieve sustained expression of factors in PwH for 5–8 years or longer, with significant clinical benefits.8,9 Among non-factor drugs, emicizumab is widely used as a prophylactic treatment for patients with HA (PwHA) and is recommended by multiple guidelines and principles.1,10
Although currently available products for hemophilia treatment improve the patient's quality of life and health equity, they have drawbacks such as poor clinical compliance or limited use for episodic and perioperative treatment in inhibitor-positive patients. The shortcomings of these products must be carefully considered when exploring strategies for developing new drugs for hemophilia. This review provides an overview of the available biopharmaceutical products for hemophilia, discusses pharmaceutical technology innovations aiding the development of hemophilia therapies, and highlights emerging biopharmaceutical products that are less burdensome with potential for market approval.
Survival and medication status of patients with hemophilia require more attention
PwH experience major physiological, psychological, emotional, economic, and employment challenges. In low- and middle-income countries, the diagnosis, registration, management, care, and treatment of PwH are not as advanced as in high-income countries. The lack of access to coagulation products makes it difficult to meet the patient's needs.11 In contrast, PwH in high-income countries predominantly struggle with inhibitors emergence owing to the frequent use of CFCs.12
Hemophilia is under-diagnosed globally
According to estimates from the World Federation of Hemophilia (WFH), most PwH worldwide (>75%) have not yet been identified and diagnosed.13 A meta-analysis using national registries determined the prevalence at birth of PwHA (24.6 per 100,000 males) and PwHB (5.0 per 100,000 males) in high-income countries (Australia, Canada, France, Italy, New Zealand, and the United Kingdom) and estimated the total number of PwH globally to be 1.125 million.14 However, the number of PwH registered with WFH worldwide in 2022 was only approximately 271,00015,16 (Figure 1A); accordingly, the number of registered PwH is expected to represent a fraction of the actual total.
Figure 1.

Global distribution of registered PwH in 2022 and the historical timeline of biopharmaceutical products for hemophilia
(A) Total number of PwH in different countries or regions. Countries and regions with no reported data for 2022 are indicated in gray. Data source: WFH Annual Global Survey © Natural Earth. Abbreviations: PwH, patients with hemophilia; WFH, World Federation of Hemophilia.
(B) Advances in pharmaceutical technology driving the development of biopharmaceutical products for hemophilia. The red background indicates the introduction of biotechnology, whereas the blue background indicates the products. Abbreviations: PwH, patients with hemophilia; pdCFCs, plasma-derived coagulation factor concentrates; rCFCs, recombinant coagulation factor concentrates; rAAV, recombinant adeno-associated virus; TFPI, tissue factor pathway inhibitor; APC, activated protein C; siRNA, small interfering RNA.
More specifically, the total number of PwH registered with WFH in China in 2022 was approximately 32,000,16 while the Hemophilia Treatment Center Collaborative Network of China estimated 65,000 to 130,000 domestic PwH cases.17 Similarly, in the United States, although approximately 19,000 PwH have been registered with the WFH,16 the Centers for Disease Control and Prevention estimates that there are more than 33,000 PwH. These disparities in status may originate from the uneven distribution of medical resources in developed countries, inadequate medical resources in low- and middle-income countries, challenges in effectively identifying PwH and delays in registering new patients with local regulatory authorities.
Hemophilia is challenging to manage, and PwH is at risk of long-term health complications due to spontaneous, traumatic, and postsurgical bleeding. The associated bleeding patterns and lesions vary widely. Without intervention, multiple organs may experience frequent bleeding, and long-term repeated bleeding in joints can lead to deformity or disability.18 Various factors influence bleeding patterns. For instance, aging and physical activity can increase the incidence of joint bleeding. Women and girls with heterozygous alleles for hemophilia may also experience heavy or prolonged menstrual and pregnancy bleeding19; therefore, the care of female patients should not be ignored.
Intracranial or extracranial bleeding is possible and can be life-threatening in severe cases. Similarly, abdominal bleeding can be challenging to detect and may involve multiple organs, resulting in symptoms, such as pain, distension, vomiting of blood, and blood in the stool. Life-threatening spontaneous or traumatic bleeding can also occur in the chest and throat.18,20 Given the substantial number of undiagnosed or unregistered PwH worldwide, including female patients who have not yet received care, this population is at a high risk of experiencing potentially life-threatening bleeding episodes without appropriate medical intervention.
Diagnostic and biopharmaceutical products for hemophilia
Personalized treatments based on diagnostic accuracy can minimize the treatment burden on PwH.21 The one-stage clotting assay (OSA) measures activated partial thromboplastin time to assess plasma coagulation factor activity (FVIII:C, FIX:C). OSA, along with the two-stage (chromogenic) assay (TSA), is widely used worldwide for diagnosing hemophiloia.22 However, the accuracy is affected by several factors, including age, ABO blood type, and laboratory differences.23 Meanwhile, individual gene sequencing technologies facilitate the accurate identification of FVIII or FIX gene mutations, and can therefore be used to screen parental carriers. Sequencing-based prenatal diagnosis can determine whether offspring carry hemophilia-causing genes, supporting early intervention and management before any bleeding occurs.24
Prior to the 1960s, researchers attempted to treat hemophilia using direct blood transfusions. The successful separation of CFCs from the plasma in the 1960s and 1970s marked the beginning of modern treatment methods for this illness.25 Over the next 50 years, plasma-derived CFCs (pdCFCs), recombinant CFCs (rCFCs), monoclonal antibodies, and gene therapies, have become available for PwH (Figure 1B). Since the 1990s, the application of recombinant protein technologies in the biopharmaceutical field has facilitated the use of rCFCs with high safety and monoclonal antibody drugs with improved clinical compliance for hemophilia treatment.26,27 In 2010, gene therapy utilizing adeno-associated virus (AAV) vectors was first explored for hemophilia treatment, yielding encouraging clinical outcomes.28 Owing to the advancement of the recombinant AAV (rAAV) vector platform, gene therapy drugs requiring only a single infusion are now available for PwH as long-term replacement treatments.29
Imbalances in drug treatment for patients with hemophilia
In 2022, the median global usage of FVIII and FIX per capita was 1.38 and 0.24 IU, respectively.16 However, disparities in the median per capita usage of traditional replacement therapy CFCs are evident among countries, as a high proportion of affected patients do not receive adequate treatment and care. In 2022, the median FVIII usage per capita was 5.17 IU in high-income countries and 0.03–1.84 IU in low- and middle-income countries (Figure S1).
In high-income countries, most PwH receive prophylactic treatment. The proportion of individuals with severe HA in the United Kingdom who receive prophylactic treatment is 90–93%.16 Similarly, in the United States, up to 75% of PwHA under 20 years of age receive continuous prophylactic treatment.30 However, the rate of prophylactic treatment for PwH is extremely low in low- and middle-income countries. In China, only 15% of PwH under 18 years of age receive routine prophylactic treatment,31 potentially causing arthropathy in 90% of boys with hemophilia.32
Prophylactic use of low-to-intermediate doses has led to clinical improvements and is recommended when there is an insufficient supply of CFCs.33,34 Low-dose prophylaxis for FVIII or FIX concentrates comprises administering 10–15 IU/kg per dose 2–3 times weekly (1,000–1,500 IU/kg per year), while intermediate-dose prophylaxis requires 15–25 IU/kg per dose thrice weekly for FVIII concentrates (1,500–4,000 IU/kg per year) or 20–40 IU/kg per dose twice weekly for FIX concentrates (2,000–4,000 IU/kg per year). High-dose prophylaxis requires 25–40 IU/kg for FVIII concentrates or 40–60 IU/kg for FIX concentrates weekly (>4,000 IU/kg for FVIII or FIX concentrates per year).1 To improve joint health and promote health equity, it is recommended to maintain FVIII or FIX activity levels in patients with severe hemophilia within the activity level range observed in patients with moderate, preferably mild, hemophilia.35
Low-income countries lack resources and access to drugs to treat hemophilia. Hence, local governments should actively work with non-profit organizations to raise awareness about hemophilia among the public and medical staff and improve early diagnosis and treatment. Organizations such as the WFH and hemophilia drug companies should gradually increase humanitarian aid for patients in low- and middle-income countries. Research and development of factor-based biosimilars should be encouraged in middle-income countries with the capacity for the biopharmaceutical production of these drugs. Additionally, the local industrial chains should be employed to reduce the terminal sales price of drugs, economic pressure on patients, and medical insurance pressure on governments.
While the supply of CFCs in high-income countries has relatively met the needs of PwH,36 a notable increased risk of inhibitor formation has arisen with repeated use. In patients who develop inhibitors, continued use of CFCs elicits only limited hemostatic effects, while high-titer inhibitors may render hemophilia replacement therapy ineffective.11,37 Hence, there is an increasing demand for effective non-factor drugs unaffected by inhibitors.
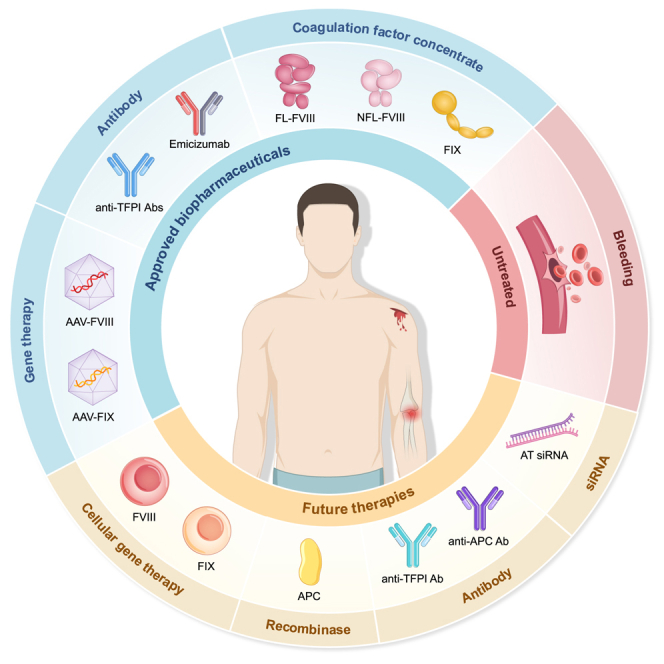
Biopharmaceuticals approved for hemophilia treatment
CFCs remain the most commonly used biopharmaceutical products for treating hemophilia. However, a new recombinant factor drug (efanesoctocog alfa), monoclonal antibody drugs, and gene therapies have demonstrated better clinical compliance than traditional replacement therapies; and the effectiveness of monoclonal antibody drugs is unaffected by inhibitors.38
Traditional plasma-derived and recombinant coagulation factor concentrates are the most commonly used therapeutics
Since the 1960s, the development of plasma multi-component separation and purification technologies has improved the concentration of coagulation factors in preparations, resulting in the increased availability of pdCFCs to treat PwH.39 To generate pdCFCs, mixed plasma is used as raw material, and subjected to a series of purification steps (centrifugation, precipitation, and chromatography); this is followed by viral inactivation and elimination processes, such as solvent/detergent (S/D) treatment, dry heat, or nanofiltration. The resulting solution is then filled and lyophilized to create a pharmaceutical preparation (Figure 2A).
