

Abstract
INTRODUCTION:
The discovery of the antide-pressant effects of ketamine is arguably the most important advance in mental health in decades. Given ketamine’s rapid and potent antidepressant activity, a great challenge in neuroscience is to understand its direct brain target(s), both at the molecular and neural circuit levels. At the molecular level, ketamine’s primary target must be a molecule that directly interacts with ketamine. A strong candidate that has the highest affinity for ketamine and has been strongly implicated in ketamine’s anti-depressant action is the N-methyl-D-aspartate receptor (NMDAR). At the neural circuit level, because NMDAR is ubiquitously expressed in the brain, it was unclear whether ketamine simultaneously acts on many brain regions or specifically on one or a few primary site(s) that sets off its antidepressant signaling cascade.
RATIONALE:
We reasoned that the primary regional target of ketamine should show an immediate response to ketamine. Specifically, if ketamine’s direct molecular target is NMDAR, then its direct regional target should be the one in which systemic ketamine treatment inhibits its NMDARs most rapidly. One clue for a possible mechanism of brain region selectivity comes from a biophysical property of ketamine: As a use-dependent NMDAR open-channel blocker, ketamine may act most potently in a brain region(s) with a high level of basal activity and consequently more NMDARs in the open state. In several whole-brain–based screens in animal models of depression, the lateral habenula (LHb), which is known as the brain’s “anti-reward center,” has stood out as one of the very few brain regions that show hyperactivity. Previously, we and others have shown that under a depressive-like state, LHb neurons are hyperactive and undergo NMDAR dependent burst firing, indicating that the LHb is a strong candidate for being ketamine’s primary regional target.
RESULTS:
In the present study, using in vitro slice electrophysiology, we found that a single systemic injection of ketamine in depressive-like mice, but not naïve mice, specifically blocked NMDAR currents in LHb neurons, but not in hippocampal CA1 neurons. In vivo tetrode recording revealed that the basal firing rate and bursting rate were much higher in LHb neurons than in CA1 neurons. LHb neural activity was significantly suppressed within minutes after systemic ketamine treatment, preceding the increase of serotonin in the hippocampus. By increasing the intrinsic activity of CA1 neurons or decreasing the activity of LHb neurons, we were able to swap their sensitivity to ketamine blockade. LHb neurons also had a smaller extrasynaptic NMDAR reservoir pool and thus recovered more slowly from ketamine blockade. Furthermore, conditional knockout of the NMDAR subunit NR1 locally in the LHb occluded ketamine’s antidepressant effects and blocked the systemic ketamine-induced increase of serotonin and brain-derived neurotrophic factor in the hippocampus.
CONCLUSION:
Collectively, these results reveal that ketamine blocks NMDARs in vivo in a brain region– and depression state–specific manner. The use-dependent nature of ketamine as an NMDAR blocker converges with local brain region properties to distinguish the LHb as a primary brain target of ketamine action. Both the ongoing neural activity and the size of the extrasynaptic NMDAR reservoir pool contribute to the region-specific effects. Therefore, we suggest that neurons in different brain regions may be recruited at different stages, and that an LHb-NMDAR–dependent event likely occurs more upstream, in the cascade of ketamine signaling in vivo. By identifying the cross-talk from the LHb to the hippocampus and delineating the primary versus secondary effects, the present work may provide a more unified understanding of the complex results from previous studies on the anti-depressant effects of ketamine and aid in the design of more precise and efficient treatments for depression.
Graphical Abstract
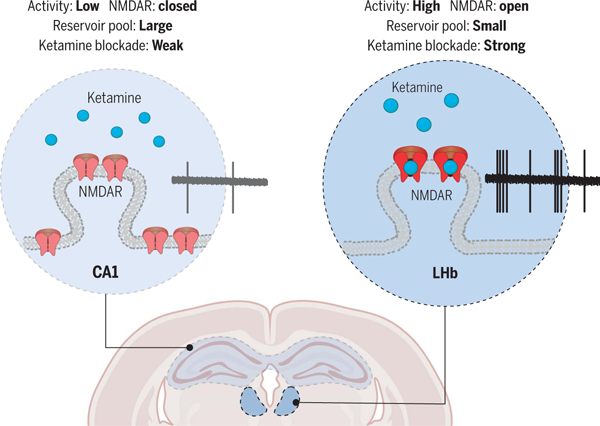
Brain region–specific action of ketamine. Model illustrating why systemic ketamine specifically blocks NMDARs in LHb neurons, but not in hippocampal CA1 pyramidal neurons, in depressive-like mice. This regional specificity depends on the use-dependent nature of ketamine as a channel blocker, local neural activity, and the extrasynaptic reservoir pool size of NMDARs.
Ketamine has been found to have rapid and potent antidepressant activity. However, despite the ubiquitous brain expression of its molecular target, the N-methyl-d-aspartate receptor (NMDAR), it was not clear whether there is a selective, primary site for ketamine’s antidepressant action. We found that ketamine injection in depressive-like mice specifically blocks NMDARs in lateral habenular (LHb) neurons, but not in hippocampal pyramidal neurons. This regional specificity depended on the use-dependent nature of ketamine as a channel blocker, local neural activity, and the extrasynaptic reservoir pool size of NMDARs. Activating hippocampal or inactivating LHb neurons swapped their ketamine sensitivity. Conditional knockout of NMDARs in the LHb occluded ketamine’s antidepressant effects and blocked the systemic ketamine–induced elevation of serotonin and brain-derived neurotrophic factor in the hippocampus. This distinction of the primary versus secondary brain target(s) of ketamine should help with the design of more precise and efficient antidepressant treatments.
Given ketamine’s rapid and potent anti-depressant activity and low addiction liability (1–4), a challenge in neuroscience is to understand its direct brain target(s) at both themolecular and neural circuit levels. At the molecular level, a few key molecules that can mediate ketamine’s effect have been identified, including brain-derived neurotrophic factor (BDNF) (5, 6), AMPAR (7–9), mammalian target of rapamycin (mTOR) (10), the potassium channel Kcnq2 (11), the translation initiation factor 4E-binding proteins (4E-BPs) (12), Menin (13), and microglia-related inflammatory cytokines (14, 15). However, because these molecules do not bind to ketamine, they cannot be its direct target and are most likely important downstream players in the signaling pathways. The initiator of the signaling cascade must be a molecule that directly interacts with ketamine. One such molecule that has been strongly implicated in ketamine’s antidepressant function is the N-methyl-D-aspartate receptor (NMDAR) (16–26).
At the neural circuit level, we reasoned that the primary regional target should show an immediate response to ketamine. Specifically, if ketamine’s direct molecular target is NMDAR, then its direct regional target should be the one in which systemic ketamine treatment most rapidly inhibits its NMDARs. Several brain regions have been heavily studied for ketamine’s anti-depressant effects. Many studies have focused on the hippocampus (5, 7, 12, 23, 27, 28) or the medial prefrontal cortex (mPFC) (10, 29, 30). Recent work also reveals a new player in ketamine’s antidepressant action, the lateral habenula (LHb), which is known as the brain’s “anti-reward center” (31–36). However, it is not clear whether these different regions all respond to ketamine at the initial stage or if they are recruited at different stages of ketamine action.
Despite the ubiquitous expression of NMDARs, one clue for a possible mechanism for selectivity comes from a biophysical property of ketamine: As a use-dependent NMDAR blocker (37, 38), ketamine may act most potently in a brain region(s) with a high level of basal activity and consequently more open NMDARs (33). Indeed, in several whole-brain-based screens in multiple animal models of depression, the LHb has stood out as the only (32, 39) or one of the very few (40) brain regions showing hyperactivity. Previously, we and others have shown that LHb neurons are activated and undergo NMDAR-dependent burst firing in a depressive-like state (31, 41–43). We thus set out to investigate the NMDAR responses of the LHb and hippocampal neurons in depressive-like mice after systemic ketamine administration.
Results
Systemic ketamine injection in depressive-like mice specifically inhibits NMDAR currents in LHb but not pyramidal neurons in hippocampal CA1
We exposed C57BL/6 mice to chronic restraint stress (CRS), a model used to induce a depressive-like state, injected mice with either saline or ketamine [10 mg/kg intraperitoneally (i.p.)], and then measured NMDAR-mediated synaptic currents in the LHb and hippocampal CA1 neurons (Fig. 1A). Because most LHb neurons are glutamatergic (44), and neurons co-expressing GABA biosynthesis enzymes (gad1/ gad2) and vesicular GABA transporter (vgat) only constitute a very small proportion (<0.8%) (45, 46), we did not distinguish neuron types in the LHb (Fig. 1, B to G). In the hippocampus, we focused on the pyramidal (PYR) neurons, which are the major output neurons of this region and can be distinguished on the basis of their morphological and electrophysiological properties (see the materials and methods) (Fig. 1, H to M). One hour after ketamine injection, brain slices were prepared, recovered, and then underwent whole-cell patch-clamp recording in ketamine-free artificial cerebrospinal fluid (ACSF) solutions (Fig. 1A). NMDAR- and AMPAR-mediated excitatory postsynaptic currents (NMDAR-eEPSCs and AMPAR-eEPSCs, respectively) were isolated on the basis of their temporal characteristics (see the materials and methods) from both saline- and ketamine-injected mice (Fig. 1, C and I). In LHb neurons of CRS mice, the ketamine-injected group showed strongly reduced amplitudes of NMDAR-eEPSCs across a range of stimulation intensities, with the reduction as large as 74% at 1.5-mA stimulation intensity (saline, 49.4 ± 11.8 pA; ketamine, 12.7 ± 2.7 pA; P = 0.002, Mann-Whitney test; Fig. 1, D and F). This continued blockade of NMDAR-eEPSCs from brain slices that had been recovered and recorded for hours in the ketamine-free ACSF was attributed to the use-dependent trapping of ketamine in the NMDAR channel (37, 47, 48). Western blot analysis revealed that the surface level of NR1, the obligatory subunit of NMDAR, was unchanged after ketamine treatment in the habenula (fig. S1C), suggesting that the suppression of NMDAR-eEPSCs was unlikely to have been caused by NMDAR endocytosis. The amplitude of AMPAR-eEPSCs, despite an insignificant trend to increase, was not different between the saline- and ketamine-treated groups (saline, 181.3 ± 34.2 pA; ketamine, 243.5 ± 48.2 pA; P = 0.36, Mann-Whitney test; Fig. 1E and fig. S1A). Correspondingly, there was a significant decrease in the NMDA/AMPA ratios in LHb neurons of CRS mice after ketamine administration (saline, 0.45 ± 0.12; ketamine, 0.09 ± 0.02; P = 0.002, Mann-Whitney test; Fig. 1G).
Fig. 1. Systemic ketamine injection in depressive-like mice specifically inhibits NMDAR currents in LHb neurons, but not hippocampal CA1 PYR neurons.
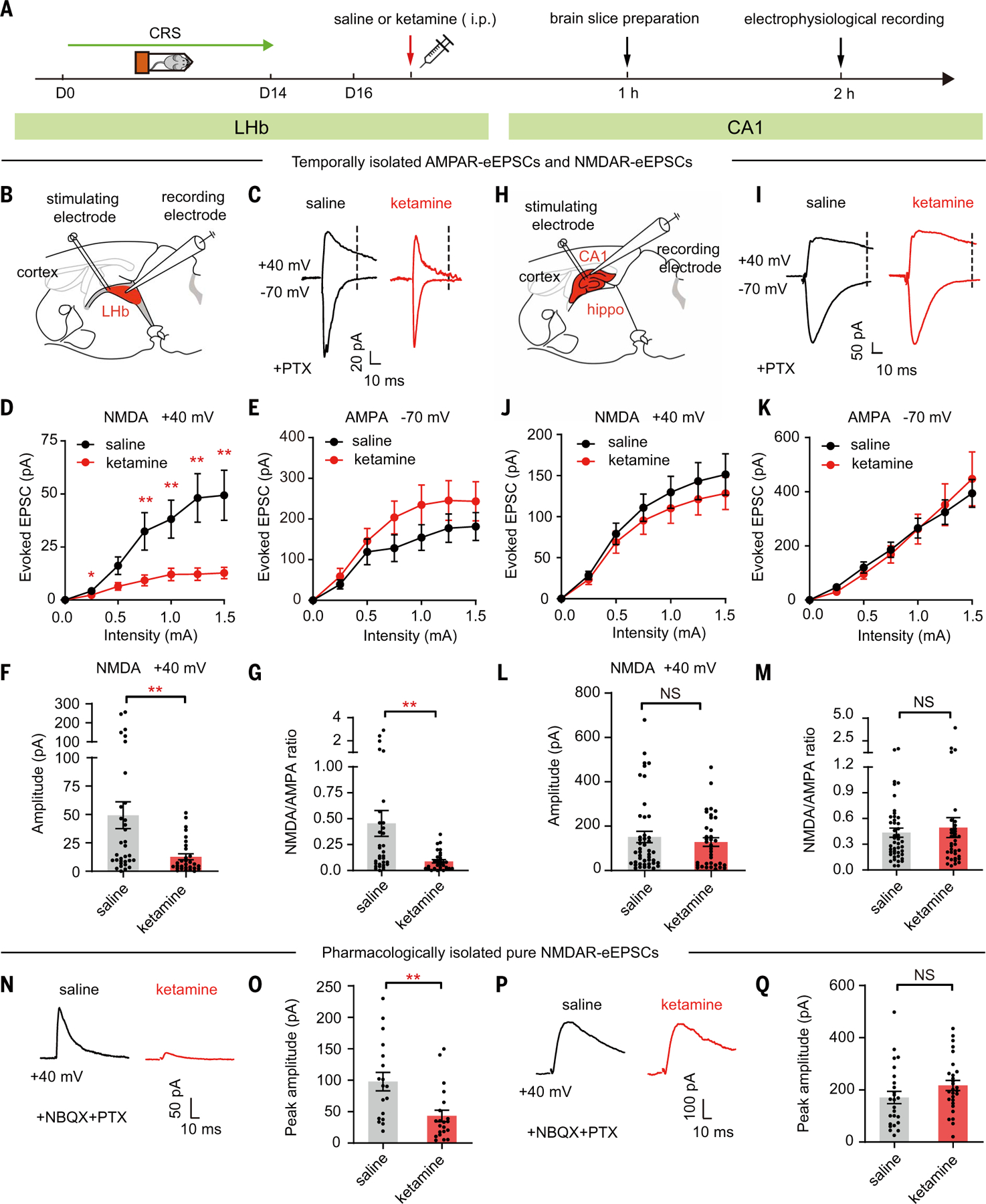
(A) Experimental paradigm. Intraperitoneal injection of ketamine (10 mg/kg) in CRS mice. (B and H) Schematic of the whole-cell recording of evoked responses (eEPSCs) in LHb (B) and hippocampal CA1 (H) slices. Hippo, hippocampus. (C and I) AMPAR-eEPSCs (–70 mV, measured at the peak) and NMDAR-eEPSCs (+40 mV, measured at 35 ms after stimulation in LHb neurons and at 60 ms after stimulation in CA1 neurons, dotted lines) in LHb (C) and CA1 PYR (I) neurons in presence of PTX. (D and J) Stimulus-response (input-output) curves of NMDAR-eEPSCs of LHb neurons (D) and CA1 PYR neurons (J). (E and K) Stimulus-response curves of AMPAR-eEPSCs of LHb neurons (E) and CA1 PYR neurons (K). (F and L) Bar graphs of NMDAR-eEPSCs of LHb neurons [P = 0.002, Mann-Whitney test (F)] and CA1 PYR neurons [P = 0.73, Mann-Whitney test (L)] recorded at 1.5-mA stimulation intensity. (G and M) Ratios of NMDAR-eEPSCs and AMPAR-eEPSCs recorded at 1.5-mA stimulation intensity in LHb neurons [P = 0.002, Mann-Whitney test (G)] and CA1 PYR neurons [P = 0.56, Mann-Whitney test (M)]. n = 33 cells in four mice in the saline group and n = 30 cells in three mice in the ketamine group for LHb data; n = 45 cells in five mice in the saline group and n = 36 cells in our mice in the ketamine group for CA1 data in (B) to (M). (N and P) NMDAR-eEPSCs (+40 mV, measured at the peak) in LHb neurons (N) and CA1 PYR neurons (P) in the presence of PTX and NBQX in brain slices prepared 1 hour after intraperitoneal injection of saline or ketamine in CRS mice. (O and Q) Bar graphs of NMDAR-eEPSCs of LHb neurons [P = 0.001, Mann-Whitney test (O)] and CA1 PYR neurons [P = 0.08, Mann-Whitney test (Q)] at 1.5-mA stimulation intensity in brain slices prepared 1 hour after intraperitoneal injection of saline or ketamine in CRS mice. n = 18 cells in two mice in the saline group and n = 21 cells in two mice in the ketamine group for LHb data; n = 25 cells in three mice in saline group and n = 28 cells in three mice in the ketamine group for CA1 data in (N) to (Q). *P < 0.05; **P < 0.01; NS, not significant. Error bars indicate SEM.