Figure 2.

Application of pharmaceutical technologies in drugs approved for hemophilia
(A) Healthy human plasma is the raw material from which blood factors (FVIII and FIX) are separated and purified. Abbreviations: pdFVIII, plasma-derived FVIII; pdFIX, plasma-derived FIX; S/D, solvent/detergent.
(B) Recombinant factors and monoclonal antibodies biosynthesized using recombinant DNA technology. Abbreviations: Abs, antibodies; rFVIII, recombinant FVIII; rFIX, recombinant FIX; S/D, solvent/detergent; TFPI, tissue factor pathway inhibitors.
(C) Viral gene therapies that introduce nucleic acids into patient somatic cells to correct disease-causing mutations. Abbreviations: Cap, capsid genes; GOI, gene of interest; ITR, inverted terminal repeat; PwHA, patients with hemophilia A; PwHB, patients with hemophilia B; rAAV, recombinant adeno-associated virus; rBV, recombinant baculovirus; Rep, replication genes.
Advances in plasma separation technologies have facilitated the conversion of cryoprecipitate and de-cryoprecipitate supernatants into CFCs with high purity and potency. CFCs can be classified into medium, high, and ultra-high.40 Medium-purity pdFVIII with a specific activity of 1–50 IU/mg is prepared by removing large amounts of impurities and microorganisms via aluminum hydroxide adsorption and polyethylene glycol (PEG) segmented precipitation or PEG glycine precipitation.41 After precipitation, pdFVIII with a specific activity of 50–200 IU/mg is prepared using ion-exchange chromatography, gel filtration chromatography, or affinity chromatography for high-purity preparation. pdFVIII preparations reaching a specific activity of 1,000–3,000 IU/mg are considered ultra-high purity.41 Although high- and ultra-high-purity FVIII may be subject to greater losses during production than medium-purity FVIII,42 they have a higher potency per unit, thereby offering a smaller injection volume and more treatment convenience.
pdFIX is found in the supernatant of plasma following cryoprecipitate removal. Owing to the limitations of separation technologies in the late 1980s, only FIX complex concentrates with relatively low pdFIX purity and other coagulation factors (FII, FVII, and FX) could be prepared.43 However, after the 1990s, ion exchange, gel filtration, and affinity chromatography enabled the preparation of high-purity pdFIX,44 reducing the risk of thromboembolic complications in PwHB receiving long-term infusions of FIX complex concentrates.45
In addition to the purity of pdCFCs concentrates, viral safety must also be considered.46 In the 1960s and 1970s, the precipitation and adsorption steps in the preparation process of pdCFCs were insufficient to eliminate all potentially transmissible pathogens posing a major safety risk of causing human infectious diseases. Studies in the 1980s reported that pdCFC use caused human immunodeficiency virus and hepatitis C virus infections in PwH.47,48 In the 1990s and early 2000s, infectious non-enveloped viruses, such as hepatitis A and parvovirus B19, were also reported in pdCFCs.49 Viral inactivation and elimination processes are crucial for ensuring the safety of pdCFC products.50 The European Medicines Agency (EMA) detailed necessary measures in its Guideline on Plasma-Derived Medicinal Products to control pathogens, including donor screening, screening of known viral markers in donors and plasma pools, and the introduction of two complementary steps to inactivate and/or remove enveloped and non-enveloped viruses during production.51 The China National Medical Products Administration also issued the Technical Methods and Validation Guidelines for Elimination/Inactivation of Viruses in Plasma-derived Products in 2002, requiring the addition of specific steps to inactivate or eliminate lipid-enveloped and non-lipid-enveloped viruses.52
The threat of plasma-derived pathogens and the development of recombinant DNA technology have propelled research and development of recombinant coagulation factor products, which have been approved for marketing since the 1990s (Table S1).
Currently, rCFCs are produced through the large-scale fermentation of mammalian cells, followed by multi-step chromatography purification, S/D treatment or low pH incubation, nanofiltration inactivation and/or virus elimination, and lyophilization with a stable formula (Figure 2B). rCFCs can be divided into three generations. First-generation products are produced using Chinese hamster (CHO) or baby hamster kidney cells, which are cultured in a medium containing animal-derived proteins with human albumin added as an excipient in the final formulation.53 Second-generation products use human-derived proteins in the cell culture medium, but the final formulation does not contain human albumin.54 Third-generation products have the same final formulation as the second-generation products, with the addition of human-derived proteins in the culture medium further removed.55
In 1992, the first rFVIII, Recombinate, was approved by the United States Food and Drug Administration (FDA), as an alternative to pdCFCs (Table S1). rCFCs avoid the risk of blood-borne pathogen transmission and are equally effective as pdCFCs with similar immunogenicity.56 Moreover, rCFCs acquisition eliminated the dependence on plasma raw materials and promoted the popularization of prophylactic treatments among PwH. Thus, the proportion of children with severe hemophilia receiving prophylaxis in the United States increased from 33% in 1995 to 52% in 2004,57 then 86% by 2018.58 rFVIII products can be classified into structure full-length rFVIII (FL-rFVIII), B-domain-truncated (BDT) rFVIII, and B-domain-deleted (BDD) rFVIII. FVIII synthesized by hepatocytes contains A1-A2-B-A3-C1-C2 domains with a molecular weight of 265 kDa, making the expression of rFVIII considerably more difficult than that of other recombinant proteins.59 The B domain is not necessary for the coagulation activity of FVIII.60 Moreover, BDT-rFVIII, BDD-rFVIII, and FL-rFVIII have similar pharmacokinetics, clinical efficacy, and safety characteristics.61 Removing the B domain increases the levels of FVIII secreted by the expressed cell line by 15- to 25-fold.62 Therefore, BDT-rFVIII and BDD-rFVIII have been selected as alternatives to FL-rFVIII.
CFCs are used for episodic treatment, routine prophylaxis, and perioperative factor management in PwH (Figure 3). The initial phase of clinical research on hemophilia focused solely on episodic treatment, wherein CFCs were transfused after bleeding to prevent the possible occurrence of severe, life-threatening hemorrhages. However, compared with episodic treatment, routine prophylaxis can prevent severe bleeding in individuals with severe hemophilia, prevent or delay the occurrence and development of joint lesions, and protect the normal function of joints.63 Unlike patients with severe hemophilia, those with mild-to-moderate hemophilia typically do not receive prophylactic treatment and exhibit a low rate of spontaneous bleeding.25 Guidelines or recommendations from various medical agencies advise that patients with severe and moderate hemophilia exhibiting severe phenotypes should undergo prophylactic treatment as early as possible,1,64,65 ideally prior to joint bleeding or before the age of three, to maintain normal joint function.66 Although long-term prophylactic treatment with CFCs can significantly improve patients’ quality of life, the treatment and financial burdens of intravenous injections 1–4 times weekly are significantly heavy. Accordingly, the long-term treatment needs of PwH have directed the pharmaceutical industry to research and promote long half-life products that would improve clinical compliance.
Figure 3.

Application scenarios for different biopharmaceutical products in patients with hemophilia
Approved biopharmaceutical products for hemophilia can be classified into three categories: coagulation factor concentrates (CFCs), recombinant monoclonal antibodies, and gene therapies. The products have different characteristics: blue font in the patient type and usage section indicates the common applications, and green indicates the unique advantages of the corresponding products. The half-life of EHL-FVIII and EHL-FIX is at least 1.3-fold and 3-fold greater than that of SHL-FVIII and SHL-FIX, respectively. Small children (SC): 0 to <6 years; older children (OC): 6 to <12 years; adolescents: 12 to <18 years; adults: 18 to <65 years; older adults (OA): ≥65 years. Abbreviations: Abs, antibodies; EHL, extended half-life; FL-rFVIII, full-length recombinant FVIII; gc, genome copies; NFL-rFVIII, non-full-length rFVIII; pdFVIII, plasma-derived FVIII; pdFIX, plasma-derived FIX; PwHA, patients with hemophilia A; PwHB, patients with hemophilia B; rFVIII, recombinant FVIII; SHL, standard half-life; TFPI, tissue factor pathway inhibitor; vg, vector genomes.
Compared with the standard half-life (SHL)-FVIII, the half-life of extended half-life (EHL)-FVIII is at least 1.3-fold longer.67 The pharmacokinetic performance of EHL-FIX is superior to that of EHL-FVIII. Compared with SHL-FIX, the half-life of EHL-FIX is 3–5 times longer (Table S1). Between 2014 and 2023, six EHL-FVIII and three EHL-FIX products were approved, increasing the available medication options for PwH. The most commonly used half-life extension technologies include fusion fragment crystallizable (Fc) protein or albumin, PEGylation, and single-chain factor (Figure 3).
After coagulation factors are fused with Fc or albumin, the fusion protein can be recycled into the circulation by binding to the neonatal Fc receptor (FcRn), delaying degradation via the lysosomal pathway and extending the pharmacokinetic half-life of the factor products.68,69 Factors with site-specific or random PEGylation can reduce their proteolysis in the blood and exposure to scavenging receptors, prolonging their survival in circulation.70 Lonoctocog alfa is a product that covalently links the light chain and BDT heavy chain of FVIII, exhibiting improved stability and high binding affinity to von Willebrand factor (vWF), which may translate into meaningful clinical outcomes.71 A matching-adjusted indirect comparison confirmed that the annual consumption of lonoctocog alfa was significantly lesser than that of octocog alfa (an FL-FVIII product) and comparable to that of efmoroctocog alfa, a BDD-FVIII product linked to the dimeric Fc domain of human immunoglobulin G1 (IgG1).69 Additionally, the annualized bleeding rate (ABR) of lonoctocog alfa was also similar to that of efmoroctocog alfa.72 Although these three products mentioned above are different, the clinical data have demonstrated the long-acting properties of lonoctocog alfa. FVIII binds to vWF in circulation; hence, the half-life of vWF limits that of EHL-rFVIII and is only 1.3–2.0 times longer than that of SHL-FVIII (Table S1). To further prolong the half-life of rFVIII, efanesoctocog alfa (Fc-vWF-XTEN fusion protein) was constructed by rFVIII fusing to the Fc fragment and adding the D′D3 domain of vWF; it is also covalently combined with two XTEN hydrophilic polypeptides.73 This design blocks FVIII binding to endogenous vWF, extending the efanesoctocog alfa half-life to 3- to 4-fold that of SHL-rFVIII supported by FcRn.74,75
Antibody drugs enrich treatment options for patients with hemophilia and improve quality of life
Recombinant DNA technology is used to express recombinant monoclonal antibodies in CHO cells to prepare emicizumab, concizumab or marstacimab. Subsequently, the fermentation supernatant is collected and prepared through protein A affinity chromatography, ion-exchange chromatography, and viral inactivation and elimination and is finally packaged into finished preparations (Figure 2B).