By contrast, ketamine did not reduce the NMDAR-eEPSCs in the PYR neurons of CA1 brain slices (Fig. 1, H to M). The NMDAR-eEPSCs (Fig. 1, J and L), AMPAR-eEPSCs (Fig. 1K and fig. S1B), and the NMDA/AMPA ratios (Fig. 1M) were indistinguishable between the saline- and ketamine-treated mice in CA1-PYR neurons. We also pharmacologically isolated the pure NMDAR currents in the presence of both the GABAAR blocker picrotoxin (PTX) and the AMPAR blocker 2,3-dioxo-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX), and confirmed that 1 hour after intraperitoneal ketamine injection in CRS mice, NMDAR-eEPSCs were specifically inhibited in the LHb, but not in hippocampal CA1 PYR neurons (Fig. 1, N to Q).
Systemic ketamine injection in depressive-like mice rapidly inhibits the activity of LHb but not CA1 neurons
To understand the basis of the brain region– specific ketamine effect, we compared the basal in vivo neural activity of LHb and hippocampal CA1 in depressive-like mice (Fig. 2, A to G, and fig. S2). Using movable tetrodes, we recorded the spontaneous neural activity of 239 LHb neurons and 147 PYR neurons in the CA1 of CRS mice (Fig. 2, D to G; see the materials and methods for classification of PYR neurons). The spontaneous firing rate (FR) of LHb neurons was 6.3-fold higher than that of PYR neurons in hippocampal CA1 (LHb, 5.7 ± 0.50 Hz; CA1, 0.90 ± 0.07 Hz; P < 0.0001, Mann-Whitney test; Fig. 2F). The bursting FR (i.e., the bursting spike number per second) was even higher in the LHb, 8-fold higher than that in hippocampal CA1 (LHb, 2.5 ± 0.4 Hz; CA1, 0.31 ± 0.03 Hz; P < 0.0001, Mann-Whitney test; Fig. 2G).
Fig. 2. Systemic ketamine injection in depressive-like mice rapidly inhibits the activity of LHb neurons, but not hippocampal CA1 PYR neurons, in vivo.
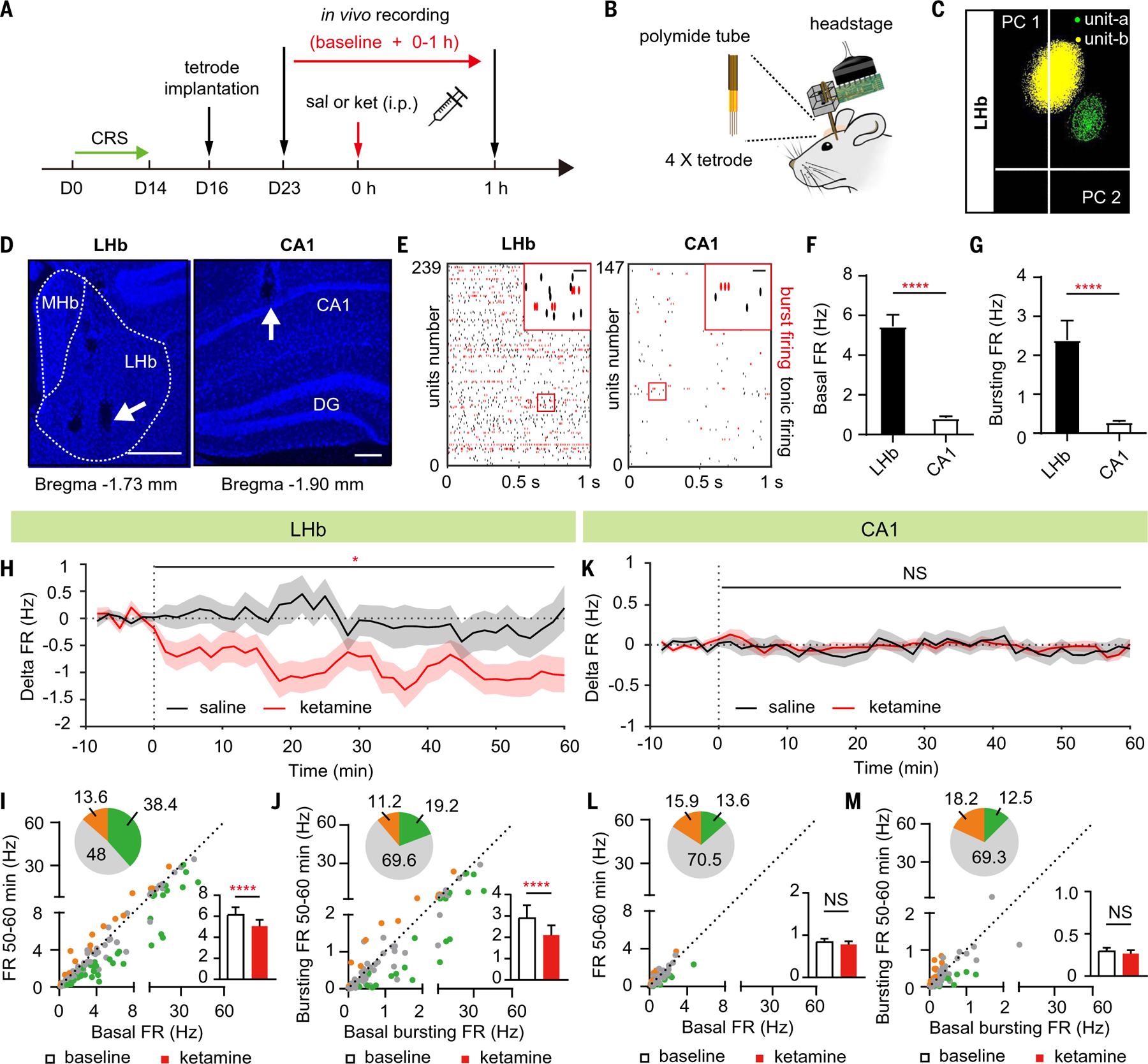
(A) Experimental paradigm for in vivo recording after intraperitoneal injection of saline or ketamine (10 mg/kg) in CRS mice. (B) Illustration of in vivo tetrode recording. (C) Principal component analysis clustering display of two well-isolated single units in LHb (yellow and green clusters). (D) Example recording sites stained with DAPI. White dotted lines demarcate the medial habenula (MHb) and LHb. White arrows indicate tetrode tracks. Scale bar, 200 µm. DG, dentate gyrus. (E) Raster of sample basal firing of all recorded LHb and CA1 PYR neurons (red indicates bursting firing). Top right: magnified images of the red square area on the left. Scale bar, 20 ms. (F and G) Bar graphs illustrating the basal FR (spike number per second) [P < 0.0001, Mann-Whitney test (F)] and bursting FR (bursting spike number per second) [P < 0.0001, Mann-Whitney test (G)] in LHb neurons and CA1 PYR neurons. n = 239 units in 15 mice in LHb; n = 147 units in 10 mice in CA1. (H and K) Delta firing rate (FRreal time – FRbaseline) in LHb neurons (H) and CA1 PYR neurons (K) after intraperitoneal injection of saline or ketamine (10 mg/kg) in CRS mice. Bin: 100 s. (I and L) Scatter plots of the FR of recorded LHb neurons (I) and CA1 PYR neurons (L) at baseline state plotted against FR at 50 to 60 min after intraperitoneal injection of ketamine. Green, gray, and orange dots indicate neurons showing significant inhibition, no change, and significant increase of FR, respectively. Pie graphs show the percentage of inhibited (green), excited (orange), and unchanged (gray) units. Bar graphs show the FR in LHb neurons [P < 0.0001, Wilcoxon matched-pairs test (I)] and CA1 PYR neurons [P = 0.43, Wilcoxon matched-pairs test (L)] in CRS mice at 50 to 60 min after intraperitoneal injection of ketamine. (J and M) Scatter plots of the bursting FR of recorded LHb neurons (J) and CA1 PYR neurons (M) at baseline state plotted against bursting FR at 50 to 60 min after intraperitoneal injection of ketamine. Pie graphs show the percentage of inhibited (green), excited (orange), and unchanged (gray) units. Bar graphs illustrate the bursting FR in LHb neurons [P < 0.0001, Wilcoxon matched-pairs test (J)] and CA1 PYR neurons [P = 0.47, Wilcoxon matched-pairs test (M)] in CRS mice at 50 to 60 min after intraperitoneal injection of ketamine. (H) to (M), n = 114 cells in 14 mice in saline group and n = 125 cells in 14 mice in the ketamine group in LHb; n = 59 cells in eight mice in the saline group and n = 88 cells in eight mice in the ketamine group in CA1. *P < 0.05; ****P < 0.0001; NS, not significant. Error bars indicate SEM.
In the slice experiments shown in Fig. 1, although neurons were recorded as early as 2 hours after intraperitoneal ketamine injection, one may still argue that other changes could have occurred within this time window to indirectly cause the observed differences. Therefore, we monitored the change of neural activity in vivo in CRS mice immediately after ketamine injection and studied its effects (Fig. 2, H to M). In the LHb, there was a rapid suppression of both the FR and bursting FR as soon as 0 to 10 min after ketamine injection (Fig. 2H and fig. S3, A and B), and this suppression lasted for at least 1 hour (Fig. 2, H to J). In the CA1, there was no significant change in either rate within 1 hour after drug injection (Fig. 2, K to M). In addition, within the LHb, proportionally more neurons with higher basal bursting FR were inhibited by ketamine (fig. S3D). For example, 0 to 10 min after ketamine injection, a majority (64.3%, 9/14) of ketamine-inhibited (bursting FR) neurons had basal bursting FR higher than 2 Hz (fig. S3D), even though the latter only constituted 21% (49/239) of total recorded neurons (fig. S3C).
Systemic ketamine injection inhibits NMDAR currents and neuronal activity in LHb of depressive-like mice but not naïve mice
Because LHb neural activity is lower in naïve mice (31, 42, 49), we investigated whether ketamine injection would inhibit LHb NMDARs in naïve mice equally well as in the depressive-like state (Fig. 3). First, we found significantly larger NMDAR-eEPSCs in LHb in depressive-like CRS mice at 1.5-mA stimulation intensity (CRS, 36.8 ± 4.2 pA; naïve, 15.9 ± 4.0 pA; P < 0.0001, Mann-Whitney test; Fig. 3, A to C) than in naïve mice. A large percentage (62.5%) of LHb neurons in naïve mice showed undetectable NMDAR responses (<10 pA), but this percentage was significantly lower in CRS mice (30.1%, P < 0.0001, chi-square test). By contrast, there was no significant difference in the LHb AMPAR-eEPSCs (Fig. 3, D and E) between the naïve and depressive-like state. Correspondingly, there were significantly larger NMDA/AMPA ratios in LHb neurons of depressive-like mice (CRS, 0.26 ± 0.04; naïve, 0.10 ± 0.02; P = 0.0006, Mann-Whitney test; Fig. 3F). Western blot analysis confirmed that membrane NR1 was up-regulated in the habenula of CRS mice (Fig. 3G). This potentiation of LHb NMDARs after depression may be explained by a recent study finding that LHb-NMDAR transmission can undergo burst-driven long-term potentiation (50) because depression induces burst firing of LHb neurons (31). The severity of depressive-like behaviors, as measured by the immobility duration in the forced swimming test (FST), which models behavioral despair or a decrease in motivated behavior, showed a strong positive correlation with the amplitude of LHb NMDAR-eEPSCs (R2 = 0.46, P = 0.004; Fig. 3H).
Fig. 3. Systemic ketamine injection inhibits NMDAR currents and neuronal activity in LHb of depressive-like, but not naïve mice.
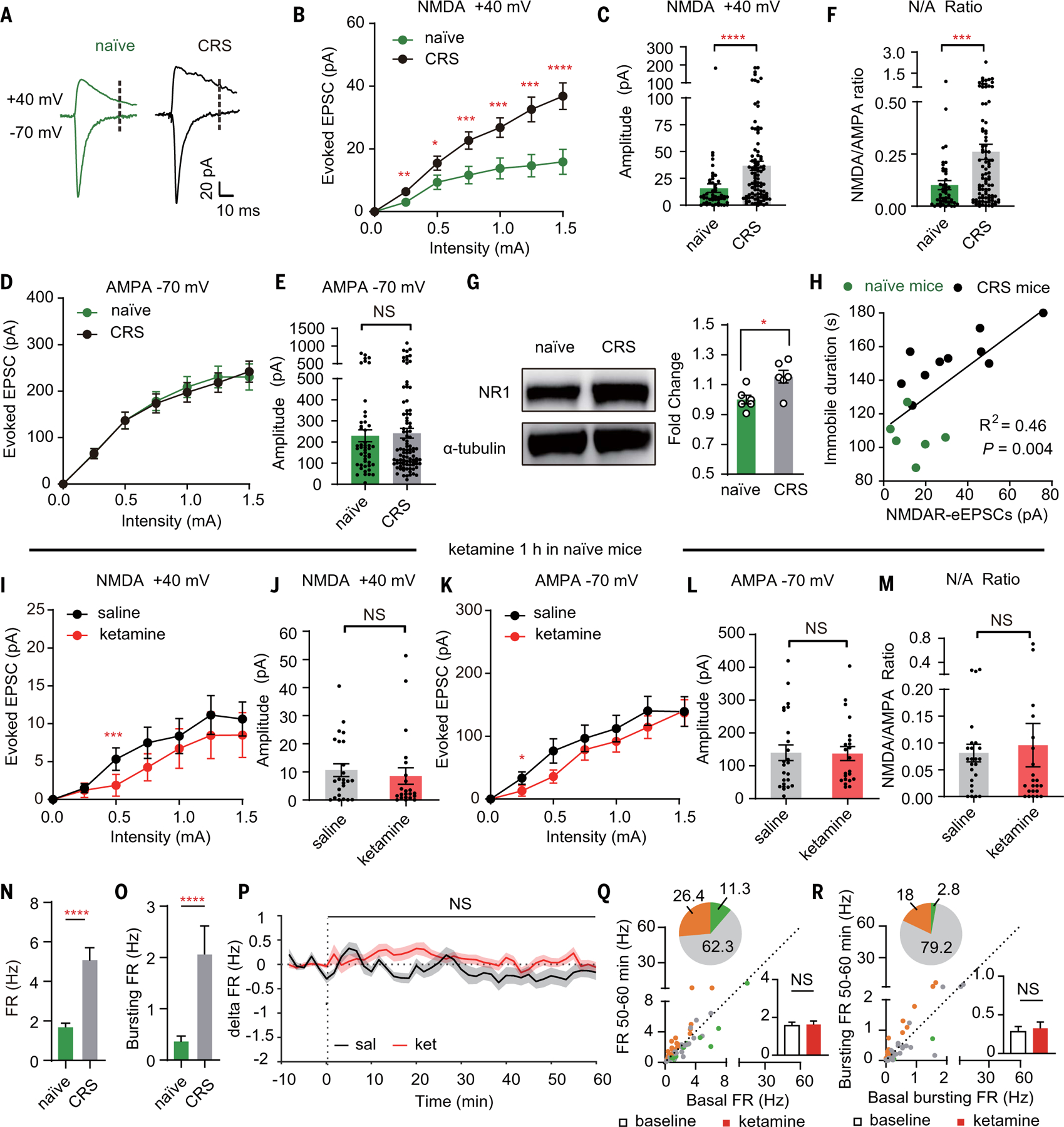
(A) AMPAR-eEPSCs (–70 mV, measured at the peak) and NMDAR-eEPSCs (+40 mV, measured at 35 ms after stimulation, dotted lines) in the presence of PTX in LHb neurons in naïve or CRS mice. (B) Stimulus-response curves of NMDAR-eEPSCs of LHb neurons in naïve or CRS mice. (C) Bar graphs of NMDAR-eEPSCs (P < 0.0001, Mann-Whitney test) of LHb neurons at 1.5-mA stimulation intensity in naïve or CRS mice. (D) Stimulus-response curves of AMPAR-eEPSCs of LHb neurons in naïve or CRS mice. (E) Bar graphs of AMPAR-eEPSCs (P = 0.92, Mann-Whitney test) of LHb neurons at 1.5-mA stimulation intensity in naïve or CRS mice. (F) Bar graphs of ratios of NMDAR-eEPSCs and AMPAR-eEPSCs (P = 0.0006, Mann-Whitney test) of LHb neurons at 1.5-mA stimulation intensity in naïve or CRS mice. (G) Western blot analysis showing up-regulation of NR1 protein in the membrane fraction of the habenula of CRS mice (P = 0.02, n = 6, 6). Tubulin was used as a loading control. (H) Correlation between averaged NMDAR-eEPSCs of recorded LHb neurons and immobile duration in FST (R2 = 0.46, black line; P = 0.004, linear regression test). Green indicates naïve mice (n = 6); black indicates CRS mice (n = 10). NMDAR-eEPSCs are averaged by all recorded LHb neurons in one animal. In (A) to (F) and (H), n = 48 cells in six naïve mice and 93 cells in 10 CRS mice. (I and K) Stimulus-response curves of NMDAR-eEPSCs (I) and AMPAR-eEPSCs (K) of LHb neurons in brain slices prepared 1 hour after intraperitoneal injection of saline or ketamine (10 mg/kg) in naïve mice. (J, L, and M) Bar graphs of NMDAR-eEPSCs [P = 0.28, Mann-Whitney test (J)] and AMPAR-eEPSCs [P = 0.84, Mann-Whitney test (L)], ratios of NMDAR-eEPSCs and AMPAR-eEPSCs [P = 0.31, Mann-Whitney test (M)] of LHb neurons at 1.5-mA stimulation intensity in brain slices prepared 1 hour after intraperitoneal injection of saline or ketamine in naïve mice. In (I) to (M), n = 24 cells in three mice in the saline group and n = 22 cells in two mice in the ketamine group. (N and O) Bar graphs illustrating the FR [P < 0.0001, Mann-Whitney test (N)] and bursting FR [P < 0.0001, Mann-Whitney test (O)] of LHb neurons in naïve or CRS mice. n = 108 cells in six mice in the naïve group and n = 114 cells in 14 mice in the CRS group. (P) Delta firing rate (FRreal time – FRbaseline) in LHb neurons after intraperitoneal injection of saline or ketamine (10 mg/kg) in naïve mice. Bin: 100 s. (Q and R) Scatter plots of the FR (Q) and bursting FR (R) of recorded LHb neurons at the baseline state plotted against FR or bursting FR at 50 to 60 min after intraperitoneal injection of ketamine. Green, gray, and orange dots indicate neurons showing significant inhibition, no change, and significant increase of FR or bursting FR, respectively. Pie graphs show the percentage of inhibited (green), excited (orange), and unchanged (gray) units. Bar graphs show the FR [P = 0.20, Wilcoxon matched-pairs test (Q)] or bursting FR [P = 0.36, Wilcoxon matched-pairs test (R)] of LHb neurons in naïve mice 50 to 60 min after intraperitoneal injection of ketamine. In (P) to (R), n = 108 cells in six mice in the saline group and n = 106 cells in six mice in the ketamine group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, not significant. Error bars indicate SEM.
To test whether ketamine injection inhibits LHb-NMDAR currents in naïve mice, we intra-peritoneally injected ketamine or saline, measured LHb synaptic responses, and found no significant difference in LHb NMDAR-eEPSCs, AMPAR-eEPSCs, or NMDA/AMPA ratios between saline- and ketamine-injected mice (Fig. 3, I to M). In vivo tetrode recording showed that the overall FR and bursting FR in the LHb were significantly higher in the CRS mice than in the naïve mice (Fig. 3, N and O), consistent with previous reports of LHb hyperactivity under a depressive-like state (31, 42). One hour after ketamine injection, the bursting activity, as well as the overall spiking activity, were decreased in the LHb of CRS mice, but not of naïve mice (Figs. 2, H to J, and 3, P to R).
Activation of hippocampal CA1 neurons increases ketamine-induced suppression of NMDARs
The above results suggest that high local endogenous neural activity may be required for ketamine to inhibit the NMDARs. To causally test this hypothesis, we used two strategies to induce neuronal activation in CA1 PYR neurons and examined the inhibitory effects of systemic ketamine on their NMDARs. First, we subjected naïve mice to a contextual fear condition learning paradigm, which elevates hippocampal neural activity (51, 52). Mice were foot shocked (five shocks within 5 min) in a novel context 5 min after intraperitoneal ketamine injection, and hippocampal brain slices were obtained 50 min afterward for whole-cell patch-clamp recording (fig. S4A). Under this condition, there was a significant decrease in CA1 NMDAR-eEPSCs in ketamine-treated mice compared with saline-treated mice (fig. S4B). However, the CA1 AMPAR-eEPSCs also showed a decrease in ketamine-treated mice, which was significant at strong stimulation intensity (fig. S4C). This concomitant change makes it difficult to dissociate whether the decreases were caused by a reduction in presynaptic release (which affects both NMDAR-eEPSCs and AMPAR-eEPSCs) or by blockade of both postsynaptic NMDARs and AMPARs.
To clarify the above results, we tested a second strategy, selectively activating a subpopulation of CA1 neurons by expressing the chemogenetic excitatory hM3D (53). We expressed AAV-hM3D-mCherry virus in the CA1 region and intraperitoneally injected mice in a novel context with clozapine-N-oxide (CNO), an exogenous ligand for hM3D, 30 min before injection of saline or ketamine (Fig. 4, A and E, figure legend, and the materials and methods). CNO effectively depolarized and induced activation of mCherry-positive (hM3D+) CA1 neurons (Fig. 4, B to D). One hour after saline or ketamine injection, hippocampal brain slices were prepared and whole-cell patch recordings were performed at the border of the viral injection site on pairs of mCherry-negative (hM3D–) and hM3D+ PYR neurons (Fig. 4, A and E to G). In ketamine-injected mice, NMDAR-eEPSCs showed a significantly smaller amplitude in hM3D+ neurons (P = 0.003, Wilcoxon matched-pairs test; Fig. 4H), whereas in saline-injected mice, hM3D+ and hM3D– CA1 neurons showed a similar amplitude of NMDAR-eEPSCs (P = 0.20, Wilcoxon matched-pairs test; Fig. 4I). By contrast, AMPAR-eEPSCs were indistinguishable between hM3D+ and hM3D– neurons in both saline- and ketamine-injected mice (Fig. 4, H and I).
Fig. 4. Activation of CA1 and inhibition of LHb swap their sensitivity to ketamine blockade.
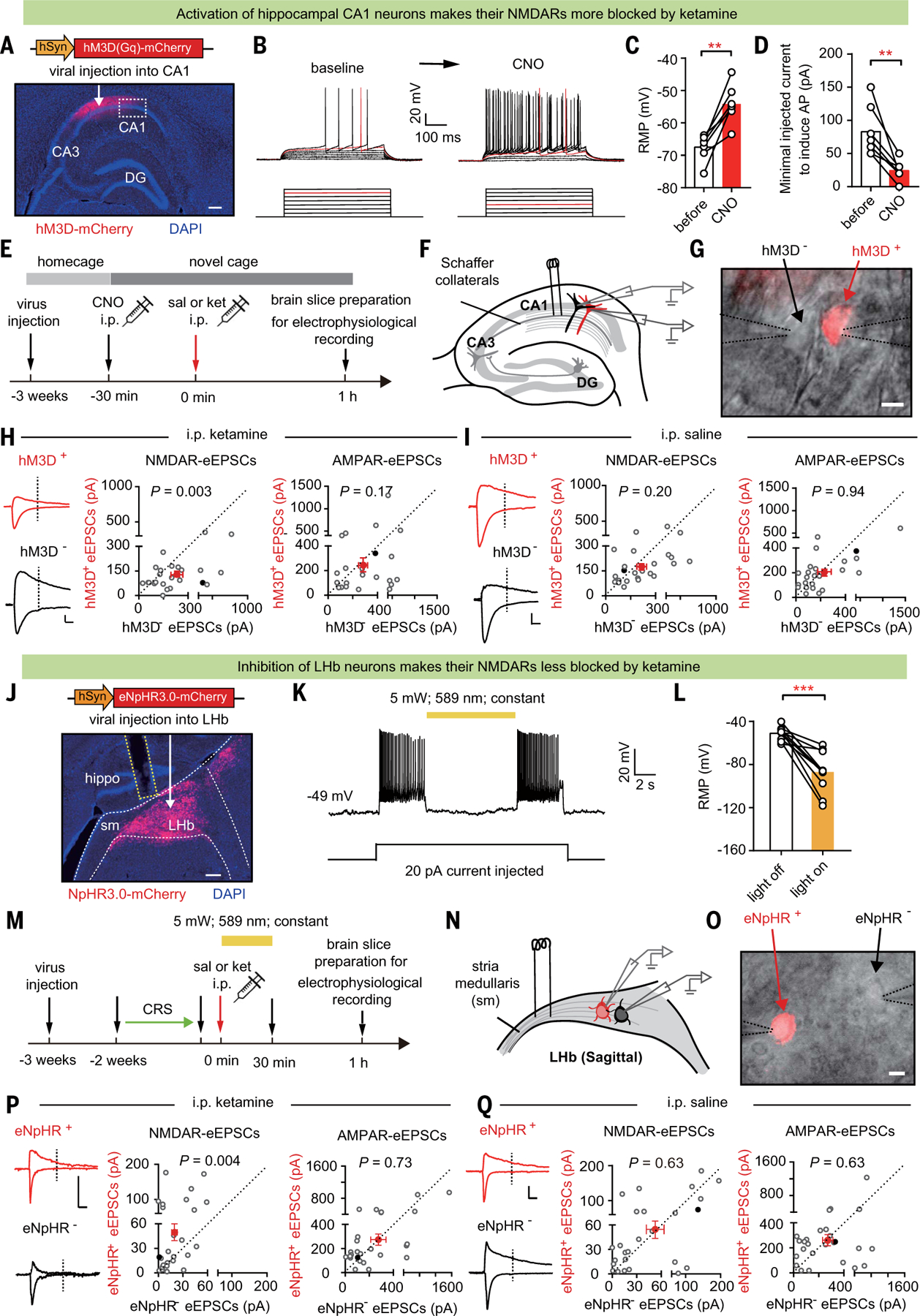
(A) Immunostaining showing the expression of hM3D in CA1. White arrow indicates site of AAV-hM3D-mCherry virus injection. White dashed box indicates the infection border for whole-cell patch recording. Scale bar, 200 μm. (B) Current-voltage relationship of an hM3D-viral-infected CA1 PYR neuron recorded before and after 5 mM CNO perfusion. Raw traces show individual voltage responses to a series of 500-ms current pulses from 0 to 140 pA in 20-pA steps. Red traces indicate the minimal current to induce action potentials. (C) Resting membrane potential (RMP) before and after 5 µM CNO perfusion (n = 7 cells; P = 0.001, paired t test). (D) Minimal injected current to induce action potential (AP) before and after 5 µM CNO perfusion (n = 7 cells; P = 0.004, paired t test). (E) Experimental paradigm recording of CA1 PYR neurons in brain slices prepared 1 hour after intraperitoneal injection of saline or ketamine (10 mg/kg) in mice expressing hM3D-mCherry in CA1 PYR neurons, with CNO (1 mg/kg) intraperitoneal injection 30 min before ketamine administration. (F) Schematic of stimulation electrode placement and paired-recording of neighboring hM3D+ (red) and hM3D– (black) PYR neurons in CA1. (G) Patchclamp recording of a pair of transfected hM3D+ and neighboring untransfected hM3D– CA1 PYR neurons under transmitted and fluorescent light microscopy. Dotted lines indicate the patch pipettes. Scale bar, 10 µm. (H and I) Left: eEPSCs in recorded hM3D+ and hM3D– CA1 PYR neuron pairs. Scale bar, 20 ms, 100 pA. Right: scatter plots of NMDAR-eEPSCs [P = 0.003, Wilcoxon matched-pairs test (H); P = 0.20, Wilcoxon matched-pairs test (I)] and AMPAR-eEPSCs [P = 0.17, Wilcoxon matched-pairs test (H); P = 0.94, Wilcoxon matched-pairs test (I)] recorded at 0.75-mA stimulation intensity in recorded hM3D+ and hM3D– CA1 PYR neuron pairs after intraperitoneal injection of saline (I) or ketamine (H) (n = 25 cell pairs in three mice in the saline group and 24 cell pairs in four mice in the ketamine group). Red dots indicate the averaged values of all recorded cells, and solid black dots indicate the example cells. (J) White arrow indicates site of AAV-eNpHR3.0-mCherry virus injection. Immunostaining showing expression of eNpHR3.0 in LHb. White dashed lines indicate the LHb. Sm: stria medullaris. Yellow dashed lines indicate the optic fiber. Scale bar, 200 μm. (K) Inhibitory effect of yellow light (589 nm) on eNpHR3.0-expressing LHb neurons. Shown is a sample trace of whole-cell recording in LHb neurons under current-clamp mode with 20-pA current injected. (L) RMP of LHb neurons during lights off and lights on (n = 10; P = 0.0003, paired t test). (M) Experimental paradigm. (N) Schematic of stimulation electrode placement and paired-recording of neighboring eNpHR+ (red) and eNpHR– (black) neurons in LHb. (O) Patch-clamp recording of a pair of transfected eNpHR+ and neighboring untransfected eNpHR– LHb neurons under transmitted and fluorescent light microscopy. Dotted lines indicate the patch pipettes. Scale bar, 10 µm. (P and Q) Left: example traces of evoked EPSCs in recorded eNpHR+ and eNpHR– LHb neuron pairs. Scale bar, 10 ms, 100 pA. Right: scatter plots of NMDAR-eEPSCs [P = 0.004, Wilcoxon matched-pairs test (P); P = 0.63, Wilcoxon matched-pairs test (Q)] and AMPAR-eEPSCs [P = 0.73, Wilcoxon matched-pairs test (P); P = 0.63, Wilcoxon matched-pairs test (Q)] recorded at 1.5-mA stimulation intensity in recorded eNpHR+ and eNpHR– LHb neuron pairs after intraperitoneal injection of saline (Q) or ketamine (P). n = 26 cell pairs in seven mice in the saline group and n = 26 cell pairs in seven mice in the ketamine group. Red dots indicate the averaged values of all recorded cells, and solid black dots indicate the example cells. **P < 0.01; ***P < 0.001; NS, not significant. Error bars indicate SEM.