Emicizumab is a recombinantly expressed bispecific human IgG4 antibody that simulates the function of activated FVIII to bridge activated FIX and FX. Since 2017, emicizumab was approved for marketing in the United States for the prophylaxis of bleeding in PwHA with or without inhibitors.76 The bleeding rates are significantly lower in individuals treated with emicizumab than in those who previously received FVIII prophylaxis among PwHA with or without inhibitors.77,78 Emicizumab has also demonstrated excellent pharmacokinetic performance and the frequency of its long-term administration can be once weekly, every 2 weeks, or every 4 weeks79
The amino acid sequence of emicizumab has been modified to resemble that of human antibodies, aiming to reduce immunogenicity. However, data from seven phase III clinical trials revealed that 5.1% of PwHA developed anti-drug antibodies (ADAs) after receiving emicizumab; 11.8% of these ADAs reduced emicizumab exposure. Moreover, over 50% of samples containing ADAs showed neutralizing effects.80 Mim8 is a new bispecific antibody that mimics FVIIIa with an action mechanism similar to that of emicizumab but differs in its amino acid sequence and does not cross-react with anti-emicizumab antibodies.81 Mim8 has demonstrated positive clinical outcomes in FRONTIER 2 trials and will be submitted for its first regulatory approval by the end of 2024,82 thus providing patients experiencing emicizumab resistance an alternative.
Concizumab, a humanized IgG4 antibody targeting tissue factor pathway inhibitor (TFPI), was approved in Canada in 2023 for prophylaxis in adolescent and adult PwHB with inhibitors.83 In phase III clinical trials of concizumab, explorer7 included PwHA and PwHB with inhibitors reported a median ABR of 0, whereas explorer8 included PwHA and PwHB without inhibitors reported a median ABR of 1.7 and 2.8 times, respectively. Both explorer7 and explorer8 reported that the ABRs of PwHA and PwHB with or without inhibitors treated with concizumab were lower than those of PwH without prophylaxis.84,85 Additionally, patients experienced thromboembolic events in both trials; accordingly, when concizumab was restarted, the initial maintenance dose was reduced from 0.25 to 0.20 mg/kg84,86
Marstacimab, a human IgG1 antibody developed by Pfizer that targets the Kunitz 2 domain of TFPI, was approved for marketing in the United States in 2024 for the prophylaxis of adolescent and adult PwHA or PwHB without inhibitors.87,88 Compared with routine prophylaxis and episodic treatment, in a key phase III clinical trial, marstacimab reduced the ABR in patients with severe HA and moderate-to-severe HB without inhibitors for treating bleeds by 35% and 92% after a 12-month active treatment period, respectively.87 Marstacimab was administered weekly with non-weight-based flat dosing as a subcutaneous 300 mg loading dose followed by 150 mg once weekly; dose adjustment to 300 mg weekly could be considered. And, no thromboembolic events were recorded in the phase III trial or long-term extension study.89
Emicizumab, concizumab, and marstacimab are administered via subcutaneous injection, which improves patient compliance (Figure 3). Although the blood concentration of intravenous CFCs immediately peaks following administration, the time to achieve the maximum plasma concentration of subcutaneous antibodies is as long as several days.90,91 This accounts for why emicizumab, concizumab, and marstacimab are difficult to use for episodic treatment. To date, consensus on whether emicizumab can be used for the perioperative management of PwHA remains lacking.92
As the antibody structure entirely differs from that of FVIII, the hemostatic effects of emicizumab, concizumab and marstacimab are theoretically unaffected by FVIII inhibitors.93 Clinical trials have demonstrated the effectiveness of emicizumab and concizumab in inhibitor-positive PwH (Table S1). In addition to monoclonal antibody drugs, siRNAs and recombinase therapeutics, such as fitusiran and serpinPC, with no structural homology with FVIII or FIX, are also expected to be adopted for treating inhibitor-positive PwH.94,95
Globally, the overall prevalence of inhibitors in PwHA or PwHB is 5–7% and 1.5–3%, respectively.96,97 The incidence of inhibitors is significantly high at 20–40% in individuals with severe HA and 9–23% in individuals with severe HB.96,98 For PwH who develop inhibitors, long-term, and frequent supratherapeutic CFC doses may establish peripheral tolerance to coagulation factors and eradicate inhibitors, a therapy known as ITI.37 Both pdCFCs and rCFCs can be used for ITI in PwH.6 The success rate of ITI is influenced by various factors.99 In PwHA, the success rate of ITI can reach 60–70%, while in PwHB, the success rate is relatively low at 13–31%.100,101 The success of ITI in PwH improves in patients under 7 years old102; the inhibitor titer is ≤10 BU/mL before initiating ITI, historical and ITI period peak inhibitor titers are ≤100 BU/mL103; the time between inhibitor diagnosis and ITI initiation is less than 2 years102 However, the risk of recurrence remains even after successful ITI, a multicenter retrospective cohort study showed a relapse rate of 20–30%.104 In cases where ITI is ineffective, recurred, or unaffordable for inhibitor-positive patients, novel non-factor replacement therapies with efficacies unaffected by inhibitors can be adopted for effective bleeding control.
Gene therapy offers long-term replacement treatment options for patients with hemophilia
If a single gene, FVIII or FIX, can be delivered into the body to achieve sustained and stable expression, a long-term increase in the activity of plasma factors can be maintained. Consequently, PwH are ideal candidates for gene therapy. Since the 1990s, clinical trials of gene therapy for hemophilia have been attempted, including the in vitro transfection of autologous fibroblasts followed by reinfusion, intravenous injection of retroviral vectors, and intravenous injection of adenoviral vectors.105,106 Although unsuccessful, this represented an important attempt at gene therapy applied for treating hemophilia. In 2010, Nathwani et al. administered the first single intravenous infusion of rAAV serotype 8 vectors containing the FIX gene in PwHB. This increased the circulating FIX levels to 1–6% of the normal value, improving the bleeding episodes of patients in the long term, and establishing the concept of gene therapy for hemophilia.28
FVIII or FIX genes can be efficiently delivered to the liver for specific expression based on rAAV vectors. Three AAV gene therapies are effective for the expression of coagulation factors and approved by the FDA and EMA for hemophilia long-term replacement treatment. The therapies are valoctocogene roxaparvovec for PwHA; etranacogene dezaparvovec, and fidanacogene elaparvovec for PwHB. AAV is a non-enveloped, non-pathogenic parvovirus with a 4.7 kb single-stranded DNA genome.107 AAV vectors boast a high safety profile, stable long-term expression, and the ability to accommodate relatively large exogenous DNA fragments (5.2 kb).108 As the size of the FL-FVIII cDNA (7.0 kb) exceeds the loading capacity of AAV vectors,109 and the B domain of FVIII does not affect its coagulation efficacy, valoctocogene roxaparvovec for PwHA only transfers the BDD-FVIII cDNA (4.97 kb) sequence.110 Meanwhile, the FIX cDNA (1.5 kb) is relatively small and simple to express. Hence, etranacogene dezaparvovec and fidanacogene elaparvovec carry the complete FIX Padua (R338L) variant gene sequence.111,112
The complete rAAV vector preparation process involves three stages: upstream culture, downstream purification, and product packaging (Figure 2C). Production of valoctocogene roxaparvovec and etranacogene dezaparvovec uses the recombinant baculovirus-Sf9 insect cell (Bac-Sf9) system to express rAAV vectors.113 In contrast, fidanacogene elaparvovec uses the HEK293 three-plasmid transfection system.114 After the recombinant baculoviruses or plasmids are introduced into the host cells, numerous rAAV vectors are released into the cell culture growth media.113 The supernatant is harvested for downstream purification, concentration, and filtering of the rAAV vectors to obtain the final product.113 Once the rAAV vectors carrying the BDD-FVIII or FIX Padua gene are administered intravenously, hepatocytes consistently express FVIII or FIX for an extended period of time, effectively improving the circulating coagulation factor levels and maintaining the same factor level as that in mild-to-moderate PwH, significantly reducing the risk of bleeding.115,116
An open-label, multicenter, single-group phase III clinical trial was conducted with valoctocogene roxaparvovec. The results showed that, after a single infusion of rAAV5 vectors with an integrated FVIII gene, the median FVIII activity level of the participants was maintained at ≥ 5 IU/dL in approximately 88% and 75% of the participants at weeks 49–52 and 104, respectively.117,118 At week 104, the average ABR and the use of FVIII concentrate decreased by approximately 77% and 98%, respectively, compared with the baseline.118 In a dose-escalating phase I/II clinical study of valoctocogene roxaparvovec, after a single infusion of valoctocogene roxaparvovec, the median FVIII activity level was maintained at > 8 IU/dL for at least 5 years119 The FVIII level in patients with severe hemophilia is less than 1 IU/dL (1% of normal); after receiving gene therapy, FVIII levels can increase to those of patients with mild hemophilia A (5–40 IU/dL), and remain constant for an extended period. The clinical symptoms of hemophilia are directly determined by the serum factor activity level. For untreated patients with severe and mild hemophilia A, up to 60 and less than one bleeding episode occur per year, respectively.46
Single-arm, open-label phase III clinical trials of etranacogene dezaparvovec and fidanacogene elaparvovec showed that compared with the use of FIX for routine prophylaxis, both gene therapies reduced the ABR and FIX usage in PwHB; the activity level of FIX remained stable for a long time.115 At 12 and 18 months after PwHB received etranacogene dezaparvovec treatment, the average FIX activity level remained at 39% and 34%, respectively, from baseline levels.115 After PwHB received fidanacogene elaparvovec treatment, changes in the FIX activity level from the baseline were 26% and 25% at 12 and 24 months, respectively.120 These clinical findings suggest that for PwHB who undergo gene therapy, the activity levels of FIX remain stable over time.
Amid the rapid development of gene therapy for hemophilia, several critical issues require attention. The liver health of patients before and after gene therapy is a crucial factor that cannot be ignored; although hepatocellular carcinoma associated with gene therapy has not been observed, there is still a hypothesized risk of malignancy owing to the integration of the vector into the genome.121 The three hemophilia gene therapies approved for marketing have shown a self-limiting increase in alanine aminotransferase (ALT) in clinical trials.8 In a phase III clinical trial of valoctocogene roxaparvovec, approximately 86% of PwHA experienced adverse reactions associated with ALT elevation and required long-term use of corticosteroids for immunosuppressive synergistic treatment.117 ALT levels may increase due to inflammatory or immune responses induced by AAV gene transfer and may be associated with a decline in transgene expression. Immunosuppressive treatment helps recover ALT levels but does not necessarily restore the initial elevated transgene levels. Additionally, no consensus has been reached on the dose, type, or duration of immunosuppressive treatment.121
Patient-specific limitations continue to pose challenges for all AAV gene therapies for hemophilia (Figure 3). These three commercially available gene therapies cannot be administered to children or women.122,123,124 Additionally, gene therapy efficacy is affected by pre-existing AAV-neutralizing antibodies in patients. Analysis of 546 serum samples of PwHA worldwide indicated that the positive rate (34.8%) of pre-existing antibodies against AAV5 was the lowest,125 possibly accounting for why rAAV5 is used as a gene delivery vector for valoctocogene roxaparvovec and etranacogene dezaparvovec.