Inhibition of LHb neurons protects their NMDARs from blockade by systemic ketamine
We next tested the inverse conclusion, that is, whether inhibition of LHb neuronal activity protects their NMDARs from blockade by ketamine (Fig. 4, J to Q). We used optogenetics to selectively inhibit populations of LHb neurons by expressing the inhibitory opsin eNpHR (54) in the LHb of CRS mice (Fig. 4J). AAV-eNpHR3.0-mCherry virus was expressed in the LHb, and constant yellow light (589 nm) was delivered through an optic fiber immediately after intraperitoneal saline or ketamine injection (Fig. 4, J and M). Yellow light caused a significant hyperpolarized membrane potential [note that the normal resting membrane potential of LHb neurons is close to –50 to –60 mV (31, 55)] and a reduction in the firing of LHb neurons expressing the eNpHR (Fig. 4, K and L). Because ketamine’s half-life is 13 min and its brain concentration drops to 2.1 μM by 30 min after intraperitoneal injection (48), we inhibited the LHb with light for 30 min (Fig. 4M). One hour after saline or ketamine injection and 30 min after light inhibition, LHb brain slices were prepared and whole-cell patch recordings were performed on pairs of neurons that were mCherry negative (eNpHR–) and positive (eNpHR+) (Fig. 4, M to O). In ketamine-injected mice, NMDAR-eEPSCs were significantly larger in eNpHR+ neurons (P = 0.004, Wilcoxon matched-pairs test; Fig. 4P), whereas in saline-injected CRS mice, eNpHR+ and eNpHR– LHb neurons showed a similar amplitude of NMDAR-eEPSCs (P = 0.63, Wilcoxon matched-pairs test; Fig. 4Q). By contrast, AMPAR-eEPSCs were indistinguishable between eNpHR+ and eNpHR– neurons in both saline- and ketamine-injected mice (Fig. 4, P and Q).
Reservoir pool size of NMDARs and recovery rate from ketamine blockade also contribute to brain region specificity
In addition to the difference in local neural activity under the depressive state, we observed another interesting difference between the NMDAR responses of CA1-PYR and LHb neurons: In response to ketamine blockade and washout, their recovery rates were different (Fig. 5A). In LHb and hippocampal brain slices, we pharmacologically isolated and continuously monitored the NMDAR-eEPSCs at –70 mV in the absence of magnesium while washing ketamine in and out (Fig. 5A). After a 5-min stable baseline recording, ketamine was perfused at a treatment-relevant dosage (10 μm) into the recording ACSF and then washed out after 10 min (Fig. 5A). Consistent with the property of a use-dependent blocker, ketamine gradually blocked NMDAR-eEPSCs, because each recording stimulus opened a portion of NMDARs. At the end of the 10-min blockade (60 stimuli), NMDAR-eEPSCs were reduced similarly in the two brain regions, by 64.5 ± 5.5% in the CA1-PYR neurons (n = 9 cells) and by 72.7 ± 4.6% in the LHb neurons (n = 9 cells) (Fig. 5A). However, in the 50 min after ketamine washout, the recovery of NMDAR-eEPSCs showed a marked difference in the two regions: Whereas CA1 NMDAR-eEPSCs quickly recovered (n = 9; P = 0.01, paired t test; Fig. 5A), LHb NMDAR-eEPSCs showed persistent block-ade (n = 9; P = 0.25, paired t test; Fig. 5A). Even when a higher concentration of ketamine (1 mM) was used to achieve a complete blockade, there was still considerable recovery in CA1-PYR neurons (n = 9; P = 0.02, Wilcoxon matched-pairs test; Fig. 5A). The recovery of NMDAR-eEPSCs from use-dependent blockade in the CA1-PYR neurons has been attributed to lateral movement of NMDARs and replacement of blocked synaptic NMDARs by unblocked extrasynaptic NMDARs (56, 57) (Fig. 5B). To confirm that lateral exchange indeed accounts for the quick recovery of NMDAR-eEPSCs in the CA1 neurons, we repeated the ketamine wash-in and washout experiment in Fig. 5A but replaced the electrical stimulation (synaptic activity– induced block) with 20 μm NMDA in perfusion (agonist-induced block) to open all surface NMDARs (Fig. 5, C and D). Under these conditions, NMDAR-eEPSCs in CA1-PYR neurons did not recover after ketamine washout (n = 5; P > 0.99, Wilcoxon matched-pairs test, Fig. 5C). Therefore, when extrasynaptic NMDARs are also blocked, NMDAR responses in CA1-PYR neurons cannot recover.
Fig. 5. Reservoir pool size of NMDARs and recovery rate from ketamine blockade also contribute to brain region specificity.
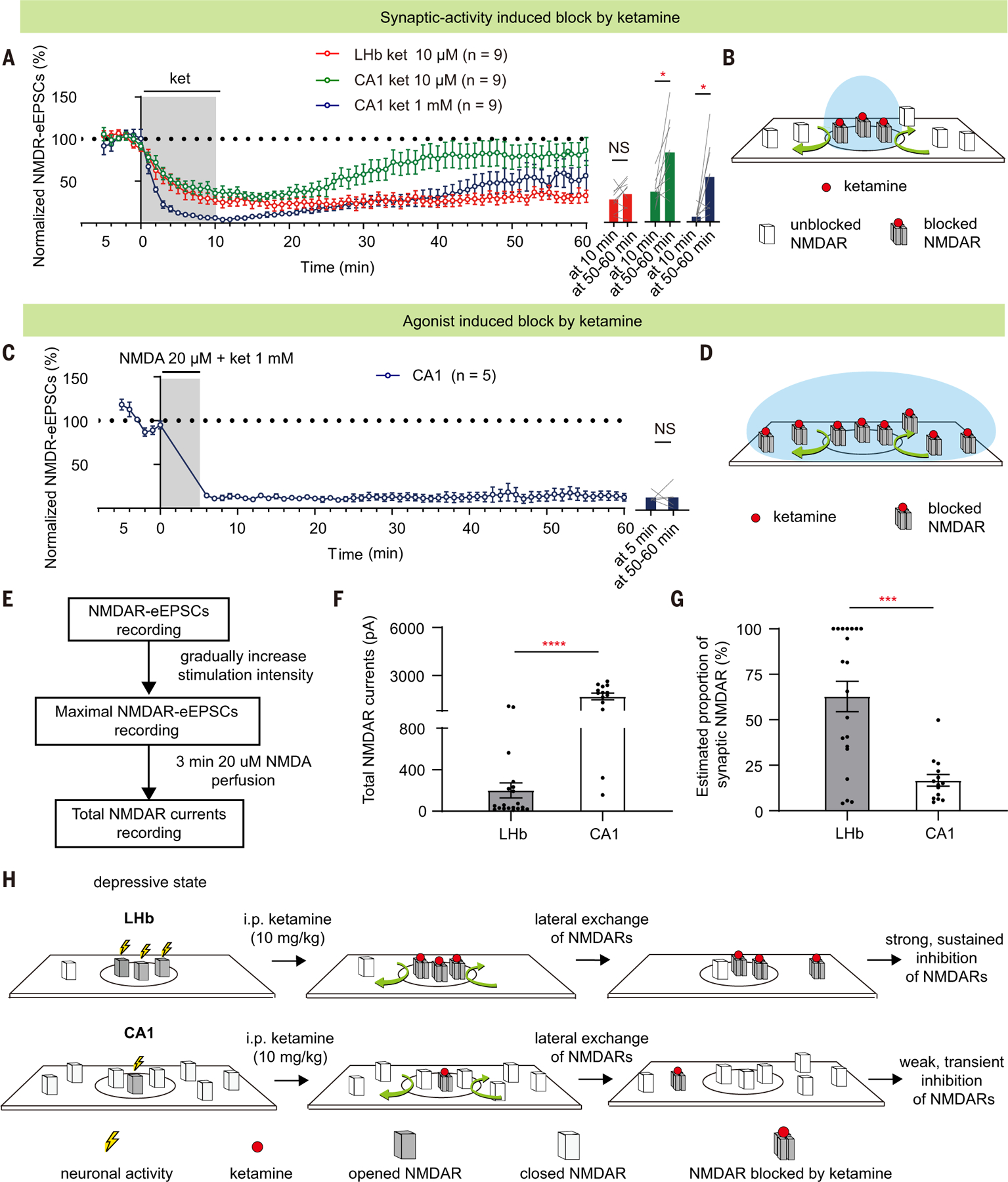
(A) NMDAR-eEPSCs (normalized by baseline) during incubation and washout of 10 µM or 1 mM ketamine in LHb or CA1 PYR neurons. Right: bar graphs showing NMDAR-eEPSCs at the end of the 10-min perfusion period and at 50 to 60 min after perfusion (LHb group: P = 0.25, paired t test; CA1 10 µM group: P = 0.01, paired t test; CA1 10 µM group: P = 0.02, Wilcoxon matched-pairs test). n = 9. (B and D) Schematics illustrating how synaptic blockade [(B) for conditions in (A)] and agonist-induced blockade [(D) for conditions in (C)] of NMDARs by ketamine are affected by lateral movement of NMDARs in and out of synapse. Black circles represent synaptic sites. Blue circles represent the area where NMDARs can be opened by corresponding treatment [synaptic stimulation in (A) or agonist perfusion in (C)]. Red dots represent ketamine. (C) NMDAR-eEPSCs (normalized by baseline) during incubation and washout of ketamine (1 mM) and NMDA (20 µM) in CA1 PYR neurons (n = 5). Right: bar graphs showing NMDAR-eEPSCs at the end of the 5-min perfusion period and at 50 to 60 min after perfusion (P > 0.99, Wilcoxon matched-pairs test). n = 5. (E) Experimental paradigm for slice recording to estimate the proportion of synaptic NMDAR-eEPSCs in total NMDAR currents. (F and G) Bar graphs illustrating the total NMDAR currents [P < 0.0001, Mann-Whitney test (F)] and estimated proportion of synaptic NMDAR [P = 0.001, Mann-Whitney test (G)] of LHb and CA1 PYR neurons. Estimated proportion of synaptic NMDAR is calculated as maximal NMDAR-eEPSCs divided by the total NMDAR currents. n = 20 in the LHb group and n = 14 cells in the CA1 group. (H) Schematic model illustrating why systemic ketamine has a stronger and more sustained blockade of NMDARs in the LHb, but not hippocampal CA1 PYR neurons, under a depressive state. The high basal activity allows LHb neurons for ketamine’s open-channel blockade, and the small reservoir pool and the trapping effect are responsible for a slow recovery of NMDAR transmission. By contrast, in hippocampal CA1 neurons, which are not as active under a depressive-like state, the available pool of open NMDARs for ketamine blockade is small to start with. After this small pool is inhibited, the large, extrasynaptic reservoir pool of NMDARs quickly exchanges with the blocked ones through lateral movement, resulting in a rapid recovery of NMDAR transmission. Circles represent synaptic areas where NMDARs can bind to synaptically released glutamate. *P < 0.05; ***P < 0.001; ****P < 0.0001; NS, not significant. Error bars indicate SEM.