Future therapies for hemophilia
Emerging targets and advanced pharmaceutical technologies signify the future of hemophilia treatment, and cannot be ignored. In addition to traditional FVIII and FIX targets, clinical trials are assessing agents that regulate the balance between anticoagulant and procoagulant effects, including TFPI (new indications), activated protein C (APC), and antithrombin. Importantly, their efficacy is not affected by FVIII or FIX inhibitors (Figure 4; Table 1). A recently approved antibody drug targeting TFPI has shown encouraging clinical results at a once-weekly dosing frequency and is being tested clinically in inhibitor-positive PwH and specifically children with hemophilia under the age of 12 years. This is expected to expand the clinically applicable populations of anti-TFPI antibody drugs, warranting further attention.
Figure 4.

Innovative treatments for hemophilia associated with coagulation pathways
Interventions targeting specific processes in the coagulation cascade have shown promising results in clinical trials. Abbreviations: Ab, antibody; APC, activated protein C; AT, antithrombin; siRNA, small interfering RNA; TF, tissue factor; TFPI, tissue factor pathway inhibitor.
Table 1.
Exploration of new targets or pharmaceutical technologies for treating hemophilia
| Product type/name | Patient population Target |
Trial No. | Administration method Frequency |
Latest clinical results | References |
|---|---|---|---|---|---|
| Antibody | |||||
| Marstacimab | PwHA and PwHB with and without inhibitors TFPI |
NCT03363321 | Subcutaneous Once-weekly |
FDA has approved marstacimab for PwHA or PwHB without inhibitors, and the BASIS clinical trial for marstacimab in inhibitor-positive PwH, as well as the BASIS KIDS clinical trial in children aged 1 to <18 years with or without inhibitors, are both ongoing. | Pfizer87 and Matino et al.89 |
| SR604 | PwHA and PwHB with and without inhibitors APC |
NCT06349473; CTR20241608 (CDE) |
Subcutaneous Once every 2 weeks (tentative) |
SR604 exhibited prophylactic and therapeutic efficacy in tail-bleeding and knee-injury mouse models of HA and HB expressing human APC (pre-clinical). CDE has approved SR604 for phase I clinical trials. | Jiang et al.126 and CDE127 |
| Recombinase | |||||
| SerpinPC | PwHA and PwHB APC |
NCT04073498; NCT05789524; NCT05789537 |
Subcutaneous Once every 2 or 4 weeks |
SerpinPC treatment reduced the median all-bleed ABR to 1.0, representing a 96% reduction from the pre-exposure baseline ABR (phase I/IIa). A phase II clinical trial is underway. | Baglin et al.95 and Centessa Pharmaceuticals128 |
| Cellular gene therapy | |||||
| Auto CD34+ cells transduced with LV to produce FVIII | PwHA without inhibitors FVIII |
NCT03818763 | Intravenous Only once |
No reports of spontaneous bleeding or a need for “on-demand” factor VIII post-infusion following the demonstration of whole blood vector copy number (phase I). | Wilcox et al.129 |
| BE-101 (autologous primary human B cells medicine engineered to insert the human FIX gene) | PwHB FIX |
Pre-clinical | Intravenous Unknown |
A single dose of BE-101 could achieve active and sustained FIX levels. The data confirmed the expected biodistribution of FIX-expressing B cells in bone marrow tissue, where they engrafted stably over time (pre-clinical). FDA has approved BE-101 to conduct phase I/II clinical trials. | Be Biopharma 130 |
| SiRNA | |||||
| Fitusiran | PwHA and PwHB with and without inhibitors Antithrombin |
NCT03417245; NCT03417102 |
Subcutaneous Once-monthly |
Fitusiran prophylaxis showed hemostatic efficacy and statistically significant reductions in ABR (phase III). | Young et.al.131and Srivastava et al.132 |
ABR, annualized bleeding rate; APC, activated protein C; CDE, the Center for Drug Evaluation of the China National Medical Products Administration; CFCs, coagulation factor concentrates; FDA, US Food and Drug Administration; FIX, factor IX; FVIII, coagulation factor VIII; HA, hemophilia A; HB, hemophilia B; LV, lentiviral vector; PwHA, patients with hemophilia A; PwHB, patients with hemophilia B; siRNA, small interfering RNA; TFPI, tissue factor pathway inhibitor.
Novel biopharmaceutical products for hemophilia, including siRNAs and cellular gene therapies, have elicited significant improvements in reducing the frequency of dosing in clinical trials (Figure 4 and Table 1). The aforementioned therapeutic drugs under development for hemophilia are designed to improve patient compliance, reduce dosing frequency, and provide potential solutions for inhibitor-positive patients.
Investigating new targets for hemophilia management and potential new indications for anti-TFPI antibody drugs
Concizumab, which targets TFPI, has been approved for marketing in Canada as a prophylactic treatment for PwHB with inhibitors. However, as its half-life is only 38 h; thus, it must be administered subcutaneously once daily.91 The necessity for frequent dosing may be a significant factor hindering its widespread marketing approval. Although concizumab can effectively treat PwHA and PwHB with inhibitors, Health Canada has not approved concizumab for PwHA with inhibitors.83
The development of antibodies against TFPI will hopefully lead to the approval of hemophilia drugs with a broad spectrum of applications. Although marstacimab has been approved for marketing in the United States, the approved indications do not include PwH with inhibitors. The phase II clinical trial of marstacimab included a small number of inhibitor-positive PwH, and was found to be safe and well tolerated; moreover, the overall ABR was quite low.88 Currently, a phase III clinical trial of marstacimab in inhibitor-positive PwH is underway, and the results may be announced by 2025.87 Additionally, the ongoing BASIS KIDS clinical trial aims to study the safety and efficacy of marstacimab in children with hemophilia aged 1 to <18 years87 These clinical trials will have greater significance for expanding the patient population in which marstacimab could be used.
Befovacimab, another monoclonal antibody that simultaneously targets the Kunitz 1 and Kunitz 2 domains of TFPI, was discontinued in a non-randomized, open-label phase II clinical trial owing to severe adverse reactions of thrombosis in three cases.133 These findings suggest that the development of antibodies targeting TFPI should focus on the risk factors of thrombosis. Future studies should focus on a safe and effective window for anti-TFPI treatment to enhance patient outcomes and minimize potential adverse effects.
APC, an anticoagulant inactivating FVa and FVIIIa, can effectively degrade FVIIIa and FVa on phospholipid membranes to inhibit coagulation.134 SerpinPC is a highly specific recombinant serine protease inhibitor that enhances thrombin activity in sites with tissue damage and shortens the coagulation time in PwH by reducing the activity of APC in circulation.135 So far, two phase II clinical trials have evaluated the efficacy, safety, tolerability, and pharmacokinetics of serpinPC in patients with severe HA and moderate-to-severe HB, with or without inhibitors (NCT05789524, NCT05789537). The humanized antibody SR604 targets APC to block its anticoagulant activity, as demonstrated in mouse models with hemophilia.126 Moreover, SR604 does not induce apparent toxic effects in mice with hemophilia and demonstrates excellent bioavailability in non-human primates via subcutaneous injection.126 Accordingly, the Center for Drug Evaluation of the China National Medical Products Administration (CDE) has approved a clinical registration trial for SR604 to prevent and treat bleeding episodes in PwHA, PwHB, and patients with congenital FVII deficiency.127
Recent advances in pharmaceutical technologies for treating hemophilia
Cellular gene therapy addresses hemophilia by editing cells in vitro and reinfusing them into the patient to produce coagulation factors (Figure 4). Currently, cellular gene therapy using lentiviral vectors is available to modify a person’s own CD34-positive cells, facilitating the production of FVIII in megakaryocytes through the platelet-specific ITGA2B gene promoter. This approach is being evaluated in a phase I clinical trial (NCT03818763).129
BE-101, a cell therapy that encodes autologous B cells to express FIX, has passed the Investigational New Drug application of the FDA and is expected to be studied in a phase I/II clinical trial in the second half of 2024 to evaluate the safety and preliminary efficacy of BE-101 in patients with moderate-to-severe hemophilia.130
Combining new targets and new pharmaceutical technologies
The rapid development of siRNA technology has provided novel options for treating various rare diseases. siRNA drugs can selectively silence the mRNA expression of disease-related genes, thereby preventing target gene expression. These drug pipelines have high clinical translatability and shorter development timelines than conventional drugs.136 The potential of siRNAs targeting antithrombin, protein S, and heparin cofactor II for treating hemophilia has been verified in animal models and clinical trials.137,138
Among these drugs, fitusiran, jointly developed by Alnylam and Sanofi, is the most advanced siRNA therapeutic for hemophilia (Figure 4). Two phase III clinical trials evaluating its prophylactic treatment efficacy and safety in patients with severe HA and HB, with or without inhibitors, are nearly complete. Clinical data show that a monthly subcutaneous injection of fitusiran significantly reduces the ABR in PwH.131,132 As of June 2024, the application for the listing of fitusiran was accepted by the FDA and CDE.139 Additionally, siRNA drugs are generally less expensive than conventional treatment modalities, making them potentially safe, effective, and economical treatment options for PwH.136
Conclusions
PwH requires lifelong treatment to limit the frequency and severity of bleeding; innovative delivery methods and biopharmaceutical products have significantly improved the quality of life and medication choices for PwH. Subcutaneous drug administration and gene therapies have alleviated the inconvenience of frequent intravenous CFC injections, and non-factor drugs allow inhibitor-positive PwH to select treatments without inhibitor restrictions.
However, there remains a risk of not effectively managing thromboembolism or thrombotic microangiopathies if non-factor hemostatic drugs are used during breakthrough bleeding or perioperative management. This risk is indicated by a black box warning on the package insert that comes with emicizumab.79 Gene therapies aim to provide effective and long-term replacement treatment options for PwH with a single intravenous treatment, eliminating the need for regular prophylaxis. However, strategies are required to reduce associated costs. Challenges with pre-existing immunity to AAV vectors and the applicability of treatment to minors and female patients remain to be addressed. Extensive clinical testing is also warranted to determine treatment longevity and safety.
To improve treatment outcomes and reduce adverse effects, researchers are developing siRNA drugs, recombinases, and cellular gene therapies. Developing innovative biopharmaceutical products and allowing clinical use may positively impact market prices and national bidding behavior, benefiting more PwH.140 In low- and middle-income countries, there remains an unmet clinical demand for traditional replacement therapies, with CFC usage per capita significantly lower than that in high-income countries.
As clinicians gain more experience with CFCs and new biopharmaceutical products, a growing interest in personalized hemophilia treatments has emerged. The pharmacokinetics of CFCs in PwH can significantly vary, with half-lives differing by 2- to 4-fold between individuals.141 Existing dosage calculations and intervals are unsuitable for all patients; and therefore, prophylactic treatment should be guided by the actual pharmacokinetics of the individual PwH.141,142,143 Non-factor gene therapies and monoclonal antibody drugs have individualized control measures for safety and effectiveness. Gene therapies require consistent long-term monitoring of multiple liver enzymes post-administration, with regimens selected based on individual results.122,123,124 Concizumab requires pre-dose plasma concentration measurements 4 weeks following treatment initiation to determine maintenance doses.91
The focus on hemophilia treatment has driven progress in biotechnology, leading to breakthroughs in gene therapy and bispecific antibody pharmaceuticals. Research and development of related drugs and personalized prophylactic treatments will offer valuable insights into treating other single-gene diseases.
Acknowledgments
The authors thank Mr. Liming Yang from the Research and Development Department of Chengdu Rongsheng Pharmaceuticals Co., Ltd for his helpful discussions regarding this article.
This work was supported by Chengdu Rongsheng Pharmaceuticals Co., Ltd.
Author contributions
D.Y. and J.Z.W. conceptualized the review. J.Z.W. and X.L.L. drafted the article. J.Z.W. X.L.L., H.C.Y., and Y.L.H. organized the data. All authors read and approved the final article.
Declaration of interests
J.Z.W., X.L.L., and D.Y. are paid employees of Chengdu Rongsheng Pharmaceuticals Co., Ltd. H.C.Y is an employee of China National Biotec Group Company Limited. D.Y. and Y.L.H are employees of Beijing Tiantan Biological Products Co., Ltd. These companies are involved in the development of rFVIII-Fc, rFVIIa, pdFVIII, and pdFIX. The views presented here should not be considered endorsements of any specific products or company.
Declaration of generative AI and AI-assisted technologies in the writing process
No AI was used in the preparation of this review.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111436.
Supplemental information
References
- 1.Srivastava A., Santagostino E., Dougall A., Kitchen S., Sutherland M., Pipe S.W., Carcao M., Mahlangu J., Ragni M.V., Windyga J., et al. WFH Guidelines for the Management of Hemophilia. Haemophilia. 2020;26:1–158. doi: 10.1111/hae.14046. [DOI] [PubMed] [Google Scholar]
- 2.Swan D., Mahlangu J., Thachil J. Non-factor therapies for bleeding disorders: A primer for the general haematologist. EJH. 2022;3:584–595. doi: 10.1002/jha2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miesbach W., Schwäble J., Müller M.M., Seifried E. Treatment options in hemophilia. Dtsch. Arztebl. Int. 2019;116:791–798. doi: 10.3238/arztebl.2019.0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waitt C., Waitt P., Pirmohamed M. Intravenous therapy. Postgrad. Med. 2004;80:1–6. doi: 10.1136/pmj.2003.010421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckhardt C.L., van Velzen A.S., Peters M., Astermark J., Brons P.P., Castaman G., Cnossen M.H., Dors N., Escuriola-Ettingshausen C., Hamulyak K., et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122:1954–1962. doi: 10.1182/blood-2013-02-483263. [DOI] [PubMed] [Google Scholar]
- 6.Collins P.W., Chalmers E., Hart D.P., Liesner R., Rangarajan S., Talks K., Williams M., Hay C.R., UK Haemophilia Centre Doctors Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). UK Haemophilia Centre Doctors Organization. Br. J. Haematol. 2013;160:153–170. doi: 10.1111/bjh.12091. [DOI] [PubMed] [Google Scholar]
- 7.US Food and Drug Administration. GUIDANCE DOCUMENT . 2020. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). Guidance for Industry 2020;II; p. 2.https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigational-new-drug [Google Scholar]
- 8.Nathwani A.C. Gene therapy for hemophilia. Hematology. Am. Soc. Hematol. Educ. Program. 2022;2022:569–578. doi: 10.1182/hematology.2022000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miesbach W., Klamroth R., Oldenburg J., Tiede A. Gene therapy for hemophilia-opportunities and risks. Dtsch. Arztebl. Int. 2022;119:887–894. doi: 10.3238/arztebl.m2022.0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escuriola-Ettingshausen C., Auerswald G., Königs C., Kurnik K., Scholz U., Klamroth R., Oldenburg J. Optimizing the management of patients with haemophilia A and inhibitors in the era of emicizumab: Recommendations from a German expert panel. Haemophilia. 2021;27:e305–e313. doi: 10.1111/hae.14010. [DOI] [PubMed] [Google Scholar]
- 11.Wu J., Zhang H., Lian T., Ding Y., Song C., Li D., Wu L., Lei T., Liang H. Biological activity of a new recombinant human coagulation factor VIII and its efficacy in a small animal model. Biochem. Biophys. Res. Commun. 2023;640:80–87. doi: 10.1016/j.bbrc.2022.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Pratap R., Misra M., N V., Morampudi S., Patil A., Reddy J. The existing scenario of haemophilia care in Canada and China - A review. Hematol. Transfus. Cell Ther. 2020;42:356–364. doi: 10.1016/j.htct.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.World Federation of Hemophilia (2024). Identification and diagnosis. https://wfh.org/identification-and-diagnosis/. Accessed 18 June 2024.
- 14.Iorio A., Stonebraker J.S., Chambost H., Makris M., Coffin D., Herr C., Germini F., Data and Demographics Committee of the World Federation of Hemophilia Establishing the prevalence and prevalence at birth of hemophilia in males: A meta-analytic approach using national registries. Ann. Intern. Med. 2019;171:540–546. doi: 10.7326/m19-1208. [DOI] [PubMed] [Google Scholar]
- 15.World Federation of Hemophilia (2022). Annual Global Survey. http://shiny.wfh.org/ags/. Accessed 9 October 2024.
- 16.World Federation of Hemophilia (2022). World Federation of Hemophilia Report on the Annual Global Survey 2022. https://www1.wfh.org/publications/files/pdf-2399.pdf. Accessed 18 June 2024.
- 17.Yang R., Poon M.C., Luke K.H., Zhao Y., Sun J., Wang X., Wu R., Chen L., Zhang X., Wu J. Building a network for hemophilia care in China: 15 years of achievement for the Hemophilia Treatment Center Collaborative Network of China. Blood Adv. 2019;3:34–37. doi: 10.1182/bloodadvances.2019GS121524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Berg H.M., De Groot P.H.G., Fischer K. Phenotypic heterogeneity in severe hemophilia. J. Thromb. Haemostasis. 2007;5:151–156. doi: 10.1111/j.1538-7836.2007.02503.x. [DOI] [PubMed] [Google Scholar]
- 19.Chaudhury A., Sidonio R., Jr., Jain N., Tsao E., Tymoszczuk J., Oviedo Ovando M., Kulkarni R. Women and girls with haemophilia and bleeding tendencies: Outcomes related to menstruation, pregnancy, surgery and other bleeding episodes from a retrospective chart review. Haemophilia. 2021;27:293–304. doi: 10.1111/hae.14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peyvandi F., Garagiola I., Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388:187–197. doi: 10.1016/s0140-6736(15)01123-x. [DOI] [PubMed] [Google Scholar]
- 21.Peyvandi F., Kenet G., Pekrul I., Pruthi R.K., Ramge P., Spannagl M. Laboratory testing in hemophilia: Impact of factor and non-factor replacement therapy on coagulation assays. J. Thromb. Haemostasis. 2020;18:1242–1255. doi: 10.1111/jth.14784. [DOI] [PubMed] [Google Scholar]
- 22.Müller J., Miesbach W., Prüller F., Siegemund T., Scholz U., Sachs U.J., Standing Commission Labor STAEKOLA of the Society of Thrombosis and Haemostasis Research GTH An update on laboratory diagnostics in haemophilia A and B. Hämostaseologie. 2022;42:248–260. doi: 10.1055/a-1665-6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham J.B., Rizza C.R., Chediak J., Mannucci P.M., Briët E., Ljung R., Kasper C.K., Essien E.M., Green P.P. Carrier detection in hemophilia A: a cooperative international study. I. The carrier phenotype. Blood. 1986;67:1554–1559. doi: 10.1182/blood.V67.6.1554.1554. [DOI] [PubMed] [Google Scholar]
- 24.Dardik R., Janczar S., Lalezari S., Avishai E., Levy-Mendelovich S., Barg A.A., Martinowitz U., Babol-Pokora K., Mlynarski W., Kenet G. Four decades of carrier detection and prenatal diagnosis in hemophilia A: Historical overview, state of the art and future directions. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms241411846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weyand A.C., Pipe S.W. New therapies for hemophilia. Blood. 2019;133:389–398. doi: 10.1182/blood-2018-08-872291. [DOI] [PubMed] [Google Scholar]
- 26.Bray G.L., Gomperts E.D., Courter S., Gruppo R., Gordon E.M., Manco-Johnson M., Shapiro A., Scheibel E., White G., 3rd, Lee M. A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood. 1994;83:2428–2435. doi: 10.1182/blood.V83.9.2428.2428. [DOI] [PubMed] [Google Scholar]
- 27.Li A., Goodfriend C., Sokol J., Kruse-Jarres R. Patterns and predictors of emicizumab adherence in people with hemophilia. Blood. 2019;134:2178. doi: 10.1182/blood-2019-128083. [DOI] [Google Scholar]
- 28.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.High K.A., Roncarolo M.G. Gene therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 30.Manco-Johnson M.J., Soucie J.M., Gill J.C., Joint Outcomes Committee of the Universal Data Collection, US Hemophilia Treatment Center Network Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: a surveillance project. Blood. 2017;129:2368–2374. doi: 10.1182/blood-2016-02-683169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song X., Zhong J., Xue F., Chen L., Li H., Yuan D., Xie J., Shi J., Zhang L., Wu E.Q., Yang R. An overview of patients with haemophilia A in China: Epidemiology, disease severity and treatment strategies. Haemophilia. 2021;27:e51–e59. doi: 10.1111/hae.14217. [DOI] [PubMed] [Google Scholar]