To explain the difference of ketamine recovery after washout, we compared the size of the reservoir pool and the synaptic proportion of NMDARs of CA1 and LHb neurons of CRS mice. We sequentially recorded the synaptically maximal NMDAR-eEPSCs and total, NMDA perfusion–induced currents in the same LHb or CA1 neurons. After whole-cell patching of a neuron, we first pharmacologically isolated its synaptic NMDAR currents and obtained the maximal NMDAR-eEPSCs by gradually increasing the stimulation intensity. We then used a 3-min perfusion of 20 μm NMDA agonist to activate all surface NMDARs to obtain its total (including both synaptic and extrasynaptic) NMDAR currents (Fig. 5E). The total NMDAR responses were ~8.4 times larger in CA1-PYR neurons than in LHb neurons (CA1, 1696 ± 206 pA; LHb, 201 ± 72 pA; P < 0.0001, Mann-Whitney test; Fig. 5F). Therefore, the synaptic proportion of NMDARs, estimated as maximal NMDAR-eEPSCs divided by total NMDAR currents, was significantly larger in the LHb than in the CA1-PYR neurons (CA1, 16.7 ± 3.2%; LHb, 62.8 ± 8.3%; P = 0.001, Mann-Whitney test; Fig. 5G).
Local knockout of NR1 in LHb is sufficient to be antidepressant and occludes ketamine’s antidepressant effects
If blockade of LHb NMDAR is critical for ketamine’s behavioral effects, then removal of NMDARs in the LHb should have an anti-depressant effect and occlude that of ketamine. We generated local knockout of NR1 in an LHb-specific manner (Fig. 6A). AAV-eGFP-Cre or AAV-eGFP virus was bilaterally injected into the LHb of NR1 fl/fl mice (58) to generate LHb-specific conditional knockout (LHb-NR1-cKO) or control mice, respectively (Fig. 6B). Two weeks after viral expression, mice were subjected to CRS and then tested for depressive-like behaviors using the FST and the sucrose preference test (SPT), which model behavioral despair and anhedonia, respectively (Fig. 6, C and D). Compared with the control eGFP-expressing mice, LHb-NR1-cKO mice showed less depressive-like behavior in both the FST (P = 0.0002, unpaired t test; Fig. 6D) and SPT (P = 0.002, Mann-Whitney test; Fig. 6D), suggesting that NR1 cKO in the LHb prevents depressive-like behaviors.
Fig. 6. Local knockout of NR1 in LHb is sufficient to have an antidepressant effect and occludes ketamine’s anti-depressant effects.
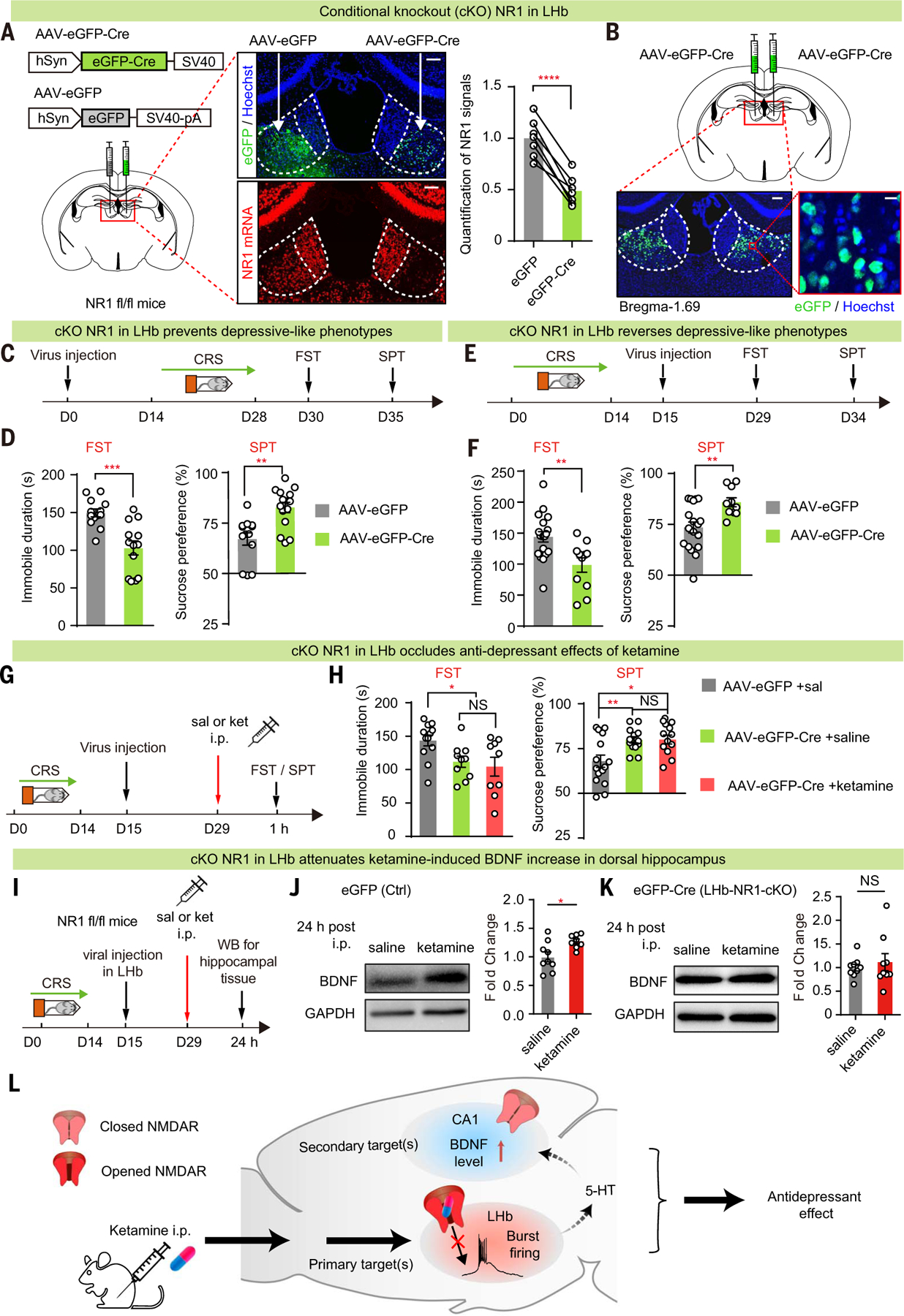
(A) Left: schematics of viral constructs and injection of AAV virus expressing eGFP on one side and Cre in the other side of the LHb of NR1 flox/flox (NR1 fl/fl) mice. Middle: viral expression (top) and RNAscope staining of NR1 (bottom) in brain slices expressing AAV-eGFP and AAV-eGFP-Cre in one of the two sides of the LHb. Scale bars, 100 µm. Right: quantification of NR1 signals to estimate knockout efficiency (n = 7 for AAV-eGFP-Cre and AAV-eGFP; P < 0.0001, paired t test). (B) Illustration of bilateral viral injection of AAV-eGFP-Cre in LHb of NR1 fl/fl mice stained with Hoechst. Scale bars, 100 µm (left) and 10 µm (right). (C, E, and G) Experimental paradigm for behavioral testing with virus injected in the LHb before (C) or after [(E) and (G)] induction of CRS. (G) Ketamine (10 mg/kg) was intraperitoneally injected 1 hour before FST or SPT. (D, F, and H) Depressivelike behaviors in FST [P = 0.0002, n = 13, 13 (D); P = 0.004, n = 19, 11 (F); n = 12, 10, 9 (H), eGFP + saline versus Cre + saline P = 0.01, Cre + saline versus Cre + ketamine P = 0.65, eGFP + saline versus Cre + ketamine P = 0.02] and SPT [P = 0.002, n = 13, 15 (D); P = 0.005, n = 18, 10 (F); n = 15, 13, 12 (H), eGFP + saline versus Cre + saline P = 0.007, Cre + saline versus Cre + ketamine P = 0.95, eGFP + saline versus Cre + ketamine P = 0.01]. (I to K) Experimental paradigm (I) and Western blot analysis [(J) and (K)] of dorsal hippocampal BDNF 24 hours after intraperitoneal injection of ketamine (10 mg/kg) in control (J) or LHb-NR1-cKO (K) mice [P = 0.02 (J); P = 0.98 (K)]. GAPDH was used as a loading control. For control, n = 9 mice in the saline group and n = 8 mice in the ketamine group; for cKO, n = 9 mice in the saline group and n = 9 mice in the ketamine group. (L) Schematic model illustrating the primary and secondary brain targets of ketamine in mediating its antidepressant effects. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, not significant. Error bars indicate SEM.
Because depressive-like behaviors induced by CRS can last to 3 weeks (59), we were able to also test whether LHb-NR1-cKO is capable of reversing depressive-like behaviors. To do this, AAV-eGFP-Cre or AAV-eGFP virus was injected into the LHb of NR1 fl/fl mice after they had gone through CRS induction (Fig. 6, E and F). Two weeks after viral injection, the LHb-NR1-cKO mice showed reduced depressive-like behaviors in both the FST (P = 0.004, Mann-Whitney test; Fig. 6F) and SPT (P = 0.005, unpaired t test; Fig. 6F) compared with the eGFP-expressing control mice, suggesting that NR1 cKO in the LHb can reverse depressive-like behaviors after they have been induced. Using the same protocol shown in Fig. 6E, if we injected ketamine or saline before the behavioral test (Fig. 6G), then the ketamine group did not show any further decrease of depressive-behaviors than the saline group (Fig. 6H), suggesting that ketamine’s anti-depressant effects had been occluded by NR1 cKO in the LHb.
Local knockout of NR1 in LHb reduces systemic ketamine–induced changes in the hippocampus
LHb-NR1-cKO mice also provided us with an opportunity to investigate how ketamine’s action in the LHb may affect other brain regions. In addition to inhibiting LHb bursting, systemic ketamine injection also induced a delayed (24 hours after but not 30 min after injection of ketamine) up-regulation of BDNF in the hippocampus (7) (fig. S5). We investigated whether there was any change in this ketamine-induced effect in the hippocampus when NR1 was locally knocked out in the LHb (Fig. 6, I to K). Western blots were performed to measure the level of hippocampal BDNF 24 hours after ketamine injection. In control mice, consistent with previous reports (7, 60, 61), there was an elevated level of BDNF (Fig. 6, I and J). This elevation was no longer significant in LHb-NR1-cKO mice (Fig. 6, I and K).
We next investigated the mechanism by which ketamine’s action in the LHb may affect hippocampal BDNF. Hippocampal BDNF has been shown to be regulated by serotonin (5-HT) (62, 63). Because the LHb can inhibit 5-HT neurons (64, 65), lesioning the LHb increases the levels of 5-HT (66), as well as BDNF in the hippocampus (67). We monitored the level of 5-HT in the hippocampus in control and LHb-NR1-cKO mice after ketamine treatment. A GRAB5-HT sensor (68) was expressed in the dorsal hippocampus of CRS mice (fig. S6A). After systemic ketamine injection, consistent with previous reports (69–71), there was a marked increase in the 5-HT signal in control mice (fig. S6, B to D). However, such an increase was absent in the LHb-NR1-cKO mice (fig. S6, E to G). In terms of temporal dynamics, the ketamine-induced increase in hippocampal 5-HT signal lagged behind the decrease in LHb firing. After ketamine treatment, while the increase of 5-HT signal became significant at 6 min, the suppression of LHb firing rate was significant as soon as 1 min after injection (fig. S6H).
Discussion
We found that in depressive-like animals, ketamine selectively inhibits NMDAR responses in LHb neurons, but not in hippocampal CA1-PYR neurons. Compared with CA1-PYR neurons, LHb neurons have much higher intrinsic activity in the depressive-like state and a much smaller extrasynaptic reservoir pool of NMDARs. By increasing the intrinsic activity of CA1 neurons or decreasing the activity of LHb neurons, we were able to swap the sensitivity of their NMDAR responses to ketamine blockade. Behaviorally, conditional knockout of the NMDAR obligatory subunit NR1 in the LHb occludes ketamine’s antidepressant effects and blocks the ketamine-induced increase in 5-HT and BDNF in the hippocampus. On the basis of the above results, we propose the following model for ketamine’s brain region–specific action (Fig. 6L). In the depressive-like state, because of the difference in basal activity in vivo, NMDARs in the LHb and hippocampus are in different open or closed states. Because ketamine is an open-channel blocker, when it is transiently elevated by intraperitoneal injection, it preferentially blocks open NMDARs in the LHb or other regions with similar properties. Blockade of the NMDAR-dependent LHb bursts can then lead to disinhibition of the downstream aminergic neurons (33, 64, 72). Through their widespread projections, aminergic neurons can have global effects in many brain regions. In particular, by blocking LHb-NMDARs, ketamine disinhibits the release of 5-HT, which can lead to increased BDNF in the hippocampus (62, 63).
Considering the region specificity of ketamine, in addition to its high basal activity, we also emphasize a distinguished feature of LHb neurons: their small reservoir of extrasynaptic NMDARs (Fig. 5). While the high basal activity shifts the LHb NMDARs to an open state, making them susceptible to ketamine blockade, the small NMDAR reservoir pool and the trapping effect (47, 48, 73) are responsible for the slow recovery of NMDAR transmission (Fig. 5, A and H). In the hippocampus, which is not as active under the depressive-like state, the pool of open NMDARs available for ketamine blockade is small to start with. After this small pool is inhibited, the large extra-synaptic reservoir pool of unblocked NMDARs quickly exchanges with the blocked NMDARs through lateral movement (56, 57) (Fig. 5H). Given the short half-life of unbound ketamine in tissue, this rapid replacement potentially leads to the recovery of NMDAR transmission in regions such as the CA1 (Fig. 5A). The significance of such replacement is directly demonstrated by activating all surface receptors with applied NMDA. When ketamine subsequently blocks all activated NMDARs, a prolonged inhibition of NMDA responses can then be achieved in the CA1 region (Fig. 5C). By contrast, for the LHb region, where the pool of extrasynaptic NMDAR is nearly 10-fold smaller than that in CA1 (Fig. 5, F and G), it is likely that there is an insufficient reserve for replacement. As a result, ketamine blockade in the LHb region is stronger and more sustained (Fig. 5H).
By describing the inherently embedded difference in their properties and delineating a temporal sequence in their responses to ketamine, we suggest that neurons in different brain regions may be recruited at different stages, and that an LHb-NMDAR-dependent event likely occurs more upstream in the cascade of ketamine signaling in vivo. Therefore, we are not arguing that the LHb is the exclusive sole target of ketamine. The primary targets of ketamine should also include other brain regions or neuron types that have similar properties as the LHb neurons. Ketamine is likely to act on multiple brain regions and multiple molecular partners, but it will be important to understand the difference in the timing of their enrollment. Overall, these primary/direct and secondary/indirect changes in different brain areas may work in concert to execute the full spectrum of ketamine’s long-term effects.