- 32.Wu R., Wu X., Zhang N., Zhao L., Zhang J., Luke K.H., Poon M.C., Peng Y., Andrea D. Joint disease status of severe and moderate haemophilia patients at the Beijing Children's Hospital: early onset and rapid increasing severity of arthropathy in 90% of patients by 6 years of age. Haemophilia. 2014;20:e227–e230. doi: 10.1111/hae.12394. [DOI] [PubMed] [Google Scholar]
- 33.Seth T., Garg K., Mandal P.K., Datta A., Verma S., Hanagavadi S., Thota U.R. Cost-effectiveness analysis of low-dose prophylaxis versus on-demand treatment for moderate-to-severe hemophilia A in India. Hematology. 2023;28 doi: 10.1080/16078454.2023.2277497. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y., Lu J., Zhou Y., Li K., Liu Y., Liu S., Li Z., Zhao Y., Poon M.C., Xiao J. Long-term joint outcomes of regular low-dose prophylaxis in Chinese children with severe haemophilia A. Haemophilia. 2021;27:237–244. doi: 10.1111/hae.14256. [DOI] [PubMed] [Google Scholar]
- 35.Malec L., Matino D. Targeting higher factor VIII levels for prophylaxis in haemophilia A: a narrative review. Haemophilia. 2023;29:1419–1429. doi: 10.1111/hae.14866. [DOI] [PubMed] [Google Scholar]
- 36.Pierce G.F., Adediran M., Diop S., Dunn A.L., El Ekiaby M., Kaczmarek R., Konkle B.A., Pipe S.W., Skinner M.W., Valentino L.A., et al. Achieving access to haemophilia care in low-income and lower-middle-income countries: expanded Humanitarian Aid Program of the World Federation of Hemophilia after 5 years. Lancet. Haematol. 2022;9:e689–e697. doi: 10.1016/s2352-3026(22)00209-5. [DOI] [PubMed] [Google Scholar]
- 37.Kempton C.L., Meeks S.L. Toward optimal therapy for inhibitors in hemophilia. Blood. 2014;124:3365–3372. doi: 10.1182/blood-2014-05-577643. [DOI] [PubMed] [Google Scholar]
- 38.Croteau S.E., Wang M., Wheeler A.P. 2021 clinical trials update: Innovations in hemophilia therapy. Am. J. Hematol. 2021;96:128–144. doi: 10.1002/ajh.26018. [DOI] [PubMed] [Google Scholar]
- 39.Kasper C.K. Judith Graham Pool and the discovery of cryoprecipitate. Haemophilia. 2012;18:833–835. doi: 10.1111/hae.12042. [DOI] [PubMed] [Google Scholar]
- 40.Gogia P., Tarantino M., Schramm W., Aledort L. New directions to develop therapies for people with hemophilia. Expert Rev. Hematol. 2023;16:417–433. doi: 10.1080/17474086.2023.2184341. [DOI] [PubMed] [Google Scholar]
- 41.Berntorp E. Why prescribe highly purified factor VIII and IX concentrates? Vox Sang. 1996;70:61–68. doi: 10.1111/j.1423-0410.1996.tb01295.x. [DOI] [PubMed] [Google Scholar]
- 42.Burnouf T. Modern plasma fractionation. Transfus. Med. Rev. 2007;21:101–117. doi: 10.1016/j.tmrv.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menache D. Prothrombin complex concentrates: clinical use. Ann. N. Y. Acad. Sci. 1981;370:747–756. doi: 10.1111/j.1749-6632.1981.tb29782.x. [DOI] [PubMed] [Google Scholar]
- 44.Kim H.C., McMillan C.W., White G.C., Bergman G.E., Horton M.W., Saidi P. Purified factor IX using monoclonal immunoaffinity technique: clinical trials in hemophilia B and comparison to prothrombin complex concentrates. Blood. 1992;79:568–575. doi: 10.1182/blood.V79.3.568.568. [DOI] [PubMed] [Google Scholar]
- 45.Coan M.H., Fournel M.A., Mozen M.M. Properties of commercial factor IX concentrates. Ann. N. Y. Acad. Sci. 1981;370:731–746. doi: 10.1111/j.1749-6632.1981.tb29781.x. [DOI] [PubMed] [Google Scholar]
- 46.Srivastava A., Brewer A.K., Mauser-Bunschoten E.P., Key N.S., Kitchen S., Llinas A., Ludlam C.A., Mahlangu J.N., Mulder K., Poon M.C., et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–e47. doi: 10.1111/j.1365-2516.2012.02909.x. [DOI] [PubMed] [Google Scholar]
- 47.Evatt B.L., Gomperts E.D., McDougal J.S., Ramsey R.B. Coincidental appearance of LAV/HTLV-III antibodies in hemophiliacs and the onset of the AIDS epidemic. N. Engl. J. Med. 1985;312:483–486. doi: 10.1056/nejm198502213120805. [DOI] [PubMed] [Google Scholar]
- 48.Mauser-Bunschoten E.P., Bresters D., van Drimmelen A.A., Roosendaal G., Cuypers H.T., Reesink H.W., van der Poel C.L., van den Berg H.M., Lelie P.N. Hepatitis C infection and viremia in Dutch hemophilia patients. J. Med. Virol. 1995;45:241–246. doi: 10.1002/jmv.1890450302. [DOI] [PubMed] [Google Scholar]
- 49.Blümel J., Schmidt I., Effenberger W., Seitz H., Willkommen H., Brackmann H.H., Löwer J., Eis-Hübinger A.M. Parvovirus B19 transmission by heat-treated clotting factor concentrates. Transfusion. 2002;42:1473–1481. doi: 10.1046/j.1537-2995.2002.00221.x. [DOI] [PubMed] [Google Scholar]
- 50.Kevane B., O'Connell N. The current and future role of plasma-derived clotting factor concentrate in the treatment of haemophilia A. Transfus. Apher. Sci. 2018;57:502–506. doi: 10.1016/j.transci.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 51.European Medicines Agency (2011). Guideline on plasma-derived medicinal products. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-plasma-derived-medicinal-products_en.pdf. Accessed 18 June 2024.
- 52.The China National Medical Products Administration (2002). Technical Methods and Validation Guidelines for Elimination/Inactivation of Viruses in Plasma-derived Products. https://www.csbt.org.cn/plus/view.php?aid=83616. Accessed 18 June 2024.
- 53.Boedeker B.G. Production processes of licensed recombinant factor VIII preparations. Semin. Thromb. Hemost. 2001;27:385–394. doi: 10.1055/s-2001-16891. [DOI] [PubMed] [Google Scholar]
- 54.Lusher J.M., Scharrer I. Evolution of recombinant factor VIII safety: KOGENATE and Kogenate FS/Bayer. Int. J. Hematol. 2009;90:446–454. doi: 10.1007/s12185-009-0435-x. [DOI] [PubMed] [Google Scholar]
- 55.Sankar A.D., Weyand A.C., Pipe S.W. The evolution of recombinant factor replacement for hemophilia. Transfus. Apher. Sci. 2019;58:596–600. doi: 10.1016/j.transci.2019.08.010. [DOI] [PubMed] [Google Scholar]
- 56.Gouw S.C., van der Bom J.G., Ljung R., Escuriola C., Cid A.R., Claeyssens-Donadel S., van Geet C., Kenet G., Mäkipernaa A., Molinari A.C., et al. Factor VIII products and inhibitor development in severe hemophilia A. N. Engl. J. Med. 2013;368:231–239. doi: 10.1056/NEJMoa1208024. [DOI] [PubMed] [Google Scholar]
- 57.Manco-Johnson M.J., Abshire T.C., Shapiro A.D., Riske B., Hacker M.R., Kilcoyne R., Ingram J.D., Manco-Johnson M.L., Funk S., Jacobson L., et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N. Engl. J. Med. 2007;357:535–544. doi: 10.1056/NEJMoa067659. [DOI] [PubMed] [Google Scholar]
- 58.Malec L.M., Cheng D., Witmer C.M., Jaffray J., Kouides P.A., Haley K.M., Sidonio R.F., Jr., Johnson K., Recht M., White G., et al. The impact of extended half-life factor concentrates on prophylaxis for severe hemophilia in the United States. Am. J. Hematol. 2020;95:960–965. doi: 10.1002/ajh.25844. [DOI] [PubMed] [Google Scholar]
- 59.Qu J., Ma C., Xu X.Q., Xiao M., Zhang J., Li D., Liu D., Konkle B.A., Miao C.H., Li L., Xiao W. Comparative glycosylation mapping of plasma-derived and recombinant human factor VIII. PLoS One. 2020;15 doi: 10.1371/journal.pone.0233576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shestopal S.A., Hao J.J., Karnaukhova E., Liang Y., Ovanesov M.V., Lin M., Kurasawa J.H., Lee T.K., McVey J.H., Sarafanov A.G. Expression and characterization of a codon-optimized blood coagulation factor VIII. J. Thromb. Haemostasis. 2017;15:709–720. doi: 10.1111/jth.13632. [DOI] [PubMed] [Google Scholar]
- 61.Ogata K., Selvaraj S.R., Miao H.Z., Pipe S.W. Most factor VIII B domain missense mutations are unlikely to be causative mutations for severe hemophilia A: implications for genotyping. J. Thromb. Haemostasis. 2011;9:1183–1190. doi: 10.1111/j.1538-7836.2011.04268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miao H.Z., Sirachainan N., Palmer L., Kucab P., Cunningham M.A., Kaufman R.J., Pipe S.W. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–3419. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- 63.Munawar Ali R., Abid M., Zafar S., Ali M.S., Nadeem R., Ahmed R., Borhany M. Management of severe hemophilia A: low-dose prophylaxis vs. on-demand treatment. Cureus. 2023;15 doi: 10.7759/cureus.41410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125:2038–2044. doi: 10.1182/blood-2015-01-528414. [DOI] [PubMed] [Google Scholar]
- 65.Chalmers E., Williams M., Brennand J., Liesner R., Collins P., Richards M., Paediatric Working Party of United Kingdom Haemophilia Doctors' Organization Guideline on the management of haemophilia in the fetus and neonate. Br. J. Haematol. 2011;154:208–215. doi: 10.1111/j.1365-2141.2010.08545.x. [DOI] [PubMed] [Google Scholar]
- 66.Berntorp E., Hermans C., Solms A., Poulsen L., Mancuso M.E. Optimising prophylaxis in haemophilia A: The ups and downs of treatment. Blood Rev. 2021;50 doi: 10.1016/j.blre.2021.100852. [DOI] [PubMed] [Google Scholar]
- 67.Mahlangu J., Young G., Hermans C., Blanchette V., Berntorp E., Santagostino E. Defining extended half-life rFVIII-A critical review of the evidence. Haemophilia. 2018;24:348–358. doi: 10.1111/hae.13438. [DOI] [PubMed] [Google Scholar]
- 68.Metzner H.J., Pipe S.W., Weimer T., Schulte S. Extending the pharmacokinetic half-life of coagulation factors by fusion to recombinant albumin. Thromb. Haemostasis. 2013;110:931–939. doi: 10.1160/th13-03-0213. [DOI] [PubMed] [Google Scholar]
- 69.Frampton J.E. Efmoroctocog Alfa: a review in haemophilia A. Drugs. 2021;81:2035–2046. doi: 10.1007/s40265-021-01615-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peters R., Harris T. Advances and innovations in haemophilia treatment. Nat. Rev. Drug Discov. 2018;17:493–508. doi: 10.1038/nrd.2018.70. [DOI] [PubMed] [Google Scholar]