One prediction from the behavior state–dependent action of ketamine is that it should have fewer effects on nondepressed individuals, whose LHb is much less active. Indeed, in a double-blind, placebo-controlled trial, ketamine administrated at a subanesthetic dosage only had mood-elevating effects in major depressive disorder patients, not in healthy controls (74). Another interesting clinical implication from the model is that cognitive activity should be avoided during and shortly after ketamine treatment to reduce the potential side effects caused by ketamine blockade of NMDARs in cognitive brain regions. In summary, our study delineates a mechanistic basis for why a pharmacologically important compound, ketamine, has differential effects on different brain loci or under different behavioral states in vivo. The combination of use-dependent block, activity levels, and receptor reserves as a mechanism for region-specific action on a ubiquitously expressed receptor may inspire new ways of thinking about region-specific issues in neuropathology and neuropharmacology. Distinction of the primary versus secondary target(s) of ketamine should help in the design of more precise and efficient treatments for depression.
Materials and methods
Animals
Male adult (8 to 16 weeks old) C57BL/6 (SLAC or Shanghai Jihui) and homozygous NMDA-receptor1flox/flox (NR1 fl/fl; B6.129S4-Grin1tm2Stl/J) mice were used. Mice were group housed four per cage and subjected to a 12-hour light-dark cycle (lights on from 7:00 a.m. to 7:00 p.m.) with free access to food and water. All animal studies and experimental procedures were approved by the Animal Care and Use Committee of the animal facility at Zhejiang University.
CRS
Mice were subjected to CRS by placing them in 50-ml conical tubes with holes for air flow for 2 to 6 hours per day for 14 consecutive days (48, 75).
Systemic drug delivery
All drugs were dissolved in 0.9% saline and administered intraperitoneally. The dosage of ketamine (Gutian Pharma Co. or Beikang Pharma Co.) was 10 mg/kg. The ketamine used in this study was a mixture of R- and S-ketamine. One hour after drug injection, animals were subjected to behavioral tests or sacrificed for in vitro electrophysiology recordings. For mice expressing hM3D(Gq) (53, 76) in CA1, CNO (Sigma) was administered intraperitoneally at a dosage of 1 mg/kg 30 min before saline or ketamine injection.
Viral vectors
AAV2/1-hSyn-HI-eGFP-Cre (1:5 dilution, 0.1 ml bilateral into LHb, University of Pennsylvania vector core), AAV2/8-hSyn-eGFP (titer: 1.28 × 1013 v.g./ml, 1:5 dilution, 0.1 ml bilateral into LHb, Taitool), AAV2/9-hSyn-hM3D(Gq)-mCherry-WPRE-pA (titer: 2.15 × 1013 v.g./ml, 1:10 dilution, 0.2 ml bilateral into CA1, Taitool), AAV2/ 9-hSyn-eNpHR 3.0-mCherry (titer: 9.56 × 1012 v.g./ml, 1:5 dilution, 0.1 ml unilateral into LHb, Taitool), and AAV2/9-hSyn-5-HT2h (titer: 7.48 × 1013 v.g./ml, 1:5 dilution, 0.2 ml unilateral into dorsal hippocampus, Vigen Biosciences) were aliquoted and stored at –80°C until use.
Surgery
Mice were deeply anesthetized with 1% sodium pentobarbital (100 mg/kg body weight; Sigma) and secured in a stereotactic frame (RWD Instruments). The virus was injected into the LHb (0.1 ml) (AP, −1.72 mm from bregma; ML, ±0.47 mm; DV, −2.62 mm from the dura) or hippocampal CA1 (0.2 ml) [AP, −1.90 mm from bregma; ML, ±1.20 mm; DV, −1.20 mm from the dura for AAV2/9-hSyn-hM3D(Gq)-mCherry-WPRE-pA virus and AP, −2.30 mm from bregma; ML, ±2.30 mm; DV, −2.0 mm from the dura for AAV2/9-hSyn-5-HT2h virus] using a pulled glass capillary with a pressure microinjector (Picospritzer III; Parker) at a rate of 0.1 ml/min. The injection needle was withdrawn 10 min after the end of the injection. After surgery, mice were allowed to recover from anesthesia on a heating pad. For optic fiber implantation, a 200-μm fiber-optic cannula was placed 200 to 400 μm above the center of viral injection site and cemented onto the skull using dental cement. To verify the virus injection sites and optic fiber or tetrodes sites, mice were sacrificed after all experiments. Brain sections were cut at a thickness of 60 μm (Leica CM1950) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) or Hoechst. Fluorescent images were acquired using the Olympus MVX10 microscope and VS120 virtual microscopy slide scanning system. Only data from mice with correct injection sites were included.
FST
The FST was used to model behavioral despair, as previously described (77). Mice were placed individually in a cylinder (12 cm diameter, 25 cm height) filled with water (23° to 25°C) and swam for 6 min under normal light conditions (30 to 35 lux). The water depth was set to ensure that animals could not touch the bottom with their tails or hind limbs. Animal behaviors were recorded from the side, and the immobile duration during the 2- to 6-min period was counted offline by an observer blinded to the animal treatment. Immobile duration was defined as the time when animals remained floating or motionless except for the movements necessary to keep their balance in the water.
SPT
The SPT was used to model anhedonia, as described in previous studies (48, 78). Mice were single housed and habituated with two bottles of water for 2 days, followed by two bottles of 2% sucrose for 2 days. Next, animals were water deprived for 24 hours and then exposed to one bottle of 2% sucrose and one bottle of water for 2 hours during the dark phase. The positions of the bottles were switched 1 hour after test started. The total consumption of each fluid was measured, and sucrose preference was defined as the average of sucrose consumption ratios during the first and second hours. The sucrose consumption ratio was calculated by dividing the total consumption of sucrose by the total consumption of both water and sucrose.
Western blot analysis
The habenular membrane fraction for NR1 detection (43) and the hippocampal total protein for BDNF detection (79) were extracted as previously described. Animals were anesthetized with isoflurane, and habenular tissue was quickly dissected from the brain and homogenized in lysis buffer [2 M sucrose (Sigma); 1 M HEPES, pH 7.4 (Sigma); pro-tease inhibitor cocktail tablets (Roche); and phosphatase inhibitor cocktail tablets (Roche)] on ice. The hippocampal tissue was dissected and homogenized in radioimmunoprecipitation assay buffer (Solarbio) on ice. After measurement of the protein concentration using the bicinchoninic acid assay, 10 to 20 mg of habenular membrane fraction protein for each lane was separated on a 10% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel and 40 mg of hippocampal total protein for each lane was separated on a 4 to 20% SDS-PAGE gel, and both were transferred for Western blot analysis. Mouse anti-GluN1 (1:2000; Millipore), rabbit anti-BDNF (1:1500; Abcam), rabbit anti-a-tubulin (1:3000; Affinity Biosciences), and mouse anti-GAPDH-HRP (1:5000; Aksomics) antibodies were used, along with high-sensitivity ECL reagent (Cytiva). All bands were analyzed using ImageJ software.
Foot-shock protocol
For the foot-shock protocol shown in fig. S4, five foot shocks of 1.0- to 1.5-mA intensity and 3-s duration were randomly delivered within 5 min after saline or ketamine administration in a fear conditioning chamber (Coulbourn Instruments).
Optogenetic light delivery and protocols
For mice expressing eNpHR 3.0 (54, 80) in LHb, a 589-nm yellow light laser (Inper) was delivered immediately after saline or ketamine injection for 30 min. The light intensity was set at 5 mW.
Brain slice preparation
Mice were anesthetized with 1% sodium pentobarbital (100 mg/kg body weight; Sigma) and then perfused with 20 ml of ice-cold artificial cerebrospinal fluid (ACSF) oxygenated with 95% O2 + 5% CO2 and containing the following (in mM): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 25 glucose, with 1 mM pyruvate added. The brain was removed as quickly as possible after decapitation and put into chilled and oxygenated ACSF. Sagittal slices containing the LHb or dorsal hippocampal CA1 were sliced into 300-μm sections in cold ACSF using the Leica VT1200S vibratome and then transferred to ACSF at 32°C for incubation and recovery. ACSF was continuously gassed with 95% O2 and 5% CO2. Slices were allowed to recover for at least 1 hour before recording.
In vitro electrophysiological recordings
Whole-cell patch-clamp recordings were obtained using pipettes with a typical resistance of 3 to 7 MΩ (3 to 5 MΩ for hippocampal neurons; 4 to 7 MΩ for LHb neurons). For recording neuronal activity under current-clamp conditions, the pipettes were filled with internal solution containing the following (in mM): 105 K-gluconate, 30 KCl, 4 Mg-ATP, 0.3 Na-GTP, 0.3 EGTA, 10 HEPES, and 10 Na-phosphocreatine, with pH adjusted to 7.25 to 7.30. For evoked EPSCs and NMDA-induced current recording under voltage-clamp conditions, the pipettes were filled with internal solution containing the following (in mM): 115 CsMeSO3, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 Na-ATP, 0.4 Na-GTP, 10 Na-phosphocreatine, 0.6 EGTA, and 5 QX-314, with pH adjusted to 7.25 to 7.30. The external ACSF solution contained the following (in mM): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4,1 MgCl2, 2 CaCl2, and 25 glucose. Cells were visualized using infrared optics on an upright microscope (BX51WI; Olympus), and electrophysiology was performed using a MultiClamp 700B amplifier controlled by a DigiData 1550 digitizer and pCLAMP10 software (Axon Instruments). The series resistance and capacitance were compensated automatically after a stable gigaseal was formed, and recordings were typically conducted 3 to 10 min after break-in.
Voltage-clamp recordings of eEPSCs were obtained at –70 mV or +40 mV by stimulating the input stria medullaris fiber in sagittal LHb slices or Schaffer collaterals in sagittal CA1 slices. A modified extracellular ACSF solution containing the GABAAR blocker PTX (100 μm; Tocris Bioscience) was used. A bipolar stimulating electrode was placed ~200 μm from the recorded cell bodies. For the LHb, the neuron types were not distinguished because most in that region are glutamatergic neurons (44). For the CA1, we selectively recorded PYR neurons on the basis of their positional, morphological, and electrophysiological properties (81, 82). In CA1, the PYR neurons are located in the PYR layer, and the interneurons are mainly in the stratum radiatum. Moreover, the input resistance of CA1 PYR neurons (~100 MΩ) is smaller than CA1 interneurons (~350 MΩ) (81). Therefore, we selectively recorded neurons with small input resistance in the PYR layer of the hippocampus. Stimulation pulse (0.25 to 1.50 mA for 0.2 ms in 0.25-mA steps) were delivered every 6 to 10 s. The cell was initially held at –70 mV to record AMPAR-eEPSCs, which were determined by the peak current amplitude at –70 mV. Subsequently, the cell was held at +40 mV to record a combination of AMPAR-eEPSCs and the slower NMDAR-mediated eEPSCs. To isolate the NMDAR-eEPSCs at +40 mV, we selected a time point after stimulation onset when AMPAR-eEPSCs had decayed to the baseline. LHb synapses are known to largely express GluR2-lacking AMPARs (calcium-permeable AMPARs) (41, 42) that deactivate much faster and exhibit stronger inward rectification than the PYR GluR2-containing AMPARs (83). The time point was chosen to be 35 ms for LHb neurons (84, 85) and 60 ms for CA1 PYR neurons (86, 87). More than three traces were averaged at each stimulation intensity and holding potential. NMDA/AMPA ratios were determined by dividing the NMDAR-eEPSCs by the AMPAR-eEPSCs at 1.5-mA stimulation intensity.
The pure NMDAR-eEPSCs were recorded at +40 mV using a pharmacological isolation method with NBQX to block AMPARs and PTX to block GABAARs in the recording ACSF. The NMDAR-eEPSCs were calculated by measuring the peak amplitude in this recording condition.
For the paired recording shown in Fig. 4, the border area of the viral injection site was selected to increase the likehood of having both fluorescently positive and negative neurons adjacent to each other for sequential whole-cell patch recording. The order of patching fluorescently positive and negative neurons was randomized. For the experiment in Fig. 4E, note that mice are placed in a novel cage to provide more presynaptic inputs to hippocampal neurons, because NMDAR opening requires both postsynaptic depolarization and presynaptic glutamate release.
To validate optogenetic and chemogenetic manipulations, LHb or CA1 neurons were recorded under current-clamp conditions (I = 0 pA). The spontaneous and current injection– induced activities of LHb neurons were recorded while yellow light laser (589 nm, 5 mW, constant) was turned on and off. Photostimulation induced robust hyperpolarization and almost entirely silenced LHb neurons. The resting membrane potential and the current-voltage relationship (500-ms current pulses from 0 to 140 pA in 10-pA steps) of hippocampal CA1 neurons were recorded before and after perfusion with 5 μm CNO (Sigma). The minimal currents required to induce an action potential were calculated.
For pharmacological washout recordings of NMDAR-eEPSCs, NMDAR-eEPSCs were recorded under voltage-clamp conditions at –70 mV in sagittal LHb or CA1 slices using a modified extracellular ACSF solution with NBQX (10 μm; Sigma) and PTX (100 μm; Tocris Bioscience). Recordings were made in ACSF without added Mg2+ to reduce the Mg2+ blockade of NMDARs. The stimulation intensity (0.1 to 0.3 ms at 0.1 to 5 mA) was adjusted for each cell to produce adequate responses. LHb neurons with an NMDAR-eEPSC <10 pA were excluded from the washout experiments. Stimulation pulses were delivered every 10 s. After a 5-min stable baseline recording, 10 μm or 1 mM ketamine was added to the recording ACSF and then washed out 10 min later to monitor the recovery of NMDAR-eEPSCs over the next 50 min. NMDAR-eEPSCs were normalized by the baseline before drug application. To fully block the NMDAR in hippocampal CA1 neurons, 20 μm NMDA (Sigma) was perfused together with 1 mM ketamine for 5 min. The normalized NMDAR-eEPSCs at the end of drug perfusion (at 10 min in Fig. 5A or at 5 min in Fig. 5C) were calculated to show the degrees of drug blockade, and the averaged normalized NMDAR-eEPSCs at 50 to 60 min were calculated to show the degrees of response recovery (Fig. 5).