- 71.Al-Salama Z.T., Scott L.J. Lonoctocog Alfa: A review in haemophilia A. Drugs. 2017;77:1677–1686. doi: 10.1007/s40265-017-0815-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bonanad S., Núñez R., Poveda J.L., Kurnik K., Goldmann G., Andreozzi V., Vandewalle B., Santos S. Matching-adjusted indirect comparison of efficacy and consumption of rVIII-single chain versus two recombinant FVIII products Used for prophylactic treatment of adults/adolescents with severe haemophilia A. Adv. Ther. 2021;38:4872–4884. doi: 10.1007/s12325-021-01853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lissitchkov T., Willemze A., Katragadda S., Rice K., Poloskey S., Benson C. Efanesoctocog alfa for hemophilia A: results from a phase 1 repeat-dose study. Blood Adv. 2022;6:1089–1094. doi: 10.1182/bloodadvances.2021006119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prezotti A.N.L., Frade-Guanaes J.O., Yamaguti-Hayakawa G.G., Ozelo M.C. Immunogenicity of current and new therapies for hemophilia A. Pharmaceuticals. 2022;15:911. doi: 10.3390/ph15080911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nolan B., Mahlangu J., Pabinger I., Young G., Konkle B.A., Barnes C., Nogami K., Santagostino E., Pasi K.J., Khoo L., et al. Recombinant factor VIII Fc fusion protein for the treatment of severe haemophilia A: Final results from the ASPIRE extension study. Haemophilia. 2020;26:494–502. doi: 10.1111/hae.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Young G., Liesner R., Chang T., Sidonio R., Oldenburg J., Jiménez-Yuste V., Mahlangu J., Kruse-Jarres R., Wang M., Uguen M., et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134:2127–2138. doi: 10.1182/blood.2019001869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mahlangu J., Oldenburg J., Paz-Priel I., Negrier C., Niggli M., Mancuso M.E., Schmitt C., Jiménez-Yuste V., Kempton C., Dhalluin C., et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N. Engl. J. Med. 2018;379:811–822. doi: 10.1056/NEJMoa1803550. [DOI] [PubMed] [Google Scholar]
- 78.Oldenburg J., Mahlangu J.N., Kim B., Schmitt C., Callaghan M.U., Young G., Santagostino E., Kruse-Jarres R., Negrier C., Kessler C., et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N. Engl. J. Med. 2017;377:809–818. doi: 10.1056/NEJMoa1703068. [DOI] [PubMed] [Google Scholar]
- 79.US Food and Drug Administration HEMLIBRA® (emicizumab-kxwh) injection, for subcutaneous use. Package Insert. 2017 [Google Scholar]
- 80.Schmitt C., Emrich T., Chebon S., Fernandez E., Petry C., Yoneyama K., Kiialainen A., Howard M., Niggli M., Paz-Priel I., Chang T. Low immunogenicity of emicizumab in persons with haemophilia A. Haemophilia. 2021;27:984–992. doi: 10.1111/hae.14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Valsecchi C., Gualtierotti R., Arcudi S., Ciavarella A., Siboni S.M., Schiavone L., Beeg M., Gobbi M., Peyvandi F. Anti-emicizumab antibodies do not cross-react with mim8 in vitro. Res. Pract. Thromb. Haemost. 2023;7 doi: 10.1016/j.rpth.2023.102161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Novo Nordisk (2024). Company announcement: Once-weekly and once-monthly Mim8 demonstrate superior reduction of treated bleeding episodes compared to on-demand and prior prophylaxis treatment in people with haemophilia A in the Frontier 2 trial. https://www.novonordisk.com/news-and-media/news-and-ir-materials/news-details.html?id=168515. Accessed 18 June 2024.
- 83.Keam S.J. Concizumab: first approval. Drugs. 2023;83:1053–1059. doi: 10.1007/s40265-023-01912-6. [DOI] [PubMed] [Google Scholar]
- 84.Matsushita T., Shapiro A., Abraham A., Angchaisuksiri P., Castaman G., Cepo K., d'Oiron R., Frei-Jones M., Goh A.S., Haaning J., et al. Phase 3 trial of concizumab in hemophilia with inhibitors. N. Engl. J. Med. 2023;389:783–794. doi: 10.1056/NEJMoa2216455. [DOI] [PubMed] [Google Scholar]
- 85.Astermark J., Apte S., Lyu C.J., Rhode Høgh Nielsen A., Saulyte Trakymiene S., Thaung Zaw J.J., Tran H., Windyga J., Eichler H. Efficacy and safety of concizumab prophylaxis in patients with hemophilia A or B without inhibitors: 56-Week cut-off results of the Phase 3 explorer8 study. Blood. 2023;142:2609. doi: 10.1182/blood-2023-173118. [DOI] [Google Scholar]
- 86.Seremetis S.V., Cepo K., Skovgaard Rasmussen J., Høyer Rose T., Tamer S., Porstmann T., Haaning J. Risk mitigation strategy for concizumab clinical trials after pause due to non-fatal thrombotic events. Blood. 2020;136:40. doi: 10.1182/blood-2020-139563. [DOI] [Google Scholar]
- 87.Pfizer (2024). U.S. FDA Approves Pfizer’s HYMPAVZI™ (marstacimab-hncq) for the Treatment of Adults and Adolescents with Hemophilia A or B Without Inhibitors. https://www.pfizer.com/news/press-release/press-release-detail/us-fda-approves-pfizers-hympavzitm-marstacimab-hncq. Accessed 12 October 2024.
- 88.Mahlangu J., Luis Lamas J., Cristobal Morales J., Malan D.R., Teeter J., Charnigo R.J., Hwang E., Arkin S. Long-term safety and efficacy of the anti-tissue factor pathway inhibitor marstacimab in participants with severe haemophilia: Phase II study results. Br. J. Haematol. 2023;200:240–248. doi: 10.1111/bjh.18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Matino D., Acharya S., Palladino A., Hwang E., McDonald R., Taylor C.T., Teeter J. Efficacy and safety of the anti-tissue factor pathway inhibitor marstacimab in participants with severe hemophilia without inhibitors: results from the phase 3 basis trial. Blood. 2023;142:285. doi: 10.1182/blood-2023-181263. [DOI] [Google Scholar]
- 90.Li H., Zhang W., Petry C., Li L., Fernandez E., Kiialainen A., Feng S., Hsu W., Li L., Wei Y., Schmitt C. Evaluation of the pharmacokinetics, pharmacodynamics, and safety of a single dose of emicizumab in healthy Chinese subjects. Clin. Pharmacol. Drug Dev. 2021;10:30–38. doi: 10.1002/cpdd.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Novo Nordisk Canada Inc. Concizumab (Alhemo™): Canadian product monograph. (2023). https://www.novonordisk.ca. Accessed 18 June 2024.
- 92.Rener K., Anžej Doma S., Fink M., Podgornik H., Preložnik Zupan I. Management and outcomes of invasive procedures in individuals with hemophilia a on emicizumab prophylaxis: a single center experience. Hematol. Rep. 2023;15:597–607. doi: 10.3390/hematolrep15040062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kitazawa T., Igawa T., Sampei Z., Muto A., Kojima T., Soeda T., Yoshihashi K., Okuyama-Nishida Y., Saito H., Tsunoda H., et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat. Med. 2012;18:1570–1574. doi: 10.1038/nm.2942. [DOI] [PubMed] [Google Scholar]
- 94.Young G., Lenting P.J., Croteau S.E., Nolan B., Srivastava A. Antithrombin lowering in hemophilia: a closer look at fitusiran. Res. Pract. Thromb. Haemost. 2023;7 doi: 10.1016/j.rpth.2023.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baglin T., Huntington J.A., Koch A., Mocanu I., Makhaldiani L. Serpin-PC in persons with severe hemophilia (PwH): updated results from a multicenter multi-part, first-in-human study. Blood. 2023;142:2619. doi: 10.1182/blood-2023-179969. [DOI] [Google Scholar]
- 96.Iorio A. Textbook of Hemophilia. 2014. Epidemiology of Inhibitors in Hemophilia; pp. 53–58. [DOI] [Google Scholar]
- 97.Santoro C., Quintavalle G., Castaman G., Baldacci E., Ferretti A., Riccardi F., Tagliaferri A. Inhibitors in hemophilia B. Semin. Thromb. Hemost. 2018;44:578–589. doi: 10.1055/s-0038-1660817. [DOI] [PubMed] [Google Scholar]
- 98.Peyvandi F., Kavakli K., El-Beshlawy A., Rangarajan S. Management of haemophilia A with inhibitors: A regional cross-talk. Haemophilia. 2022;28:950–961. doi: 10.1111/hae.14638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berntorp E. Immune tolerance induction in development. Blood. 2023;141:1901–1902. doi: 10.1182/blood.2022019465. [DOI] [PubMed] [Google Scholar]
- 100.Benson G., Auerswald G., Elezović I., Lambert T., Ljung R., Morfini M., Remor E., Salek S.Z. Immune tolerance induction in patients with severe hemophilia with inhibitors: expert panel views and recommendations for clinical practice. Eur. J. Haematol. 2012;88:371–379. doi: 10.1111/j.1600-0609.2012.01754.x. [DOI] [PubMed] [Google Scholar]
- 101.Malec L., Van Damme A., Chan A.K.C., Spasova M., Jain N., Sensinger C., Dumont J., Lethagen S., Carcao M., Peyvandi F. Recombinant factor VIII Fc fusion protein for first-time immune tolerance induction: final results of the verITI-8 study. Blood. 2023;141:1982–1989. doi: 10.1182/blood.2022017780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Escuriola Ettingshausen C., Vdovin V., Zozulya N., Svirin P., Andreeva T., Benedik-Dolničar M., Jiménez-Yuste V., Kitanovski L., Zupancic-Šalek S., Pavlova A., et al. Immune tolerance induction (ITI) with a pdFVIII/VWF concentrate (octanate) in 100 patients in the observational ITI (ObsITI) study. TH Open. 2022;6:e124–e134. doi: 10.1055/s-0042-1748756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oomen I., Camelo R.M., Rezende S.M., Voorberg J., Mancuso M.E., Oldenburg J., Carcao M., Matino D., Lillicrap D., Fischer K., et al. Determinants of successful immune tolerance induction in hemophilia A: systematic review and meta-analysis. Res. Pract. Thromb. Haemost. 2023;7 doi: 10.1016/j.rpth.2022.100020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Antun A., Monahan P.E., Manco-Johnson M.J., Callaghan M.U., Kanin M., Knoll C., Carpenter S.L., Davis J.A., Guerrera M.F., Kruse-Jarres R., et al. Inhibitor recurrence after immune tolerance induction: a multicenter retrospective cohort study. J. Thromb. Haemostasis. 2015;13:1980–1988. doi: 10.1111/jth.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang J., Yang D. Big stride in gene therapy for hemophilia B in China. Blood Sci. 2023;5:138–139. doi: 10.1097/bs9.0000000000000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaczmarek R., Miesbach W., Ozelo M.C., Chowdary P. Current and emerging gene therapies for haemophilia A and B. Haemophilia. 2024;30:12–20. doi: 10.1111/hae.14984. [DOI] [PubMed] [Google Scholar]
- 107.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu Z., Yang H., Colosi P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roberts S.A., Dong B., Firrman J.A., Moore A.R., Sang N., Xiao W. Engineering factor viii for hemophilia gene therapy. J. Genet. Syndr. Gene Ther. 2011;1:S1-006–S006. doi: 10.4172/2157-7412.S1-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bunting S., Zhang L., Xie L., Bullens S., Mahimkar R., Fong S., Sandza K., Harmon D., Yates B., Handyside B., et al. Gene therapy with BMN 270 results in therapeutic levels of FVIII in mice and primates and normalization of bleeding in hemophilic mice. Mol. Ther. 2018;26:496–509. doi: 10.1016/j.ymthe.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Anguela X.M., High K.A. Hemophilia B and gene therapy: a new chapter with etranacogene dezaparvovec. Blood Adv. 2024;8:1796–1803. doi: 10.1182/bloodadvances.2023010511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dhillon S. Fidanacogene elaparvovec: first approval. Drugs. 2024;84:479–486. doi: 10.1007/s40265-024-02017-4. [DOI] [PubMed] [Google Scholar]
- 113.Dobrowsky T., Gianni D., Pieracci J., Suh J. AAV manufacturing for clinical use: insights on current challenges from the upstream process perspective. Curr. Opin. Biomed. Eng. 2021;20 doi: 10.1016/j.cobme.2021.100353. [DOI] [Google Scholar]
- 114.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J., et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pipe S.W., Leebeek F.W.G., Recht M., Key N.S., Castaman G., Miesbach W., Lattimore S., Peerlinck K., Van der Valk P., Coppens M., et al. Gene therapy with etranacogene dezaparvovec for Hemophilia B. N. Engl. J. Med. 2023;388:706–718. doi: 10.1056/NEJMoa2211644. [DOI] [PubMed] [Google Scholar]
- 116.Fong S., Handyside B., Sihn C.R., Liu S., Zhang L., Xie L., Murphy R., Galicia N., Yates B., Minto W.C., et al. Induction of ER stress by an AAV5 BDD FVIII construct is dependent on the strength of the hepatic-specific promoter. Mol. Ther. Methods Clin. Dev. 2020;18:620–630. doi: 10.1016/j.omtm.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ozelo M.C., Mahlangu J., Pasi K.J., Giermasz A., Leavitt A.D., Laffan M., Symington E., Quon D.V., Wang J.D., Peerlinck K., et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N. Engl. J. Med. 2022;386:1013–1025. doi: 10.1056/NEJMoa2113708. [DOI] [PubMed] [Google Scholar]
- 118.Mahlangu J., Kaczmarek R., von Drygalski A., Shapiro S., Chou S.C., Ozelo M.C., Kenet G., Peyvandi F., Wang M., Madan B., et al. Two-year outcomes of valoctocogene roxaparvovec therapy for hemophilia A. N. Engl. J. Med. 2023;388:694–705. doi: 10.1056/NEJMoa2211075. [DOI] [PubMed] [Google Scholar]
- 119.Pasi K.J., Laffan M., Rangarajan S., Robinson T.M., Mitchell N., Lester W., Symington E., Madan B., Yang X., Kim B., et al. Persistence of haemostatic response following gene therapy with valoctocogene roxaparvovec in severe haemophilia A. Haemophilia. 2021;27:947–956. doi: 10.1111/hae.14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Klamroth R., Cuker A., Alzahrani H., Astermark J., Frenzel L., Katsarou O., Kavakli K., Matino D., Shapiro A., Wang J.D., et al. Efficacy and safety of fidanacogene elaparvovec in adults with moderately severe or severe hemophilia b: results from the phase 3 BENEGENE-2 gene therapy trial. Hämostaseologie. 2024;44:S81–S82. doi: 10.1055/s-0044-1779185. [DOI] [Google Scholar]
- 121.Miesbach W., Foster G.R., Peyvandi F. Liver-related aspects of gene therapy for hemophilia: need for collaborations with hepatologists. J. Thromb. Haemostasis. 2023;21:200–203. doi: 10.1016/j.jtha.2022.11.026. [DOI] [PubMed] [Google Scholar]
- 122.US Food and Drug Administration (2023). ROCTAVIAN (valoctocogene roxaparvovec-rvox) suspension for intravenous infusion. Package Insert.
- 123.US Food and Drug Administration (2022). HEMGENIX (etranacogene dezaparvovec-drlb) suspension, for intravenous infusion. Package Insert.
- 124.Pfizer Canada ULC. PrBEQVEZTM (fidanacogene elaparvovec): Canadian product monograph. (2023). https://health-products.canada.ca/dpd-bdpp/info?lang=eng&code=103268. Accessed 18 June 2024.
- 125.Klamroth R., Hayes G., Andreeva T., Gregg K., Suzuki T., Mitha I.H., Hardesty B., Shima M., Pollock T., Slev P., et al. Global seroprevalence of pre-existing immunity against AAV5 and other AAV serotypes in people with hemophilia A. Hum. Gene Ther. 2022;33:432–441. doi: 10.1089/hum.2021.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jiang M., Yang F., Jiang Y., Cheng L., Han J., Yi J., Zuo B., Huang L., Ma Z., Li T., et al. Safety and efficacy of an anti-human APC antibody for prophylaxis of congenital factor deficiencies in preclinical models. Blood. 2023;142:1071–1081. doi: 10.1182/blood.2023020005. [DOI] [PubMed] [Google Scholar]
- 127.Center for Drug Evaluation of the China National Medical Products Administration. CTR20241608. 2024. http://www.chinadrugtrials.org.cn/clinicaltrials.searchlist.dhtml
- 128.Centessa Pharmaceuticals. (2023). Centessa Pharmaceuticals Announces New Data from an Additional 52-Weeks of Continuous Treatment from Third Year (Part 5) of Ongoing Phase 2a Study of SerpinPC for the Treatment of Hemophilia. https://investors.centessa.com/news-releases/news-release-details/centessa-pharmaceuticals-announces-new-data-additional-52-weeks. Accessed 9 November 2024.
- 129.Wilcox D.A., Armant M., Malec L., Shi Q., Johnson B.D., Xu H., Du L.M., Duffy L.J., Bushman F.D., Jerkins J.H., et al. Gene therapy expressing platelet-derived factor viii for correction of hemophilia α with a history of inhibitors. Blood. 2023;142:2249. doi: 10.1182/blood-2023-172845. [DOI] [Google Scholar]
- 130.Be Biopharma. (2024). Be Biopharma Announces FDA Clearance of IND Application for BE-101 in Hemophilia B. https://be.bio/be-biopharma-announces-fda-clearance-of-ind-application-for-be-101-in-hemophilia-b/. Accessed 18 June 2024.
- 131.Young G., Srivastava A., Kavakli K., Ross C., Sathar J., You C.W., Tran H., Sun J., Wu R., Poloskey S., et al. Efficacy and safety of fitusiran prophylaxis in people with haemophilia A or haemophilia B with inhibitors (ATLAS-INH): a multicentre, open-label, randomised phase 3 trial. Lancet. 2023;401:1427–1437. doi: 10.1016/s0140-6736(23)00284-2. [DOI] [PubMed] [Google Scholar]
- 132.Srivastava A., Rangarajan S., Kavakli K., Klamroth R., Kenet G., Khoo L., You C.W., Xu W., Malan N., Frenzel L., et al. Fitusiran prophylaxis in people with severe haemophilia A or haemophilia B without inhibitors (ATLAS-A/B): a multicentre, open-label, randomised, phase 3 trial. Lancet. Haematol. 2023;10:e322–e332. doi: 10.1016/s2352-3026(23)00037-6. [DOI] [PubMed] [Google Scholar]
- 133.Mancuso M.E., Ingham S.J.M., Kunze M. Befovacimab, an anti-tissue factor pathway inhibitor antibody: Early termination of the multiple-dose, dose-escalating Phase 2 study due to thrombosis. Haemophilia. 2022;28:702–712. doi: 10.1111/hae.14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dahlbäck B., Villoutreix B.O. Regulation of blood coagulation by the protein C anticoagulant pathway: novel insights into structure-function relationships and molecular recognition. Arterioscler. Thromb. Vasc. Biol. 2005;25:1311–1320. doi: 10.1161/01.Atv.0000168421.13467.82. [DOI] [PubMed] [Google Scholar]
- 135.Baglin T., Koch A., Mocanu I., Makhaldiani L., Huntington J.A. Serpinpc in persons with severe hemophilia (PwH): updated results from a multi-center, multi-part, first-in-human study. Blood. 2022;140:460–461. doi: 10.1182/blood-2022-159631. [DOI] [Google Scholar]
- 136.Tang Q., Khvorova A. RNAi-based drug design: considerations and future directions. Nat. Rev. Drug Discov. 2024;23:341–364. doi: 10.1038/s41573-024-00912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Prince R.E., Schaeper U., Dames S., Calzavarini S., Quarroz C., Reina Caro M.D., Chanfon Bätzner A., Löffner K., Eisermann M., Angelillo-Scherrer A. Targeting protein s using small interfering RNA is well tolerated and protects mice with hemophilia a from acute hemarthrosis. Blood. 2020;136:20–21. doi: 10.1182/blood-2020-138692. [DOI] [Google Scholar]
- 138.Lin W.Y., Zhu R., Zhang Z., Lu X., Wang H., He W., Hu Y., Tang L. RNAi targeting heparin cofactor II promotes hemostasis in hemophilia A. Mol. Ther. Nucleic Acids. 2021;24:658–668. doi: 10.1016/j.omtn.2021.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hemophilia News Today (2024). Fitusiran reviewed for hemophilia A, B, with or without inhibitors. https://hemophilianewstoday.com/news/fda-reviewing-fitusiran-application-hemophilia-a-b-with-without-inhibitors/. Accessed 9 November 2024.
- 140.Mancuso M.E., Mahlangu J.N., Pipe S.W. The changing treatment landscape in haemophilia: from standard half-life clotting factor concentrates to gene editing. Lancet. 2021;397:630–640. doi: 10.1016/s0140-6736(20)32722-7. [DOI] [PubMed] [Google Scholar]
- 141.Hazendonk H.C.A.M., van Moort I., Mathôt R.A.A., Fijnvandraat K., Leebeek F.W.G., Collins P.W., Cnossen M.H., OPTI-CLOT study group Setting the stage for individualized therapy in hemophilia: What role can pharmacokinetics play? Blood Rev. 2018;32:265–271. doi: 10.1016/j.blre.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 142.Poon M.C., Lee A. Individualized prophylaxis for optimizing hemophilia care: can we apply this to both developed and developing nations? Thromb. J. 2016;14:32. doi: 10.1186/s12959-016-0096-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dirzu N., Hotea I., Jitaru C., Brinza M., Urian L., Peters M.C., Gal K., Popescu L., Blag C., Marian M., et al. Mobile health technology for the personalized therapy of hemophilia. Front. Med. 2021;8 doi: 10.3389/fmed.2021.711973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.