To compare the sizes of the NMDAR reservoir pool and to quantify the proportion of synaptic NMDAR-eEPSCs in the total NMDAR currents of LHb and CA1 neurons, we recorded the maximal NMDAR-eEPSCs and total NMDAR currents in the same cell. First, we recorded the NMDAR-eEPSCs of LHb neurons and CA1 neurons under voltage-clamp conditions at –70 mV in a modified ACSF with NBQX (10 μm; Sigma) and PTX (100 μm; Tocris Bioscience) and zero Mg2+. We then gradually increased the stimulation intensity until reaching the maximal responses, which we calculated as the synaptic NMDAR-eEPSCs. Next, we recorded the NMDA-induced currents (3 min of 20 μm NMDA perfusion) in the same neuron as the total NMDAR currents. The proportion of synaptic NMDAR-eEPSCs in total NMDAR currents was calculated as maximal NMDAR-eEPSCs divided by NMDA-induced currents. Because of the desensitization of NMDARs and the presence of NMDARs tonically activated by ambient glutamate, the NMDA-induced currents could be underestimated. Therefore, the proportions sometimes exceeded 100% in LHb neurons, which were uniformly calculated as 100%.
In vivo electrophysiology
For in vivo single-unit recording experiments, a custom-made screw-driven microdrive consisting of a 4 × 4 platiniridium alloy wire [size 0 (0.0007”), Platinum 10; California Fine Wire Company] tetrode was implanted into the unilateral LHb (AP, –1.72 mm; ML, 0.47 mm; DV, –2.50 mm from the brain surface) or CA1 (AP, –1.90 mm; ML, 1.20 mm; DV, –1.10 mm from the brain surface) in mice. Four wires wounded into one strand were used as a unit, and each wire represented one channel. Silver wires were attached to two screws on the skull as ground. The microdrive was secured to the skull with dental cement. After >1 week of recovery, mice were allowed to adapt to the recording headstage for 30 min before recording. Spontaneous spiking activity was recorded by a neural recording system (Plexon Inc.) and digitized at 40 kHz with a gain of 1000×. Spontaneous spiking signals were band-pass filtered between 300 and 6000 Hz. The common average reference was assigned as a digital reference. The amplitude threshold for spike capture was adjusted for each unit according to the signal-to-noise ratio. Spontaneous spiking signals of the mice were recorded for 10 min after habituation in their home cages as the baseline. Spiking signals were then continuously recorded for 1 hour after saline or ketamine treatment (10 mg/kg, i.p.) with the head stage unremoved. The tetrodes were lowered in steps of 62.5 μm in LHb, 31.2 μm in CA1 after each recording session, followed by at least a 3-day recovery. If mice received a second ketamine injection, an interval of at least 3 to 7 days was introduced before the next recording session. The CRS animals that showed high immobile duration (>120 s) and naïve animals that showed low immobile duration (<120 s) in the FST were used in the in vivo recordings. The positions of the tetrodes were verified at the end of all experiments, and only data from mice with correctly positioned tetrodes were used.
Spike sorting
All waveforms recorded from each tetrode were imported into Offline Sorter V3/4 (Plexon Inc.). Single units were manually identified by threshold crossing and principal component analysis. Spikes with an interspike interval less than the refractory period (1.0 ms) were excluded. Cross-correlograms were plotted to ensure that no cell was discriminated more than once on overlapping tetrodes. Because most LHb neurons are glutamatergic (44), they were not subdivided in our study. Hippocampal CA1 neurons were further divided into three cell types on the basis of their spike width and spontaneous firing frequency (88–90). Neurons with a spike width larger than 400 ms were identified as putative PYR neurons, whereas neurons with a spike width shorter than 400 ms and firing frequency higher than 8 Hz were identified as fast-spiking putative interneurons (also known as putative parvalbumin-positive neurons). The remaining neurons were identified as non–fast-spiking putative interneurons. Only putative PYR neurons with an average basal firing rate of at least 0.1 Hz were included for analysis in this study (91).
Data analysis
Data were analyzed using NeuroExplorer 4/5 (Plexon Inc.) and MATLAB. We defined in vivo bursting as clusters of spikes beginning with a maximal interspike interval of 20 ms and ending with a maximal interspike interval of 50 ms. The minimal intraburst interval was set at 50 ms, and the minimal number of spikes in a burst was set at 2. FR and bursting FR were further analyzed using NeuroExplorer 4/5 and Excel 2013. For the ketamine or saline treatment data, the inhibited or excited units were statistically analyzed by -score transformation of FR or bursting FR. The postinjection -score of each unit was calculated as: , where is the mean of all the 100-s-bin values during the postinjection period (i.e., 50 to 60 min after drug injection) and and are the mean and SD of all the 100-s-bin values during baseline period (i.e., 10 min before drug injection).
The inhibition or excitation of a unit was identified when its postinjection -score of firing rate was ≤ –1.67 or ≥ 1.67 (P < 0.05) (92), respectively.
RNAscope HiPlex assay
RNAscope HiPlex assays were performed following the manufacturer’s protocol (Advanced Cell Diagnostics) and as described in a previous study (93). Brain slices were 15 μm thick and obtained using a slicer (Leica CM1950). The slices were immersed in phosphate-buffered saline twice for 10 min each at room temperature, followed by dehydration with 50%, 70%, and 100% ethanol. Subsequently, the slices were immersed in H2O2 for 10 min and then incubated with rabbit anti-GFP (Invitrogen) at a 1:100 dilution overnight. The slices were immersed in 4% formalin for 30 min at room temperature and digested with protease plus for 30 min at 40°C. After hybridization with the designed probes Mm-Grin1 (Advanced Cell Diagnostics, catalog no. 431611) for 3 hours at 40°C, the sections were treated with HiPlex Amp 1–3 for 30 min at 40°C, followed by treatment with HiPlex Fluoro C1 for 15 min at 40°C and TSA Plus Fluorescein 690-C1 buffer (1:500) for 30 min at 40°C. HRP blocker was then applied for 30 min at 40°C. The slices were imaged using a Nikon A1 confocal microscope equipped with laser lines (405, 488, and 650 nm) and a 10× objective. All images were analyzed using ImageJ software.
Fiber photometry recording
Fiber photometry experiments were conducted in accordance with methodologies described previously (94). Using the fiber photometry apparatus provided by Thinker Tech Nanjing Biotech Co., Ltd., a 488-nm laser beam was directed toward the dorsal hippocampus for the purpose of exciting and capturing the green fluorescent signals emitted by the 5-HT sensor. In an effort to mitigate the photobleaching of the 5-HT sensor, the laser’s intensity was meticulously adjusted to a minimal level of 20 µW at the optic fiber’s tip, and the sampling frequency was set at 50 Hz. The experimental protocol included a baseline recording period of 15 min, followed by a 1-hour post-intraperitoneal injection (either saline or ketamine at a dosage of 10 mg/kg) recording phase. Data were analyzed by the codes (OpSignal, from Thinker Tech Nanjing Biotech Co., Ltd.) based on MATLAB. The analysis focused on the fluorescence responses, denoted as ΔF/F, calculated by the formula (F – F0)/F0, where F0 represents the mean baseline fluorescence intensity quantified during a period of 400 s immediately preceding the administration of the drug through intraperitoneal injection. Furthermore, the area under the curve was determined by summing the fluorescence changes observed between 500 and 3000 s after the intraperitoneal injection of either saline or ketamine (10 mg/kg). The results, including DF/F, are depicted as heatmaps and as average plots, with the SEM represented by a shaded area surrounding the plots.
Statistical analysis
Required sample sizes were estimated on the basis of our previous experience performing similar experiments. Mice were randomly assigned to treatment groups, and all behavioral experiments were performed in a blinded manner. Statistical analyses were conducted using GraphPad Prism software v7/8. Values were excluded from the analysis if the tetrode sites were outside of the target brain area according to pre-established criteria. All statistical tests were two-tailed, and significance was assigned at P < 0.05. Normality and equal variances between group samples were assessed using the D’Agostino-Pearson omnibus normality test and Brown-Forsythe tests, respectively. For samples with normality and equal variance, we used paired or unpaired t test, and for samples with unequal variance, we used Mann-Whitney test or Wilcoxon matchedpairs test. The linear regression test was used in the appropriate situations. The data shown in fig. S6H were smoothed with a zero-phase Gaussian filter, and P values were further corrected for multiple comparisons using the false discovery rate. Further details are provided in table S1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Q. Li and C. Zarate for stimulating discussions; H. Li and X. Xiang for advice on tetrode recording; and X. Jia, Q. Xin, Y. Jiang, S. Wang, and J. Wang for assistance with experiments.
Funding:
This work was supported by the STI2030-Major Projects (2021ZD0203000 and 2021ZD0203001), the National Natural Science Foundation of China (32130042, 31830032, and 82288101), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SN-ZJU-SIAS-002), the New Cornerstone Science Foundation, Key R&D Program of Zhejiang (2024SSYS0016), the Fundamental Research Funds for the Central Universities (2023ZFJH01-01 and 2024ZFJH01-01), the Project for Hangzhou Medical Disciplines of Excellence, and Key Project for Hangzhou Medical Disciplines (All to H.H.). S.M. is supported by the Postdoctoral Fellowship Program of the China Postdoctoral Science Foundation (GZB20230640). C.J.L. is supported in part by the National Institute of General Medical Sciences of the National Institutes of Health (R35GM118114).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
All data are available in the main manuscript or the supplementary materials.
REFERENCES AND NOTES
- 1.Berman RM et al. , Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354 (2000). doi: 10.1016/S0006-3223(99)00230-9; [DOI] [PubMed] [Google Scholar]
- 2.Zarate CA Jr et al. , A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864 (2006). doi: 10.1001/archpsyc.63.8.856; [DOI] [PubMed] [Google Scholar]
- 3.Krystal JH, Abdallah CG, Sanacora G, Charney DS, Duman RS, Ketamine: A paradigm shift for depression research and treatment. Neuron 101, 774–778 (2019). doi: 10.1016/j.neuron.2019.02.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simmler LD et al. , Dual action of ketamine confines addiction liability. Nature 608, 368–373 (2022). doi: 10.1038/s41586-022-04993-7; [DOI] [PubMed] [Google Scholar]
- 5.Autry AE et al. , NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95 (2011). doi: 10.1038/nature10130; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun HL et al. , Role of hippocampal p11 in the sustained antidepressant effect of ketamine in the chronic unpredictable mild stress model. Transl. Psychiatry 6, e741 (2016). doi: 10.1038/tp.2016.21; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zanos P et al. , NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486 (2016). doi: 10.1038/nature17998; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeng S et al. , Cellular mechanisms underlying the antidepressant effects of ketamine: Role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 63, 349–352 (2008). doi: 10.1016/j.biopsych.2007.05.028; [DOI] [PubMed] [Google Scholar]
- 9.Zaytseva A et al. , Ketamine’s rapid antidepressant effects are mediated by Ca2+-permeable AMPA receptors. eLife 12, e86022 (2023). doi: 10.7554/eLife.86022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li N et al. , mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 (2010). doi: 10.1126/science.1190287; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez JP et al. , Ketamine exerts its sustained antidepressant effects via cell-type-specific regulation of Kcnq2. Neuron 110, 2283–2298.e9 (2022). doi: 10.1016/j.neuron.2022.05.001; [DOI] [PubMed] [Google Scholar]
- 12.Aguilar-Valles A et al. , Antidepressant actions of ketamine engage cell-specific translation via eIF4E. Nature 590, 315–319 (2021). doi: 10.1038/s41586-020-03047-0; [DOI] [PubMed] [Google Scholar]
- 13.Leng L et al. , Menin reduces parvalbumin expression and is required for the anti-depressant function of ketamine. Adv. Sci 11, e2305659 (2024). doi: 10.1002/advs.202305659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan S, Wang Y, Chen K, Long Z, Zou J, Ketamine alleviates depressive-like behaviors via down-regulating inflammatory cytokines induced by chronic restraint stress in mice. Biol. Pharm. Bull 40, 1260–1267 (2017). doi: 10.1248/bpb.b17-00131; [DOI] [PubMed] [Google Scholar]
- 15.Jia X, Gao Z, Hu H, Microglia in depression: Current perspectives. Sci. China Life Sci 64, 911–925 (2021). doi: 10.1007/s11427-020-1815-6; [DOI] [PubMed] [Google Scholar]
- 16.Wong EHF et al. , The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc. Natl. Acad. Sci. U.S.A 83, 7104–7108 (1986). doi: 10.1073/pnas.83.18.7104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsons CG et al. , Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology 34, 1239–1258 (1995). doi: 10.1016/0028-3908(95)00092-K; [DOI] [PubMed] [Google Scholar]
- 18.Anis NA, Berry SC, Burton NR, Lodge D, The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br. J. Pharmacol 79, 565–575 (1983). doi: 10.1111/j.1476-5381.1983.tb11031.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison NL, Simmonds MA, Quantitative studies on some antagonists of N-methyl D-aspartate in slices of rat cerebral cortex. Br. J. Pharmacol 84, 381–391 (1985). doi: 10.1111/j.1476-5381.1985.tb12922.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomson AM, West DC, Lodge D, An N-methylaspartate receptor-mediated synapse in rat cerebral cortex: A site of action of ketamine? Nature 313, 479–481 (1985). doi: 10.1038/313479a0; [DOI] [PubMed] [Google Scholar]
- 21.Wang LY, Orser BA, Brautigan DL, MacDonald JF, Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature 369, 230–232 (1994). doi: 10.1038/369230a0; [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y et al. , Structural basis of ketamine action on human NMDA receptors. Nature 596, 301–305 (2021). doi: 10.1038/s41586-021-03769-9; [DOI] [PubMed] [Google Scholar]
- 23.Su T, Lu Y, Fu C, Geng Y, Chen Y, GluN2A mediates ketamine-induced rapid antidepressant-like responses. Nat. Neurosci 26, 1751–1761 (2023). doi: 10.1038/s41593-023-01436-y; [DOI] [PubMed] [Google Scholar]
- 24.Gerhard DM et al. , GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J. Clin. Invest 130, 1336–1349 (2020). doi: 10.1172/JCI130808; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller OH et al. , GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife 3, e03581 (2014). doi: 10.7554/eLife.03581; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trullas R, Skolnick P, Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur. J. Pharmacol 185, 1–10 (1990). doi: 10.1016/0014-2999(90)90204-J; [DOI] [PubMed] [Google Scholar]
- 27.Casarotto PC et al. , Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313.e19 (2021). doi: 10.1016/j.cell.2021.01.034; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang C, Yang J, Luo A, Hashimoto K, Molecular and cellular mechanisms underlying the antidepressant effects of ketamine enantiomers and its metabolites. Transl. Psychiatry 9, 280 (2019). doi: 10.1038/s41398-019-0624-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moda-Sava RN et al. , Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364, eaat8078 (2019). doi: 10.1126/science.aat8078; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carreno FR et al. , Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol. Psychiatry 21, 1298–1308 (2016). doi: 10.1038/mp.2015.176; [DOI] [PubMed] [Google Scholar]
- 31.Yang Y et al. , Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554, 317–322 (2018). doi: 10.1038/nature25509; [DOI] [PubMed] [Google Scholar]
- 32.Andalman AS et al. , Neuronal dynamics regulating brain and behavioral state transitions. Cell 177, 970–985.e20 (2019). doi: 10.1016/j.cell.2019.02.037; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui Y, Hu S, Hu H, Lateral habenular burst firing as a target of the rapid antidepressant effects of ketamine. Trends Neurosci. 42, 179–191 (2019). doi: 10.1016/j.tins.2018.12.002; [DOI] [PubMed] [Google Scholar]
- 34.Proulx CD, Hikosaka O, Malinow R, Reward processing by the lateral habenula in normal and depressive behaviors. Nat. Neurosci 17, 1146–1152 (2014). doi: 10.1038/nn.3779; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu H, Cui Y, Yang Y, Circuits and functions of the lateral habenula in health and in disease. Nat. Rev. Neurosci 21, 277–295 (2020). doi: 10.1038/s41583-020-0292-4; [DOI] [PubMed] [Google Scholar]
- 36.Hikosaka O, Habenula (Scholarpedia, 2007). doi: 10.4249/scholarpedia.2703 [DOI] [Google Scholar]
- 37.MacDonald JF, Miljkovic Z, Pennefather P, Use-dependent block of excitatory amino acid currents in cultured neurons by ketamine. J. Neurophysiol 58, 251–266 (1987). doi: 10.1152/jn.1987.58.2.251; [DOI] [PubMed] [Google Scholar]
- 38.Lodge D, Johnson KM, Noncompetitive excitatory amino acid receptor antagonists. Trends Pharmacol. Sci 11, 81–86 (1990). doi: 10.1016/0165-6147(90)90323-Z; [DOI] [PubMed] [Google Scholar]
- 39.Caldecott-Hazard S, Mazziotta J, Phelps M, Cerebral correlates of depressed behavior in rats, visualized using 14C-2-deoxyglucose autoradiography. J. Neurosci 8, 1951–1961 (1988). doi: 10.1523/JNEUROSCI.08-06-01951.1988; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirrione MM et al. , Increased metabolic activity in the septum and habenula during stress is linked to subsequent expression of learned helplessness behavior. Front. Hum. Neurosci 8, 29 (2014). doi: 10.3389/fnhum.2014.00029; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B et al. , Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 470, 535–539 (2011). doi: 10.1038/nature09742; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerniauskas I et al. , Chronic stress induces activity, synaptic, and transcriptional remodeling of the lateral habenula associated with deficits in motivated behaviors. Neuron 104, 899–915.e8 (2019). doi: 10.1016/j.neuron.2019.09.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li K et al. , bCaMKII in lateral habenula mediates core symptoms of depression. Science 341, 1016–1020 (2013). doi: 10.1126/science.1240729; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aizawa H, Kobayashi M, Tanaka S, Fukai T, Okamoto H, Molecular characterization of the subnuclei in rat habenula. J. Comp. Neurol 520, 4051–4066 (2012). doi: 10.1002/cne.23167; [DOI] [PubMed] [Google Scholar]
- 45.Quina LA, Walker A, Morton G, Han V, Turner EE, GAD2 expression defines a class of excitatory lateral habenula neurons in mice that project to the raphe and pontine tegmentum. eNeuro 7, ENEURO.0527-19.2020 (2020). doi: 10.1523/ENEURO.0527-19.2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hashikawa Y et al. , Transcriptional and spatial resolution of cell types in the mammalian habenula. Neuron 106, 743–758. e5 (2020). doi: 10.1016/j.neuron.2020.03.011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mealing GA, Lanthorn TH, Murray CL, Small DL, Morley P, Differences in degree of trapping of low-affinity uncompetitive N-methyl-D-aspartic acid receptor antagonists with similar kinetics of block. J. Pharmacol. Exp. Ther 288, 204–210 (1999). [PubMed] [Google Scholar]
- 48.Ma S et al. , Sustained antidepressant effect of ketamine through NMDAR trapping in the LHb. Nature 622, 802–809 (2023). doi: 10.1038/s41586-023-06624-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lecca S et al. , Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat. Med 22, 254–261 (2016). doi: 10.1038/nm.4037; [DOI] [PubMed] [Google Scholar]
- 50.Nuno-Perez A, Mondoloni S, Tchenio A, Lecca S, Mameli M, Biophysical and synaptic properties of NMDA receptors in the lateral habenula. Neuropharmacology 196, 108718 (2021). doi: 10.1016/j.neuropharm.2021.108718; [DOI] [PubMed] [Google Scholar]
- 51.Whitlock JR, Heynen AJ, Shuler MG, Bear MF, Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097 (2006). doi: 10.1126/science.1128134; [DOI] [PubMed] [Google Scholar]
- 52.Kim JJ, Fanselow MS, Modality-specific retrograde amnesia of fear. Science 256, 675–677 (1992). doi: 10.1126/science.1585183; [DOI] [PubMed] [Google Scholar]
- 53.Dong S, Allen JA, Farrell M, Roth BL, A chemical-genetic approach for precise spatio-temporal control of cellular signaling. Mol. Biosyst 6, 1376–1380 (2010). doi: 10.1039/c002568m; [DOI] [PubMed] [Google Scholar]
- 54.Gradinaru V, Thompson KR, Deisseroth K, eNpHR: A Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol 36, 129–139 (2008). doi: 10.1007/s11068-008-9027-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiss T, Veh RW, Morphological and electrophysiological characteristics of neurons within identified subnuclei of the lateral habenula in rat brain slices. Neuroscience 172, 74–93 (2011). doi: 10.1016/j.neuroscience.2010.10.047; [DOI] [PubMed] [Google Scholar]
- 56.Tovar KR, Westbrook GL, Mobile NMDA receptors at hippocampal synapses. Neuron 34, 255–264 (2002). doi: 10.1016/S0896-6273(02)00658-X; [DOI] [PubMed] [Google Scholar]
- 57.McQuate A, Barria A, Rapid exchange of synaptic and extrasynaptic NMDA receptors in hippocampal CA1 neurons. J. Neurophysiol 123, 1004–1014 (2020). doi: 10.1152/jn.00458.2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Billingslea EN et al. , Parvalbumin cell ablation of NMDA-R1 causes increased resting network excitability with associated social and self-care deficits. Neuropsychopharmacology 39, 1603–1613 (2014). doi: 10.1038/npp.2014.7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu X et al. , Distinct thalamocortical circuits underlie allodynia induced by tissue injury and by depression-like states. Nat. Neurosci 24, 542–553 (2021). doi: 10.1038/s41593-021-00811-x; [DOI] [PubMed] [Google Scholar]
- 60.Ribeiro ACR et al. , Molecular mechanisms for the antidepressant-like effects of a low-dose ketamine treatment in a DFP-based rat model for Gulf War Illness. Neurotoxicology 80, 52–59 (2020). doi: 10.1016/j.neuro.2020.06.011; [DOI] [PubMed] [Google Scholar]
- 61.Choi M, Lee SH, Park MH, Kim YS, Son H, Ketamine induces brain-derived neurotrophic factor expression via phosphorylation of histone deacetylase 5 in rats. Biochem. Biophys. Res. Commun 489, 420–425 (2017). doi: 10.1016/j.bbrc.2017.05.157; [DOI] [PubMed] [Google Scholar]
- 62.Nibuya M, Morinobu S, Duman RS, Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci 15, 7539–7547 (1995). doi: 10.1523/JNEUROSCI.15-11-07539.1995; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nibuya M, Nestler EJ, Duman RS, Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci 16, 2365–2372 (1996). doi: 10.1523/JNEUROSCI.16-07-02365.1996; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang RY, Aghajanian GK, Physiological evidence for habenula as major link between forebrain and midbrain raphe. Science 197, 89–91 (1977). doi: 10.1126/science.194312; [DOI] [PubMed] [Google Scholar]
- 65.Stern WC, Johnson A, Bronzino JD, Morgane PJ, Effects of electrical stimulation of the lateral habenula on single-unit activity of raphe neurons. Exp. Neurol 65, 326–342 (1979). doi: 10.1016/0014-4886(79)90102-X; [DOI] [PubMed] [Google Scholar]
- 66.Yang LM, Hu B, Xia YH, Zhang BL, Zhao H, Lateral habenula lesions improve the behavioral response in depressed rats via increasing the serotonin level in dorsal raphe nucleus. Behav. Brain Res 188, 84–90 (2008). doi: 10.1016/j.bbr.2007.10.022; [DOI] [PubMed] [Google Scholar]
- 67.Wang Y et al. , Association between increased inflammatory cytokine expression in the lateral habenular nucleus and depressive-like behavior induced by unpredictable chronic stress in rats. Exp. Neurol 349, 113964 (2022). doi: 10.1016/j.expneurol.2021.113964; [DOI] [PubMed] [Google Scholar]
- 68.Fei Deng JW et al. , Dual-color GRAB sensors for monitoring spatiotemporal 1 serotonin release in vivo. BioRxiv (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lorrain DS et al. , Group II mGlu receptor activation suppresses norepinephrine release in the ventral hippocampus and locomotor responses to acute ketamine challenge. Neuropsychopharmacology 28, 1622–1632 (2003). doi: 10.1038/sj.npp.1300238; [DOI] [PubMed] [Google Scholar]
- 70.Rivera-García MT, López-Rubalcava C, Cruz SL, Preclinical characterization of toluene as a non-classical hallucinogen drug in rats: Participation of 5-HT, dopamine and glutamate systems. Psychopharmacology 232, 3797–3808 (2015). doi: 10.1007/s00213-015-4041-8; [DOI] [PubMed] [Google Scholar]
- 71.Witt CE et al. , Serotonin is a common thread linking different classes of antidepressants. Cell Chem. Biol 30, 1557–1570.e6 (2023). doi: 10.1016/j.chembiol.2023.10.009; [DOI] [PubMed] [Google Scholar]
- 72.Matsumoto M, Hikosaka O, Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 447, 1111–1115 (2007). doi: 10.1038/nature05860; [DOI] [PubMed] [Google Scholar]
- 73.Mealing GA et al. , Structural modifications to an N-methyl-D-aspartate receptor antagonist result in large differences in trapping block. J. Pharmacol. Exp. Ther 297, 906–914 (2001). [PubMed] [Google Scholar]
- 74.Nugent AC et al., Ketamine has distinct electrophysiological and behavioral effects in depressed and healthy subjects. Mol. Psychiatry 24, 1040–1052 (2019). doi: 10.1038/s41380-018-0028-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim KS, Han PL, Optimization of chronic stress paradigms using anxiety- and depression-like behavioral parameters. J. Neurosci. Res 83, 497–507 (2006). doi: 10.1002/jnr.20754; [DOI] [PubMed] [Google Scholar]
- 76.Sudarov A et al. , Mature hippocampal neurons require LIS1 for synaptic integrity: Implications for cognition. Biol. Psychiatry 83, 518–529 (2018). doi: 10.1016/j.biopsych.2017.09.011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Porsolt RD, Le Pichon M, Jalfre M, Depression: A new animal model sensitive to antidepressant treatments. Nature 266, 730–732 (1977). doi: 10.1038/266730a0; [DOI] [PubMed] [Google Scholar]
- 78.Monleon S et al. , Attenuation of sucrose consumption in mice by chronic mild stress and its restoration by imipramine. Psychopharmacology 117, 453–457 (1995). doi: 10.1007/BF02246218; [DOI] [PubMed] [Google Scholar]
- 79.Kim JW et al. , Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat. Neurosci 24, 1100–1109 (2021). doi: 10.1038/s41593-021-00868-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gradinaru V et al. , Targeting and readout strategies for fast optical neural control in vitro and in vivo. J. Neurosci 27, 14231–14238 (2007). doi: 10.1523/JNEUROSCI.3578-07.2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martina M, Krasteniakov NV, Bergeron R, D-Serine differently modulates NMDA receptor function in rat CA1 hippocampal pyramidal cells and interneurons. J. Physiol 548, 411–423 (2003). doi: 10.1113/jphysiol.2002.037127; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Freund TF, Buzsáki G, Interneurons of the hippocampus. Hippocampus 6, 347–470 (1996). doi: ; [DOI] [PubMed] [Google Scholar]
- 83.Geiger JR et al. , Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15, 193–204 (1995). doi: 10.1016/0896-6273(95)90076-4; [DOI] [PubMed] [Google Scholar]
- 84.Maroteaux M, Mameli M, Cocaine evokes projection-specific synaptic plasticity of lateral habenula neurons. J. Neurosci 32, 12641–12646 (2012). doi: 10.1523/JNEUROSCI.2405-12.2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mameli M, Bellone C, Brown MT, Lüscher C, Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat. Neurosci 14, 414–416 (2011). doi: 10.1038/nn.2763; [DOI] [PubMed] [Google Scholar]
- 86.DeNardo LA, de Wit J, Otto-Hitt S, Ghosh A, NGL-2 regulates input-specific synapse development in CA1 pyramidal neurons. Neuron 76, 762–775 (2012). doi: 10.1016/j.neuron.2012.10.013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schnell E, Bensen AL, Washburn EK, Westbrook GL, Neuroligin-1 overexpression in newborn granule cells in vivo. PLOS ONE 7, e48045 (2012). doi: 10.1371/journal.pone.0048045; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin L et al. , Large-scale neural ensemble recording in the brains of freely behaving mice. J. Neurosci. Methods 155, 28–38 (2006). doi: 10.1016/j.jneumeth.2005.12.032; [DOI] [PubMed] [Google Scholar]
- 89.Fox SE, Ranck JB Jr., Electrophysiological characteristics of hippocampal complex-spike cells and theta cells. Exp. Brain Res 41, 399–410 (1981). doi: 10.1007/BF00238898; [DOI] [PubMed] [Google Scholar]
- 90.Ranck JB Jr., Studies on single neurons in dorsal hippocampal formation and septum in unrestrained rats. I. Behavioral correlates and firing repertoires. Exp. Neurol 41, 462–531 (1973). doi: 10.1016/0014-4886(73)90290-2; [DOI] [PubMed] [Google Scholar]
- 91.MacDonald CJ, Lepage KQ, Eden UT, Eichenbaum H, Hippocampal “time cells” bridge the gap in memory for discontiguous events. Neuron 71, 737–749 (2011). doi: 10.1016/j.neuron.2011.07.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Courtin J et al. , Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature 505, 92–96 (2014). doi: 10.1038/nature12755; [DOI] [PubMed] [Google Scholar]
- 93.Zhao Q et al. , A multidimensional coding architecture of the vagal interoceptive system. Nature 603, 878–884 (2022). doi: 10.1038/s41586-022-04515-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wan J et al. , A genetically encoded sensor for measuring serotonin dynamics. Nat. Neurosci 24, 746–752 (2021). doi: 10.1038/s41593-021-00823-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main manuscript or the supplementary materials.